

Abstract
Despite complications were significantly reduced due to the popularity of percutaneous coronary intervention (PCI) in clinical trials, reperfusion injury and chronic cardiac remodeling significantly contribute to poor prognosis and rehabilitation in AMI patients. We revealed the effects of HSP47 on myocardial ischemia–reperfusion injury (IRI) and shed light on the underlying molecular mechanism. We generated adult mice with lentivirus-mediated or miRNA (mi1/133TS)-aided cardiac fibroblast-selective HSP47 overexpression. Myocardial IRI was induced by 45-min occlusion of the left anterior descending (LAD) artery followed by 24 h reperfusion in mice, while ischemia-mediated cardiac remodeling was induced by four weeks of reperfusion. Also, the role of HSP47 in fibrogenesis was evaluated in cardiac fibroblasts following hypoxia–reoxygenation (HR). Extensive HSP47 was observed in murine infarcted hearts, human ischemic hearts, and cardiac fibroblasts and accelerated oxidative stress and apoptosis after myocardial IRI. Cardiac fibroblast-selective HSP47 overexpression exacerbated cardiac dysfunction caused by chronic myocardial IRI and presented deteriorative fibrosis and cell proliferation. HSP47 upregulation in cardiac fibroblasts promoted TGFβ1–Smad4 pathway activation and Smad4 deubiquitination by recruiting ubiquitin-specific peptidase 10 (USP10) in fibroblasts. However, cardiac fibroblast specific USP10 deficiency abolished HSP47-mediated fibrogenesis in hearts. Moreover, blockage of HSP47 with Col003 disturbed fibrogenesis in fibroblasts following HR. Altogether, cardiac fibroblast HSP47 aggravates fibrosis post-myocardial IRI by enhancing USP10-dependent Smad4 deubiquitination, which provided a potential strategy for myocardial IRI and cardiac remodeling.
KEY WORDS: Heat shock protein 47, Myocardial ischemia–reperfusion injury, Ubiquitin-specific protease 10, Cardiac fibrosis, Smad4, Fibroblast, Cell proliferation, Cardiac dysfunction
Graphical abstract
Cardiac fibroblast HSP47 inspired TGFβ1–Smad4 pathway by USP10 dependent deubiquitination in fibrogenesis under hypoxia. HSP47 suppression by chemicals with Col003 may have therapeutic potential for alleviating ischemic cardiac fibrosis.
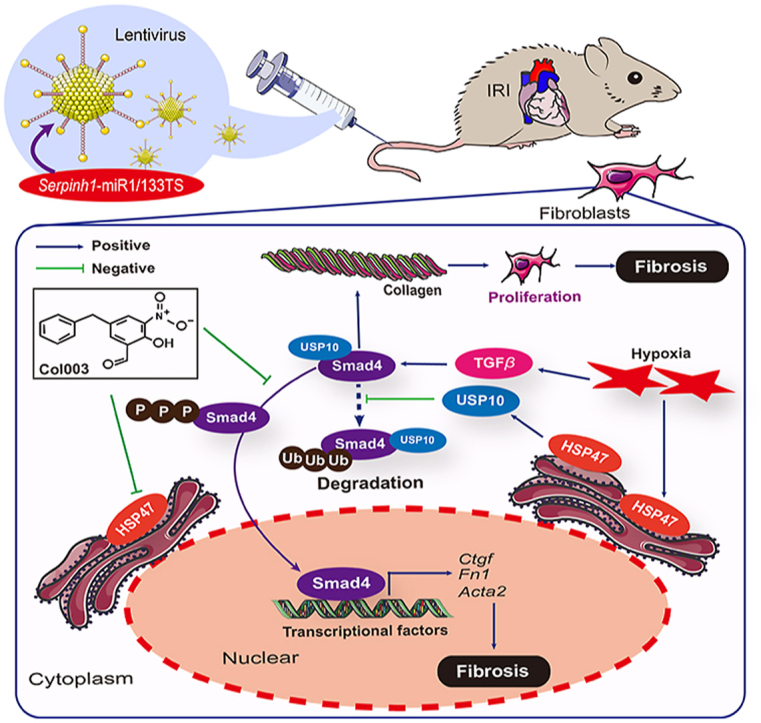
1. Introduction
Cardiovascular diseases (CVDs) contribute to significant health risks worldwide, leading to 17 million deaths per year globally1. Despite the rapid growth of therapeutic strategies, number of people who suffer from CVDs continues to grow as the population ages. Ischemic cardiomyopathy, in particular, has been identified as the leading cause of death in CVD patients2. Cardiac remodeling induced by chronic hypoxia contributed largely to poor prognosis except for ischemia-mediated acute cardiac damage. Unfortunately, ischemic hearts pose a serious threat to public health, but there are no effective therapeutic strategies so far.
Cardiac fibrosis, characterized by excessive deposition of extracellular matrix (ECM) proteins and collagen in the myocardium, disturbs the architecture of the myocardium, promotes cardiac dysfunction progression, and impacts the clinical course and outcome in patients with heart failure3. Upon ischemic injury, multiple pathological changes were observed in heart tissues, such as apoptotic cardiomyocytes and oxidative stress at the onset of acute injury4,5. Subsequently, immune cells infiltrated the damaged myocardium and released extensive profibrotic cytokines, eventually activating cardiac fibroblasts to produce ECM proteins such as collagen and fibronectin in heart tissues6. As part of the proliferative phase of cardiac repair, cardiac fibroblasts transform into synthetic myofibroblasts, forming stress fibers and expressing contractile proteins like α smooth muscle actin (α-SMA) and the embryonal isoform of smooth muscle myosin in the damaged area7,8. Moreover, massive collagen secreted by myofibroblasts generated a network. They deposited post-myocardial ischemia–reperfusion injury (IRI) for healing and scar formation, all of which accelerated cardiac structural remodeling and restricted contraction9,10. Previous evidence suggested that poor outcomes in patients with ischemic hearts were associated with low type I collagen crosslinking and high type I collagen deposition in pathological tissues, which led to a difficult field for eliminating lesions11. Hence, it is urgent to implement treatment strategies for patients with diffused cardiac fibrosis.
Transforming growth factor β (TGFβ) is well-known for its role in fibrosis, as it stimulates collagen production in dermal fibroblasts via the fibroblast-to-myofibroblast transition12. In addition, TGFβ/Smad signaling pathway is a critical response mediated by TGFβ13. TTGFβ binds to the type II serine/threonine kinase receptor (TGFBR II) and substantially induces the type I serine/threonine kinase receptor (TGFBR I). TGFBR I then phosphorylates the cytoplasmic transduction factors Smad2 and Smad3 and subsequently forms an active nuclear transcription complex with Smad4, all of which enter the nucleus to regulate the transcription of downstream target genes14,15. Moreover, TGFβ/Smad signaling pathway has been uncovered to contribute to fibrosis development in post-myocardial infarction (post-MI)16.
Among the molecular chaperones in mammals, heat shock protein 47 (HSP47) has been identified to interact with pro-collagens directly and is associated with various fibrotic diseases17. TGFβ1 and IL1β have been reported to favor nuclear localization of heat shock factor 1 (HSF1) and enhance HSF1 trimerization, leading to an increase in HSF1 binding to its specific gene response element, including the one in the HSP47 promoter18. Previous studies revealed that HSP47 promotes cancer cell colonization by enhancing cell-platelet interactions19. Furthermore, recently, HSP47 has been reported to regulate osteogenesis imperfecta (OI) by binding to KDELR220. Moreover, HSP47 ablation in cardiac fibroblasts is associated with a substantial reduction in cardiac fibrosis in cardiac remodeling caused by pressure overload21. However, the role and mechanism of HSP47 in myocardial IRI and pathological fibrosis remain unknown.
In the current study, we explored the effects of HSP47 on ischemia-associated fibrosis in hearts and uncovered that HSP47 inspired TGFβ1–Smad4 pathway in fibrogenesis under hypoxia. Moreover, extensive HSP47 contributed to the enrichment of Smad4 by ubiquitin-specific peptidase 10 (USP10)-dependent deubiquitination. Our findings revealed that targeting HSP47 might be a potential strategy for myocardial IRI and cardiac remodeling.
2. Materials and methods
2.1. Human heart samples
All human heart samples measurement conformed to the principles outlined in the Declaration of Helsinki (Version. 2020) and were approved by the Renmin Hospital of Wuhan University Review Board. The Patients with end-stage heart failure attributed by ischemic cardiomyopathy were involved. Left ventricular (LV) tissue was discarded after heart transplantation, and unmatched LV tissue from healthy donors were also collected for histological and molecular analysis in this experiment. Informed consent was signed by all donors and their families.
2.2. Animals
All animals were cared for in a specific pathogen-free facility and were approved by the Animal Ethics Committee at Renmin Hospital of Wuhan University (No. WDRM20210102A). Eight to ten-week-old C57BL/6J mice purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China) were housed in a quarantine room for at least a week, as described22. Cardiac fibroblast specific USP10 deficient mice were generated by crossing Col1a2Cre+ and USP10fl/fl mice. All rodents subjected to lentivirus (LV) vectors were undergone sham or 45 min occlusion of the left anterior descending (LAD) artery followed by 24 h reperfusion, during four weeks reperfusion in ischemia-mediated cardiac remodeling. All mice were euthanized by an overdose of pentobarbital sodium (200 mg/kg), as previously illustrated23, before collecting peripheral blood and hearts. To maintain myocardial tissue heterogeneity, the intact heart area was dissected and fixed in 10% neutral buffered formalin for 48 h before being embedded in paraffin and sectioned at 4 μm. Hematoxylin and eosin staining were used. In addition, the corresponding myocardial tissue was quickly frozen in liquid nitrogen and stored at −80 °C for RNA and protein analysis. The protocol was presented in Supporting Information Fig. S1.
2.3. Lentiviral vectors construction and administration
Recombinant LV vectors containing HSP47 with two tandem target sequences for mmu-miR1a-3p and mmu-mir-133a-3p introduced at their 3′ end (LV-HSP47-miR1/133TS) were constructed following manufacturer's instructions. Complementary oligonucleotides contained 2 tandem targeting sequences for mmu-miR1a-3p and mmu-miR133a3p: 5′-ataaATACATACTTCTTTACATTCCAcgtaATACATACTTCTTTACATTCCAacgcgtCAGCTGGTTGAAGGGGACCAAAtcacCAGCTGGTTGAAGGGGACCAAAC-3′. For HSP47-mir-1-mir-133TS vectors, mir-1 and mir-133TS were annealed and ligated into sites of 3′ UTR end of HSP47 expression cassette with EcoRI. Next, HSP47-miR1/133TS were amplified by the following pairs of primers: 5′- ATGCGCTCCTCCTGCTTCTCAGCGCCCTTCAGGTGGAGGTCGAATCG-3′; 5′- CTGGCTGGATGCCAAGCCTGACTGAGCCATTGACAAGAACCAGACTTGC-3′. The resulting PCR products were subcloned into a LV-pUbi-MCS-psSV40-EGFP vector (using AgeI/NheI restriction sites) under the control of a pUbi promoter. LV-Control-miR1/133TS vectors replace HSP47 vectors with negative control vectors. All final LV constructs were confirmed by sequencing, purified, dialyzed and titrated in 293T cells. Cardiac fibroblasts (CFs) and cardiomyocytes (CMs) were isolated from 1-week-old C57BL/6J mouse. LV-vectors were transduced into CFs at a final concentration of 50 PFU/cell24. After 72 h, HSP47 expression was evaluated by Western blot. Two weeks before surgery, both vectors were administered via tail vein injection to C57BL/6J mice (dose 3 × 108 PFU per mouse).
2.4. Generation of myocardial IRI model
The mouse model of myocardial IRI was constructed as described25. Briefly, all rodents were anesthetized with pentobarbital serum (50 mg/kg, i.p. once). Afterward, mice undergoing skin preparation were intubated and connected to a small animal ventilator. Subsequently, the fourth left intercostal space was dissected with surgical scissors to expose the heart fully. After that, the left anterior descending coronary artery (LAD) was ligated with a 6-0 silk suture at 2.0 mm below the left atrial appendage. All animals were exposed to LAD occlusion for 45 min and then perfused for 24 h. In contrast, four weeks of reperfusion in ischemia-mediated cardiac remodeling at the same time, a small animal electrocardiogram (ECG) monitoring system was utilized to detect ST-segment elevation.
2.5. Echocardiography
After reperfusion for four weeks, rodents were anesthetized with 1.5% isoflurane inhalation to receive echocardiography monitoring, as previously described25. A MyLab 30CV system (Biosound Esaote, Inc.) equipped with a 15 MHz probe was used to monitor cardiac structure and function by a small animal ultrasound instrument. Subsequently, the parameters were obtained by more than three beats and then took the average. Left ventricular ejection fraction (LVEF) and global longitudinal strain (GLS) were measured.
2.6. Hemodynamics analysis
A 1.4-French catheter tip micromanometer catheter (SPR-839; Millar Instruments, Houston, TX, USA) was used to obtain hemodynamic parameters, which was inserted into the left ventricle (LV) through the right carotid artery, as previously described25. Subsequently, ARIA pressure-volume (PV) conductance system (MPVS-300 signal conditioner, Millar Instruments, Houston, TX, USA) coupled with Power Lab/4SPA/D converter was used to measure PV parameters and display PV curve. All hemodynamic parameters were analyzed by the laboratory chart 8.0.
2.7. Histological analysis
Histopathology analysis was performed as previously reported25. Briefly, heart tissues were fixed, embedded, and sectioned into 5 μm thick slices. For collagen deposition analysis, sections were stained with Masson's trichrome (collagen, blue; cytoplasm, red/pink) and quantitated blue area in the heart with Image-Pro Plus 6.0. The sections were stained with wheat germ agglutinin (WGA) to assess cellular hypertrophy. The myocyte cross-sectional area (CSA) was measured with a quantitative digital image analysis system (Image-Pro Plus 6.0) using images that were captured from WGA-stained sections. More than 100 myocytes in the examined sections were outlined for each group of mice. For immunohistochemistry, heart sections were incubated with antibodies against collagen I, vimentin, and 4-hydroxynonenal (4-HNE) at 4 °C overnight, then with secondary antibodies at 4 °C for 1 h and detected with 3,3′-diaminobenzidine, and sections were counterstained with hematoxylin. TUNEL assay was performed to evaluate apoptosis. Immunohistochemistry images were captured by a Leica Aperio VERSA 8.0 and analyzed by a person blinded to treatment using Image-Pro Plus 6.0. For immunofluorescence, frozen heart sections were labeled with primary antibodies HSP47, collagen I, Flag, HA, and GFP, then incubated with Alexa Fluor 568- or 488-conjugated secondary antibody (ab175471 and ab150077, Abcam). Immunofluorescence images were captured with Leica ST8 laser scanning confocal microscope. To quantify positive fluorescence area, images were analyzed positive score ratio with Image-Pro Plus 6.0.
2.8. Evans blue and TTC staining
At the end of reperfusion, LAD was ligated again, and Evans blue (0.25% phosphate-buffered saline; Sigma–Aldrich; Merck Kgaa) was injected from the orbital vein. The non-ischemic area is stained blue, presenting the site at risk (AR, non-blue area). Then, the heart was frozen at −20 °C for 30 min and cut into five pieces (2 mm/slice). Next, the sections were incubated in triphenyl tetrazolium hydrogen chloride (TTC; 1% in phosphate buffer; Sigma–Aldrich; Merck Kgaa) at 37 °C for 10 min then immersed in 4% paraformaldehyde at room temperature for 4 h. TTC staining can separate the infarct size (white area) area into the non-infarct area at risk (AR: red area). These heart slices are only used for Evans-Blue-TTC staining. Finally, the slices are arranged from vertices into bases and taken digitally. The slices' digital images were analyzed using ImageJ-Pro Plus 6.0 software.
2.9. Adult mouse cardiac fibroblasts (AMCFs) isolation, culture and administration
Cardiac fibroblasts (CFs) were isolated from adult mice by enzymatic digestion and cultured in a DMEM medium, as previously described26. CFs in HR group were treated with hypoxia for 24 h and reoxygenation for 6 h with 5% CO2, 94% N2, and 1% O2 at 37 °C in tri-gas incubators for 48 h. CFs were infected with Ad-LacZ or Ad-HSP47-GFP for 24 h and treated with HR or normoxia for another 48 h. CFs were pretreated with HSP47 inhibitor (Col003, 50 μmol/L) or DMSO for 48 h in the absence or presence of normoxia. CFs activation was evaluated via α-SMA and collagen I protein expression.
2.10. Western blot and coimmunoprecipitation
As previously described, cells were lysed with RIPA lysis buffer (Sevicebio, China) and protease inhibitors26. The protein concentration was measured with a BCA protein determination kit (Thermo Scientific, 23225). Afterward, an equal amount of protein was loaded and separated on a 10% SDS-PAGE gel and then transferred to a PVDF membrane (Merck Millipore, Germany). After blocking in 5% skimmed milk, the membrane overnight was incubated at 4 °C with the primary antibody (Supporting Information Table S1), followed by a horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:10,000; RRID: AB_2099233; Cell Signaling Technology), incubated together, and left at room temperature for 1 h. Immunoblotting was detected using Chemidoc Touching Imaging System (BIO-RAD, USA).
The coimmunoprecipitation test was performed according to manufacturer's protocol (Protein A/G Magnetic Beads, HY-K0202, MCE), as already described26. Briefly, the tissues (n = 3) of hearts and CFs (three samples per pool) were lysed in octyl-d-glucoside (ODG, 2%) buffer. Afterward, the supernatant protein (500 μg) and anti-USP10 antibody (Thermo Scientific, A300-900A) were spun overnight at 4 °C, then spun with Protein A/G Magnetic Beads 4 °C for 4 h. Subsequently, the sample was washed four times in Tris-buffer. Furthermore, the protein is eluted with Laemmli sample buffer. After that, the sample was boiled at 100 °C for 10 min to detect USP10 and Smad4 by Western blotting. Reverse verification was performed as well. The results are expressed as the ratio between the expression intensity of Smad4 and the expression intensity of immunoprecipitated USP10.
2.11. RNA isolation and relative quantitative RT-PCR
Total RNA was extracted using Trizol (Invitrogen, 15596-026). cDNA was synthesized from 1 μg of RNA with a Prime Script RT reagent Kit (04897030001, Roche Diagnostics, Basel, Switzerland) in a 20 μL reaction volume. qPCR was conducted in Step-one Plus Real-time PCR System using iTaq Universal SYBR Green Supermix (Roche, 04707516001). Cycle parameters: 95 °C for 10 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. β-Tubulin served as the endogenous reference gene. The primer sequences are listed in Supporting Information Table S2.
2.12. RNA-sequencing
Total RNA from CFs of mice in different treatment groups was used for transcriptome sequencing. Three biological replicates were performed in each group. Mini RNeasy Mini kit (15055-50, QIAGEN) isolated total RNA from all samples. The Agilent 2100 RNA Nano 6000 Assay Kit (Agilent Technologies, CA, USA) was used to check the integrity of RNA, and then oligo (dT) beads were used to enrich the mRNA. A fragmentation buffer degrades the purified mRNA to obtain short mRNA fragments. Use random primers and related buffers to synthesize cDNA, and then make a purification by QIA-Quick PCR kit and EB buffer. The purified cDNA was modified and amplified by PCR to construct a cDNA library. Each cDNA library is sequenced on Illumina HiSeq2000 instrument.
2.13. Immunofluorescent staining
AMCFs were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% Triton X-100 for 20 min, and then blocked with 5% BSA in PBS-Tween for 1 h. Subsequently, AMCFs were diluted with α-SMA antibody (Abcam, ab5694) or HSP47 antibody (Abcam, ab109117) in 5% BSA at 4 °C overnight. DAPI was used to counterstain the nuclei.
2.14. Statistical analysis
All data are presented as mean ± standard error of mean (SEM, n ≥ 3). Student's t test was used to analyze the significance of differences between two groups. One-way or two-way analysis of variance (ANOVA) was used for statistical comparisons between more than two groups, followed by Tukey's post hoc test. Statistical analysis was performed by GraphPad Prism 8.0 (GraphPad Software, Inc.). P ≤ 0.05 indicates a statistically significant difference.
3. Results
3.1. Upregulation of HSP47 in cardiac fibroblast cells is associated with ischemic cardiac pathogenesis
Given that HSP47 was closely associated with multiple organ fibrosis in patients17, we thus explored levels of HSP47 in the mouse model of myocardial IRI for four weeks. We firstly assessed the production of HSP47 in heart tissues. HSP47 showed substantial enrichment in the infarcted area and a modest increase in the non-infarcted area (Fig. 1A and Supporting Information Fig. S2A). Immunohistochemistry analyses targeting HSP47 corroborated these data by presenting a higher density of HSP47 in infarcted areas (Fig. 1B). To determinate the effective cells generating HSP47, we investigated HSP47 expression in isolated AMCMs and AMCFs under HR conditions. Expectedly, cardiac fibroblasts contributed largely to HSP47 production but not cardiomyocytes, endothelial cells, and macrophages (Fig. 1C, D and Fig. S2B). Consistently, immunoblot results pointed towards the same trend (Fig. 1E). After that, we explored HSP47 in patients with end-stage heart failure due to ischemic cardiomyopathy (ICM). Consistent with animal experiments, HSP47 significantly increased in hearts obtained from patients with ICM compared with LV tissue from healthy donors (Fig. 1F and Fig. S2C). Notably, HSP47 showed substantial upregulated in the infarcted area and a modest increase in the non-infarcted area, all of which were paralleled with collagen I production in LV tissues (Fig. 1G). These data indicate that cardiac fibroblast HSP47 markedly upregulated in ischemic cardiac pathogenesis.
Figure 1.

HSP47 is upregulated in CFs after myocardial IRI. (A, B) C57BL/6J mice underwent myocardial IRI for 4 weeks. (A) Heart tissue homogenate from the special area (sham, non-infarcted area, and infarcted area) was harvested for protein isolation, and immunoblotting was used for HSP47 analyses and then offered quantitative analysis by image lab software. They were normalized to β-tubulin, n = 4 per group. Differences were assessed by one-way ANOVA and Tukey's multiple comparison test. (B) Heart slices from sham and I/R mice were collected for immunohistochemistry (IHC) analyses of HSP47 and then offered quantitative analysis by image pro plus 6.0 software. n = 6 mice per group (23/21/19 images in sham/non-infarcted area/infarcted area). Scale bar 0.5 cm (up) and 200 μm (down). Differences were assessed by one-way ANOVA and Tukey's multiple comparison test. (C–E) AMCFs obtained from adult C57BL/6J male mice were treated with Normoxia or HR. (C) Immunofluorescence analyses targeting HSP47 were performed. n = 3 independent experiments. Scale bar 20 μm. (D). Total RNA isolated from AMCFs was utilized for qPCR analyses and normalized to β-tubulin (n = 4 per group). Differences assessed by Student's t-test. (E) Proteins obtained from AMCFs, and immunoblotting was used for HSP47 analyses and then offered quantitative analysis by image lab software. They were normalized to β-tubulin, n = 4 per group. Differences assessed by Student's t-test. (F, G) Heart tissues from donors and patients with ICM were collected. (F) Heart homogenate was harvested for protein isolation, and immunoblotting was used for HSP47 analyses and then offered quantitative analysis by image lab software. They were normalized to β-tubulin, n = 4 per group. Differences assessed by Student's t-test. (G) Heart slices underwent immunofluorescence (IF) of HSP47 and IHC of collagen I (n = 4 per group). 18/22/25 images for IF and 22/16/20 images for IHC in donor/non-infarcted area/infarcted area. Scale bar is 100 μm. Differences were assessed by one-way ANOVA and Tukey's multiple comparison test. Data are presented as the mean ± SEM, with each point representing a heart or a cell sample.
3.2. Extensive HSP47 in fibroblasts accelerates acute myocardial injury post ischemia reperfusion
To examine the effects of cardiac fibroblast HSP47 in ICM, we next established a miRNA-aided/lentivirus-mediated system, as previously reported24,27, markedly enriched in HSP47-miR1/133TS transfected cardiac fibroblasts and disturbed HSP47 overexpression in cardiomyocytes (Fig. 2A and B). We designed a HSP47 expression cassette driven by a Ubi promoter containing 2 tandem copies of a 22-bp complementary target sequence for the mir-1a-3p and mir-133a-3p, both of which avoid HSP47 overexpression in cardiomyocytes, strands at the 3ʹ end of the vector cassette. Firstly, we investigated whether HSP47 overexpression in CFs impacts acute myocardial injury post-ischemia–reperfusion. Evans blue/TTC double staining showed that HSP47 in CFs substantially enlarged infarct area following reperfusion for 24 h compared with the control vector, suggesting HSP47 promoted myocardial injury in ischemic hearts (Fig. 2C and D). Also, HSP47 contributed to plasma TnT and CK-MB content, implying that HSP47 agglutinated cardiac damage in response to hypoxia in mice (Fig. 2E and F). Previous studies have reported that acute myocardial injury induced by ischemia–reperfusion is characterized by diffused apoptotic monocytes and extensive ROS28,29. We further investigated the effects of HSP47 in myocardial IRI. HE staining showed apparent cytoplasmic vacuolation, whereas HSP47 worsened vacuolation in cardiomyocytes (Fig. 2G and Supporting Information Fig. S3A). Similarly, more apoptotic monocytes were observed in mice that received HSP47 vectors than in control vectors (Fig. 2G and Fig. S3B). Western blot analyses corroborated these data by presenting a noticeable enhancement of Bax and cleaved caspase-3 and a substantial reduction of Bcl2 in HSP47 pretreated heart samples (Fig. S3C). On the other hand, HSP47 overexpression markedly promoted ROS production in heart tissues compared with mice subjected to control vectors, as identified by increased 4HNE and DHE (Fig. 2H, Fig. S3D and S3E). In parallel, Western blot analyses revealed that HSP47 facilitated the expression of Gp91 phox and P47 phox while suppressing SOD2 in heart tissue homogenate (Fig. S3F). These data suggest that HSP47 accelerates myocardial injury post-ischemia–reperfusion with increased apoptosis and oxidative stress.
Figure 2.

HSP47 overexpression in CFs accelerates acute myocardial injury post-ischemia–reperfusion. (A) Schematic representation of HSP47 expression cassette in the lentivirus vectors engineered to overexpress exogenous HSP47 or negative control in CFs expression mir-1a-3p and mir-133a-3p. (B) C57BL/6J mice were injected with Con-miR1/133TS or HSP47-miR1/133TS, and HSP47 expression was assessed in isolated AMCFs or AMCMs by immunoblotting two weeks later and normalized to β-tubulin. n = 3 independent experiments. (C, D). Left ventricular tissue sections of both Con-miR1/133TS and HSP47-miR1/133TS pretreated mice stained with Evans blue and TTC at 24 h after myocardial IRI to delineate the area at risk (AAR) and infarcted region (IR) (n = 6 per group). Scale bar = 0.5 cm. Differences assessed by Student's t-test. (E, F) The enzyme activity of CK-MB and TnT in serum were accessed in mice that underwent sham or IR operation for 24 h by Elisa assay. n = 6 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (G) Heart slices from sham and I/R mice with Con-miR1/133TS or HSP47-miR1/133TS were collected for cytoplasmic vacuolation and apoptosis analyses by H&E and TUNEL staining, respectively (n = 6 per group). 23/19/21/25 images for HE and 22/19/24/26 images for Tunel in sham-con/sham-HSP47/IR-con/IR-HSP47. Scale bar = 100 μm. (H) Heart slices were collected for IHC detection of 4-HNE and DHE staining (n = 6 per group). 16/16/22/18 images for 4-HNE and 23/19/21/18 images for DHE in sham-con/sham-HSP47/IR-con/IR-HSP47. Scale bar = 200 μm. Data are presented as the mean ± SEM, with each point representing a mouse.
3.3. Overexpressed HSP47 in fibroblasts aggravates heart failure following ischemia reperfusion in myocardium
To determine whether HSP47 overexpression in fibroblasts protected against or contributed to heart failure, we, therefore, detected cardiac function and remodeling in ischemic hearts. First, HSP47 overexpression in cardiac fibroblasts aggravated cardiac dysfunction following myocardial IRI for four weeks, as evaluated by a substantial reduction in LV ejection fraction (LVEF) and GLS (Fig. 3A–C). Likewise, cardiac fibroblast HSP47 upregulation led to a decrease in LV compliance in response to myocardial IRI for four weeks, as assessed by PV curve shifting right and increased LV volume in systole and diastole (Fig. 3D and E). Furthermore, a markedly hypertrophic heart was observed in the non-infarct area pretreated with HSP47 vectors, as shown by the increased cross-section area of cardiomyocytes (Fig. 3F). In addition, HSP47 in cardiac fibroblasts activated higher pro-hypertrophic genes, including Anp and Bnp, and disturbed transcription of Myh6 in the non-infarct area compared with rodents in control vectors (Fig. 3G). These results reveal that overexpressed HSP47 in fibroblasts aggravates heart failure and cardiac hypertrophy in myocardial IRI for four weeks.
Figure 3.

Extensive HSP47 in fibroblasts aggravates heart failure following ischemia–reperfusion. (A–E) Echocardiography and hemodynamics analyses were performed in mice injected with Con-miR1/133TS or HSP47-miR1/133TS and underwent IR surgery for four weeks. (A) Representative M-mode and ventricular wall motion echocardiography were recorded. (B) LV ejection fraction (LVEF) was analyzed by the equation LVEF= (LVEDD-LVESD)/LVEDD. n = 12 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (C) Global longitudinal strain (GLS) was measured by spot tracking technology. n = 8 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (D) Representative PV loops of Con-miR1/133TS or HSP47-miR1/133TS pretreated mice following I/R for four weeks and (E) measurement of end-systolic volume (ESV) and end-diastolic volume (EDV). n = 10 per group. Differences assessed by Student's t-test. (F) Heart slices from the non-infract area were detected by WGA staining and then quantitatively analyzed cross-sectional heart area in indicated mice. n = 6 per group. 21/18/18/24 images for WGA in sham-con/sham-HSP47/IR-con/IR-HSP47. Scar bar = 80 μm. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (G) Total RNA from the non-infract area was performed to measure Anp, Bnp, and Myh6 by qPCR and normalized to β-tubulin. n = 4 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. Data are presented as the mean ± SEM, with each point representing a mouse.
3.4. Increased HSP47 in cardiac fibroblasts exacerbates fibrosis and cell proliferation in ischemic hearts
It is increasingly recognized that HSP47 is one of the molecular chaperones in mammals that directly interacts with pro-collagens and is associated with various fibrotic diseases30. We next determined the effects of HSP47 on cardiac fibrosis following myocardial IRI for four weeks. In concordance with previous evidence, HSP47 co-located on collagen in the infarct area (Fig. 4A). However, there was no discernible statistical difference between the groups regarding mortality rate or statistics (Supporting Information Fig. S4A). Further analyses upon Masson staining revealed that HSP47 overexpression in cardiac fibroblasts resulted in more collagen production and deposition in both perivascular and interstitial area in chronic IRI hearts (Fig. 4B, C and Fig. S4C). Western blot analyses corroborated these data by presenting a noticeable enhancement of collagen I, α-SMA, and fibronectin in HSP47 vector pretreated heart samples (Fig. 4D and E). Fibroblast proliferation has been reported to be involved in the progression of fibrosis31. Subsequently, we evaluated cell proliferation in myocardial IRI. We found that HSP47 upregulation in CFs contributed to cell proliferation, as assessed by substantial production of vimentin, and increased Ki67 in the heart tissues (Fig. 4F and G). Results of immunofluorescence show that HSP47 overexpression in CFs markedly promoted co-location of vimentin and Ki67 in non-ischemic hearts (Fig. 4H and Fig. S4B). These data suggest increased HSP47 in cardiac fibroblasts exacerbates fibrosis and cell proliferation in chronic ischemic hearts.
Figure 4.

Increased HSP47 in cardiac fibroblasts exacerbates fibrosis and cell proliferation in ischemic hearts. (A) Heart slices from indicated area were used for immunofluorescence of HSP47, collagen I and DAPI. Scale bar 200 μm. 18/16 images for IF in non-infarcted area/infarcted area. (B, C) Representative Masson staining of heart slices from Con-miR1/133TS or HSP47-miR1/133TS pretreated mice following I/R for four weeks and quantitative analyses of LV collagen volume. n = 6 (22 images) per group. Scale bar = 0.5 cm (up) and 200 μm (down). (D, E) Con-miR1/133TS or HSP47-miR1/133TS pretreated mice were subjected to sham or IR for four weeks. Then, the heart tissue homogenate was harvested for protein isolation, and immunoblotting was used for collagen I, α-SMA, and fibronectin analyses and subsequently offered quantitative analysis by image lab software. They were normalized to β-tubulin; n = 4 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (F, G) Heart tissue homogenate from indicated mice was harvested for protein isolation, and immunoblotting was used for Vimentin and Ki67 analyses and subsequently offered quantitative analysis by image lab software. They were normalized to β-tubulin. n = 4 per group. (H) Immunofluorescence images showing the fibroblast proliferation in heart tissue. n = 3 per group. 18/16/22/25 images for IF in sham-con/sham-HSP47/IR-con/IR-HSP47. Vimentin staining is showed in green, Ki67 is showed in red, and positive results were indicated by white arrow (Scale bar = 80 μm). Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. Data are presented as the mean ± SEM, with each point representing a mouse.
3.5. HSP47 promotes activation of TGFβ1/Smad4 in fibroblasts subjected to hypoxia and reoxygenation
To further investigate how HSP47 impacts ischemic cardiac fibrosis, we next isolated AMCFs from rodents under myocardial IRI for four weeks and performed RNA sequencing. 118 DEGs were screened out to be regulated by HSP47 (Fig. 5A–C). Expectedly, HSP47 overexpression in CFs contributed to the activation of TGFβ pathway and led to obvious fibrosis in ischemic hearts (Fig. 5D). These findings were supported by significant transcription of pro-fibrogenic molecular markers such as Tgfβ1, Tgfβ3, Fibulin2, Postn, Loxl2, Nupr1, and Cola1a3 in hearts exposed to IRI for four weeks (Fig. 5E). We evaluated TGFβ–Smads signaling in isolated CFs from indicated mice by immunoblotting to ascertain the precise mechanism. We uncovered that HSP47 induced fibrogenesis by enhancing the activation of TGFβ1–Smad4 pathway instead of Smad2/3 in CFs (Fig. 5F and G), which was paralleled with previous publication32. These data indicate that HSP47 promoted TGFβ1/Smad4 in fibroblasts subjected to hypoxia and reoxygenation.
Figure 5.

HSP47 promotes activation of TGFβ1/Smad4 in fibroblasts subjected to hypoxia and reoxygenation. (A–D) Con-miR1/133TS or HSP47-miR1/133TS pretreated mice were subjected to sham or IR for four weeks, and AMCFs were isolated to RNA sequencing; n = 3 per group. (A) The volcano map was shown in HSP47-miR1/133TS-IR vs. Con-miR1/133TS-IR. (B) A heat map was presented in HSP47-miR1/133TS-IR vs. Con-miR1/133TS-IR. (C) Differentially expressed genes (DEGs) were subjected to Venn diagram analyses. (D) Top 250 DEGs were undergone gene ontology (GO) analyses. (E) Total RNA of AMCFs was collected for qPCR and mRNA expression related to cardiac fibrosis in isolated AMCFs and normalized to β-tubulin; n = 4 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (F, G) AMCFs from indicated mice were harvested for protein isolation, and immunoblotting was used for TGFβ1, p-Smad3, t-Smad3, p-Smad4, and t-Smad4 analyses and subsequently offered quantitative analysis by image lab software. They were normalized to β-tubulin (n = 4 per group). Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. Data are presented as the mean ± SEM, with each point representing an AMCFs sample from a mouse.
3.6. HSP47 reinforces deubiquitination of Smad4 by recruiting ubiquitin specific peptidase 10 (USP10) in fibroblasts
To identify the mechanism of HSP47 acting on Smad4, we further detected expression of Smad4 by transfecting with adenovirus (Ad)-HSP47-GFP or Ad-LacZ in isolated NRCFs (Supporting Information Fig. S5A and S5B). As expected, HSP47 overexpression contributed to the activation of the TGFβ1–Smad4 pathway in CFs that underwent HR, as evaluated by increased protein level (Fig. 6A and B). Interestingly, HSP47 did not affect the transcription of Smad4, which conflicted with the findings in protein measurement (Fig. 6C). We thus conjectured that HSP47 might impact the degradation of Smad4 in response to HR. Given that ubiquitination of Smad4 has been identified to regulate fibrosis33,34, we next investigated the effects of HSP47 on the ubiquitination of Smad4 in HR-treated CFs. Moreover, we revealed that HSP47 overexpression in CFs markedly suppressed ubiquitination and degradation of Smad4, which led to the aggregation of Smad4 in CFs (Fig. 6D and E). Subsequently, we assessed whether HSP47 directly targeted Smad4 and found that HSP47 indeed showed no affinity for Smad4 (Fig. S5C). Previous studies have suggested that USP10 contributes to the deubiquitination of Smad4 and hepatocellular carcinoma metastasis35. Then, we uncovered that USP10 directly interacted with Smad4 in NRCFs (Fig. 6F). Consistently, the confocal images demonstrated that USP10 and Smad4 were co-localized in HEK 293T cells (Fig. 6G and H). In addition, USP10-mediated post-translational modification of Smad4 was verified by using USP10 siRNA in NRCFs (Fig. S5D). USP10 knockdown in NRCFs disturbed deubiquitination of Smad4 and promoted Smad4 degradation (Fig. S5E). Taken together, these results show that HSP47 substantially increased Smad4 by recruiting USP10 in CFs, which reinforces Smad4 deubiquitination.
Figure 6.

HSP47 reinforces deubiquitination of Smad4 by recruiting ubiquitin-specific peptidase 10 (USP10) in fibroblasts. NRCFs were transfected with adenovirus (Ad)-HSP47-GFP or Ad-LacZ for 24 h and then cultured with Normoxia or HR. (A, B) Protein isolation and immunoblotting were used for TGFβ1 and t-Smad4 analyses and subsequently offered quantitative analysis by image lab software. They were normalized to β-tubulin; n = 4 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (C) Total RNA of NRCFs was collected for qPCR and mRNA expression of Smad4 and normalized to β-tubulin. n = 4 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (D, E) Ubiquitination of Smad4 was detected by immunoprecipitation in NRCFs, and subsequently offered quantitative analysis by image lab software. n = 4 independent experiments. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (F) NRCF lysates were immunoprecipitated with anti-Smad4 or USP10 antibodies and probed with anti-USP10 or anti-Smad4 antibodies. n = 3 independent experiments. (G) Flag-agarose beads or HA agarose were incubated with lysates of HEK 293 cells transfected with either Flag-tagged Smad4 or together with HA-tagged USP10, and immunoprecipitation was probed with anti-HA or Flag antibody. n = 3 independent experiments. (H) Immunofluorescence images present Smad4 and USP10 (merge, yellow) colocation in HEK 293 cells. Scar bar = 10 μm. Data are presented as the mean ± SEM, with each point representing a cell sample.
3.7. HSP47 encouraged IR-induced fibrogenesis and cardiac dysfunction in USP10 dependent manner in vivo
To confirm the significance of USP10 in HSP47–Smad4 mediated fibrosis under chronic myocardial IRI, we used cardiac fibroblast-specific USP10 deficient mice with HSP47-mi1/133TS in IRI model for 24 h or four weeks (Supporting Information Fig. S6A). Evans blue/TTC double staining showed that genetic depletion of USP10 abrogated cardiac fibroblast HSP47 overexpression induced enlarged infarct area following reperfusion 24 h (Fig. 7A–C). Furthermore, ablation of USP10 modestly reduced HSP47-mediated upregulation of plasma TnT and CK-MB, implying inhibition of USP10 alleviated HSP47-induced cardiac damage in response to hypoxia in mice (Fig. S6B and S6C). Expectedly, suppression of USP10 in vivo ameliorated HSP47-mediated reduction of LVEF and heart failure in chronic IRI mice (Fig. 7D and E), and exerted no effect in sham mice (Fig. S6D). In addition, genetic depletion of USP10 in CFs impeded HSP47-induced reduction of GLS (Supporting Information Fig. S7A and S7B) and LV compliance (Fig. S7C and S7D) under hypoxia condition, as assessed by speckle tracking echocardiography and PV curve, respectively. In parallel, USP10 deficiency mitigated diffuse fibrosis in both perivascular and interstitial area due to HSP47 overexpression in CFs (Fig. 7F and G), and showed no alteration in sham mice (Fig. S6E and S6F). RT-PCR analyses were corroborated by the drop in mRNA of Mmp9, Mmp2, Col1a1, Atac1, and Ctgf in heart tissues (Fig. S6G and S6H). Mechanically, USP10 knockout dampened deubiquitination of Smad4 and contributed to Smad4 degradation in AMCFs (Fig. 7H). Therefore, these results collectively reveal that cardiac fibroblast-specific USP10 deficiency abolished HSP47-mediated cardiac damage and fibrogenesis in IRI hearts.
Figure 7.

Cardiac fibroblast specific USP10 deficiency abolished HSP47-mediated cardiac dysfunction and fibrogenesis in chronic ischemic hearts. (A–C) Con-miR1/133TS or HSP47-miR1/133TS was administered via tail vein injection to the USP10fl/fl or USP10fl/fl-Cre+ mice (dose 3 × 108 PFU per mouse) for two weeks before surgery, and reperfused for 24 h. (A) Heart slices from indicated mice were stained with Evans blue and TTC at 24 h after myocardial IRI to determine (B, C) the area at risk (AAR) and infarcted region (IR). n = 6 per group. Scale bar = 0.5 cm. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (D–H) Con-miR1/133TS or HSP47-miR1/133TS was administered via tail vein injection to the USP10fl/fl or USP10fl/fl-Cre+ mice (dose 3 × 108 PFU per mouse) for two weeks before surgery, and continuously reperfused for 4 weeks. (D) Representative M-mode echocardiography was recorded. (E) LV ejection fraction (LVEF) was analyzed. n = 11–12 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (F, G) Representative Masson staining of heart slices from indicated mice and quantitative analyses of LV collagen volume. n = 6 per group. Scar bar = 80 μm. (H) Ubiquitination of Smad4 was detected by immunoprecipitation in isolated cardiac fibroblasts. n = 4 independent experiments. Data are presented as the mean ± SEM, with each point representing a mouse.
3.8. Targeting HSP47 disturbs fibrogenesis in fibroblasts following hypoxia and reoxygenation
To further identify the role of HSP47 inhibition in HR-induced collagen production, we treated isolated AMCFs with HSP47 inhibitor Col003. AMCFs were obtained from C57BL/6J mice in the sham group. Col003 inhibited HR-induced fibrogenesis in vitro (Fig. 8A); this response was similar to that observed in immunofluorescence of α-SMA when CFs were cultured in the presence of HR stress (Fig. 8B). On the other hand, HR increased the expression of pro-fibrogenic factors, including collagen I and CTGF in AMCFs, while blockage of HSP47 significantly mitigated this response (Fig. 8C). Furthermore, immunoblotting analyses showed that HR treatment substantially promoted activation of TGFβ1–Smad4 pathway, whereas the HSP47 inhibitor impeded the TGFβ1–Smad4 pathway in CFs in the presence of HR stress (Fig. 8D–E). These data suggest that targeting HSP47 hinders fibrogenesis in fibroblasts following hypoxia and reoxygenation.
Figure 8.

Targeting HSP47 disturbs fibrogenesis in CFs following hypoxia and reoxygenation. Cultured AMCFs were treated with Normoxia or HR for 24 h and co-treated with HSP47 inhibitor (Col003, 50 μmol/L) or DMSO. (A) Collagen deposition evaluation was conducted with Sirius Red/fast green staining (Chondrex/Amsbio, 9046); n = 6 per group. Differences assessed by Student's t-test. (B) Representative image of immunofluorescence of α-SMA in AMCFs treated with HR. (C) Total RNA of AMCFs was collected for qPCR and mRNA expression of Col1a1 and Ctgf, and normalized to β-tubulin. n = 4 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. (D, E) Protein isolation and immunoblotting were used for HSP47, TGFβ1, p-Smad4, and t-Smad4 analyses and subsequently offered quantitative analysis by image lab software. They were normalized to β-tubulin. n = 4 per group. Differences were assessed by two-way ANOVA and Tukey's multiple comparison test. Data are presented as the mean ± SEM, with each point representing a cell sample.
4. Discussions
Heart failure is one of the main threats that threaten people's health worldwide. It is mainly characterized by high morbidity and mortality36. Cardiac remodeling induced by long-term myocardial ischemia contributes largely to the poor prognosis in patients with AMI, which is mainly characterized by extensive myocardial fibrosis and reduced left ventricular (LV) compliance37. However, there has been no effective treatment for this pathological response. Based on previous research evidence, our study regarded HSP47 as the point of penetration to deeply explore the relationship between HSP47 in cardiac fibroblasts and fibrosis following myocardial ischemia. It was clarified that HSP47 accelerated the process of cardiac fibrosis after chronic myocardial IRI. In the current study, we revealed that HSP47 promoted deubiquitination of Smad4 by recruiting USP10 and facilitated activation of TGFβ/Smad4 signaling in fibroblasts, ultimately leading to diffused fibrosis. This study systematically explained the role of HSP47 in myocardial fibrosis following IRI and uncovered its specific mechanism, providing a potential strategy for treating patients with ischemic cardiomyopathy (ICM) in clinical practice.
Cardiac fibrosis is mainly manifested as collagen fiber deposition and cell proliferation, in which fibroblasts play an important role in these pathological changes. However, cardiac fibrosis results from a repair process activated by cardiomyocyte injury38. After a transmural infarction, fibrous scars are essential to protect the heart from rupture by maintaining the integrity of the ventricular structure and preventing dilated remodeling39. Many other pathophysiological conditions cause more insidious interstitial or perivascular fibrosis forms, usually without significant replacement of cardiomyocytes. Disturbance of the fibrotic heart's interstitial collagen network may also cause contractile dysfunction through several different mechanisms40. First, fibrosis may disrupt the coordination of myocardial excitation and contraction coupling. Second, vascular fibrosis may cause microvascular dysfunction and reduce myocardial cell perfusion under stress conditions. Third, the interstitial collagen deposition in the fibrotic area can stimulate the protease-dependent pathway, leading to the degradation of fibrous collagen, thereby destroying the connection between the matrix and the muscle fiber contraction device and causing systolic dysfunction. The current study showed that HSP47 overexpression in cardiac fibroblasts contributed to acute cardiac injury following myocardial IRI for 24 h and prolonged chronic cardiac remodeling and heart failure in response to myocardial IRI for four weeks.
Since the endoplasmic reticulum (ER) is a key organelle for synthesizing membrane-bound proteins and secreted proteins, it has a complex mechanism to promote protein maturation and folding, including a set of specific molecular chaperones41. HSP47 is one of these molecular chaperones and has unique characteristics42. Mouse embryos lacking HSP47 cannot survive 11.5 days after conception. They exhibit major growth defects, developmental delays, significant reduction of type IV collagen, destruction of the basement membrane, and a lack of mature type I collagen and fibrils in the mesenchymal tissue43. Col2a1-Cre transgenic mice expressing Cre recombinase in chondrocytes confirmed the role of HSP47 in collagen maturation44. In vitro, HSP47 deficient cells revealed abnormal collagen processing and impaired folding into a triple helix, as demonstrated by the sensitivity of procollagen from HSP47 knockout cells compared with procollagen from wild-type cells45. In summary, these findings emphasized the impact of HSP47 on several stages of collagen production, including procollagen triple helix formation, intracellular transport of procollagen, and extracellular fibril aggregation. In particular, HSP47 in cardiac fibroblasts has been confirmed to be associated with cardiac fibrosis in cardiac remodeling caused by pressure overload without reflecting the precise molecular mechanism21. We further identified that cardiac fibroblast HSP47 contributed to ischemia-related fibrogenesis in mice. In addition, misfolded procollagen accumulation in ER leads to ER stress signal activation, causing apoptosis and oxidative stress46. Consistently, we also suggested that HSP47 led to apoptosis and oxidative stress in acute myocardial ischemia in vivo.
Likewise, TGF-β is part of the related growth factor superfamily. Mammalian TGF- subtypes (TGFβ-1, TGFβ-2, and TGFβ-3) have been identified47. TGFβ-1 is a potent inducer of mesenchymal gene expression programs, capable of transforming epithelial, endothelial, and intrinsic renal fibroblasts into myofibroblasts expressing -smooth muscle actin (α-SMA)48. Although TGFβ-1 plays a leading role in the occurrence and development of renal fibrosis, its effector Smad proteins (Smad2, Smad3, and Smad4) play different or even opposite functions in fibrosis regulation. An important difference between Smad proteins is that Smad3 can directly bind to the Smad binding element in the gene promoter to enhance transcription. However, neither Smad2 nor Smad4 has a DNA binding domain but acts as a regulator of gene transcription based on Smad349,50. Smad4 is a common component of TGFβ and bone morphogenetic proteins (BMPs) signaling pathways, promoting nuclear translocation of Smad2/3 and Smad1/5/8 complexes in response to TGFβ and BMPs, respectively51. The lack of Smad4 in mesangial cells in vitro significantly inhibits the type I collagen promoter52. Here, we suggested that HSP47 promoted activation of the TGFβ1/Smad4 pathway instead of Smad2/3 in cardiac fibroblasts to result in fibrosis. We also revealed that HSP47 indeed showed no affinity for Smad4, and HSP47 did not affect the transcription of Smad4, which conflicted with the findings in upregulated protein levels. Furthermore, a previous study has suggested that USP10 contributed to the deubiquitination of Smad4 and hepatocellular carcinoma metastasis35. Subsequently, we uncovered that USP10 directly interacted with Smad4 and caused the deubiquitination of Smad4 to sustain Smad4 activity in NRCFs. However, USP4 was also identified to regulate the deubiquitination of Smad433, which was excluded in the current study due to the content of USP4 in fibroblasts being much lower than USP10. Besides MMP-2/-9 regulated cardiac fibrosis, cathepsins S has been reported to improve cardiac inflammation and fibrosis induced by Ang II53, and cathepsins K deficiency impairs mouse cardiac function after myocardial infarction54. Hence, both of cathepsins S and K probably warrants further testing. Furthermore, lipid nanoparticles delivering siRNA targeting HSP47 treat fibrosis and tumor metastasis55, 56, 57; however, small-molecule inhibitors of HSP47 used in vivo remain in the early stages of development, and more research is required in the future.
5. Conclusion
In summary, we explored the effects of HSP47 on ischemia-associated fibrosis in hearts and uncovered that HSP47 inspired the TGFβ1–Smad4 pathway in fibrogenesis under hypoxia. Moreover, extensive HSP47 contributed to the enrichment of Smad4 by USP10-dependent deubiquitination. The presented study filled the gaps and suggested aspects of HSP47-mediated cardiac fibrosis missing. Meanwhile, suppression of HSP47 by chemicals such as Col003 may have therapeutic potential for alleviating ischemic cardiac fibrosis. Our findings reveal that targeting HSP47 might be a potential strategy for myocardial IRI and chronic cardiac remodeling.
Acknowledgments
The work of the authors is supported by grants from the National Key R&D Program of China (2018YFC1311300); the Key Project of the National Natural Science Foundation of China (No. 81530012); the National Natural Science Foundation of China (Nos. 81860080, 82170245 and 81700254); and the Fundamental Research Funds for the Central Universities (2042018kf1032, China); the Development Center for Medical Science and Technology National Health and Family Planning Commission of the People's Republic of China (The prevention and control project of cardiovascular disease, 2016ZX-008-01); Science and Technology Planning Projects of Wuhan (2018061005132295).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.07.022.
Author contributions
Qizhu Tang and Saiyang Xie make formulation of overarching research goals and aims, Min Zhang, Saiyang Xie, Wenke Shi and Yun Xing conduct a research and investigation process, Tong Zhang and Wenxi Fang analyze or synthesize study data, Shiqiang Liu, MengYa Chen, and Si Chen verificate data, Xiaofeng Zeng and Shasha Wang write the initial draft, Wei Deng, Qizhu Tang and Saiyang Xie revised the manuscript. All authors discussed and approved the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Appendix A. Supporting information
The following is the Supplementary data to this article:
References
- 1.Thomas H., Diamond J., Vieco A., Chaudhuri S., Shinnar E., Cromer S., et al. Global atlas of cardiovascular disease 2000–2016: the path to prevention and control. Glob Heart. 2018;13:143–163. doi: 10.1016/j.gheart.2018.09.511. [DOI] [PubMed] [Google Scholar]
- 2.Sun H.J., Wu Z.Y., Nie X.W., Wang X.Y., Bian J.S. An updated insight into molecular mechanism of hydrogen sulfide in cardiomyopathy and myocardial ischemia/reperfusion injury under diabetes. Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.651884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li L., Zhao Q., Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018;68–69:490–506. doi: 10.1016/j.matbio.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 4.Dumont E.A., Hofstra L., van Heerde W.L., van den Eijnde S., Doevendans P.A., DeMuinck E., et al. Cardiomyocyte death induced by myocardial ischemia and reperfusion: measurement with recombinant human annexin-V in a mouse model. Circulation. 2000;102:1564–1568. doi: 10.1161/01.cir.102.13.1564. [DOI] [PubMed] [Google Scholar]
- 5.Frangogiannis N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Travers J.G., Kamal F.A., Robbins J., Yutzey K.E., Blaxall B.C. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prabhu S.D., Frangogiannis N.G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner N.A., Porter K.E. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair. 2013;6:5. doi: 10.1186/1755-1536-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tallquist M.D., Molkentin J.D. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. 2017;14:484–491. doi: 10.1038/nrcardio.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bacmeister L., Schwarzl M., Warnke S., Stoffers B., Blankenberg S., Westermann D., et al. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol. 2019;114:19. doi: 10.1007/s00395-019-0722-5. [DOI] [PubMed] [Google Scholar]
- 11.Ravassa S., Lopez B., Querejeta R., Echegaray K., San Jose G., Moreno M.U., et al. Phenotyping of myocardial fibrosis in hypertensive patients with heart failure. Influence on clinical outcome. J Hypertens. 2017;35:853–861. doi: 10.1097/HJH.0000000000001258. [DOI] [PubMed] [Google Scholar]
- 12.Kasuya A., Tokura Y. Attempts to accelerate wound healing. J Dermatol Sci. 2014;76:169–172. doi: 10.1016/j.jdermsci.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y. New insights into epithelial–mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coulouarn C., Factor V.M., Thorgeirsson S.S. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology. 2008;47:2059–2067. doi: 10.1002/hep.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haider C., Hnat J., Wagner R., Huber H., Timelthaler G., Grubinger M., et al. Transforming growth factor-beta and Axl induce CXCL5 and neutrophil recruitment in hepatocellular carcinoma. Hepatology. 2019;69:222–236. doi: 10.1002/hep.30166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue K., Zhang J., Li C., Li J., Wang C., Zhang Q., et al. The role and mechanism of transforming growth factor beta 3 in human myocardial infarction-induced myocardial fibrosis. J Cell Mol Med. 2019;23:4229–4243. doi: 10.1111/jcmm.14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ito S., Nagata K. Roles of the endoplasmic reticulum-resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease. J Biol Chem. 2019;294:2133–2141. doi: 10.1074/jbc.TM118.002812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellaye P.S., Burgy O., Bonniaud P., Kolb M. HSP47: a potential target for fibrotic diseases and implications for therapy. Expert Opin Ther Targets. 2021;25:49–62. doi: 10.1080/14728222.2021.1861249. [DOI] [PubMed] [Google Scholar]
- 19.Xiong G., Chen J., Zhang G., Wang S., Kawasaki K., Zhu J., et al. Hsp47 promotes cancer metastasis by enhancing collagen-dependent cancer cell-platelet interaction. Proc Natl Acad Sci U S A. 2020;117:3748–3758. doi: 10.1073/pnas.1911951117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Dijk F.S., Semler O., Etich J., Kohler A., Jimenez-Estrada J.A., Bravenboer N., et al. Interaction between KDELR2 and HSP47 as a key determinant in osteogenesis imperfecta caused by bi-allelic variants in KDELR2. Am J Hum Genet. 2020;107:989–999. doi: 10.1016/j.ajhg.2020.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khalil H., Kanisicak O., Vagnozzi R.J., Johansen A.K., Maliken B.D., Prasad V., et al. Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight. 2019;4 doi: 10.1172/jci.insight.128722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie S., Qi X., Wu Q., Wei L., Zhang M., Xing Y., et al. Inhibition of 5-lipoxygenase is associated with downregulation of the leukotriene B4 receptor 1/interleukin-12p35 pathway and ameliorates sepsis-induced myocardial injury. Free Radic Biol Med. 2021;166:348–357. doi: 10.1016/j.freeradbiomed.2021.02.034. [DOI] [PubMed] [Google Scholar]
- 23.Ma Z.G., Yuan Y.P., Zhang X., Xu S.C., Kong C.Y., Song P., et al. C1q-tumour necrosis factor-related protein-3 exacerbates cardiac hypertrophy in mice. Cardiovasc Res. 2019;115:1067–1077. doi: 10.1093/cvr/cvy279. [DOI] [PubMed] [Google Scholar]
- 24.Li Y., Li Z., Zhang C., Li P., Wu Y., Wang C., et al. Cardiac fibroblast-specific activating transcription factor 3 protects against heart failure by suppressing MAP2K3–p38 signaling. Circulation. 2017;135:2041–2057. doi: 10.1161/CIRCULATIONAHA.116.024599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M., Wei L., Xie S., Xing Y., Shi W., Zeng X., et al. Activation of Nrf2 by lithospermic acid ameliorates myocardial ischemia and reperfusion injury by promoting phosphorylation of AMP-activated protein kinase alpha (AMPKalpha) Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.794982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saiyang X., Qingqing W., Man X., Chen L., Min Z., Yun X., et al. Activation of Toll-like receptor 7 provides cardioprotection in septic cardiomyopathy-induced systolic dysfunction. Clin Transl Med. 2021;11:e266. doi: 10.1002/ctm2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown B.D., Venneri M.A., Zingale A., Sergi Sergi L., Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. 2006;12:585–591. doi: 10.1038/nm1398. [DOI] [PubMed] [Google Scholar]
- 28.Chouchani E.T., Pell V.R., Gaude E., Aksentijevic D., Sundier S.Y., Robb E.L., et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhai M., Li B., Duan W., Jing L., Zhang B., Zhang M., et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res. 2017;63 doi: 10.1111/jpi.12419. [DOI] [PubMed] [Google Scholar]
- 30.Bellaye P.S., Burgy O., Causse S., Garrido C., Bonniaud P. Heat shock proteins in fibrosis and wound healing: good or evil?. Pharmacol Ther. 2014;143:119–132. doi: 10.1016/j.pharmthera.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Nieto M.A., Huang R.Y., Jackson R.A., Thiery J.P. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 32.Wei S., Wang Q., Zhou H., Qiu J., Li C., Shi C., et al. miR-455-3p alleviates hepatic stellate cell activation and liver fibrosis by suppressing HSF1 expression. Mol Ther Nucleic Acids. 2019;16:758–769. doi: 10.1016/j.omtn.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou F., Xie F., Jin K., Zhang Z., Clerici M., Gao R., et al. USP4 inhibits SMAD4 monoubiquitination and promotes activin and BMP signaling. EMBO J. 2017;36:1623–1639. doi: 10.15252/embj.201695372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang Q., Tang C., Tang M., Zhang Q., Gao Y., Ge Z. TRIM47 is up-regulated in colorectal cancer, promoting ubiquitination and degradation of SMAD4. J Exp Clin Cancer Res. 2019;38:159. doi: 10.1186/s13046-019-1143-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan T., Chen Z., Yan F., Qian M., Luo H., Ye S., et al. Deubiquitinating enzyme USP10 promotes hepatocellular carcinoma metastasis through deubiquitinating and stabilizing Smad4 protein. Mol Oncol. 2020;14:197–210. doi: 10.1002/1878-0261.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziaeian B., Fonarow G.C. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13:368–378. doi: 10.1038/nrcardio.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frangogiannis N.G. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. 2019;65:70–99. doi: 10.1016/j.mam.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Baehr A., Umansky K.B., Bassat E., Jurisch V., Klett K., Bozoglu T., et al. Agrin promotes coordinated therapeutic processes leading to improved cardiac repair in pigs. Circulation. 2020;142:868–881. doi: 10.1161/CIRCULATIONAHA.119.045116. [DOI] [PubMed] [Google Scholar]
- 39.Cheng L., Sun X., Zhao X., Wang L., Yu J., Pan G., et al. Surface biofunctional drug-loaded electrospun fibrous scaffolds for comprehensive repairing hypertrophic scars. Biomaterials. 2016;83:169–181. doi: 10.1016/j.biomaterials.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Iwanaga Y., Aoyama T., Kihara Y., Onozawa Y., Yoneda T., Sasayama S. Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats. J Am Coll Cardiol. 2002;39:1384–1391. doi: 10.1016/s0735-1097(02)01756-4. [DOI] [PubMed] [Google Scholar]
- 41.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 42.Ito S., Nagata K. Biology of Hsp47 (Serpin H1), a collagen-specific molecular chaperone. Semin Cell Dev Biol. 2017;62:142–151. doi: 10.1016/j.semcdb.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Nagai N., Hosokawa M., Itohara S., Adachi E., Matsushita T., Hosokawa N., et al. Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J Cell Biol. 2000;150:1499–1506. doi: 10.1083/jcb.150.6.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ovchinnikov D.A., Deng J.M., Ogunrinu G., Behringer R.R. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146. [PubMed] [Google Scholar]
- 45.Matsuoka Y., Kubota H., Adachi E., Nagai N., Marutani T., Hosokawa N., et al. Insufficient folding of type IV collagen and formation of abnormal basement membrane-like structure in embryoid bodies derived from Hsp47-null embryonic stem cells. Mol Biol Cell. 2004;15:4467–4475. doi: 10.1091/mbc.E04-01-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawasaki K., Ushioda R., Ito S., Ikeda K., Masago Y., Nagata K. Deletion of the collagen-specific molecular chaperone Hsp47 causes endoplasmic reticulum stress-mediated apoptosis of hepatic stellate cells. J Biol Chem. 2015;290:3639–3646. doi: 10.1074/jbc.M114.592139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 48.Margadant C., Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11:97–105. doi: 10.1038/embor.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dennler S., Itoh S., Vivien D., ten Dijke P., Huet S., Gauthier J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piek E., Ju W.J., Heyer J., Escalante-Alcalde D., Stewart C.L., Weinstein M., et al. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945–19953. doi: 10.1074/jbc.M102382200. [DOI] [PubMed] [Google Scholar]
- 51.Meng X.M., Chung A.C., Lan H.Y. Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond) 2013;124:243–254. doi: 10.1042/CS20120252. [DOI] [PubMed] [Google Scholar]
- 52.Tsuchida K., Zhu Y., Siva S., Dunn S.R., Sharma K. Role of Smad4 on TGF-beta-induced extracellular matrix stimulation in mesangial cells. Kidney Int. 2003;63:2000–2009. doi: 10.1046/j.1523-1755.2003.00009.x. [DOI] [PubMed] [Google Scholar]
- 53.Pan L., Li Y., Jia L., Qin Y., Qi G., Cheng J., et al. Cathepsin S deficiency results in abnormal accumulation of autophagosomes in macrophages and enhances Ang II-induced cardiac inflammation. PLoS One. 2012;7 doi: 10.1371/journal.pone.0035315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fang W., He A., Xiang M.X., Lin Y., Wang Y., Li J., et al. Cathepsin K-deficiency impairs mouse cardiac function after myocardial infarction. J Mol Cell Cardiol. 2019;127:44–56. doi: 10.1016/j.yjmcc.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Morry J., Ngamcherdtrakul W., Gu S., Goodyear S.M., Castro D.J., Reda M.M., et al. Dermal delivery of HSP47 siRNA with NOX4-modulating mesoporous silica-based nanoparticles for treating fibrosis. Biomaterials. 2015;66:41–52. doi: 10.1016/j.biomaterials.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamakawa T., Ohigashi H., Hashimoto D., Hayase E., Takahashi S., Miyazaki M., et al. Vitamin A-coupled liposomes containing siRNA against HSP47 ameliorate skin fibrosis in chronic graft-versus-host disease. Blood. 2018;131:1476–1485. doi: 10.1182/blood-2017-04-779934. [DOI] [PubMed] [Google Scholar]
- 57.Yoneda A., Minomi K., Tamura Y. HSP47 promotes metastasis of breast cancer by interacting with myosin IIA via the unfolded protein response transducer IRE1alpha. Oncogene. 2020;39:4519–4537. doi: 10.1038/s41388-020-1311-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.