

Abstract
Background
Treatment of newly diagnosed acute myeloid leukaemia (AML) is based on combination chemotherapy with cytarabine (ara‐C) and anthracyclines. Five‐year overall survival is below 30%, which has partly been attributed to cytarabine resistance. Preclinical data suggest that the addition of hydroxyurea potentiates cytarabine efficacy by increasing ara‐C triphosphate (ara‐CTP) levels through targeted inhibition of SAMHD1.
Objectives
In this phase 1 trial, we evaluated the feasibility, safety and efficacy of the addition of hydroxyurea to standard chemotherapy with cytarabine/daunorubicin in newly diagnosed AML patients.
Methods
Nine patients were enrolled and received at least two courses of ara‐C (1 g/m2/2 h b.i.d. d1‐5, i.e., a total of 10 g/m2 per course), hydroxyurea (1–2 g d1‐5) and daunorubicin (60 mg/m2 d1‐3). The primary endpoint was safety; secondary endpoints were complete remission rate and measurable residual disease (MRD). Additionally, pharmacokinetic studies of ara‐CTP and ex vivo drug sensitivity assays were performed.
Results
The most common grade 3‐4 toxicity was febrile neutropenia (100%). No unexpected toxicities were observed. Pharmacokinetic analyses showed a significant increase in median ara‐CTP levels (1.5‐fold; p = 0.04) in patients receiving doses of 1 g hydroxyurea. Ex vivo, diagnostic leukaemic bone marrow blasts from study patients were significantly sensitised to ara‐C by a median factor of 2.1 (p = 0.0047). All nine patients (100%) achieved complete remission, and all eight (100%) with validated MRD measurements (flow cytometry or real‐time quantitative polymerase chain reaction [RT‐qPCR]) had an MRD level <0.1% after two cycles of chemotherapy. Treatment was well‐tolerated, and median time to neutrophil recovery >1.0 × 109/L and to platelet recovery >50 × 109/L after the start of cycle 1 was 19 days and 22 days, respectively. Six of nine patients underwent allogeneic haematopoietic stem‐cell transplantation (allo‐HSCT). With a median follow‐up of 18.0 (range 14.9–20.5) months, one patient with adverse risk not fit for HSCT experienced a relapse after 11.9 months but is now in second complete remission.
Conclusion
Targeted inhibition of SAMHD1 by the addition of hydroxyurea to conventional AML therapy is safe and appears efficacious within the limitations of the small phase 1 patient cohort. These results need to be corroborated in a larger study.
Keywords: acute myeloid leukaemia, cytarabine, hydroxyurea, precision medicine, SAMHD1, targeted therapy
Introduction
Acute myeloid leukaemia (AML) has a yearly incidence of 3–5 per 100,000 and occurs at a median age of ∼70 years [1, 2]. The overall prognosis is poor but varies with risk group and age. In patients fit for intensive chemotherapy, the 2‐year overall survival (OS) is ∼50% in patients <65 years but only 25% in those ≥65 years [1, 2]. This stems both from challenges of administering intensive chemotherapy in frail patients and from differences in disease biology, with more myelodysplasia‐related features in the elderly [3]. The standard‐of‐care for non‐promyelocytic AML consists of intensive induction and consolidation chemotherapy with cytarabine (ara‐C) and an anthracycline, frequently daunorubicin [4, 5]. Multiple attempts have been made to improve the outcome by adding novel components to the daunorubicin + ara‐C backbone, but with little success. Important exceptions are the FLT3 inhibitor midostaurin and gemtuzumab ozogamicin in patients with somatic mutations in FLT3 and core‐binding factor AML (CBF‐AML), respectively [4, 6–8]. Risk‐adapted allogeneic haematopoietic stem‐cell transplantation (allo‐HSCT) should be considered after achieving complete remission [4, 6, 9].
High‐dose ara‐C is a critical part of AML therapy, particularly during post‐remission consolidation [4, 6, 10, 11]. Ara‐C is a deoxycytidine analogue that is transported into leukemic cells by the equilibrative nucleoside transporter 2 (hENT2), and is subsequently mono‐, di‐ and triphosphorylated by deoxycytidine kinase (dCK), UMP/CMP kinase and nucleotide diphosphate kinases 1 and 2 (NDPK1/2), respectively. Ara‐C and ara‐C monophosphate (ara‐CMP) can be inactivated through deamination by cytidine deaminase (CDA) and dCMP deaminase, and ara‐CMP can be dephosphorylated by cytosolic 5′‐nucleotidases. Ara‐C triphosphate (ara‐CTP) is a substrate for DNA polymerases and can be incorporated into DNA during replication, perturbing further DNA elongation and triggering DNA damage responses [12]. Failure to accumulate the active metabolite ara‐CTP in leukemic blasts is correlated to inferior clinical response [13, 14, 15, 16, 17, 18]. We and others have recently shown that the deoxynucleoside triphosphate triphosphohydrolase SAMHD1 can hydrolyse ara‐CTP, and expression levels of SAMHD1 in leukaemic blasts were shown to negatively correlate with event‐free and overall survival following high‐dose ara‐C‐containing regimens [19, 20, 21, 22, 23]. Drug screening to alleviate SAMHD1‐mediated resistance to ara‐C resulted in the identification of the non‐competitive ribonucleotide reductase inhibitor (RNRi) hydroxyurea as a potent synergistic drug with ara‐C with the ability to increase intracellular ara‐CTP accumulation [24, 25]. Mechanistically, we have shown that hydroxyurea causes dNTP imbalances by reducing dATP and dGTP levels without affecting dCTP. In parallel, dCK is activated [24]. Hence, neither allosteric inhibition of dCK nor substrate competition at DNA polymerase by dCTP are expected effects of hydroxyurea treatment. Hydroxyurea has pleiotropic effects [26], with potent SAMHD1 inhibition already at concentrations of 20 μM [14], while reversible S‐phase cell‐cycle arrest is increasingly seen at concentrations in the millimolar range [27]. The plasma levels reached in adults dosed with 1 g hydroxyurea is expected to fully inhibit SAMHD1 while only having a limited effect on cell cycling [28].
In support of the addition of RNRi to ara‐C, the competitive RNRi fludarabine had been reported to clinically increase ara‐CTP accumulation [29], which led to the establishment of AML treatment strategies combining fludarabine and ara‐C [30]. However, more recent data suggest that fludarabine, in contrast to hydroxyurea, does not increase the ara‐C sensitivity or the ara‐CTP levels in cellular models of AML [24]. Another previously employed strategy to increase leukaemic ara‐CTP concentrations was inhibition of the ara‐C deaminating enzyme CDA [31]. However, a clinical trial in relapsed AML patients incorporating a CDA inhibitor did not achieve significant inhibition of ara‐C deamination [32].
Therefore, in this phase 1 trial, we evaluated the feasibility, safety and efficacy of hydroxyurea to enhance ara‐C‐based AML‐directed conventional therapy in newly diagnosed AML patients by the addition of hydroxyurea. Analysis of the phase 1 run‐in part reported here was preplanned and specified in the protocol (see Methods and Supplementary Materials). Whereas hydroxyurea monotherapy has a long‐standing role in cytoreductive and palliative treatment [33], toxicity in combination with intensive chemotherapy including high‐dose ara‐C and daunorubicin in treatment‐naïve patients has not been studied. Rather than a dose escalation to a maximum tolerated dose, this phase 1 trial was designed as a run‐in for a subsequent phase 2 trial evaluating the tolerability of hydroxyurea in the context of highly intensive chemotherapy at doses expected to yield SAMHD1 inhibition based on preclinical data [24] and known pharmacokinetics of hydroxyurea [28].
While definite conclusions regarding enhanced efficacy of hydroxyurea‐augmented ara‐C therapies will require larger patient cohorts, this phase 1 trial incorporated translational endpoints of in vivo ara‐CTP measurements and ex vivo drug treatment to provide a proof of principle alongside the reported clinical outcomes (HEAT‐AML trial; EudraCT‐number: 2018‐004050‐16).
Methods
Clinical study design and eligibility
This national open‐label, phase 1 trial is part of a phase 1/2 trial and was run at two sites (Karolinska University Hospital and Uppsala University Hospital, Sweden). The aim was to evaluate the feasibility, safety, and efficacy of adding hydroxyurea to standard AML‐directed therapy according to national guidelines. Eligibility criteria included age >18 years, newly diagnosed non‐promyelocytic AML and fitness for intensive chemotherapy. Patients with CBF‐AML eligible for treatment with gemtuzumab ozogamicin were excluded. Treatment comprised 2 to 4 cycles of ara‐C 1 g/m2/2 h i.v. b.i.d. on days 1–5 during all four cycles and daunorubicin 60 mg/m2/8 h i.v. q.d. on days 1–3 during cycles 1 and 2, and on days 1–2 during cycle 3. Patients with FLT3‐mutated AML received midostaurin 50 mg b.i.d. on days 8–21 of each cycle, but no maintenance was given after completed consolidation, in line with national guidelines. Risk‐adapted allo‐HSCT was performed at the discretion of the treating haematologist. Hydroxyurea was given 1 h prior to the start of the ara‐C infusion b.i.d. on days 1–5, and the dose was escalated in a 3 + 3 design: 500 mg b.i.d (level 1), 1000 mg q.d. + 500 mg q.d (level 2) and 1000 mg b.i.d. (level 3). Full inclusion/exclusion criteria and details regarding cycle intervals/dose reductions are provided in the protocol in the Supplementary Material.
Clinical study oversight
The clinical study protocol was designed by the study investigators. The sponsor of the clinical study was Karolinska University Hospital. The study was monitored by the Center for Clinical Cancer Studies, Karolinska University Hospital. Study oversight was performed by an independent data and safety monitoring committee. Data were collected and analysed by the study investigators. The protocol was reviewed by national and institutional ethics and regulatory bodies (Swedish Ethical Review Authority Dnr 5.1‐2019‐4650, Swedish Medical Products Agency, Research Council at Karolinska University Hospital). The study has been conducted in accordance with the Declaration of Helsinki and International Council for Harmonisation on Good Clinical Practice guidelines, and all patients provided written informed consent.
Endpoints
Primary endpoints were safety and tolerability (frequency and severity of toxicities according to CTCAE v5), including time to haematopoietic recovery (absolute neutrophil count [ANC] 0.5 and 1.0 × 109/L; platelets 50 × 109/L) after each chemotherapy cycle, defined as the time from the start of the cycle until recovery.
Secondary endpoints were response according to European LeukemiaNet (ELN) criteria [4] and measurable residual disease (MRD) <0.1% following treatment cycle 2, and ara‐CTP accumulation in peripheral mononuclear cells during the first chemotherapy cycle with or without hydroxyurea.
An additional explorative endpoint not predefined in the protocol was ex vivo drug screening of patient AML cells.
Comparison of SAMHD1 gene expression in healthy bone marrow and AML blasts
SAMHD1 gene expression and annotations in healthy bone marrow cells and AML samples were accessed using the publicly available BloodSpot portal [34]. The following datasets were retrieved: Human Normal Hematopoiesis—GSE42519 and Human AML cells—GSE13159, GSE15434, GSE61804, GSE14468, The Cancer Genome Atlas. Human AML cells were grouped into cytogenetic risk groups according to ELN criteria [4]. Data were visualised using Qlucore Omics Explorer version 3.8(1).
SAMHD1 protein expression
Expression of SAMHD1 was assessed using a double immunostaining method for SAMHD1/CD68 at diagnosis or SAMHD1/CD34 at remission, an autostainer system (BenchMark Ultra, Ventana, Rotkreuz, Switzerland) and previously validated protocols [21]. CD68+/SAMHD1+ histiocytes (macrophages) served as internal controls in the diagnostic bone marrow biopsies assessed. CD34 costaining at remission allowed assessment of SAMHD1 expression in haematopoietic stem and progenitor cells (HSPCs). The percentage of SAMHD1‐positive blasts was calculated by counting at least 500 blasts in each case, assessed by two haematopathologists independently.
MRD measurement
MRD was measured by flow cytometry after cycle 2 (according to the EuroFlow and NOPHO‐AML protocols, Table S1) or with real‐time quantitative polymerase chain reaction (RT‐qPCR) if validated genetic markers were available (such as mutated NPM1 or DEK::NUP214 fusion transcript, Table S3) [35, 36, 37]. To ensure additional information on mutational clearance, patients without a standard validated genetic marker for MRD were analysed by sensitive deep sequencing after cycle 2, as previously described [36].
Pharmacokinetic study
On day 1 of treatment cycle 1, patients received hydroxyurea 1 h prior to one of two ara‐C infusions. Patients 1–3 received 500 mg hydroxyurea prior to the first, patients 4–6 received 1000 mg hydroxyurea prior to the first and patients 7–9 received 1000 mg hydroxyurea prior to the second ara‐C infusion. The reason to allocate hydroxyurea either prior to the first or second administration of ara‐C was to control for possible accumulation of ara‐CTP after two doses of ara‐C, which might introduce a bias towards higher ara‐CTP levels following the second ara‐C infusion. In that case, the efficacy of hydroxyurea to increase ara‐CTP might be overestimated. Eighteen millilitres of peripheral blood was drawn directly following the first and second infusion of ara‐C, and mononuclear cells (MNCs) were isolated using Lymphoprep (STEMCELL Technologies, Cambridge, UK). Following methanol extraction, ara‐CTP was quantified using chromatography tandem mass spectrometry [24] with a modified liquid chromatography (LC) method (column: Hypercarb [100 × 2.1 mm, 5 μm, Thermo Scientific]; mobile phase A: 5 mM hexylamine and 0.4% dimethylhexylamine [v/v], pH 10; mobile phase B: 50 % acetonitrile; LC gradient: mobile phase B increased from 5% to 50% in 13 min, 50% to 80% in 1 min and then returned to the initial condition in 1 min; flow rate 0.4 ml/min).
Ex vivo drug sensitivity analysis
AML MNCs, healthy CD34+ haematopoietic stem and progenitor cell (HSPC) donor cells and THP‐1 SAMHD1‐wt or THP‐1 SAMHD1‐knockout cells [19] were dispensed in 384‐well tissue culture plates (Corning) at a concentration of 2.5 × 105 cells per millilitre reconstituted in RPMI 1640 media (ThermoFisher) supplemented with penicillin, streptomycin, 10% foetal calf serum and 12.5% conditioned cell culture supernatant (obtained from confluent HS‐5 cells [CRL‐11882, ATCC]) [38]. Cells were incubated for 72 h with the drugs hydroxyurea (10–100,000 nM) or gemcitabine (0.1–1000 nM) together with ara‐C (1–10,000 nM) in five combinations. For analysis of drug combinations, all concentrations of hydroxyurea or gemcitabine were tested against all concentrations of ara‐C in duplicates. The drugs were preplated using acoustic dispensing (Echo 550, Labcyte). Following incubation for 72 h, cell viability was measured using CellTiter‐Glo (Promega). Data were collected on an EnSight (PerkinElmer) system. Data on each plate were normalised to a plate‐specific negative control (vehicle) and a positive control (100 μM benzethonium chloride). Quality control and calculation of half‐maximal inhibitory concentrations (IC50) and zero interaction potency (ZIP) [39] and data analysis was performed using custom scripts in R and Breeze pipeline (breeze.fimm.fi) [40].
Ex vivo ara‐CTP measurements
AML MNCs were cultured in Iscove Modified Dulbecco Medium (IMDM) supplemented with penicillin, streptomycin, 10% foetal calf serum and 20 ng/ml interleukin 3, 20 ng/ml interleukin 6 (both R&D Systems), 20 ng/ml human recombinant Granulocyte‐monocyte colony‐stimulating factor and 100 μg/ml human recombinant thrombopoietin (both Stem Cell Technologies) and treated with 500 nM ara‐C with or without 60 μM hydroxyurea for 24 h. Samples were further processed and analysed as described above (Pharmacokinetic Study).
Statistical analysis
For ara‐CTP measurements from MNCs and ara‐C IC50 values in diagnostic bone marrow mononuclear cells with or without prior hydroxyurea, paired two‐tailed Student's t‐tests were performed prior to normalization. For comparison of ara‐CTP levels or ara‐C IC50 values with respect to SAMHD1 expression levels, Kruskal–Wallis tests were performed (Prism 9.2.0, GraphPad Software, San Diego, CA, USA).
Role of the funding sources
The funding sources had no role in the study design; in the collection, analysis and interpretation of data; in the writing of the report and in the decision to submit the paper for publication.
Results
Patients
In total, nine patients were enrolled between October 2020 and March 2021 (Table 1 and Fig. 1). During the recruitment period, a total of 31 patients with newly diagnosed AML received intensive chemotherapy at our institution. Of the 22 patients who were not screened for inclusion, the main reasons were frailty (such as age >75 years with comorbidities) that precluded full‐dose induction and hyperleukocytosis requiring pretreatment with hydroxyurea, which was not allowed in the phase 1 part of the study. In addition, two patients were ineligible due to having CBF‐AML.
Table 1.
Patient characteristics and outcome
| Patient | Karyotype | Mutations, fusions | ELN category | SAMHD1 IHC | CR cycle 1/2 | MRD after cycle 2, flow cytometry/RT‐qPCR/deep sequencing | Allo‐HSCT | Time to relapse (months) | Overall survival (months) |
|---|---|---|---|---|---|---|---|---|---|
|
1101 Female 26 years |
47,XX,t(8;19)(p21;p10),+der(8)t(8;19) [14] /46,idem,‐7 [3] /48,idem,+21 [2] /46,XX [1] | RUNX1 | Adverse | 25%–75% | Yes/Yes | 0.02%/NA/RUNX1 8.9% | Yes | NA | 20.5+ |
|
1102 Female 27 years |
46,XX [20] | NRAS, U2AF1 | Intermediate | <25% | Yes/Yes | 0.07%/NA/U2AF1 0.4%, NRAS <0.02% | Yes | NA | 20.1+ |
|
1103 Male 26 years |
46,XY [20] | FLT3‐TKD, RUNX1, IKZF1 | Adverse | 25%–75% | Yes/Yes | 0.09%/NA/RUNX1 0.74%, FLT3‐TKD 0.049% | Yes | NA | 19.2+ |
|
1104 Female 26 years |
46,XX,t(6;9)(p22;q34) [17] /46,XX [3] | DEK::NUP214 | Adverse | <25% | Yes/Yes | NA/<0.01%/NA | Yes | NA | 18.6+ |
|
1105 Female 48 years |
46,XX [20] | NPM1, FLT3‐TKD, IDH2, DNMT3A | Favourable | >75% | Yes/Yes | NA/<0.001%/NA | No | NA | 18.0+ |
|
1106 Male 76 years |
46,XY [20] | RUNX1, BCORL1, BCOR, FLT3‐TKD | Adverse | 25%–75% | No a /Yes | 0.004%/NA/RUNX1 10.5%, FLT3‐TKD 0.004% | No | 11.9 | 16.8+ |
|
1107 Male 52 years |
46,XY [20] | NPM1, FLT3‐ITD low ratio, IDH2 | Favourable | <25% | Yes/Yes | NA/0.0017/NA | No | NA b | 16.7+ |
|
1108 Female 61 years |
45,X,‐X [10]/46,XX [10] | RUNX1, SF3B1, CBL | Adverse | <25% | Yes/Yes | NA c /NA/RUNX1 0.79%, CBL 0.77% | Yes | NA | 16.0+ |
|
1109 Female 61 years |
46,XX t(5;6)(q31;q25) [6] /46,XX [16] | NPM1, FLT3‐ITD low ratio, TET2 | Favourable d | <25% | Yes/Yes | NA/0.017%/NA | Yes | NA | 14.9+ |
Abbreviations: Allo‐HSCT, allogeneic haematopoietic stem cell transplantation; BCOR, BCL6 Corepressor; BCORL1, BCL6 Corepressor Like 1; CBL, Cbl Proto‐Oncogene; CR, complete remission; DEK::NUP214, fusion of DEK Proto‐Oncogene and Nucleoporin 214; DNMT3A, DNA Methyltransferase 3α; ELN, European LeukemiaNet; FLT3‐ITD, Fms Related Receptor Tyrosine Kinase 3 internal tandem duplication; FLT3‐TKD, Fms Related Receptor Tyrosine Kinase 3 tyrosine kinase domain; IDH2, Isocitrate Dehydrogenase 2; IHC, immunohistochemistry; IKZF1, IKAROS Family Zinc Finger 1; MRD, measurable residual disease; NA, not available; NPM1, Nucleophosmin 1; NRAS, Neuroblastoma RAS viral oncogene homolog; RUNX1, Runt‐related transcription factor 1; SF3B1, Splicing Factor 3b Subunit 1; TET2, Tet Methylcytosine Dioxygenase 2; U2AF1, U2 Small Nuclear RNA Auxiliary Factor 1.
Blasts 5.5%; Granulocyte colony‐stimulating factor use prior to sampling may have increased the morphologic blast count.
At 9 months, MRD NPM1 switched from negative to positive at 0.00024%, when double‐checked then negative. Prior to receiving the confirmatory MRD result, the patient was put on AZA‐VEN and received three cycles.
Patient 1108 lacked a leukaemia‐specific phenotype for flow cytometry.
This patient had a prior history of myeloproliferative neoplasia with osteosclerosis and extramedullary haematopoiesis, and therefore better resembled a higher‐risk secondary AML.
Fig. 1.
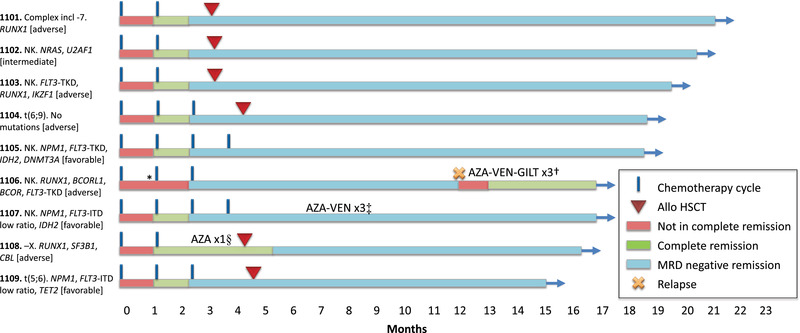
Treatment outcome. The nine patients have been followed for 14.9–20.5 months. Six patients have undergone allogeneic haematopoietic stem cell transplantation (allo‐HSCT). Measurable residual disease (MRD) was performed in accordance with national guidelines after cycle 2 and cycle 4 or prior to allo‐HSCT. In non‐transplanted patients with NPM1, MRD by real‐time quantitative polymerase chain reaction (RT‐qPCR) was performed every 3 months. After allo‐HSCT, patients were routinely monitored every 3 months either by RT‐qPCR (NPM1) or flow cytometry (all others). Eight of nine patients remain in MRD negative remission (as defined by MRD <0.1% by flow cytometry or RT‐qPCR). *, Patient 1106 had bone marrow blasts 5.5% after cycle 1 and did not fulfil complete remission (CR) criteria until after cycle 2. However, blasts may have been elevated by granulocyte colony‐stimulating factor (G‐CSF) usage prior to sampling; †, patient 1106 relapsed after 11.9 months and was treated with a combination of azacitidine–venetoclax–gilteritinib (AZA‐VEN‐GILT) and is now in a second CR; ‡, in patient 1107, NPM1 RT‐qPCR MRD increased from negative to 0.00024%, was rechecked and was then negative again. Before receiving the result of the confirmatory MRD, the patient was put on azacitidine–venetoclax (AZA‐VEN), and three cycles were given as an additional consolidation due to the inconsistent results; §, patient 1108 received one cycle of AZA as bridging while waiting for allo‐HSCT.
The patients were of all ELN risk categories: favourable (n = 3), intermediate (n = 1) and high (n = 5). FLT3 mutation was seen in five patients (two ITD, three TKD) and they all received midostaurin 50 mg b.i.d. days 8–21 after each cycle of chemotherapy. All patients received at least two cycles of therapy according to protocol. For patient 1106, the dose of ara‐C was reduced to 80% during cycle 2 due to high age (76 years) and severe sepsis after cycle 1.
Safety and tolerability
There were no non‐haematological grade 4 toxicities. The most common grade 3 adverse event was febrile neutropenia, occurring in all patients. Other grade 3 infections were sepsis (n = 2), catheter‐related infection (n = 3) and wound infection (n = 1). Gastrointestinal grade 3 events included colitis (n = 1), rectal pain (n = 1) and bleeding (n = 1). The remaining adverse events of grade 3 were back pain (n = 1), epistaxis (n = 1), bronchial obstruction (n = 1) and urinary tract obstruction (n = 1). Grade 1 and 2 events were as expected for patients undergoing intensive chemotherapy.
Specific adverse events known to be related to ara‐C were monitored in detail. No cerebellar or other central nervous adverse events occurred. Two patients, both concomitantly treated with midostaurin according to national guidelines, had grade 1 (patient 1109) or 2 (patient 1105) palmar–plantar erythrodysesthaesia. Both patients continued treatment according to protocol, and the skin lesions were successfully treated with topical steroids. Conjunctivitis was not reported above grade 1.
To estimate the likelihood of a negative impact of hydroxyurea‐mediated SAMHD1 inhibition on haematological recovery, we analysed SAMHD1 expression in a large set of publicly available AMLs and healthy bone‐marrow cells. While SAMHD1 expression is similar in different cytogenetic risk groups of AML, the HSC and other bone marrow progenitor cells with the exception of the granulocyte–monocyte progenitor expressed much lower levels of SAMHD1 (Fig. S1). This was also reflected by the general negativity for SAMHD1 in CD34+ HSCPs in the immunohistochemical evaluation of the patients’ bone marrow during remission (see below). The time‐to‐neutrophil recovery with ANC ≥0.5 and ≥1.0 × 109/L was similar, in median 19 (range 16–23) days after cycle 1. The rapid increase in ANC was likely attributed to the fact that all nine patients received G‐CSF support during the neutropenic phase, in accordance with local routines during the COVID‐19 pandemic. Time to platelet recovery ≥50 × 109/L was in median 22 (17–25) days after the first cycle. Similar haematological recovery was seen during the subsequent cycles (Table S2).
No dose‐limiting toxicities were found for the evaluated hydroxyurea doses, and the recommended phase 2 dose (RP2D) for the subsequent phase 2 part of the study was therefore set to 1000 mg b.i.d.
Response and leukaemic clearance
All nine patients achieved complete remission (CR) after cycle 2 (eight of nine already after cycle 1). One patient who received G‐CSF had 5.5% blasts after cycle 1 and hence was in partial remission per definition, despite full peripheral regeneration. Validated MRD measurements after cycle 2 were negative (<0.1%) in eight of eight evaluable patients (four of four by flow cytometry, four of four by RT‐qPCR). Patient 1108 lacked a specific leukaemia‐associated immunophenotype (LAIP) and MRD could therefore not be robustly determined with routine methods.
Targeted deep sequencing was used to further evaluate the mutational clearance after cycle 2 in the four patients for whom MRD was determined by flow cytometry. This demonstrated a reduction in variant allele frequency (VAF) to below 0.02 and up to 10.5% depending on the mutation analysed. In patient 1108 who lacked an LAIP, the VAF for RUNX1 was reduced to 0.79% after cycle 1. FLT3‐TKD was observed in two of four patients with mutated RUNX1, and the FLT3‐mutated clone was cleared <0.05% (corresponding to an MRD <0.1%) in both patients after cycle 2 (Fig. 2).
Fig. 2.
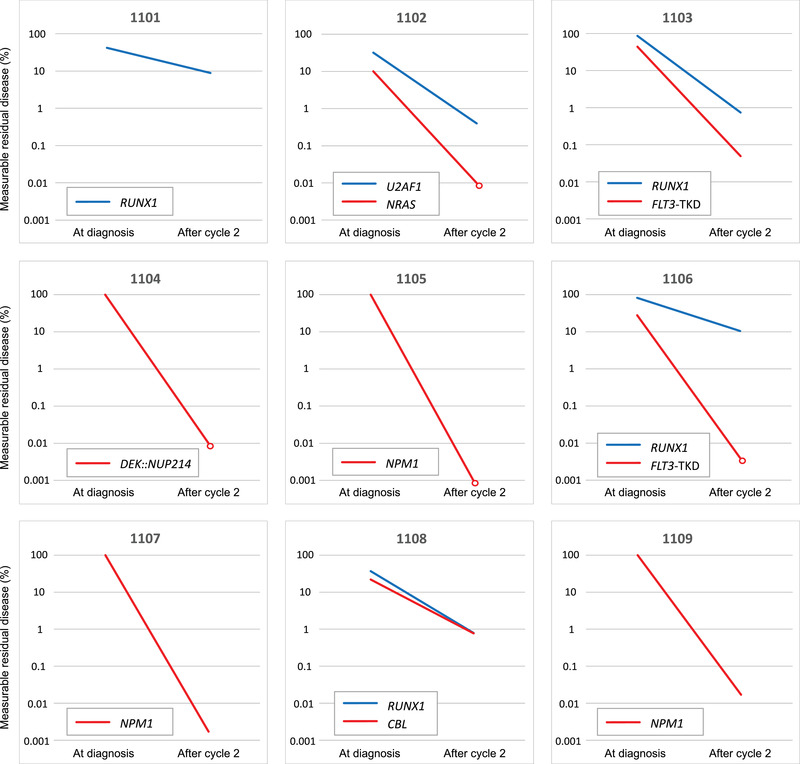
Leukemic clearance after cycle 2. Validated real‐time quantitative polymerase chain reaction (RT‐qPCR) markers for NPM1 and DEK::NUP214, respectively, were analysed in four patients, and all showed mutational clearance <0.1% (relative to the diagnostic level) after cycle 2. In the remaining five patients, one or two mutations were monitored for measurable residual disease (MRD) using deep sequencing (reporting the variant allele frequency). Mutation in RUNX1 was seen in four patients and was reduced to 0.74%–10.5% after cycle 2. In two of four patients with RUNX1, a FLT3‐TKD was also present, but was reduced below 0.05% after two cycles of treatment. After cycle 2, MRD levels below the detection limit for the method are indicated with open circles.
Patient 1107 showed a low‐level MRD positivity for NPM1 at month nine. However, resampling could not confirm detectable MRD. While the confirmatory results were pending, the patient received azacitidine and venetoclax, and received in total three cycles as additional consolidation.
Allo‐HSCT was performed in six of nine patients, after cycle 2 (n = 2), cycle 3 (n = 2) or after cycle 2 with one cycle of azacitidine as bridging (n = 1; patient 1108).
The median follow‐up was 18.0 (range 14.9–20.5) months. One elderly patient with adverse risk and not fit for HSCT relapsed after 11.9 months. The patient initially only received three cycles of chemotherapy, with cycle 3 being dose‐reduced. At the time of relapse, the patient received azacitidine–venetoclax–gilteritinib and is now in second complete remission (Fig. 1).
Enhanced ara‐CTP levels in leukaemic blasts in vivo and increased sensitivity to ara‐C ex vivo
Protein expression levels of SAMHD1 in diagnostic bone marrow biopsies varied with low (<25%), intermediate (25‐75%) and high expression (>75%) in five, three and one patient, respectively (Table 1 and Fig. 3a). At remission, SAMHD1 was largely absent particularly in CD34+ HSPCs (with the exception of histiocytes and reactive lymphocytes) (Fig. 3b). In the absence of hydroxyurea, there was no significant association of SAMHD1 expression levels with ara‐CTP levels in vivo (n = 9, p = 0.83) or ara‐C IC50 values ex vivo (n = 7; p = 0.13) (Fig. S2).
Fig. 3.
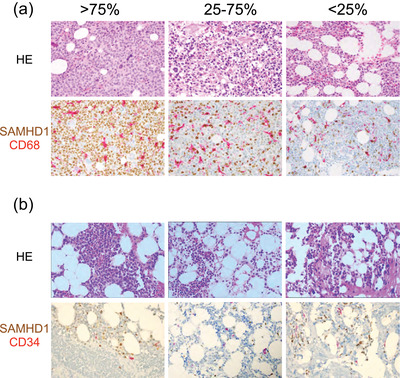
SAMHD1 protein expression in acute myeloid leukaemia (AML) blasts and in remission bone marrow. (a) Upper images show haematoxylin and eosin routine staining in diagnostic bone marrow biopsies. The lower images show the corresponding SAMHD1‐CD68 double immunohistochemical staining. SAMHD1 (in brown) is mainly expressed in the nucleus of the AML blasts, while CD68+ macrophages (magenta) are strongly positive for SAMHD1 and serve as an internal positive control. Low, intermediate and high expression levels were defined as <25%, 25%–75% and >75% positive cells, respectively, as previously described [21]. (b) Upper images show haematoxylin–eosin routine staining in remission bone marrow biopsies from patients 1105 (left), 1101 (middle) and 1104 (right), respectively, with cellularity within normal range and morphology suggestive of reactive bone marrow. Lower images show SAMHD1/CD34 double immunostaining without double‐positive blasts. Occasionally, SAMHD1+/CD34− normal blasts are present. The SAMHD1+/CD34− cells are morphologically mostly histiocytes and reactive lymphocytes. All images are shown at 400× magnification.
Paired comparisons of ara‐CTP levels in peripheral blood mononuclear cells (PBMCs) [41, 42, 43, 44, 45] treated with or without hydroxyurea showed no significant differences at a dose of 500 mg (n = 3; p = 0.45, Fig. S3). The median percentage of blasts in the PBMC fraction was 40% (range 0%–92% [Table S4]). At a dose of 1 g hydroxyurea (given either prior to the first or the second ara‐C infusion to control for increases in ara‐CTP that might stem from accumulation rather than effects of hydroxyurea), median ara‐CTP levels increased to 150% (n = 6; p = 0.04, range 100%–311%; Fig. 4). In addition, we measured ara‐CTP levels in bone marrow‐derived blasts ex vivo treated with ara‐C plus/minus hydroxyurea. A modest but significant increase of ara‐CTP could be detected (n = 8; p = 0.02, range 99%–139%; Fig. S4). Bone marrow–derived MNCs from seven patients could be subjected to ex vivo drug testing. The addition of hydroxyurea decreased the IC50 values of ara‐C by a median factor of 2.1 (p = 0.0047, range 1.6–8.3; Fig. 4). Similar results were obtained for gemcitabine, another non‐competitive inhibitor of RNR, with a factor of 1.6 (p = 0.01, range 1.3–5.4, Fig. S5). As a comparison, hydroxyurea decreased IC50 values of ara‐C by a factor of 8.9 in the SAMHD1‐positive THP‐1 cell line but had no effect in a SAMHD1‐knockout THP‐1‐derivative or in healthy CD34+ bone marrow cells (Fig. S6). The median ZIP in patient cells as another measure of ara‐C synergy with hydroxyurea and gemcitabine was 2.6 and 3.6, respectively, in line with the ZIP of 3.3 and 4.7 in the AML cell line THP‐1 (Fig. S7).
Fig. 4.
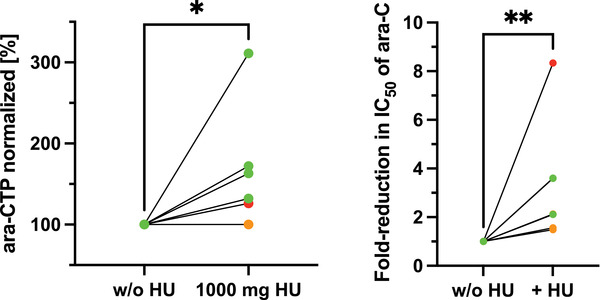
Effect of hydroxyurea on peak ara‐C triphosphate (ara‐CTP) levels in circulating mononuclear cells and ex vivo sensitivity to ara‐C. The left panel shows ara‐CTP levels measured in circulating mononuclear cells without hydroxyurea (w/o HU) as compared to ara‐CTP with hydroxyurea 1000 mg taken p.o. 1 h prior to start of ara‐C infusion, normalised to without hydroxyurea (n = 6). The right panel shows the fold reduction in IC50 values of ara‐C in diagnostic bone marrow mononuclear cells treated ex vivo in the presence or absence of 100 μM hydroxyurea, normalised to the absence of hydroxyurea (n = 7). Individual dots correspond to individual patients; colours represent levels of SAMHD1 expression at diagnosis (green, <25%, orange 25%–75% and red >75%).
Discussion
Based on preclinical data suggesting that the addition of hydroxyurea potentiates cytarabine efficacy by increasing ara‐CTP levels through inhibition of SAMHD1, this trial was designed to assess the feasibility from a toxicity perspective and to investigate the efficacy of such addition of hydroxyurea to conventional daunorubicin/ara‐C (DA)‐based chemotherapy. In parallel, the effects of hydroxyurea on ara‐CTP pharmacokinetics and ex vivo ara‐C sensitivity were assessed to evaluate the study rationale translationally [24, 27, 28]. The nine patients were representative of the general AML population eligible for intensive chemotherapy, with a median age of 48 years (range 26–76), albeit with a skewing towards adverse risk. Partially, this skewing can be explained by exclusion of CBF‐AML. Adverse genetics according to ELN were seen in five of nine (56%) patients, and FLT3‐mutation was present in five of nine (56%). The corresponding figures according to the Swedish population‐based AML registry were 30%–40% and 25%–30%, respectively [46].
It is conceivable that the addition of hydroxyurea potentiates the risk of ara‐C‐related adverse events. However, the observed toxicities and time to haematopoietic recovery were in line with expected outcomes for patients undergoing intensive chemotherapy for AML [47] and did not delay subsequent cycles of chemotherapy or time to allo‐HSCT. This might at least partially be explained by the low expression levels of SAMHD1 in healthy bone marrow progenitors including HSPC. The lack of excess myelosuppression together with our previous reports on the negative role of SAMHD1 during ara‐C‐based consolidation courses [19, 21] hence justifies the addition of hydroxyurea even in post‐remission treatments. Two patients experienced palmar–plantar erythrodysesthaesia, a well‐known side effect of ara‐C. Both patients were on concomitant midostaurin, and both were able to continue treatment according to protocol (four cycles and three cycles + allo‐HSCT, respectively). Toxicities were consistent with previous studies in which both hydroxyurea and ara‐C were administered at varying schedules and dosing to a heterogenous population of relapsed or refractory patients (summarised in [24]). Time to recovery of neutrophils and platelets was on par with what is expected after intensive chemotherapy according to the Swedish AML Registry.
Albeit the limitations of the small phase 1 cohort, the efficacy appeared highly promising, with all nine patients achieving CR, including five of five with high‐risk genetics. All eight patients evaluable for MRD assessment with flow cytometry or RT‐qPCR reached negativity (defined as <0.1%) after cycle 2. According to data from the Swedish AML Registry, the expected CR/MRD‐negativity rates with an identical chemotherapy regimen but without hydroxyurea is 92%/80% in favourable‐risk, 84%/64% in intermediate‐risk and 71%/60% in adverse‐risk patients [48]. The treatment efficacy will be further evaluated in the phase 2 extension part of the study, where an additional 60 patients with AML will be included. Addition of midostaurin for FLT3‐mutated AML patients contributes to therapeutic efficacy [7] and consequently is recommended by national guidelines for patients fit for intensive chemotherapy. While this might confound interpretation of the effects of added hydroxyurea in our study, real‐world data in FLT3‐mutated patients treated with or without the addition of midostaurin did not show any difference of complete remission rates, consistent with the seminal trial [7, 49]. This argues against the notion that midostaurin would confound the early surrogate endpoints of complete remission and MRD negativity.
To gain a deeper understanding of the degree of molecular clearance after cycle 2 in the five patients lacking validated genetic MRD markers, targeted deep sequencing was performed on selected driver mutations. VAF reduction varied from five‐fold to 8000‐fold with remaining VAFs from <0.02% to 10.5%. However, selecting relevant genetic markers for MRD in AML can be challenging as they may occur in premalignant clones. The two patients with detectable driver mutations above 1% after cycle 2 (patients 1101 and 1106) had flow cytometry based MRD <0.1%. Importantly, both patients had genetic alterations characteristic for MDS‐AML [50]—RUNX1, in combination with complex karyotype including monosomy 7 and multiple mutations including RUNX1 and BCOR, respectively, where incomplete mutational clearance is often observed [51]. In both patients with RUNX1‐mutation and concomitant FLT3‐TKD (1103 and 1106), the FLT3‐mutated clone was cleared below 0.1%. Taken together, the data indicate that the leukaemic clones may be more potently eradicated than premalignant clones.
Neither absolute peak intracellular ara‐CTP levels nor ara‐C IC50 values differed significantly with respect to SAMHD1 expression, which is consistent with a previous report in primary AML cells [19]. It is a limitation of the study that no sequential ara‐CTP measurements were performed, as this would have allowed calculation of half‐life and area under the curve, both of which correlate with clinical responses whereas peak ara‐CTP levels do not [17]. However, administering 1000 mg of hydroxyurea 1 h prior to ara‐C infusion significantly increased relative peak ara‐CTP levels in circulating patient blasts by a factor of 1.5, and those results could be confirmed by treating diagnostic patient blasts with ara‐C and hydroxyurea ex vivo. As hydroxyurea was either given before the first or the second administration of ara‐C (12 h apart), we controlled for ara‐CTP accumulation caused by accumulation due to repetitive dosing of ara‐C. Furthermore, ex vivo supplementation of hydroxyurea reduced IC50 values for ara‐C by a factor of 2.1. While more studies are needed, we suggest using ex vivo drug testing rather than ara‐CTP measurements as a putative predictor for treatment benefit of ara‐C/hydroxyurea combinations. This strategy is faster, less expensive and requires at least 100‐fold fewer cells per tested condition. The latter allows testing of a wide range of concentrations, which is necessary to correctly predict synergy of ara‐C and hydroxyurea [24].
A recent publication suggests that treatment of AML with nucleoside analogues might induce differentiation through hyperactivation of ribonucleotide reductase, leading to a dNTP pool imbalance that could be further aggravated by depletion of SAMHD1 [52]. Hence, ara‐C‐mediated differentiation that is increased by SAMHD1 inhibition might be an alternative explanation for the observed efficacy to achieve MRD negativity in our trial. However, in our previous studies, we neither observed effects of SAMHD1 depletion on protein levels of RNR subunits nor on dNTP pool balances [19, 24].
In conclusion, the high rate of complete remission and MRD negativity together with the pharmacokinetic and ex vivo evidence suggest that the efficacy of cytarabine‐based AML treatment can be enhanced by the addition of hydroxyurea as a targeted inhibitor of SAMHD1. Importantly, orally administered hydroxyurea may provide a safe, inexpensive and broadly accessible strategy to improve outcome in AML. These results need to be validated in a larger patient cohort.
Conflicts of interest
T. S. is employed by Heidelberg ImmunoTherapeutics, not relevant to this work. J. I. H. is a consultant for SOBI, not relevant to this work. The other authors declare no conflicts of interest.
Author contributions
Martin Jädersten: Conceptualization; Data curation; Formal analysis; Investigation; Project administration; Supervision; Visualization; Writing – original draft; Writing – review and editing. Ingrid Lilienthal: Data curation; Formal analysis; Investigation; Methodology; Writing – original draft; Writing – review and editing. Nikolaos Tsesmetzis: Data curation; Investigation; Methodology; Writing – review and editing. Sofia Bengtzén: Investigation; Methodology; Resources; Writing – review and editing. Anna Bohlin: Methodology; Resources; Writing – review and editing. Cornelia Arnroth: Formal analysis; Investigation; Writing – review and editing. Tom Erkers: Formal analysis; Investigation; Methodology; Writing – review and editing. Brinton Seashore‐Ludlow: Data curation; Investigation; Methodology; Writing – original draft; Writing – review and editing. Géraldine Giraud: Investigation; Resources; Writing – review and editing. Giti S. Barkhordar: Investigation; Methodology; Writing – review and editing. Sijia Tao: Data curation; Formal analysis; Investigation; Methodology; Writing – review and editing. Linda Fogelstrand: Formal analysis; Investigation; Methodology; Resources; Writing – original draft; Writing – review and editing. Leonie Saft: Formal analysis; Investigation; Methodology; Writing – review and editing. Päivi Östling: Investigation; Methodology; Resources; Visualization. Raymond F. Schinazi: Investigation; Methodology; Resources; Writing – review and editing. Baek Kim: Data curation; Investigation; Methodology; Resources; Writing – original draft; Writing – review and editing. Torsten Schaller: Conceptualization; Methodology; Supervision; Writing – original draft; Writing – review and editing. Gunnar Juliusson: Investigation; Methodology; Validation; Writing – review and editing. Stefan Deneberg: Conceptualization; Investigation; Supervision; Validation; Writing – review and editing. Sören Lehmann: Methodology; Resources; Writing – original draft; Writing – review and editing. Georgios Z. Rassidakis: Formal analysis; Methodology; Visualization; Writing – original draft; Writing – review and editing. Martin Höglund: Conceptualization; Investigation; Methodology; Supervision; Writing – original draft; Writing – review and editing. Jan‐Inge Henter: Conceptualization; Investigation; Project administration; Resources; Validation; Writing – original draft; Writing – review and editing. Nikolas Herold: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Visualization; Writing – original draft; Writing – review and editing.
Supporting information
Figure S1. In silico analysis of SAMHD1 mRNA expression in healthy bone marrow progenitors and AML blasts
Figure S2. Ara‐CTP levels in circulating blasts and ex vivo sensitivity to ara‐C with respect to SAMHD1 expression
Figure S3. Effect of 500 mg hydroxyurea on peak ara‐CTP levels in circulating mononuclear cells
Figure S4. Effect of 60 µM hydroxyurea on ara‐CTP levels in diagnostic bone‐marrow mononuclear cells ex vivo
Figure S5. Effect of gemcitabine on ex vivo sensitivity to ara‐C
Figure S6. Effect of hydroxyurea on ex vivo sensitivity to ara‐C in THP‐1 cells and healthy CD34+ HPSCs
Figure S7. Zero Interaction Potency of ara‐C and hydroxyurea in diagnostic bone‐marrow mononuclear cells ex vivo and THP‐1 cells
Table S1. Antibody panels for flow cytometry
Table S2. Haematologic recovery
Table S3. Mutations assessed with deep sequencing
Table S4. White blood cell composition at time of ara‐CTP measurements
Supplementary References
Acknowledgements
The authors would like to express their gratitude to Henrik Hasle, Björn Wahlin and Per‐Ola Andersson for their critical data and safety assessment. The authors want to thank Katja Wiklund and Yvonne Larsén for monitoring the study. The authors are grateful to Janelle Cederlund for support with trial patient management and Indranil Sinha for assistance with visualization of gene expression data. This work would not have been possible without the support of the following sources: CIMED 2019 (to M. J.), Barncancerfonden (TJ2018‐0128, PR2019‐0100, TJ2021‐0080, OA2011‐0001, KP2018‐0005, PR2018‐0016, PR2020‐0077 and TJ2019‐0072 to M. L., C. A., B. S. L., T. E., P. Ö., J. I. H. and N. H.), Knut and Alice Wallenberg Stiftelse (KAW 2015.0291 to C. A., B. S. L., T. E. and P. Ö.), Stiftelsen för Strategisk Forskning (SSF SB16‐0058 to C. A., B. S. L., T. E. and P. Ö.), Swedish Research Council (2017‐06095, 2020‐01184 to C. A., B. S. L., T. E., P. Ö. and N. H.), NIH (AI162633, AI136581 and MH116695 to B. K. and R. F. S.), Cancerfonden (21 1494 Pj and CAN 2017/517 to N. H.), Jeanssons Stiftelser (to N. H.), Märta and Gunnar V Philipson Foundation (to N. H.), Region Stockholm (20200246 and K2892‐2016 to N. H.), Sjöbergstiftelsen (2020‐008 to N. H.), Radiumhemmets Forskningsfonder (191112 and 211143 to N. H.) and Svenska Läkaresällskapet (SLS‐875361 and SLS‐961737 to N. H.).
Jädersten M, Lilienthal I, Tsesmetzis N, Lourda M, Bengtzén S, Bohlin A, et al. Targeting SAMHD1 with hydroxyurea in first‐line cytarabine‐based therapy of newly diagnosed acute myeloid leukaemia: Results from the HEAT‐AML trial. J Intern Med. 2022;292:925–940.
Contributor Information
Martin Jädersten, Email: martin.jadersten@regionstockholm.se.
Nikolas Herold, Email: nikolas.herold@ki.se.
Data availability statement
The clinical trial protocol is included in the Supplementary information. Individual participant data that underlie the results reported in this article, after de‐identification (text, tables, figures and appendices), will be made available upon request.
References
- 1. Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001–2007. Blood. 2012;119(1):34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Juliusson G, Antunovic P, Derolf A, Lehmann S, Möllgård L, Stockelberg D, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood. 2009;113(18):4179–87. [DOI] [PubMed] [Google Scholar]
- 3. Juliusson G. Older patients with acute myeloid leukemia benefit from intensive chemotherapy: an update from the Swedish Acute Leukemia Registry. Clin Lymphoma Myeloma Leuk. 2011;11(Suppl 1):S54–9. [DOI] [PubMed] [Google Scholar]
- 4. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361(13):1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dinardo CD, Wei AH. How I treat acute myeloid leukemia in the era of new drugs. Blood. 2020;135(2):85–96. [DOI] [PubMed] [Google Scholar]
- 7. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borthakur G, Kantarjian H. Core binding factor acute myelogenous leukemia‐2021 treatment algorithm. Blood Cancer J. 2021;11(6):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cornelissen JJ, Van Putten WLJ, Verdonck LF, Theobald M, Jacky E, Daenen SMG, et al. Results of a HOVON/SAKK donor versus no‐donor analysis of myeloablative HLA‐identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle‐aged adults: benefits for whom? Blood. 2007;109(9):3658–66. [DOI] [PubMed] [Google Scholar]
- 10. Mayer RJ, Davis RB, Schiffer CA, Berg DT, Powell BL, Schulman P, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med. 1994;331(14):896–903. [DOI] [PubMed] [Google Scholar]
- 11. Lowenberg B. Sense and nonsense of high‐dose cytarabine for acute myeloid leukemia. Blood. 2013;121(1):26–8. [DOI] [PubMed] [Google Scholar]
- 12. Tsesmetzis N, Paulin CBJ, Rudd SG, Herold N. Nucleobase and nucleoside analogues: resistance and re‐sensitisation at the level of pharmacokinetics, pharmacodynamics and metabolism. Cancers. 2018;10(7):240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liliemark JO, Plunkett W, Dixon DO. Relationship of 1‐beta‐D‐arabinofuranosylcytosine in plasma to 1‐beta‐D‐arabinofuranosylcytosine 5'‐triphosphate levels in leukemic cells during treatment with high‐dose 1‐beta‐D‐arabinofuranosylcytosine. Cancer Res. 1985;45(11 Pt 2):5952–7. [PubMed] [Google Scholar]
- 14. Yamauchi T, Kawai Y, Kishi S, Goto N, Urasaki Y, Imamura S, et al. Monitoring of intracellular 1‐beta‐D‐arabinofuranosylcytosine 5'‐triphosphate in 1‐beta‐D‐arabinofuranosylcytosine therapy at low and conventional doses. Jpn J Cancer Res. 2001;92(5):546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Estey E, Plunkett W, Dixon D, Keating M, McCredie K, Freireich EJ. Variables predicting response to high dose cytosine arabinoside therapy in patients with refractory acute leukemia. Leukemia. 1987;1(8):580–3. [PubMed] [Google Scholar]
- 16. Plunkett W, Heinemann V, Estey E, Keating M. Pharmacologically directed design of leukemia therapy. Haematol Blood Transfus. 1990;33:610–3. [DOI] [PubMed] [Google Scholar]
- 17. Plunkett W, Iacoboni S, Estey E, Danhauser L, Liliemark JO, Keating MJ. Pharmacologically directed ara‐C therapy for refractory leukemia. Semin Oncol. 1985;12(2 Suppl 3):20–30. [PubMed] [Google Scholar]
- 18. Riva‐Lavieille C, Ressayre C, Barra Y, Carcassonne Y, Iliadis A. Calculation of individual dosage regimen of cytosine arabinoside (ara‐C) based on metabolite levels in leukemic cells. Ther Drug Monit. 1994;16(4):375–9. [DOI] [PubMed] [Google Scholar]
- 19. Herold N, Rudd SG, Ljungblad L, Sanjiv K, Myrberg IH, Paulin CBJ, et al. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nature Medicine. 2017;23(2):256–63. [DOI] [PubMed] [Google Scholar]
- 20. Herold N, Rudd SG, Sanjiv K, Kutzner J, Myrberg IH, Paulin CBJ, et al. With me or against me: tumor suppressor and drug resistance activities of SAMHD 1. Exp Hematol. 2017;52:32–9. [DOI] [PubMed] [Google Scholar]
- 21. Rassidakis GZ, Herold N, Myrberg IH, Tsesmetzis N, Rudd SG, Henter J‐I, et al. Low‐level expression of SAMHD1 in acute myeloid leukemia (AML) blasts correlates with improved outcome upon consolidation chemotherapy with high‐dose cytarabine‐based regimens. Blood Cancer J. 2018;8(11):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rudd SG, Schaller T, Herold N. SAMHD1 is a barrier to antimetabolite‐based cancer therapies. Mol Cell Oncol. 2017;4(2):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schneider C, Oellerich T, Baldauf H‐M, Schwarz S‐M, Thomas D, Flick R, et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat Med. 2017;23(2):250–5. [DOI] [PubMed] [Google Scholar]
- 24. Rudd SG, Tsesmetzis N, Sanjiv K, Paulin CBJ, Sandhow L, Kutzner J, et al. Ribonucleotide reductase inhibitors suppress SAMHD1 ara‐CTPase activity enhancing cytarabine efficacy. EMBO Mol Med. 2020;12(3):e10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herold N. Pharmacological strategies to overcome treatment resistance in acute myeloid leukemia: increasing leukemic drug exposure by targeting the resistance factor SAMHD1 and the toxicity factor Top2β. Expert Opin Drug Discov. 2021;16(1):7–11. [DOI] [PubMed] [Google Scholar]
- 26. Musiałek MW, Rybaczek D. Hydroxyurea—the good, the bad and the ugly. Genes (Basel). 2021;12(7):1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yataganas X, Strife A, Perez A, Clarkson B. Cell kill kinetics with hydroxyurea. Med Pediatr Oncol. 1976;2(1):39–54. [DOI] [PubMed] [Google Scholar]
- 28. de Montalembert M, Bachir D, Hulin A, Gimeno L, Mogenet A, Bresson JL, et al. Pharmacokinetics of hydroxyurea 1,000 mg coated breakable tablets and 500 mg capsules in pediatric and adult patients with sickle cell disease. Haematologica. 2006;91(12):1685–8. [PubMed] [Google Scholar]
- 29. Gandhi V, Estey E, Keating MJ, Plunkett W. Fludarabine potentiates metabolism of cytarabine in patients with acute myelogenous leukemia during therapy. J Clin Oncol. 1993;11(1):116–24. [DOI] [PubMed] [Google Scholar]
- 30. Ossenkoppele GJ. The value of fludarabine in addition to ARA‐C and G‐CSF in the treatment of patients with high‐risk myelodysplastic syndromes and AML in elderly patients. Blood. 2004;103(8):2908–13. [DOI] [PubMed] [Google Scholar]
- 31. Fridland A, Verhoef V. Mechanism for ara‐CTP catabolism in human leukemic cells and effect of deaminase inhibitors on this process. Semin Oncol. 1987;14(2 Suppl 1):262–8. [PubMed] [Google Scholar]
- 32. Marsh JH, Kreis W, Barile B, Akerman S, Schulman P, Allen SL, et al. Therapy of refractory/relapsed acute myeloid leukemia and blast crisis of chronic myeloid leukemia with the combination of cytosine arabinoside, tetrahydrouridine, and carboplatin. Cancer Chemother Pharmacol. 1993;31(6):481–4. [DOI] [PubMed] [Google Scholar]
- 33. Mamez A‐C, Raffoux E, Chevret S, Lemiale V, Boissel N, Canet E, et al. Pre‐treatment with oral hydroxyurea prior to intensive chemotherapy improves early survival of patients with high hyperleukocytosis in acute myeloid leukemia. Leuk Lymphoma. 2016;57(10):2281–8. [DOI] [PubMed] [Google Scholar]
- 34. Bagger FO, Kinalis S, Rapin N. BloodSpot: a database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Research. 2018;47(D1):D881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tobal K, Frost L, Liu Yin JA. Quantification of DEK‐CAN fusion transcript by real‐time reverse transcription polymerase reaction in patients with t(6;9) acute myeloid leukemia. Haematologica. 2004;89(10):1267–9. [PubMed] [Google Scholar]
- 36. Delsing Malmberg E, Rehammar A, Pereira MB, Abrahamsson J, Samuelsson T, Ståhlman S, et al. Accurate and sensitive analysis of minimal residual disease in acute myeloid leukemia using deep sequencing of single nucleotide variations. J Mol Diagn. 2019;21(1):149–62. [DOI] [PubMed] [Google Scholar]
- 37. Rosso A, Juliusson G, Lorenz F, Lehmann S, Derolf Ã, Deneberg S, et al. Is there an impact of measurable residual disease as assessed by multiparameter flow cytometry on survival of AML patients treated in clinical practice? A population‐based study. Leuk Lymphoma. 2021;62(8):1973–81. [DOI] [PubMed] [Google Scholar]
- 38. Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, Szwajda A, et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013;3(12):1416–29. [DOI] [PubMed] [Google Scholar]
- 39. Yadav B, Wennerberg K, Aittokallio T, Tang J. Searching for drug synergy in complex dose‐response landscapes using an interaction potency model. Comput Struct Biotechnol J. 2015;13:504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Potdar S, Ianevski A, Mpindi J‐P, Bychkov D, Fiere CM, Ianevski P, et al. Breeze: an integrated quality control and data analysis application for high‐throughput drug screening. Bioinformatics. 2020;36(11):3602–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamauchi T, Ueda T, Nakamura T. A new sensitive method for determination of intracellular 1‐beta‐D‐arabinofuranosylcytosine 5'‐triphosphate content in human materials in vivo. Cancer Res. 1996;56(8):1800–4. [PubMed] [Google Scholar]
- 42. Yamauchi T, Negoro E, Kishi S, Takagi K, Yoshida A, Urasaki Y, et al. Intracellular cytarabine triphosphate production correlates to deoxycytidine kinase/cytosolic 5′‐nucleotidase II expression ratio in primary acute myeloid leukemia cells. Biochem Pharmacol. 2009;77(12):1780–6. [DOI] [PubMed] [Google Scholar]
- 43. Gandhi V, Xu YZ, Estey E. Accumulation of arabinosyluracil 5'‐triphosphate during arabinosylcytosine therapy in circulating blasts of patients with acute myelogenous leukemia. Clin Cancer Res. 1998;4(7):1719–26. [PubMed] [Google Scholar]
- 44. Plunkett W, Hug V, Keating MJ, Chubb S. Quantitation of 1‐beta‐D‐arabinofuranosylcytosine 5'‐triphosphate in the leukemic cells from bone marrow and peripheral blood of patients receiving 1‐beta‐D‐arabinofuranosylcytosine therapy. Cancer Res. 1980;40(3):588–91. [PubMed] [Google Scholar]
- 45. Estey E, Plunkett W, Gandhi V, Rios MB, Kantarjian H, Keating MJ. Fludarabine and arabinosylcytosine therapy of refractory and relapsed acute myelogenous leukemia. Leuk Lymphoma. 1993;9(4–5):343–50. [DOI] [PubMed] [Google Scholar]
- 46. Juliusson G, Jädersten M, Deneberg S, Lehmann S, Möllgård L, Wennström L, et al. The prognostic impact of FLT3‐ITD and NPM1 mutation in adult AML is age‐dependent in the population‐based setting. Blood Adv. 2020;4(6):1094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marras T, Dettori M, Caocci G, La Nasa G, Sotgiu G, Saderi L, et al. White blood cell count nadir and duration of aplasia do not associate with treatment outcome in adult patients with acute myeloid leukemia undergoing intensive chemotherapy. Chemotherapy. 2020;65(3–4):110–4. [DOI] [PubMed] [Google Scholar]
- 48. Juliusson G, Hagberg O, Lazarevic VLJ, Ölander E, Antunovic P, Cammenga J, et al. Improved survival of men 50 to 75 years old with acute myeloid leukemia over a 20‐year period. Blood. 2019;134(18):1558–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bazzell BG, Marini BL, Benitez LL, Bixby D, Burke P, Pettit K, et al. Real world use of FLT3 inhibitors for treatment of FLT3+ acute myeloid leukemia (AML): a single center, propensity‐score matched, retrospective cohort study. J Oncol Pharm Pract. 2021. 10.1177/10781552211020815. [DOI] [PubMed] [Google Scholar]
- 50. Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jongen‐Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189–99. [DOI] [PubMed] [Google Scholar]
- 52. Wang H, He X, Zhang L, Dong H, Huang F, Xian J, et al. Disruption of dNTPs homeostasis by ribonucleotide reductase hyperactivation overcomes AML differentiation blockade. Blood. 2022:3752–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. In silico analysis of SAMHD1 mRNA expression in healthy bone marrow progenitors and AML blasts
Figure S2. Ara‐CTP levels in circulating blasts and ex vivo sensitivity to ara‐C with respect to SAMHD1 expression
Figure S3. Effect of 500 mg hydroxyurea on peak ara‐CTP levels in circulating mononuclear cells
Figure S4. Effect of 60 µM hydroxyurea on ara‐CTP levels in diagnostic bone‐marrow mononuclear cells ex vivo
Figure S5. Effect of gemcitabine on ex vivo sensitivity to ara‐C
Figure S6. Effect of hydroxyurea on ex vivo sensitivity to ara‐C in THP‐1 cells and healthy CD34+ HPSCs
Figure S7. Zero Interaction Potency of ara‐C and hydroxyurea in diagnostic bone‐marrow mononuclear cells ex vivo and THP‐1 cells
Table S1. Antibody panels for flow cytometry
Table S2. Haematologic recovery
Table S3. Mutations assessed with deep sequencing
Table S4. White blood cell composition at time of ara‐CTP measurements
Supplementary References
Data Availability Statement
The clinical trial protocol is included in the Supplementary information. Individual participant data that underlie the results reported in this article, after de‐identification (text, tables, figures and appendices), will be made available upon request.