

Abstract
The dnaK gene of Vibrio cholerae was cloned, sequenced, and used to construct a dnaK insertion mutant which was then used to examine the role of DnaK in expression of the major virulence factors of this important human pathogen. The central regulator of several virulence genes of V. cholerae is ToxR, a transmembrane DNA binding protein. The V. cholerae dnaK mutant grown in standard laboratory medium exhibited phenotypes characteristic of cells deficient in ToxR activity. Using Northern blot analysis and toxR transcriptional fusions, we demonstrated a reduction in expression of the toxR gene in the dnaK mutant strain together with a concomitant increase in expression of a htpG-like heat shock gene that is located immediately upstream and is divergently transcribed from toxR. This may be due to increased heat shock induction in the dnaK mutant. In vivo, however, although expression from heat shock promoters in the dnaK mutant was similar to that observed in vitro, expression of both toxR and htpG was comparable to that by the parental strain. In both strains, in vivo expression of toxR was significantly higher than that observed in vitro, but no reciprocal decrease in htpG expression was observed. These results suggest that the modulation of toxR expression in vivo may be different from that observed in vitro.
Vibrio cholerae, a noninvasive, gram-negative bacterium, is the causative agent of the diarrheal disease cholera. For successful infection of the human host, orally ingested V. cholerae cells must colonize the intestine and produce cholera toxin (CT), a potent enterotoxin that causes the severe watery diarrhea characteristic of the disease. A toxin-coregulated pilus (TCP) coordinately expressed with CT greatly enhances colonization of the intestinal epithelium by the bacterium. Additional factors, including those necessary for survival of V. cholerae cells in the intestine, those required for evasion of the host defense system, adhesins, and accessory colonization factors, and other potential toxins, may also contribute to the virulence of this important human pathogen (17).
One regulatory pathway, controlling the expression of a subset of virulence factors of V. cholerae, that has been most extensively characterized is the ToxR-ToxT system (10). ToxR, a transmembrane DNA binding protein, directly activates expression of toxT, and the resulting enhanced level of ToxT leads to increased expression of other genes of the ToxR regulon, including those coding for CT and TcpA, the major subunit of TCP. Thus, in the virulence regulatory cascade of V. cholerae, ToxR is at the top of the hierarchy, ToxT is at the next level, and a number of virulence genes under control of ToxT are at the lowest level (10). However, it has recently been shown that production of OmpU, an osmoregulated outer membrane porin of V. cholerae, is independent of ToxT, although OmpU is a member of the ToxR regulon (5). Interestingly, in addition to its role as a transcriptional activator, ToxR can also function as a negative regulator, as suggested by the increased motility (12) and higher levels of production of an outer membrane protein OmpT in toxR mutants (28).
It has been hypothesized that the ToxR protein, probably by virtue of its location in the cytoplasmic membrane (29), can sense certain environmental parameters, leading to modulation of the ToxR-dependent expression of virulence genes in response to the external environment of the bacteria (40). It is now evident that a common strategy among pathogenic organisms is the exploitation of physical and chemical parameters that distinguish host from external environments as signals for the coordinate expression of virulence factors. This regulation presumably allows the bacteria to avoid unnecessary expenditure of energy resources to produce virulence factors under conditions where they would not be required. Thus, in most pathogens environmental conditions characteristic of the physiological sites of infection activate central regulators of virulence determinants (6, 23). However, in the case of V. cholerae biotype classical, a paradoxical situation exists, in that the intestinal environment may be presumed to display parameters similar to the nonpermissive conditions for induction of the ToxR regulon. The ToxR regulon is maximally expressed in cells grown at 30°C in media with a starting pH of 6.6 and osmolarity equivalent to 66 mM NaCl (28, 40). In the intestinal lumen, the temperature is 37°C, pH is alkaline, and osmolarity is thought to be equivalent to 300 mM NaCl or higher (42), conditions that repress the expression of ToxR-activated virulence factors in the laboratory. Although there is no doubt that ToxR is essential for successful infection, the mechanism for activation of the ToxR regulon in vivo is not clear. The possibility remains that the intestinal environment induces the production of additional virulence regulatory factors which could not be detected during in vitro growth of the cells simply because the conditions necessary for their induction are not known and hence could not be reproduced in the laboratory. Recently, a factor which is induced specifically after infection of the small intestine has been identified, although its precise role in virulence has not been determined (21).
In the course of the transition from the external environment to the human body, V. cholerae cells are exposed to a series of environmental changes, some of which are known to be stressful for bacteria, such as a sudden increase in temperature or heat shock, low pH in the stomach, bile salts in the intestine, and also perhaps anaerobiosis and starvation. Several of these stressful stimuli are known to trigger the enhanced synthesis of the evolutionarily conserved and abundant heat shock protein DnaK, a member of the Hsp70 family (22). Although primarily induced by heat shock (2), increased DnaK synthesis has also been reported in response to oxidative stress (30), osmotic stress (24), and starvation (13). In addition to its fundamental role as a molecular chaperone in protein folding with the functional cooperation of two other heat shock proteins, DnaJ and GrpE, DnaK is involved in DNA replication, RNA synthesis, ribosome assembly, protein transport, and cell division (15). In Escherichia coli, DnaK, DnaJ, and GrpE negatively modulate the induction of the heat shock response by decreasing the stability, and perhaps also the synthesis, of the heat shock sigma factor ς32, which is specifically required for transcription from heat shock promoters (4, 46). Of particular interest in the study of microbial pathogenesis is evidence that DnaK may be involved in the survival of bacterial pathogens in their hosts and pathogenicity in vivo. It has been reported that DnaK is required for multiplication of the intracellular bacterium Brucella suis in a human macrophage-like cell line (19). DnaK has also been shown to be among the dominant antigens recognized in immune response to a broad spectrum of pathogens, including V. cholerae (34, 45). It was in this context that the V. cholerae dnaK gene was cloned and a dnaK mutant was constructed and characterized in the present study to elucidate a potential role of DnaK in the survival of V. cholerae in the intestinal environment and in the regulation of expression of virulence determinants.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
All bacterial strains and plasmids used in this study are listed in Table 1. The V. cholerae and E. coli strains were maintained at −70°C in LB medium containing 20% (vol/vol) glycerol. E. coli cells were grown in LB medium, and V. cholerae cells were grown in LB, M9, or Syncase (11) medium. Ampicillin (100 μg ml−1), streptomycin (150 μg ml−1), chloramphenicol (30 μg ml−1), and kanamycin (30 μg ml−1) were used where appropriate. Plasmids were introduced into V. cholerae strains either by transformation (32) or by triparental mating using E. coli MM294(pRK2013) as a donor of mobilization factors.
TABLE 1.
Bacterial strains and plasmids used in the study
| Strain or plasmid | Relevant genotype or phenotype | Source or reference |
|---|---|---|
| V. cholerae | ||
| O395 | Smr | Laboratory collection |
| O395K1 | O395 dnaK::pGP704 Smr | This study |
| 569B | Wild type | Laboratory collection |
| JJM43 | ΔtoxR | 16 |
| E. coli | ||
| CG800 | dnaK103 thr::Tn10 | 41 |
| GW4813 | ΔdnaK | 31 |
| CG2682 | C600 dnaJ259 thr::Tn10 | 38 |
| K165 | rpoH(Am) | 7 |
| Plasmids | ||
| pACYC177 | Cloning vector, Apr Knr | Laboratory collection |
| pACYC184 | Cloning vector, Tcr Cmr | Laboratory collection |
| pSC350 | pACYC177 derivative with 3.5-kb HindIII fragment carrying 5′ part of V. cholerae dnaK gene | This study |
| pSC510 | pACYC184 derivative with 5.1-kb NcoI fragment carrying 3′ part of V. cholerae dnaK gene | This study |
| pSC830 | pACYC177 derivative with 8.3-kb fragment carrying entire V. cholerae dnaK and dnaJ genes | This study |
| pUC18/19 | Cloning vector, Apr | Boehringer Mannheim |
| pKK232.8 | Promoter probe vector carrying a MCS at 5′ end of promoterless cat gene | Pharmacia |
| pSChs | pKK232.8 derivative with 550-bp EcoRI-HincII fragment of pSC350 carrying dnaK heat shock promoter | This study |
| pNS1 | 220-bp BamHI-EcoRV fragment of pVM7 carrying htpG-toxR intergenic region cloned at BamHI-HindIII site of pKK232.8 | This study |
| pNS2 | 220-bp EcoRV-BamHI fragment of pVM7 cloned at SmaI-SalI site of pKK232.8 | This study |
| pTAC3734 | Promoter probe vector with MCS inserted between promoterless lacZ and phoA genes | N. C. Mandal, Bose Institute, Calcutta, India |
| pSCGR | pTAC3734 derivative with 220-bp BamHI-EcoRV fragment of pVM7 inserted at BamHI-StuI site | This study |
| pKP31 | dnaK gene of E. coli in pBR322 | 31 |
| pVM7 | toxR gene of V. cholerae in pBR322 | 27 |
| pSC18.1 | tcpA gene of V. cholerae in pTZ18U | 39 |
| pCVD15 | ctxAB genes of V. cholerae in pBR325 | 18 |
| pGP704 | oriR6K mobRP4 Apr | 28 |
Subgenomic libraries were constructed in plasmid pACYC177, and subcloning for sequencing was done with plasmid pUC18 or pUC19. Plasmid pKK232.8, containing a promoterless cat gene, and plasmid pTAC3704, in which a multiple cloning site (MCS) is located between promoterless lacZ and phoA genes, were used to construct promoter reporter gene fusions. Plasmid pKP31, containing the E. coli dnaK gene, was used to prepare dnaK gene probes and also as a positive control in experiments involving complementation of dnaK mutants. The suicide vector pGP704 was used for site-directed mutagenesis. Plasmids pCVD15, pSC18.1, and pVM7 were the source of ctxAB, tcpA, and toxR gene fragments used as probes. All plasmids were maintained and amplified in E. coli DH5α except pGP704, which was maintained in E. coli SM10 (28).
DNA preparation and manipulation.
Genomic DNA from V. cholerae 569B was prepared from proteinase K-digested and cetyltrimethylammonium bromide-precipitated cell lysates by using standard procedures (1). Plasmid DNA was prepared by the alkaline lysis method (36). Restriction enzymes and DNA-modifying enzymes were purchased from New England Biolabs Inc. (Beverly, Mass.) and used according to the manufacturer’s recommendation. For Southern blotting, restriction enzyme-digested chromosomal or plasmid DNA was electrophoresed on 1% agarose gels and blotted onto nylon membranes (Nytran; Schleicher & Schuell), using 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) (36). The blots were hybridized with DNA fragments labelled with [32P]dCTP (Amersham International, Amersham, United Kingdom) prepared by using a random-primed labelling kit (New England Biolabs). Subgenomic libraries of V. cholerae DNA were constructed in plasmid pACYC177. Genomic DNA was digested with an appropriate restriction enzyme, and the fragments within the size range of interest were excised from agarose gels, purified, ligated with linearized and dephosphorylated plasmid pACYC177, and transformed into E. coli DH5α according to standard recombinant DNA techniques (36).
Construction of a V. cholerae dnaK mutant strain.
A 0.6-kb HincII fragment of the V. cholerae dnaK gene (Fig. 1) was cloned at the EcoRV site of the suicide vector pGP704 and transformed into a λ pir lysogen of E. coli SM10 (28). Ampicillin-resistant transformants containing recombinant plasmids were selected and mated with V. cholerae O395 (Smr) on LB agar plates. Transconjugants resistant to both ampicillin and streptomycin were selected, and Southern blot analysis was used to confirm that integration had occurred into the chromosomal dnaK gene.
FIG. 1.

Cloning and physical map of the dnaK locus of V. cholerae. (A) Construction of plasmid pSC830 by ligation at the NcoI site of two overlapping clones, pSC350 and pSC510. All plasmids are derivatives of pACYC177. Only insert sequences are shown. Complementation of temperature-sensitive and phage λ-resistant phenotypes of E. coli CG800 and CG2682 by plasmids pSC350, pSC510, and pSC830 is indicated. (B) Physical map of the dnaK locus. The region of plasmid pSC830 within the hatched lines, which contains the complete dnaK gene and terminal portions of the dnaJ and grpE genes, was sequenced. Organization of the genes is indicated. The restriction sites shown are BamHI (B), EcoRI (E), EcoRV (EV), HincII (Hi), HindIII (H), HpaI (Hp), NcoI (N), and PstI (P).
Construction of transcriptional fusions to promoterless reporter genes.
The heat shock promoter of the V. cholerae dnaK gene was isolated as a 550-bp EcoRI-HincII fragment from plasmid pSC350 (Fig. 1) and cloned at the BamHI-HindIII site of plasmid pKK232.8 by using appropriate linkers to give plasmid pSChs. A 220-bp BamHI-EcoRV fragment carrying the V. cholerae htpG-toxR intergenic region was excised from plasmid pVM7 (27) and cloned at the BamHI-StuI site of plasmid pTAC3704 to give plasmid pSCGR (Fig. 8). To construct a toxR-cat transcriptional fusion, the BamHI-EcoRV fragment of plasmid pVM7 was cloned at the BamHI-HindIII site of plasmid pKK232.8 with appropriate linkers, to give plasmid pNS1. To construct htpG-cat transcriptional fusion, the BamHI-EcoRV fragment was cloned in an inverse direction at the SmaI-SalI site of plasmid pKK232.8 to give plasmid pNS2.
FIG. 8.

Transcriptional fusion plasmids for analysis of htpG and toxR expression. (A) The top diagram shows the organization of the htpG and toxR genes in the V. cholerae O395 chromosome. A 220-bp BamHI (B)-EcoRV (EV) fragment containing the intergenic region was inserted at the BamHI-Stu1 (S) sites between the phoA and lacZ genes in the promoter probe vector pTAC3734, to give plasmid pSCGR. The arrows denote directions of transcription. (B) β-Galactosidase and alkaline phosphatase activities in V. cholerae O395 containing plasmid pTAC3734 or pSCGR grown in vitro (LB, pH 7.2, 37°C) or in vivo. Results (means ± standard deviation of five independent experiments) are expressed as enzyme activities in Miller units, corresponding to 109 CFU.
RNA isolation and analysis.
For isolation of RNA, cells were grown to the late logarithmic phase in LB (pH 6.6) at 30°C, conditions optimum for production of CT, TcpA, and ToxR. Total RNA was extracted and purified by using guanidinium isothiocyanate as described elsewhere (1). RNA samples (15 to 25 μg/well) were electrophoresed in duplicate in 1% agarose–2.1 M formaldehyde-morpholinepropanesulfonic acid gels, and one part was stained with ethidium bromide and visualized with UV light to confirm equal loading of all samples. The other part of the gel was blotted onto nylon membranes by using 20× SSC and hybridized with labelled probes as described elsewhere (14).
Assays for β-galactosidase, alkaline phosphatase, and CAT activities.
β-Galactosidase and alkaline phosphatase activities were assayed in permeabilized cells or culture supernatants by measuring the hydrolysis of o-nitrophenyl galactopyranoside or p-nitrophenyl phosphate, respectively (26). Chloramphenicol acetylphosphatase (CAT) activity was measured in sonicated cell lysates by using a Quan-T-CAT kit (Amersham) according to the manufacturer’s instructions and expressed as milliunits of enzyme. The results presented represent the average of at least five independent experiments.
Isolation of outer membrane proteins.
V. cholerae O395 and O395K1 were grown in LB medium to about 109 CFU ml−1, and cell lysates were separated into subcellular fractions as described previously (34). The outer membrane fraction was solubilized, and outer membrane proteins were analyzed by sodium dodecyl sulfate–12.5% polyacrylamide gel electrophoresis (SDS-PAGE) followed by staining with Coomassie blue (20).
Swarm plate assay.
V. cholerae cells were stabbed into semisolid agar plates containing LB (pH 6.6 or 8.6) with 0.3% Bacto Agar (Difco). The plates were incubated at 30 or 37°C for 16 to 18 h, and swarm diameters were measured.
GM1-ganglioside dependent ELISA for assay of CT.
CT production was assayed in V. cholerae culture supernatants, sonicated cell pellets, or fluid collected from rabbit ileal loops by GM1 enzyme-linked immunosorbent (ELISA) using polyclonal rabbit serum directed against purified CT. Dilutions of purified CT of known concentration (Sigma) were used to estimate the amount of CT in the samples.
Western blot procedure.
Western immunoblot analysis was performed by the electrophoretic separation of total cellular proteins by SDS-PAGE followed by transfer to nitrocellulose membranes in a Transblot apparatus (Bio-Rad) (44). The DnaK protein was detected by using rabbit anti-E. coli DnaK polyclonal antisera and the Proto Blot system (Promega).
DNA sequencing.
Nucleotide sequence was determined with double-stranded plasmid DNA as the template either by the dideoxy-chain termination method (37) or by cycle sequencing using the Applied Biosystems Prism dye system (Perkin-Elmer). For dideoxy sequencing, [α-35S]dATP and a Sequenase 2.0 DNA sequencing kit (U.S. Biochemical Corp.) were used according to the manufacturer’s instructions. In some experiments, oligonucleotides designed from sequences obtained previously were used as primers.
Ligated rabbit ileal loop model.
In vivo growth and CT production by strains O395 and O395K1 were assayed by using the ligated rabbit ileal loop model essentially as described by De and Chatterjee (9). Briefly, rabbits fasted for 48 h were anesthetized, and the small intestine was tied into consecutive 6- and 2-cm segments proximal to the mesoappendix. An inoculum of 0.5 ml of V. cholerae strains containing about 5 × 106 CFU was introduced into each 6-cm segment, while one loop was inoculated with saline as a negative control. The intestine was returned to the peritoneal cavity, and the incision was closed. After 16 to 18 h, the animals were sacrificed and the small intestine was removed. Fluid accumulated in each loop was separately collected and measured, after which the loops were slit open and scraped. The fluid and scrapings were centrifuged (8,000 × g, 5 min) to collect the bacteria as a pellet, washed twice with normal saline, and finally resuspended in saline. Bacterial count in the suspension was measured by plating on thiosulfate-citrate-bile salt-sucrose (TCBS) agar plates containing appropriate antibiotics. β-Galactosidase, alkaline phosphatase, and CAT activities in a measured amount of the in vivo-grown cells were assayed as described above. CT in the fluid was measured by GM1 ELISA. Although no fluid accumulated in the loops inoculated with saline, these loops were also washed and scraped, and CFU, β-galactosidase, alkaline phosphatase, CAT, and CT were assayed in the scrapings and washings to determine whether the intestine contained any inhabitant flora and also to estimate if the normal intestinal content interfered with the enzymatic assays or GM1 ELISA. At least two loops in the same animal were inoculated with each bacterial strain, and each strain was tested in at least five individual animals.
Statistical analysis.
Comparison among results obtained from in vitro and in vivo experiments were made by the two-sample t test.
Nucleotide sequence accession number.
The nucleotide and deduced amino acid sequences of the V. cholerae dnaK gene appear in EMBL, GenBank, and DDBJ databases under accession no. Y14237.
RESULTS
The dnaK gene of V. cholerae: cloning, nucleotide sequence, and complementation of E. coli mutants.
The V. cholerae dnaK gene was cloned by screening a subgenomic library, using an E. coli dnaK gene fragment (31) as a probe. Construction of the plasmid carrying the V. cholerae dnaK region is shown in Fig. 1. Plasmid pSC830 could functionally complement the temperature-sensitive and phage-resistant phenotypes of E. coli dnaK mutant strains CG800 and GW4813 (31, 41) and also the E. coli dnaJ mutant CG2682 (Fig. 1) (38). The recombinant V. cholerae DnaK protein coded by plasmid pSC830 was detected by immunoblot analysis using anti-E. coli DnaK sera. In heat-shocked cell lysates of E. coli GW4813 (ΔdnaK) carrying plasmid pSC830, anti-E. coli DnaK sera cross-reacted with a 70-kDa protein that comigrated with the E. coli and native V. cholerae DnaK proteins (Fig. 2). No immunoreactive band was detected in the lysates of strain GW4813 carrying vector plasmid alone (Fig. 2, lane a).
FIG. 2.
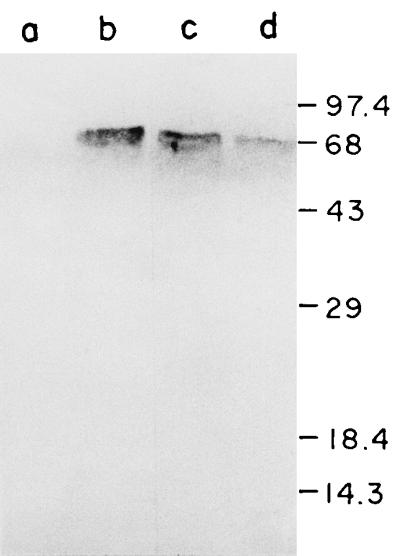
Identification of V. cholerae DnaK by Western blot analysis. E. coli GW4813 (ΔdnaK) cells containing plasmid pACYC177 (lane a), pKP31 (lane b), or pSC830 (lane c) and V. cholerae O395 cells (lane d) were heat shocked; then total cellular proteins were separated by SDS-PAGE (12.5% acrylamide), transferred to nitrocellulose membranes, and probed with anti-E. coli DnaK serum. Sizes are indicated in kilodaltons.
Using pUC18/19 subclones and oligonucleotide primers, we determined the nucleotide sequence of the 2,738-bp region of plasmid pSC830 (Fig. 1B). The sequence contained an open reading frame (ORF) of 1,907 nucleotides which has about 76% homology with the E. coli dnaK gene (2). The ORF is capable of coding for a protein of 635 amino acids whose predicted amino acid sequence is about 85% identical to that of the E. coli DnaK protein. A putative ribosome binding site was centered about 10 nucleotides upstream of the translation initiation site. Two potential ς32-specific promoters P1 and P2 were identified by comparison with the consensus −35 and −10 sequences of E. coli heat shock promoters (Fig. 3) (8). The 13-bp spacer region between the −35 and −10 sequences in both promoters is characteristic of promoters of heat shock genes. Downstream of the dnaK gene, we detected the 5′ end of an ORF which has about 64% homology with the dnaJ gene of E. coli over a stretch of 256 nucleotides at the 5′ end (3). However, the noncoding intergenic region was not conserved and was larger (200 nucleotides) than in E. coli (88 nucleotides). A weak promoter-like sequence and ribosome binding site were detected in the intergenic sequence. Analysis of the sequence upstream of the dnaK gene revealed a region coding for 44 amino acids which has significant homology with the C-terminal end of GrpE proteins. Thus, the genomic organization of the dnaK, dnaJ, and grpE genes in V. cholerae is grpE-dnaK-dnaJ (Fig. 1B).
FIG. 3.

Sequences of two putative promoters, P1 and P2, of the V. cholerae dnaK gene. Positions of the −35 and −10 regions were assigned by alignment with the consensus ς32 recognition sequence.
Construction of V. cholerae dnaK mutant.
Site-directed insertion mutation in the V. cholerae dnaK gene was constructed by chromosomal integration of the mobilizable suicide plasmid pGP704 (28), containing an internal 0.6-kb HincII fragment (Fig. 1) of the cloned dnaK gene. That the mobilized plasmid had integrated into the genome at the desired site in the dnaK gene was confirmed by Southern blot analysis (data not shown).
Similar to the E. coli dnaK mutants (31), the dnaK mutant of V. cholerae designated O395K1 was temperature sensitive for growth at 42°C, and prolonged incubation at 42°C rendered the mutant nonviable, as it was no longer capable of growth even at 30°C. The growth defects of the mutant could be complemented by plasmid pSC830, carrying the V. cholerae dnaK gene and also by plasmid pKP31, carrying the E. coli dnaK gene. The mutant could not be complemented by plasmid pSC510, carrying the V. cholerae dnaJ gene (Fig. 1). Unlike E. coli dnaK null mutants, in which growth at 30 and 37°C has been reported to be slower than in the wild-type strains (31), the growth rate of the V. cholerae dnaK insertion mutant at 30 and 37°C was comparable to that of the parental strain.
CT and TcpA production is reduced in the V. cholerae dnaK mutant.
To examine if DnaK has any role in virulence of V. cholerae, production of CT, the major virulence factor of the organism, was examined in the dnaK mutant strain O395K1. When grown in LB medium (pH 6.6) at 30°C, conditions optimum for CT production (28), although the parental strain O395 produced about 1 mg of CT per unit of optical density at 600 nm, only about 100 ng of CT could be detected in culture supernatants of the dnaK mutant strain. Practically no CT was detected in culture supernatants of V. cholerae toxR mutant strain JJM43 (16), used as control. Thus, CT in culture supernatants of strain O395K1 was reduced by more than 90% compared to the parental strain O395 when both strains were grown under conditions favorable for optimum CT production (Fig. 4). Similar results were obtained when the cells were grown in M9 or Syncase medium. Upon growth at 37°C, only about 20% of the amount of CT produced at 30°C was detected in strain O395, whereas a negligible amount of CT was produced by strain O395K1 (Fig. 4). The amount of CT in sonicated cell pellets of the parental and mutant strains was determined, and in both cases no CT was detected in the cell pellets by GM1 ELISA, indicating that production and not secretion of CT is drastically reduced in the dnaK mutant, even under conditions that normally promote high levels of CT production.
FIG. 4.

CT production in V. cholerae O395 ▨ and O395K1 ▥. Cells were grown in LB (pH 6.6) at 30°C, in LB (pH 8.6) at 37°C, or in rabbit ileal loops, and CFU per milliliter was assayed. CT was measured in culture supernatants or intestinal fluids corresponding to 109 CFU and expressed as a percentage of the amount obtained in culture supernatants of strain O395 grown at pH 6.6, 30°C.
To determine if the inhibition of CT production in the dnaK mutant was at the level of transcription, Northern blot analysis was carried out with RNA isolated from the parental and dnaK mutant strains and probed with a 1.9-kb HindIII-XbaI fragment of plasmid pCVD15 carrying the ctxAB genes (18). Although a strong and specific signal was detected in the RNA isolated from the parental strain O395 (Fig. 5A, lane a), very little ctx-specific message was detected in strain O395K1 (Fig. 5A, lane b). Since ctxAB gene expression is coordinately regulated with expression of the tcpA gene (43), transcription of tcpA was also examined in the V. cholerae dnaK mutant. A tcpA-specific transcript was barely detectable in the mutant cells with a tcpA gene fragment (39) used as a probe (Fig. 5B, lane b), although the probe did hybridize to RNA isolated from the parental cells (Fig. 5B, lane a). Thus, a mutation in the dnaK gene represses transcription of the two major virulence genes of the ToxR regulon.
FIG. 5.
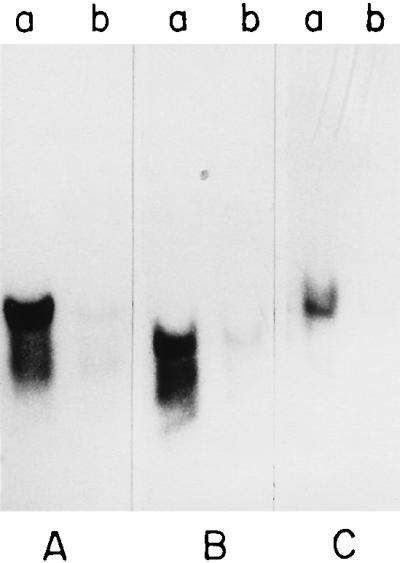
Northern blot analysis. V. cholerae O395 (lane a) and O395K1 (lane b) were grown in LB (pH 6.6) at 30°C, total RNA was isolated, and Northern blots were prepared and probed with [32P]dCTP-labelled fragments of ctxAB (A), tcpA (B), and toxR (C) genes; 15 μg of RNA was loaded per well.
Outer membrane proteins.
The two major outer membrane porins of V. cholerae are OmpU (38 kDa) and OmpT (40 kDa). OmpU, together with CT and TcpA, is positively regulated by ToxR, whereas OmpT expression is apparently under negative regulation of ToxR (28). Since expression of both ctxAB and tcpA is significantly reduced in the dnaK mutant of V. cholerae, the levels of OmpU and OmpT were examined in the mutant. SDS-PAGE of outer membrane proteins indicate that OmpU is the major porin in strain O395, and OmpT was not detectable in the outer membrane of this strain under conditions used in this study (Fig. 6). Under the same conditions, however, we detected a substantial amount of OmpT in the dnaK mutant (Fig. 6). The other outer membrane proteins were unaffected in the dnaK mutant strain except for a 23-kDa minor protein, the level of which was slightly increased. A 23-kDa minor outer membrane located protein has previously been identified as a heat shock protein (34).
FIG. 6.
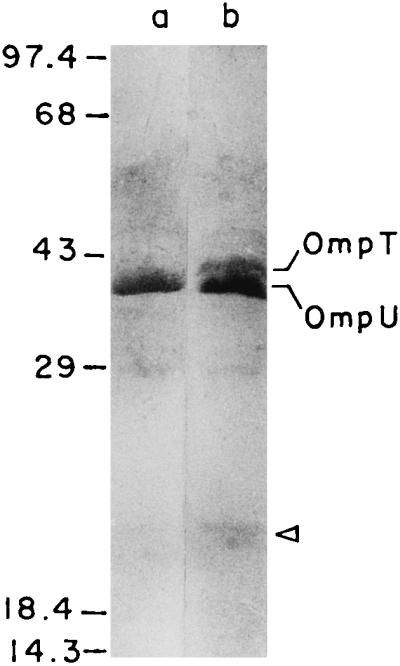
Outer membrane proteins of V. cholerae O395 (lane a) and O395K1 (lane b) analyzed by SDS-PAGE and Coomassie blue staining. The arrowhead denotes the position of the 23-kDa outer membrane-located heat shock protein. The mobilities of protein molecular mass standards (in kilodaltons) are shown to the left.
Swarm plate assays.
It has recently been reported that expression of the major virulence factors and motility are reciprocally regulated in V. cholerae (12). Since production of both CT and TcpA is drastically reduced in the dnaK mutant of V. cholerae, the swarming ability of these cells in semisolid motility medium was examined. Under ToxR-inducing conditions at 30°C, the parental strain O395 displayed little motility in the swarm plates (Fig. 7A). However, the dnaK mutant strain O395K1 was significantly more motile, and a 150% increase in swarm diameter was observed (Fig. 7A). The motility of strain O395 was somewhat higher at 37°C than at 30°C. A further 150% increase in swarm diameter was observed in strain O395K1 (Fig. 7B).
FIG. 7.

Swarming behavior of V. cholerae O395 and O395K1 on motility agar plates at 30°C (A) and 37°C (B).
CT production in vivo.
In view of the fact that a mutation in the dnaK gene significantly reduced expression of the major virulence genes of V. cholerae cells grown in the laboratory, we used the ligated rabbit ileal loop model (9) to examine the in vivo effect of the mutation. A major advantage of this model is that it allows comparative studies between parental and mutant strains simultaneously in the same animal, thus avoiding variations among individual animals. Strains O395 and O395K1 were injected separately into ligated ileal loops (about 5 × 106 CFU), and after 16 to 18 hours, the animals were sacrificed and the loops were examined. Almost equal amounts of fluid accumulated in loops inoculated with either strain O395 or O395K1. The contents of each loop were separately collected, and the number of bacteria as well as amounts of CT present in each loop was determined as described in Materials and Methods. Both strains grew to a density of 2 × 109 to 3 × 109 CFU ml−1 the ileal loops. In contrast to the results obtained from in vitro experiments, estimation of CT in intestinal fluids indicate that on a per-cell basis, the amounts of CT produced in the intestinal loops by strains O395 and O395K1 were almost equal and about 160% of the amount produced by strain O395 in LB medium (pH 6.6) at 30°C (Fig. 4).
Expression of toxR in vitro and in vivo.
To investigate the mechanism behind the dramatic difference in CT production by the V. cholerae dnaK mutant under in vitro and in vivo conditions, the expression of toxR was examined in cells grown in vitro or in vivo. Northern blot analysis revealed that expression of the toxR gene was substantially reduced in the dnaK mutant strain O395K1 grown in vitro even under ToxR-inducing conditions (Fig. 5C), which may account for the decrease in CT production observed in vitro (Fig. 4). Since Northern blot experiments with RNA isolated from cells grown in the rabbit intestine did not give clear results, a reporter plasmid (pNS1) carrying a toxR-cat transcriptional fusion was constructed as described in Materials and Methods. The plasmid as well as the control vector pKK232.8 were conjugally transferred into V. cholerae O395 and O395K1, and CAT activity was assayed in the transconjugants grown either in LB medium or in rabbit ileal loops. In agreement with a previous report that an increase in growth temperature is accompanied by a decrease in toxR expression (33), in LB medium the parental strain O395 carrying plasmid pNS1 (toxR-cat) showed 1.5-fold-lower CAT activity at 37°C than at 30°C. A twofold decrease in toxR expression was observed between cells of strain O395K1 and strain O395 grown in LB medium (pH 7.2, 37°C [Table 2]), although a higher magnitude of reduction was observed by Northern blot analysis (Fig. 5C). In contrast, when CAT activity was assayed in strains O395 and O395K1 harboring plasmid pNS1 (toxR-cat) grown in rabbit ileal loops, no statistically significant difference in toxR expression was observed between the two strains (P ≥ 0.2 [Table 2]). Furthermore, a comparison of CAT activity between in vivo- and in vitro-grown cells clearly indicated a statistically significant (P = 0.001) increase in toxR expression in cells grown in vivo. On a per-cell basis, we observed nearly 2.5-fold-higher CAT activity in cells of strain O395 grown in LB medium (pH 7.2) at 37°C than in cells grown in rabbit ileal loops. In strain O395K1, the increase in toxR expression between cells grown in vitro and in vivo was even higher (Table 2).
TABLE 2.
CAT activities in V. cholerae O395 and O395K1 containing htpG, toxR, or a heat shock promoter fused to a reporter cat gene
| Plasmid | CAT activity (mean mU/109 CFU ± SD) in straina:
|
|||
|---|---|---|---|---|
| O395
|
O395K1
|
|||
| In vitro | In vivo | In vitro | In vivo | |
| pKK232.8 | 12 | 15 | 14 | 9 |
| pNS1(toxR cat) | 108.6 ± 8 | 271 ± 31 | 50 ± 4 | 256 ± 28 |
| pNS2(htpG cat) | 426 ± 30 | 500 ± 69 | 1,022 ± 90 | 656 ± 86 |
| pSChs(phsb-cat) | 267 ± 9 | 254 ± 26 | 503 ± 39 | 565 ± 63 |
V. cholerae O395 and O395K1 were grown in LB (pH 7.2) at 37°C (in vitro) or in ligated rabbit ileal loops (in vivo), bacterial count (CFU per milliliter) was determined, and CAT activity per 109 CFU was measured.
phs, heat shock promoter.
The heat shock gene htpG has been shown to be located upstream from and in opposite orientation to toxR. It has been proposed that following heat shock induction, the divergent transcription of the htpG gene has a coordinate and reciprocal effect on expression of the toxR gene (33). Since mutation in the dnaK gene of V. cholerae decreased toxR expression in vitro, expression of htpG was examined in the V. cholerae dnaK mutant and also in the parental strain, particularly to evaluate if htpG expression was increased in the dnaK mutant and also if there is any difference in htpG expression between cells grown in vitro and cells grown in vivo. A reporter plasmid carrying a htpG-cat transcriptional fusion (pNS2) was constructed, and CAT activity was assayed in V. cholerae O395 and O395K1 carrying plasmid pNS2 grown in vitro or in vivo. As expected, in the dnaK mutant strain O395K1, htpG expression was about 2.5-fold higher than in the parental strain O395 when both strains were grown in LB medium at 37°C (Table 2). This difference was statistically significant (P = 0.001). However, in cells grown in rabbit ileal loops, little increase in CAT activity was observed in the dnaK mutant strain O395K1 compared to strain O395 (P ≥ 0.025). On a per-cell basis, both cells O395 and O395K1 expressed comparable amounts of CAT (Table 2).
Results were similar in assays using a reporter plasmid pSCGR in which the htpG-toxR intergenic region was cloned in the promoter probe vector pTAC3734, which has an MCS between promoterless phoA and lacZ genes. Plasmid pSCGR was constructed in such a manner that the promoter of the toxR gene directs transcription of the lacZ gene whereas phoA expression is under the control of the htpG promoter (Fig. 8A). With this plasmid, the relative amounts of toxR and htpG expression could be determined simultaneously under different conditions. Alkaline phosphatase and β-galactosidase activities were measured in cells carrying plasmid pSCGR (htpG-phoA toxR-lacZ) grown at either 37 or at 30°C in LB medium. We observed about 2-fold-higher alkaline phosphatase activity but 1.5-fold-lower β-galactosidase activity in cells grown at 37°C than in cells grown at 30°C. Thus, the temperature-dependent coordinate and reciprocal effect on expression from toxR and htpG promoters reported previously (33) was clearly demonstrated with plasmid pSCGR. However, such coordinated expression of toxR and htpG was not observed in vivo. When cells were grown in rabbit ileal loops, although htpG expression was only slightly higher than that observed in vitro at 37°C, toxR expression was found to be about threefold higher (Fig. 8B). The differences were statistically significant (P ≤ 0.001).
These results indicate that the ratio of toxR to htpG expression is increased in cells grown in the intestine. Furthermore, the increase in htpG expression observed in the dnaK mutant strain O395K1 under in vitro conditions could not be detected in cells grown in vivo. Since htpG expression is dependent on induction of the heat shock response, it was necessary to examine the level of heat shock induction in cells grown in vivo or in vitro.
Heat shock induction in cells grown in vitro or in vivo.
To examine the induction of the heat shock response in cells grown under in vitro or in vivo conditions, we constructed a reporter plasmid, pSChs, containing a heat shock promoter derived from the V. cholerae dnaK gene, fused to a promoterless cat gene in plasmid pKK232.8. Very little CAT activity could be detected in the E. coli rpoH mutant strain K165 (7) containing plasmid pSChs, indicating that expression from the heat shock promoter in plasmid pSChs is dependent on ς32. The plasmid was transferred to V. cholerae O395 or O395K1 by conjugation, and CAT activity was assayed in the transconjugants grown either in LB medium at 37°C or in rabbit ileal loops. A twofold increase in CAT activity was observed between cells of strain O395 and O395K1 grown in LB medium at 37°C. Identical results were obtained when the cells were grown in rabbit ileal loops (Table 2). Thus, the universal heat shock response was not repressed in cells grown in the intestine, nor did we observe any significant increase in heat shock induction in these cells.
DISCUSSION
In this study, we present evidence that the highly conserved and abundant heat shock protein DnaK influences the expression of toxR and ToxR-regulated virulence factors of V. cholerae grown in standard laboratory media but has no effect on toxR expression in cells grown in the intestine. The dnaK gene was cloned from V. cholerae 569B, and the identity of the cloned gene was confirmed by (i) phenotypic complementation of E. coli dnaK mutants (Fig. 1), (ii) immunological reactivity of the gene product with anti-E. coli DnaK sera (Fig. 2), and (iii) comparison of the nucleotide sequence with those of other dnaK and hsp70 genes. To study the role of DnaK in V. cholerae virulence, a site-directed insertion mutation in the dnaK gene of the wild-type strain O395 was constructed. The mutation produced phenotypes characteristic of dnaK mutants, including sensitivity to elevated growth temperatures and increased expression from promoters of heat shock genes (Table 2) (31). Furthermore, the following evidence suggests that mutation in dnaK has profound effects on the expression of ToxR-regulated virulence factors: (i) transcription of the ToxR-activated genes ctxAB and tcpA (10) is drastically reduced in the dnaK mutant strain O395K1 (Fig. 5), (ii) the level of the ToxR-repressed outer membrane porin OmpT (28) is significantly higher in the mutant than in the parental strain (Fig. 6), and (iii) motility of the cells which is negatively regulated by ToxR (12) is also higher in the dnaK mutant (Fig. 7). Analysis of the expression of toxR-specific mRNA and of a plasmid carrying a toxR-cat transcriptional fusion in the wild-type O395 and mutant O395K1 strains confirmed that expression of toxR was substantially reduced in the dnaK mutant (Fig. 5; Table 2). It is attractive to hypothesize that the control exerted by DnaK on transcription of the toxR gene is indirect and possibly due to the negative modulatory effect of DnaK on the production and stabilization of ς32, the heat shock sigma factor (4, 46). The increased expression of a reporter cat gene fused to a consensus heat shock promoter in the V. cholerae dnaK mutant strain O395K1 suggested that as in E. coli dnaK mutants, the level of active ς32 in the V. cholerae dnaK mutant strain O395K1 might be higher than in the parental strain. It has previously been shown that an increase in the level of ς32 resulted in reduced toxR expression (33). This effect was postulated to be due to an increase in the divergent transcription of a htpG-like heat shock gene by ς32-RNA polymerase, which leads to a proportionate decrease in the expression of the toxR gene by ς70-RNA polymerase, due to competition between RNA polymerase containing ς32 or ς70 for access to the short region between htpG and toxR (33). Consistent with this hypothesis, the higher levels of active ς32 in the dnaK mutant strain of V. cholerae may be correlated with increased htpG and reduced toxR expression in the mutant compared to the wild-type strain. The increase in htpG expression was almost proportional to the decrease in expression of toxR, further confirming the coordinate but reciprocal relationship between toxR and htpG expression proposed by Parsot and Mekalanos (33).
Although this model very elegantly explains the low levels of CT observed in cells grown in standard laboratory media at 37°C, it raises fundamental questions regarding expression of the ToxR regulon in vivo. In this study we demonstrate that toxR expression is significantly higher during growth at 37°C within the intestinal environment than in laboratory media (Table 2; Fig. 8). What could be a possible mechanism for increased toxR expression in vivo? Although many factors affect ToxR activity, the only parameter known to control expression of toxR is temperature, and it is only a decrease in the level of ς32 that is known to produce an increase in toxR expression, and vice versa. To address the possibility that the level of ς32 at 37°C is actually lower in cells grown in vivo than in cell grown in vitro, we have measured expression from a standard heat shock promoter in cells grown either in LB medium at 37°C or within rabbit ileal loops. Since it has been shown that expression of V. cholerae heat shock genes is dependent on ς32 (33, 35), the results obtained clearly indicate that there is no difference in ς32 levels between cells grown to the steady state at 37°C in vitro or in vivo (Table 2). Furthermore, when cells were grown in the intestine, expression from the heat shock promoter was about two times higher in the dnaK mutant strain than in the wild-type strain, which is comparable to the results obtained from in vitro studies (Table 2). From these results, it may be concluded that there is no mechanism that can specifically antagonize the heat shock response during intraintestinal growth of V. cholerae which may account for the increased toxR expression observed in these cells.
We next considered the possibility that access of ς32-RNA polymerase to the htpG-toxR intergenic region is specifically reduced during intraintestinal growth of V. cholerae. Using reporter gene fusions to the htpG-toxR intergenic region, we have shown that under in vitro conditions, expression from the htpG promoter was significantly greater in the V. cholerae dnaK mutant O395K1 than in the parental strain O395, consistent with the increased ς32 level in strain O395K1. In vivo, however, although the ς32 level remained higher in strain O395K1 than in strain O395, expression from the htpG promoter in strain O395K1 was lower than that observed in vitro. These results indicate that the access of ς32-RNA polymerase to the htpG promoter was specifically reduced in cells grown in vivo. It is likely that this may be mediated by as yet unknown factors induced specifically during in vivo growth of the cells. It may be noted that an essential feature of the Parsot and Mekalanos model for heat shock control of toxR expression is that any change in the level of toxR expression should be coordinated with a reciprocal alteration in htpG expression. We observed that although toxR expression was significantly increased between cells grown in vitro and in vivo, the increase in toxR expression is not accompanied by a reciprocal decrease in htpG expression (Table 2; Fig. 8). We do not know whether, in addition to increased expression, activity of ToxR is also altered during intraintestinal growth of V. cholerae. Since experiments were performed with cells grown overnight in rabbit ileal loops, we have not been able to determine at which stage of the infection process the increase in toxR expression actually occurs. Also, only the steady-state level of heat shock induction in cells grown in vitro or in vivo was considered. It is likely that immediately after infection, stressful intestinal conditions may strongly induce the heat shock response. Even if this were the case, it is doubtful whether the higher level of heat shock induction could reduce toxR expression in the early stages of infection, since in the dnaK mutant, constitutively higher levels of heat shock induction had no effect on toxR expression in vivo. If expression of the ToxR-activated genes is indeed turned off at the early stages of infection, as has been proposed, it is likely to be due to a decrease in ToxR activity rather than a reduction in toxR expression. In this context it may be mentioned that bile, a major constituent of the intestinal lumen, drastically decreases the expression of several ToxR-activated genes without affecting expression of toxR itself (14). Similar results have been reported for growth of V. cholerae cells in media of alkaline pH or high osmolarity, parameters characteristic of the intestinal environment (28).
Examination of virulence regulatory processes in many pathogenic bacteria has revealed that results obtained from laboratory analysis are often difficult to explain in the context of the in vivo situation (23, 25). In this study, we show that the model proposed for heat shock regulation of toxR expression from in vitro studies cannot be extrapolated to the in vivo situation. These studies reinforce a growing awareness that the regulation of virulence gene expression or environmental modulation of the regulatory processes in the complex and largely undefined environment of the animal body may be different from that observed under laboratory conditions.
ACKNOWLEDGMENTS
We are grateful to J. Das, Indian Institute of Chemical Biology, for generous advice and all members of the biophysics division for helpful discussions. We thank John J. Mekalanos, Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, Mass., for the generous gift of plasmids pGP704, pVM7, and pSC 18.1 and V. cholerae JJM43; Costa Georgopoulos, Department of Medicine, University of Geneva, Geneva, Switzerland, for kindly providing E. coli CG2682 and CG800; and Graham Walker, Biology Department, Massachusetts Institute of Technology, Cambridge, for the kind gift of E. coli GW4813, plasmid pKP31, and anti-E. coli DnaK antisera. We thank I. Guhathakurta for excellent technical support.
The work was supported by research grants SP/SO/D-56/96 from the Department of Science and Technology and BT/PRO411/Med/09/086/96 from the Department of Biotechnology, Government of India. S.C. is grateful to the Council of Scientific and Industrial Research for a research fellowship.
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley and Sons; 1989. [Google Scholar]
- 2.Bardwell J C A, Craig E. Major heat shock gene of Drosophila and the Escherichia coli heat inducible dnaK gene are homologous. Proc Natl Acad Sci USA. 1984;81:848–852. doi: 10.1073/pnas.81.3.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardwell J C A, Tilly K, Craig E, King J, Zylicz M, Georgopoulos C. The nucleotide sequence of the Escherichia coli dnaJ gene. J Biol Chem. 1986;261:1782–1785. [PubMed] [Google Scholar]
- 4.Bukau B. Regulation of the Escherichia coli heat shock response. Mol Microbiol. 1993;9:671–680. doi: 10.1111/j.1365-2958.1993.tb01727.x. [DOI] [PubMed] [Google Scholar]
- 5.Champion G A, Neely M N, Brennan M A, DiRita V J. A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol Microbiol. 1997;23:323–331. doi: 10.1046/j.1365-2958.1997.2191585.x. [DOI] [PubMed] [Google Scholar]
- 6.Chowdhury R, Sahu G K, Das J. Stress response in pathogenic bacteria. J Biosci. 1996;21:149–160. [Google Scholar]
- 7.Cooper S, Ruettinger T. A temperature sensitive nonsense mutation affecting the synthesis of a major protein of Escherichia coli K12. Mol Gen Genet. 1975;139:167–176. doi: 10.1007/BF00264696. [DOI] [PubMed] [Google Scholar]
- 8.Cowing D W, Bardwell J C A, Craig E A, Woolford C, Hendrix R W, Gross C A. Consensus sequence of Escherichia coli heat shock gene promoters. Proc Natl Acad Sci USA. 1985;82:2679–2683. doi: 10.1073/pnas.82.9.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De S N, Chatterjee S N. An experimental study of the mechanisms of action of Vibrio cholerae on the intestinal mucous membrane. J Pathol Bacteriol. 1953;46:559–562. doi: 10.1002/path.1700660228. [DOI] [PubMed] [Google Scholar]
- 10.DiRita V J. Co-ordinate expression of virulence genes by ToxR in Vibrio cholerae. Mol Microbiol. 1992;6:451–458. doi: 10.1111/j.1365-2958.1992.tb01489.x. [DOI] [PubMed] [Google Scholar]
- 11.Finkelstein R A, Athasampunna P, Chulasamaya P, Charunmethee P. Pathogenesis of experimental cholera: biological activities of purified procholeragen. J Immunol. 1966;96:440–449. [PubMed] [Google Scholar]
- 12.Gardel C L, Mekalanos J J. Alterations in Vibrio cholerae motility phenotypes correlate with changes in virulence factor expression. Infect Immun. 1996;64:2246–2255. doi: 10.1128/iai.64.6.2246-2255.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groat R G, Schultz J E, Zychlinsky E, Bockman A, Matin A. Starvation proteins in Escherichia coli: kinetics of synthesis and role in starvation survival. J Bacteriol. 1986;168:486–493. doi: 10.1128/jb.168.2.486-493.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta S, Chowdhury R. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun. 1997;65:1131–1134. doi: 10.1128/iai.65.3.1131-1134.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hendrick J P, Hartl F U. Molecular chaperone function of heat shock proteins. Annu Rev Biochem. 1993;62:349–384. doi: 10.1146/annurev.bi.62.070193.002025. [DOI] [PubMed] [Google Scholar]
- 16.Herrington D A, Hall R H, Losonsky G, Mekalanos J J, Taylor R K, Levine M M. Toxin, toxin co-regulated pili and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med. 1988;168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaper J B, Morris J G, Levine M M. Cholera. Clin Microbiol Rev. 1995;8:48–86. doi: 10.1128/cmr.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaper J B, Lockman H, Baldini M M, Levine M M. A recombinant live oral cholera vaccine. Bio/Technology. 1984;2:345–349. [Google Scholar]
- 19.Kohler S, Teyssier J, Cloeckaert A, Rouot B, Liautard J P. Participation of the molecular chaperone DnaK in intracellular growth of Brucella suis within U937 derived phagocytes. Mol Microbiol. 1996;20:701–712. doi: 10.1111/j.1365-2958.1996.tb02510.x. [DOI] [PubMed] [Google Scholar]
- 20.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Lee S H, Angelichio M J, Mekalanos J J, Camilli A. Nucleotide sequence and spatiotemporal expression of the Vibrio cholerae vieSAB genes during infection. J Bacteriol. 1998;180:2298–2305. doi: 10.1128/jb.180.9.2298-2305.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindquist S, Craig E A. The heat shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 23.Mekalanos J J. Environmental signals controlling expression of virulence determinants in bacteria. J Bacteriol. 1992;174:1–7. doi: 10.1128/jb.174.1.1-7.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meury J, Kohiyama M. Role of heat shock protein DnaK in osmotic adaption of Escherichia coli. J Bacteriol. 1991;173:4404–4410. doi: 10.1128/jb.173.14.4404-4410.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller J F, Mekalanos J J, Falkow S. Coordinate regulation and sensory transduction in the control of bacterial virulence. Science. 1989;243:916–922. doi: 10.1126/science.2537530. [DOI] [PubMed] [Google Scholar]
- 26.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 27.Miller V L, Mekalanos J J. Synthesis of cholera toxin is positively regulated at the transcriptional level by toxR. Proc Natl Acad Sci USA. 1984;81:3471–3475. doi: 10.1073/pnas.81.11.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller V L, Mekalanos J J. A novel suicide vector and its use in the construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller V L, Taylor R K, Mekalanos J J. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell. 1987;48:271–278. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 30.Morgan R W, Christman M F, Jacobson F S, Storz G, Ames B. Hydrogen peroxide-inducible proteins in Salmonella typhimurium overlap with heat shock and other stress proteins. Proc Natl Acad Sci USA. 1986;83:8059–8063. doi: 10.1073/pnas.83.21.8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paek K H, Walker G C. Escherichia coli dnaK null mutants are inviable at high temperature. J Bacteriol. 1987;169:283–290. doi: 10.1128/jb.169.1.283-290.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Panda D, Dasgupta U, Das J. Transformation of Vibrio cholerae by plasmid DNA. Gene. 1991;105:107–111. doi: 10.1016/0378-1119(91)90520-l. [DOI] [PubMed] [Google Scholar]
- 33.Parsot C, Mekalanos J J. Expression of ToxR, the transcriptional activator of the virulence factors in Vibrio cholerae, is modulated by the heat shock response. Proc Natl Acad Sci USA. 1990;87:9898–9902. doi: 10.1073/pnas.87.24.9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahu G K, Chowdhury R, Das J. Heat shock response and heat shock protein antigens of Vibrio cholerae. Infect Immun. 1994;62:5624–5631. doi: 10.1128/iai.62.12.5624-5631.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahu G K, Chowdhury R, Das J. The rpoH gene encoding a sigma 32 homolog of Vibrio cholerae. Gene. 1997;189:203–207. doi: 10.1016/s0378-1119(96)00849-9. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 37.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sell S M, Eisen C, Ang D, Zylicz M, Georgopoulos C. Isolation and characterization of dnaJ null mutants of Escherichia coli. J Bacteriol. 1990;172:4827–4835. doi: 10.1128/jb.172.9.4827-4835.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaw C E, Taylor R K. Vibrio cholerae O395 tcpA pilin gene sequence and comparison of predicted protein structural features to those of type 4 pilins. Infect Immun. 1990;58:3042–3049. doi: 10.1128/iai.58.9.3042-3049.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skorupski K, Taylor R K. Control of ToxR virulrnce regulon in Vibrio cholerae by environmental stimuli. Mol Microbiol. 1997;25:1003–1009. doi: 10.1046/j.1365-2958.1997.5481909.x. [DOI] [PubMed] [Google Scholar]
- 41.Spence J, Cegleshka A, Georgopoulos C. Role of Escherichia coli heat shock proteins DnaK and HtpG (C62.5) in response to nutritional deprivation. J Bacteriol. 1990;172:7157–7166. doi: 10.1128/jb.172.12.7157-7166.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spiro H M. Clinical gastroenterology. New York, N.Y: Macmillan Publishing Co., Inc.; 1983. [Google Scholar]
- 43.Taylor R K, Miller V L, Furlong D B, Mekalanos J J. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci USA. 1987;84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Towbin H, Staehelin J, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young R A, Elliot T J. Stress proteins, infection and immune surveillance. Cell. 1989;59:5–8. doi: 10.1016/0092-8674(89)90861-1. [DOI] [PubMed] [Google Scholar]
- 46.Yura T, Nagai H, Mori H. Regulation of the heat shock response in bacteria. Annu Rev Microbiol. 1993;47:321–350. doi: 10.1146/annurev.mi.47.100193.001541. [DOI] [PubMed] [Google Scholar]