

Abstract
Purpose of the review:
Over the past decade, lipoprotein(a) [Lp(a)] made it to several consensus and guideline documents. This review aims to summarize the literature which underlies the various recommendations and compares recent European and North American consensus and guideline documents of the recent 3–4 years.
Recent findings:
Multiple large epidemiological and genetic studies have provided strong evidence for a causal association between Lp(a) concentrations and atherosclerotic cardiovascular disease (ASCVD) and aortic valve stenosis. There is a dose-dependent linear relationship between Lp(a) and ASCVD risk advocating to consider Lp(a) on a continuous scale rather than using thresholds. The best way to implement this in the clinic is by individualizing the Lp(a)-related risk using tools such as the “Lp(a) risk calculator” (http://www.lpaclinicalguidance.com) that takes into account the Lp(a) level in the context of an individual’s traditional risk factors and global risk for ASCVD. There is growing agreement across the guidelines regarding the clinical utility of measuring Lp(a) and more recent expert groups advocate for a general screening approach applied to all adults. As long as the cardiovascular outcomes trials for specific Lp(a)-lowering drugs are in progress, the current management of patients with high Lp(a) should focus on the comprehensive management of all other modifiable ASCVD risk factors which can be therapeutically addressed as per guideline recommendations.
Summary:
Since the contribution of high Lp(a) concentrations to global ASCVD risk has been underestimated in the past, a clear recommendation to measure Lp(a) at least once in a person’s lifetime is imperative. Recent expert consensus recommendations provide clinicians with direction on how to manage the excess risk associated with elevated Lp(a) concentration by comprehensive and individualized management of modifiable ASCVD risk factors while awaiting the results of clinical trials of Lp(a) targeted therapies.
Keywords: Lp(a), atherosclerotic cardiovascular disease, consensus, guidelines, risk factor
Introduction
When reviewing the various clinical guidelines and consensus documents which consider Lp(a), one might get the impression of being unable to see the forest for the trees. This depends very much whether the documents include considerations of Lp(a) as only one aspect out of many related to lipid metabolism or whether the main focus is on Lp(a); which society from which part of the world with its socioeconomic background has written the document; and - probably most important - when the document was written. This paper reviews the most recent evidence which has resulted in the newest Lp(a) consensus papers and explains how and why some of the recommendations might be different compared to other statements.
Epidemiological and genetic evidence on Lp(a) as a risk factor for ASCVD
During the last 5–10 years the number of publications and the extent of knowledge on Lp(a) has increased tremendously, mainly due to the very large epidemiological and genetic studies with more than 100.000 individuals that have evaluated Lp(a) and risk of ASCVD. The most recent 2022 consensus paper from the European Atherosclerosis Society used these very large studies in order to refine recommendations on how to incorporate Lp(a) into daily patient management [1*].
What endpoints are related to Lp(a)?
An association between high Lp(a) concentrations and coronary heart disease has already been recognized since approximately 40 years. During the last decade several studies also reported associations with aortic valve stenosis and calcification, a disease still lacking an effective treatment apart from valve replacement (either surgically or catheter-based). Comparative studies demonstrated the strongest association of Lp(a) with myocardial infarction and aortic valve stenosis. Higher concentrations were found to be required for significant associations with stroke, peripheral arterial disease or heart failure [1*–4**]. Most studies did not find associations with non-cardiovascular diseases [5]. Despite a wide array of in vitro studies on the potential thrombogenic nature of Lp(a), large observational and genetic studies could not substantiate any evidence that genetically increased Lp(a) concentrations are associated with venous thromboembolism [1*, 6]. This, however, does not exclude a potential impact of Lp(a) on arterial thrombogenic potential, as attested by the observation that (very) high Lp(a) associates with early arterial stroke in children [7–9]. A meta-analysis included in the recent EAS Lp(a) consensus paper supported that lifelong very low Lp(a) concentrations may associate with future risk of diabetes; the mechanism behind is currently not well understood [1*].
Is the association between Lp(a) and outcomes continuous or is there a risk threshold?
Studies with hundreds of thousands of participants have paved the way for a much clearer picture on Lp(a) as a causal risk factor for ASCVD. Studies with approximately 1000 participants were considered large in the 1990s. These studies often allowed only a dichotomous comparison of groups with high versus low Lp(a) concentrations resulting in thresholds of Lp(a) above which the ASCVD risk was increased. The 2010 EAS Lp(a) consensus paper introduced the 50 mg/dL threshold, which simply reflected the 80th percentile in the Danish population (Figure 1A). Since then these two thresholds were often used in clinical practice. A stepwise increase in the sample size of studies has now resulted in studies with 100,000 and >400,000 subjects as seen in the Copenhagen studies and the UK Biobank, respectively, which has tremendously increased the statistical power. From these data we observe with increasing Lp(a) concentrations a continuous increase in risk rather than a threshold effect (Figure 1B) [1*, 10]. From a biological standpoint a threshold is also not plausible.
Figure 1.
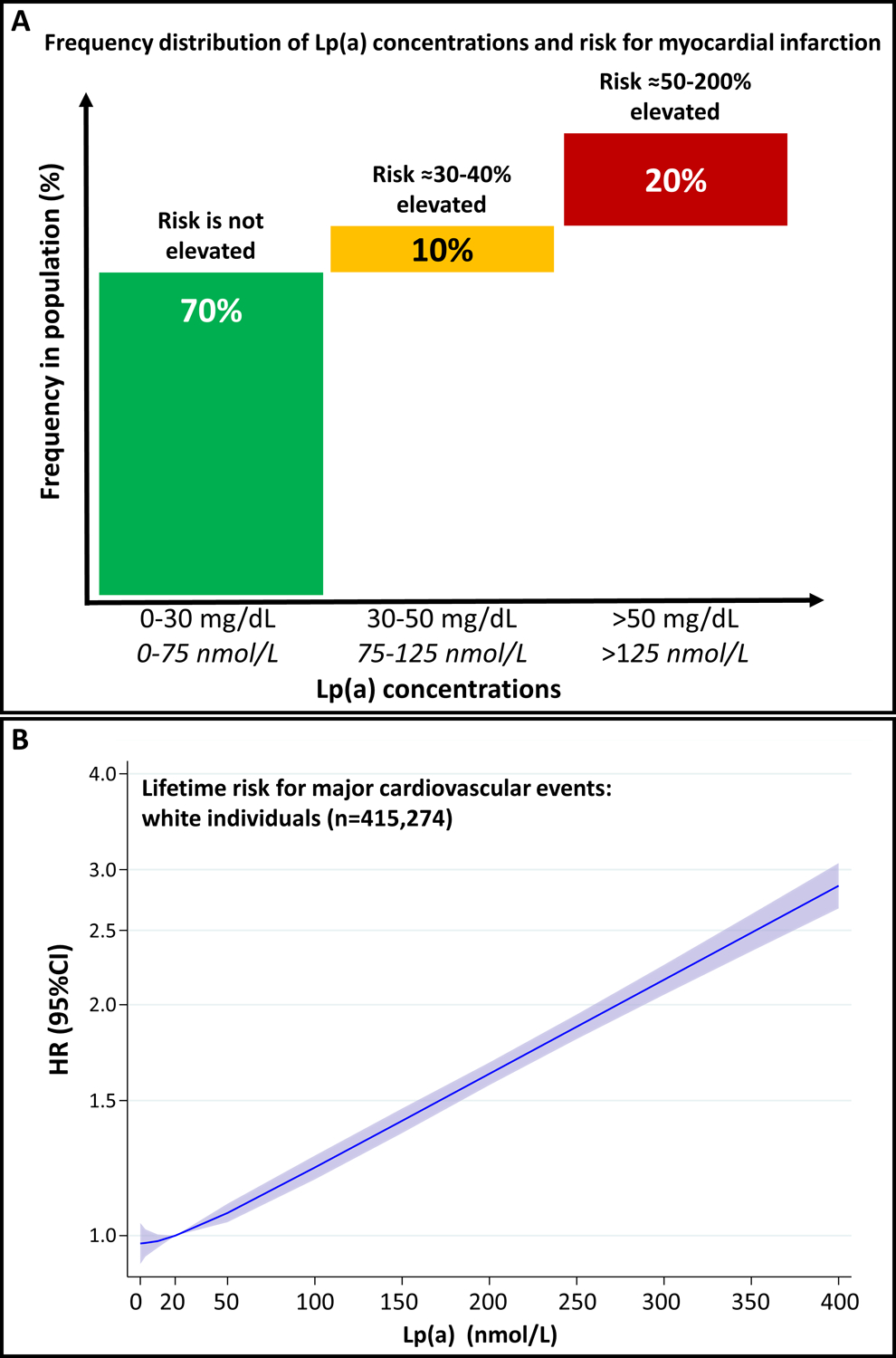
Panel A: Frequency distribution of Lp(a) concentrations and risk for myocardial infarction. Data are derived and extrapolated from Kamstrup et al. [13]. Reproduced with permission of Florian Kronenberg. Panel B: Data from the UK Biobank show the linear relationship of Lp(a) concentration with risk for major cardiovascular events in White individuals. Given are the smoothed adjusted hazard ratio (HR) and 95% confidence interval (95%CI) for lifetime risk for major cardiovascular events for a given Lp(a) concentration relative to the median Lp(a) in the population (19.7 nmol/L). These data were estimated using a Cox proportional hazards regression model adjusted for age at enrolment, sex, and the first 10 principle components of ancestry and modelled using cubic natural splines. Figure is taken with permission from the recent EAS Lp(a) Consensus paper and is based on data from the UK Biobank provided by Prof. Brian Ference and Prof. Alberico L. Catapano [1*].
Conversely, clinical practice prefers the use of thresholds for therapeutic decisions; hence, the selection of a threshold for increased risk very much depends on considerations which amount of risk increase is seen as clinically relevant. For example, compared to individuals with an Lp(a) concentration of 7 mg/dL (median of a typical White population of the UK Biobank), individuals with 30 mg/dL and 50 mg/dL have a 1.22- and 1.40-fold increased risk for ASCVD. Individuals with 100 mg/dl and 150 mg/dL have a 1.95 and 2.72-fold increased risk, respectively [1]. This tremendous increase in risk was not only seen in White but also in Black as well as Asian individuals [1, 10] making measurement of Lp(a) concentrations of global relevance.
Is Lp(a) causally associated with ASCVD?
It has been a long-lasting debate whether Lp(a) is a causal risk factor for ASCVD or whether the association of high Lp(a) with ASCVD is only based on reverse causation. Early genetic studies based on apo(a) isoforms [11] and later studies based on K-IV repeats [12] or sum of K-IV repeats [13] and single nucleotide polymorphisms (SNP) [1*, 14] have provided convincing evidence for a causal relationship. These genetic studies have resurrected the Lp(a) field and encouraged the development of Lp(a)-lowering drugs [15]. The so-called Mendelian randomization studies follow the idea that genetic variants which are associated with high Lp(a) concentrations are also associated with ASCVD outcomes in case Lp(a) is causally related to the outcome [16]. This has been repeatedly demonstrated with different Lp(a)-increasing variants (Figure 2A). Besides the apo(a) size polymorphism, SNPs such as rs10455872 or rs3798220 described by Clarke and colleagues [14] and also other variants [17] were shown to be associated with ASCVD. We discussed recently [18*] that most of them are not causally related with Lp(a) concentrations but are in linkage disequilibrium with isoforms of certain K-IV repeat numbers or other causally related variants [18*]. On the other hand, rare genetic variants which result in loss of function [19, 20] or certain very common splice sites variants [21, 22**] with pronounced Lp(a)-lowering effects, were found to protect people against the development of ASCVD (Figure 2B). The last piece in the puzzle that is currently missing, are clinical outcomes trials which demonstrate that specific lowering of Lp(a) also results in lowering the risk of ASCVD events. RNA-targeting therapies are under trial and have been shown to lower Lp(a) up to more than 80% [23, 24*, 25*]. But as usual in a puzzle with the last missing piece, we are inclined to predict, based on the genetic data, what the final picture will look like.
Figure 2:
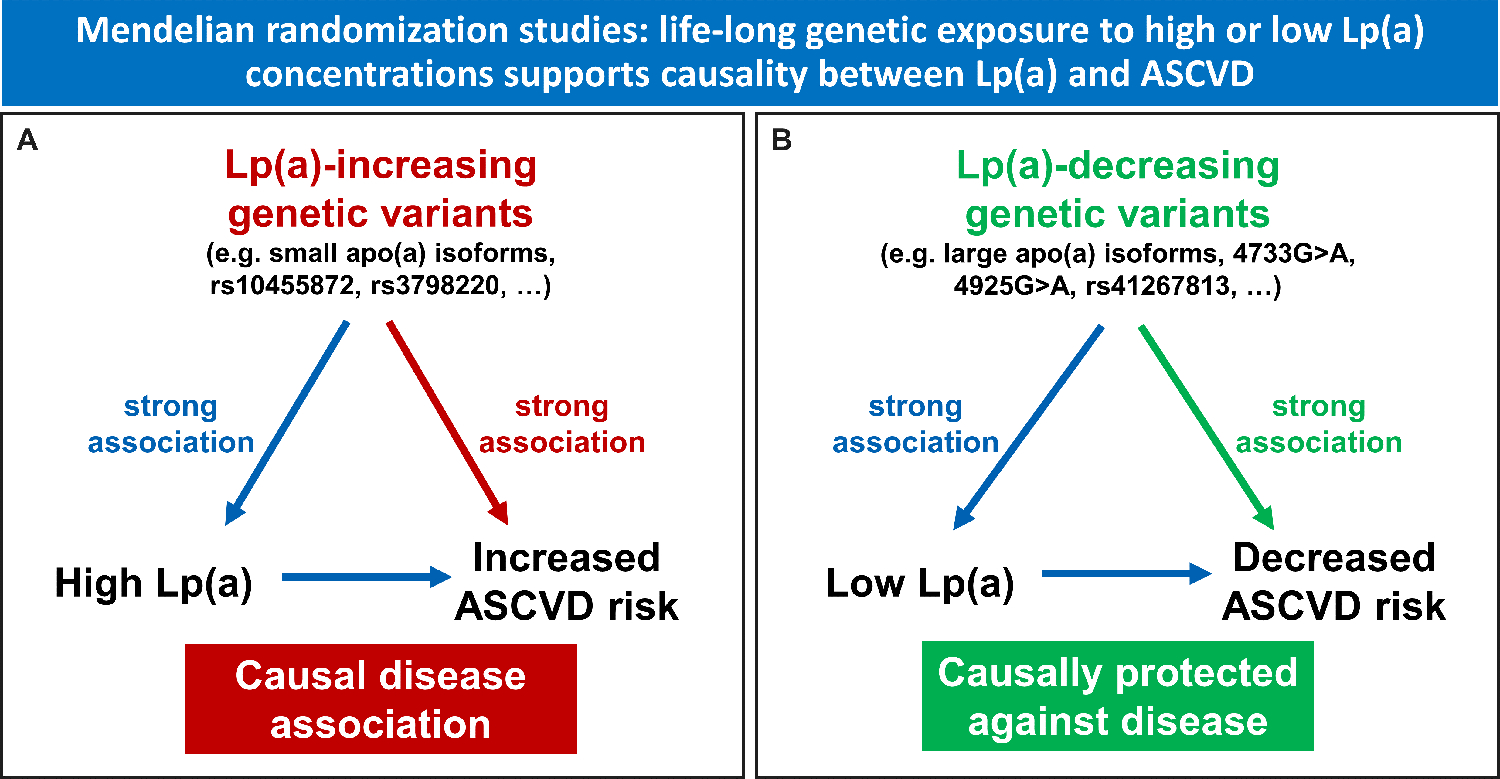
Principle of Mendelian randomization studies demonstrating that a lifelong genetic exposure to high or low Lp(a) concentrations supports causality between Lp(a) concentrations and atherosclerotic cardiovascular disease (ASCVD). Panel A shows Lp(a)-increasing variants such as small apo(a) isoforms characterized by a low number of K-IV repeats [11, 12], or a low sum of K-IV repeats of both alleles [13] or single nucleotide polymorphisms such as rs10455872 and rs3798220 [14] which show a pronounced association with high Lp(a) concentrations are also significantly associated with ASCVD outcomes. In this case the association between Lp(a) concentrations and ASCVD is strongly supported to be causal. Panel B illustrates the Lp(a)-decreasing variants such as large apo(a) isoforms or the splice site variants 4733G>A [22**] and 4925G>A [21] within the kringle-IV type 2 or the missense variant rs41267813 are associated with low Lp(a) and concentrations and a lower ASCVD risk supporting a protective role of low Lp(a) concentrations against ASCVD.
How to incorporate Lp(a) in clinical practice?
A primary requisite to incorporate Lp(a) in clinical practice is to start to measure Lp(a) and make use of the knowledge we have gained about Lp(a) as a risk factor for ASCVD.
Start by measuring Lp(a)
Earlier guidelines or consensus statements on Lp(a) had quite complicated rules in whom and when Lp(a) should be measured (Table 1). They recommended that Lp(a) should be measured once in all subjects at intermediate or high risk of CVD/CHD followed by a large number of conditions [26–28]. This has changed with the 2019 ESC/EAS dyslipidemia guidelines which made it much easier to follow the simpler recommendation which was that “Lp(a) measurement should be considered at least once in each adult person’s lifetime” [29]. The Canadian Cardiovascular Society in 2021 even added that this should be done “as a part of the initial lipid screening” [30]. The 2022 EAS Lp(a) consensus paper is in line with the ESC/EAS dyslipidemia guideline and recommends measuring Lp(a) at least once in an adult’s lifetime [1*].
Table 1:
Comparison of the most recent Guideline and Consensus papers in which Lp(a) has been addressed.
| 2022 EAS Lp(a) consensus [1*] | 2021 Canadian Cardiovascular Society Dyslipidemia Guidelines [30] | 2019 ESC/EAS dyslipidemia guidelines [29] | 2019 HEART UK consensus statement on Lp(a) [28] | 2019 ACC/AHA Guideline Primary Prevention of CVD [42] | 2019 Scientific Statement of the National Lipid Association [27, 36]# | |
|---|---|---|---|---|---|---|
| In whom should Lp(a) be measured? |
|
All individuals | In each adult person. |
|
A relative indication for its measurement is family history of premature ASCVD | Testing is reasonable in:
Testing may be reasonable to:
|
| How often? | At least once to identify those with high cardiovascular risk | Once in a person’s lifetime as a part of the initial lipid screening |
At least once in a person’s lifetime | Once, unless a secondary cause of Lp(a) elevation is suspected or specific treatment is instituted | ||
| Assay considerations |
|
|
|
|||
| Risk threshold |
|
180 mg/dL: often misunderstood as threshold but it is considered as a risk equivalent of heterozygous FH |
Risk increase: Minor: 32–90 nmol/L; Moderate: 90–200 nmol/L High: 200–400 nmol/L Very high: >400 nmol/L |
Lp(a) ≥50 mg/dL or ≥125 nmol/L constitutes a risk- enhancing factor, especially at higher levels of Lp(a) | Lp(a) ≥50 mg/dL or ≥125 nmol/L | |
| Management of patients with high Lp(a) |
|
|
In case of Lp(a) >90 nmol/L):
|
In case of Lp(a) >50 mg/dL:
|
This document has been reprinted in 2022 due to errors in the references but without any changes in the content from the 2019 document [27].
High risk = Individuals with clinical ASCVD including those with MI, ACS, stable or unstable angina, coronary or other arterial revascularization, stroke, transient ischemic attack, or peripheral artery disease including aortic aneurysm, all of atherosclerotic origin.
Very high risk = Individuals with a history of multiple major ASCVD events or 1 major ASCVD event and multiple high-risk conditions.
Abbreviations:ASCVD, atherosclerotic cardiovascular disease; FH, familial hypercholesterolemia; FRS, Framingham Risk Score;
One of the most widely argued reasons why physicians do not measure Lp(a) is “why should I measure Lp(a) when I cannot lower it?”. However, this would also be valid for other factors such as age, sex, ethnicity, HDL cholesterol, and other factors which we nevertheless take into account in the estimation of risk. Genetic factors in terms of polymorphisms or polygenic risk scores will become more and more important in the upcoming decade, although we cannot yet modify them. One of the pillars of precision or personalized medicine is to account for differences in people’s genes, environments, and lifestyles resulting in a more precise prediction, prevention and treatment of diseases. Neglecting a frequent and potent risk factor such as high Lp(a) will invariably result in major misclassifications of cardiovascular risk [31, 32*].
How to incorporate the knowledge on Lp(a) in clinical risk estimation
As we showed in the newest EAS Lp(a) consensus document, the risk attributable to Lp(a) which contributes to the global ASCVD risk can be tremendous. Figure 3 clearly demonstrates that with increasing Lp(a) concentrations the risk for ASCVD increase 1.22-fold, 1.40-fold, 1.65-fold, 1.95-fold and 2.72-fold for individuals who have Lp(a) concentrations of 30, 50, 75, 100 and 150 mg/dL, respectively, when compared to individuals who have 7 mg/dL (median of a White population). This relative increase is the same in each estimated baseline lifetime risk category based on traditional ASCVD risk factors. For example (see Figure 3), if a person has a baseline estimated lifetime risk for 10% and has an Lp(a) concentration of 75 mg/dL, the risk increases by further 6.5% to 16.5% compared to a person who has Lp(a) concentration below the median of 7 mg/dL. The risk increases by 43.1% to 68.1% in a person with a 25% baseline risk and an Lp(a) concentration of 150 mg/dL. The conclusions drawn from Figure 3 are the following: firstly, if the Lp(a) concentration is not measured and included in the risk estimation, the absolute risk might be underestimated substantially in case of high and very high Lp(a) concentrations. Secondly, a measured Lp(a) concentrations has always to be seen in the context of the other risk factors of an individual since the absolute global risk for a person with e.g. 50 mg/dL is different depending on the other risk factors: if the baseline risk without Lp(a) is only 5%, the 50 mg/dL Lp(a) increase the risk to 7%. However, if the baseline risk is 25%, the 50 mg/dL Lp(a) increase the absolute global risk to almost 35%.
Figure 3:
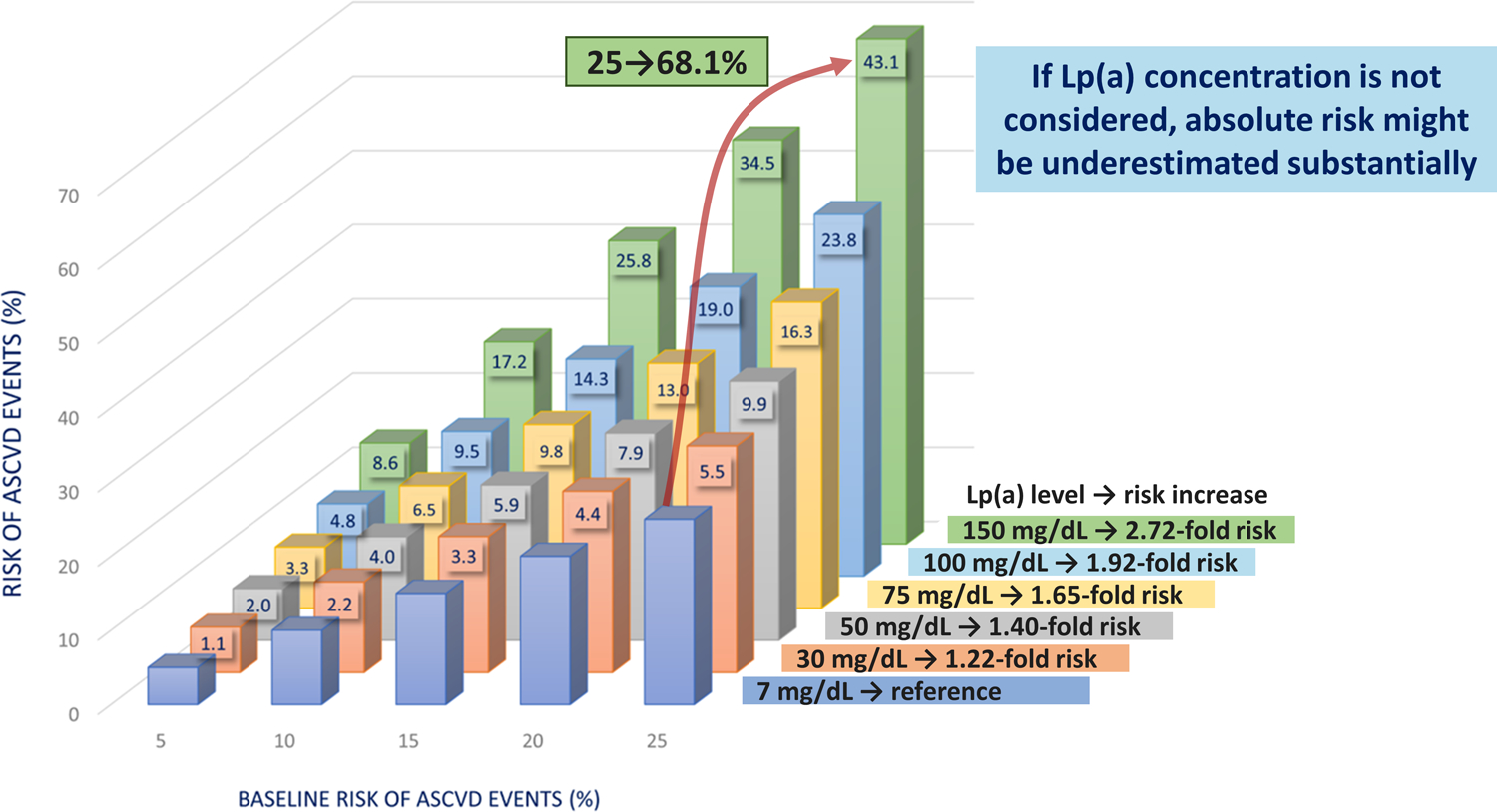
This Figure shows the estimated remaining lifetime risk of a major atherosclerotic cardiovascular events (ASCVD) among 415,274 participants of European ancestry in the UK Biobank. Participants are divided into categories of baseline estimated lifetime risk (5%, 10%, 15%, 20%, and 25%) calculated using the Joint British Societies (JBS3) Lifetime Risk Estimating algorithm (derived from a similar UK population). Within each baseline risk category, participants are then further divided into categories defined by baseline measured Lp(a) concentration. The incremental increase in risk caused by higher Lp(a) concentrations from 30 to 150 mg/dL (75 from 375 nmol/L) was estimated by adding Lp(a) as an independent exposure to the JBS3 risk estimating algorithm. The numbers at the upper end of each bar represent the increment of increased absolute risk above the estimated baseline risk caused by Lp(a). For example, for a person with a baseline risk of 25% and an Lp(a) concentration of 150 mg/dL the absolute risk of a major cardiovascular event increases by 43.1% to 68.1% (versus a person with an Lp(a) of 7 mg/dL). Figure is taken and adapted with permission from the recent EAS Lp(a) Consensus paper and is based on data from the UK Biobank provided by Prof. Brian Ference and Prof. Alberico L. Catapano [1*].
What can be done in case of a high Lp(a) concentration?
The worst recommendation would be to willingly neglect the information. As long as we have no specific Lp(a)-lowering therapies (and even thereafter), physicians should advise patients with high Lp(a) concentrations to reduce their other risk factors as strictly as possible along the lines of the various guidelines [1*]. One of the best indications from observational studies came from the population-based EPIC-Norfolk Study which followed more than 14,000 study participants for 11.5 years [33]. In this project, a so-called “cardiovascular health score” of seven therapeutically modifiable variables was formed for each participant, including body mass index, healthy diet, physical activity, smoking status, high blood pressure, diabetes and cholesterol concentration. When participants with Lp(a) concentrations above 50 mg/dL were then stratified into three groups with ideal, moderate and poor “cardiovascular health score”, those participants with a low number of risk factors had only about one third of cardiovascular risk for the subsequent 11.5 years compared to those with a high number of risk factors, despite all participants having an Lp(a) concentration above 50 mg/dL with very similar median concentrations in each of the three groups [33]. Based on the even larger UK Biobank, an “Lp(a) risk calculator” (http://www.lpaclinicalguidance.com) has been prepared in the context of the EAS Lp(a) consensus paper [1*] which calculates the ASCVD risk based on traditional risk factors once without considering Lp(a) and thereafter with considering Lp(a) concentrations. Interestingly, the algorithm also takes into account how the global ASCVD risk decreases when the patient reduces his LDL cholesterol or blood pressure for a given number. Based on these calculations it has been extrapolated that the start of an early intervention is key to prevent events: the later these modifiable risk factors are treated, the more intensified the treatment has to be. This calculator can be used to illustrate patients (and doctors) how the global risk can be mitigated if recommendations for therapeutic interventions are followed. Of course, in case the main part of the global ASCVD risk is attributable to very high Lp(a), the other risk-reducing measures will be important but insufficient to substantially lower the entire global risk. For those situations, we urgently require specific Lp(a)-lowering drugs. Meanwhile, in some countries lipoprotein apheresis is available for use in selected patients with high Lp(a) and progressive cardiovascular disease despite optimal management of risk factors.
Differences between various Lp(a) recommendations
Table 1 compares key points of the various consensus and guideline statements on Lp(a). At the first glance this overview gives the impression to be unable to see the ‘trees through the forest’. However, at a second glance they are following more or less the similar lines with some differences that are highlighted below.
In whom and when to measure Lp(a)?
There is currently a movement in the direction that Lp(a) should be measured at least in all adults [1*, 29, 30] independent whether they have a history of (premature) ASCVD, a family history of ASCVD or high Lp(a), a familial hypercholesterolemia or an indication for an intermediate or high risk for ASCVD. There are many reasons for a simplified scheme of action [34]: 1) recommendations are not followed when they are too complicated with many ifs and buts; 2) it would by cynic to wait till the first events do occur since a large number of first events is fatal; 3) only measuring Lp(a) allows to figure out how much of the global risk is derived from Lp(a). Especially Figure 3 shows that high and very high Lp(a) concentrations contribute at least as much to the absolute ASCVD risk as all other traditional risk factors combined. 4) Lp(a) testing is relatively cheap and generally widely available in clinical chemistry laboratories, is done in the majority of subjects only once and costs less than a COVID-19 test which most of the people nowadays have undergone dozens of time. Besides adults, some statements also give advice for young people and kids to measure Lp(a) mainly in those with a history of stroke or a family history of premature ASCVD or high Lp(a) and in situations of cascade testing for the before mentioned conditions [1*, 27].
How often should Lp(a) be measured?
Those documents which make a statement on this, are quite uniform and recommend testing only once since Lp(a) is genetically determined and therefore relatively stable over time [1*, 28–30]. One recent paper from the UK Biobank found that in roughly 16.000 patients with two Lp(a) measurements more than 4 years apart only 10% and 5% showed an at least 25 nmol/L increase and decrease of Lp(a) over time, respectively [35]. An exception might be secondary diseases (e.g. the development of kidney impairment, acute infectious episodes) or therapeutic intervention which may affect Lp(a) concentrations.
Considerations on Lp(a) assays
Statements from the HEART UK [28], the National Lipid Association [27, 36] and a scientific statement from the American Heart Association [37] recommend to use assays which measure Lp(a) independent from apo(a) isoforms and calibrated against the WHO/IFCCLM secondary reference material. However, this reference material is running out of stock and major efforts are under way to create new reference material and reference measurement methods [38, 39]. Concentrations should be measured in molar units which is not easy to accomplish since assays used in clinical practice use polyclonal antibodies that are likely to be directed against repetitive epitopes. As discussed recently [18*, 40*], the range of Lp(a) concentrations can vary in each apo(a) isoform group up to 200-fold which makes it difficult to select apo(a) isoform-characterized calibrators for the various concentration strata. Therefore, the request to report in molar terms is not necessarily in line with reality since the clinically available assays can at best come close to a molar measurement which is by definition hardly possible with the used antibodies. Therefore, we stated in the recent EAS Lp(a) consensus paper that measurement should be in molar units if available. If not, the units in which the assay is calibrated should be used for reporting. Keeping in mind that Lp(a) assays are not yet optimally standardized we recommended to have kind of a grey zone between 30 and 50 mg/dL and values below 30 mg/dL as a rule out zone, where the risk derived from Lp(a) might be neglectable, and values above 50 mg/dL as rule in zone for increased ASCVD risk (of course with a further increase in risk with increasing Lp(a) concentrations). Another recommendation is that Lp(a) values should not be converted from mg/dL to nmol/L and vice versa. However, we have to face reality with two different reporting units depending on the assay used. Already available measurements, especially when they are in a clear rule-in or rule-out zone are still better than nothing and have not necessarily to be repeated. The conversion factors from mg/dL to nmol/L often range from 1 : 2.0 to 2.5 [1*, 40*]. An often-raised question is whether the assays available on the market are already useful for clinical purposes? Yes, they are; even if there is still room for improvement which will be hopefully accomplished by ongoing standardization efforts [38, 39]. In some situations quite some bias might be observed [41] although this will not necessarily result in major changes of the risk classification.
Risk thresholds
As mentioned, the new EAS Lp(a) consensus clearly demonstrates a continuous increase of ASCVD risk with increasing Lp(a) concentrations. We therefore mentioned <30 and >50 mg/dL as rule out and rule in zones, respectively. The HEART UK consensus uses groups from mild to very high risk [28]. Some other statements use 50 mg/dL as a threshold [26, 27, 42]. The 2019 ESC/EAS dyslipidemia guideline created some misunderstanding since it introduced 180 mg/dL as a new “threshold” but it intended to show that these very high concentrations are simply considered as a risk equivalent of heterozygous FH [29]. This recommendation has also been adopted by the Chinese Guideline on the Primary Prevention of Cardiovascular Diseases [43].
Management of patients with high Lp(a)
Those consensus or guideline documents, which give advice on the management of patients with high Lp(a), target on the management on other treatable risk factors to reduce the overall ASCVD risk [1*, 27, 28, 30]. The use of scores such as the Framingham Risk Score [30] or other scoring systems [27] allow to give a more or less granular advice on lifestyle modifications and especially lipid-lowering drugs. For example a scientific statement for the American Heart Association echoed that the Lp(a) level can be used as a risk-enhancing factor and based on the data from Patel et al. [10], the clinician could recalibrate the 10-year risk estimate of the current American College of Cardiology/American Heart Association guidelines based on the following formula to provide an approximate updated 10-year risk estimate: predicted 10-year risk × [1.11(patient’s Lp(a) level in nmol/L/50)]. If Lp(a) is measured, the updated risk estimate might favor statin initiation among individuals at borderline (5%–7.4%) or intermediate (7.5%–19.9%) 10-year predicted risk for ASCVD [37].
A more convenient approach is recommended by the most recent EAS Lp(a) consensus paper which introduces the Lp(a) risk calculator (http://www.lpaclinicalguidance.com). This calculator does not only consider the individual traditional risk factors but also the risk derived from Lp(a) and how e.g. a certain amount of LDL-C or blood pressure lowering mitigates the overall ASCVD risk. This allows individualized counselling of patients which show the estimated expected benefits from interventions [1*].
Conclusions
The contribution of high Lp(a) concentrations to an individual’s global ASCVD risk can be substantial, sometimes higher that the risk derived from all traditional risk factors combined. Therefore, starting early to measure Lp(a) is recommended. Without measuring Lp(a) one might markedly underestimate the individual’s global risk. In case of high and very high Lp(a) concentrations, a strict and comprehensive management of all ASCVD risk factors is currently the focus as we await further data from ongoing cardiovascular outcome trials on specific Lp(a)-lowering drugs.
Key points.
The relationship between Lp(a) concentrations and atherosclerotic cardiovascular disease is continuous.
Measuring Lp(a) is essential for reliable estimation of an individual’s global ASCVD risk.
Measurement of Lp(a) should be performed in each adult at least once in a person’s lifetime; under certain circumstances Lp(a) should even be measured in young persons below the age of 18 years.
In case of high Lp(a) concentrations, a strict and comprehensive management of all ASCVD risk factors is currently the focus.
An Lp(a) risk calculator is available (http://www.lpaclinicalguidance.com/) which helps to illustrate the contribution of Lp(a) concentration to the individual’s risk and to plan interventional strategies aimed at mitigating the Lp(a)-induced increased risk.
Acknowledgements:
We are grateful for the many researchers and patients who provided the evidence and basis for the various consensus and guideline papers.
Financial support and sponsorship:
Florian Kronenberg received research support from the Austrian Research Fund (W-1253), the Austrian Genome Project “GOLD”, the Austrian Heart Fund and the “Jubiläumsfond” of the Austrian National Bank. Samia Mora was supported by research grants from the National Institute of Diabetes and Digestive and Kidney Diseases (DK112940), National Heart, Lung, and Blood Institute (R01 HL160799, R01HL134811, R01HL134168, R01HL143227, K24 HL136852). The funding sources had no role in the design and conduct of this study or the interpretation of the data. The opinions expressed in the manuscript are those of the study authors.
Conflicts of interests:
Florian Kronenberg has received consulting or lecture fees from Novartis, Amgen, Kaneka and CRISPR therapeutics. Samia Mora has served as a consultant to Pfizer for work unrelated to Lp(a). Erik Stroes has received adboard/lecturing fees, paid to the institution, from Amgen, Sanofi, Akcea/Ionis, Merck, Esperion, Novo-Nordisk, Silence-therapeutics, Astra-Zeneca and Daiichi-Sankyo.
References
- [1].Kronenberg F, Mora S, Stroes ESG et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J 2022; 43:3925–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This most recent consensus paper on Lp(a) clearly demonstrates that in case of high Lp(a) concentrations the global ASCVD risk might be markedly underestimated if Lp(a) is not considered. Based on that, an Lp(a) risk calculator has been developed which considers the risk from traditional risk factors as well as Lp(a) concentrations: http://www.lpaclinicalguidance.com
- [2].Welsh P, Welsh C, Celis-Morales CA et al. Lipoprotein(a) and cardiovascular disease: prediction, attributable risk fraction, and estimating benefits from novel interventions. Eur J Prev Cardiol 2022; 28:1991–2000. [DOI] [PubMed] [Google Scholar]
- [3].Erqou S, Kaptoge S, Perry PL et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009; 302:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arsenault BJ, Kamstrup PR. Lipoprotein(a) and cardiovascular and valvular diseases: A genetic epidemiological perspective. Atherosclerosis 2022; 349:7–16. [DOI] [PubMed] [Google Scholar]; ** Very comprehensive overview on epidemiological studies on Lp(a) concentrations and various cardiovascular outcomes.
- [5].Langsted A, Nordestgaard BG, Kamstrup PR. Low lipoprotein(a) levels and risk of disease in a large, contemporary, general population study. Eur Heart J 2021; 42:1147–1156. [DOI] [PubMed] [Google Scholar]
- [6].Nordestgaard BG, Langsted A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res 2016; 57:1953–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].deVeber G, Kirkham F, Shannon K et al. Recurrent stroke: the role of thrombophilia in a large international pediatric stroke population. Haematologica 2019; 104:1676–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nowak-Göttl U, Sträter R, Heinecke A et al. Lipoprotein (a) and genetic polymorphisms of clotting factor V, prothrombin, and methylenetetrahydrofolate reductase are risk factors of spontaneous ischemic stroke in childhood. Blood 1999; 94:3678–3682. [PubMed] [Google Scholar]
- [9].Sultan SM, Schupf N, Dowling MM et al. Review of lipid and lipoprotein(a) abnormalities in childhood arterial ischemic stroke. Int. J. Stroke 2014; 9:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Patel AP, Wang M, Pirruccello JP et al. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights From a Large National Biobank. Arterioscler Thromb Vasc Biol 2021; 41:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sandholzer C, Saha N, Kark JD et al. Apo(a) isoforms predict risk for coronary heart disease: a study in six populations. Arterioscler. Thromb 1992; 12:1214–1226. [DOI] [PubMed] [Google Scholar]
- [12].Kraft HG, Lingenhel A, Köchl S et al. Apolipoprotein(a) Kringle IV repeat number predicts risk for coronary heart disease. Arterioscler. Thromb. Vasc. Biol 1996; 16:713–719. [DOI] [PubMed] [Google Scholar]
- [13].Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA 2009; 301:2331–2339. [DOI] [PubMed] [Google Scholar]
- [14].Clarke R, Peden JF, Hopewell JC et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med 2009; 361:2518–2528. [DOI] [PubMed] [Google Scholar]
- [15].Kronenberg F, Utermann G. Lipoprotein(a) - resurrected by genetics. J. Intern. Med 2013; 273:6–30. [DOI] [PubMed] [Google Scholar]
- [16].Kronenberg F. Human genetics and the causal role of lipoprotein(a) for various diseases. Cardiovasc. Drugs Ther 2016; 30:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mack S, Coassin S, Rueedi R et al. A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J. Lipid Res 2017; 58:1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Coassin S, Kronenberg F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: The complexity of genetic variation in the LPA gene. Atherosclerosis 2022; 349:17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Very comprehensive overview on the genetic architecture of Lp(a) concentrations.
- [19].Lim ET, Wurtz P, Havulinna AS et al. Distribution and Medical Impact of Loss-of-Function Variants in the Finnish Founder Population. PLoS. Genet 2014; 10:e1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gudbjartsson DF, Thorgeirsson G, Sulem P et al. Lipoprotein(a) Concentration and Risks of Cardiovascular Disease and Diabetes. J. Am. Coll. Cardiol 2019; 74:2982–2994. [DOI] [PubMed] [Google Scholar]
- [21].Coassin S, Erhart G, Weissensteiner H et al. A novel but frequent variant in LPA KIV-2 is associated with a pronounced Lp(a) and cardiovascular risk reduction. Eur. Heart J 2017; 38:1823–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schachtl-Riess JF, Kheirkhah A, Grüneis R et al. Frequent LPA KIV-2 Variants Lower Lipoprotein(a) Concentrations and Protect Against Coronary Artery Disease. J Am Coll Cardiol 2021; 78:437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study shows that two very common functional variants in the previously inaccessible LPA KIV-2 region cooperate in determining Lp(a) variance and coronary artery disease risk. Even a moderate but lifelong genetic Lp(a) reduction translates to a noticeable CAD risk reduction.
- [23].Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med 2020; 382:244–255. [DOI] [PubMed] [Google Scholar]
- [24].Koren MJ, Moriarty PM, Baum SJ et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med 2022; 28:96–103. [DOI] [PubMed] [Google Scholar]; * This paper validates the approach of using hepatocyte-targeted siRNA to potently lower Lp(a) concentrations in individuals with elevated Lp(a) concentration.
- [25].Nissen SE, Wolski K, Balog C et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals With Elevated Plasma Lipoprotein(a) Levels. JAMA 2022; 327:1679–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Phase 1 study of a further siRNA approach which demonstrated a dose-dependent lowering of plasma Lp(a) concentrations.
- [26].Nordestgaard BG, Chapman MJ, Ray K et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur. Heart J 2010; 31:2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wilson DP, Jacobson TA, Jones PH et al. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J. Clin. Lipidol 2019; 13:374–392. [DOI] [PubMed] [Google Scholar]
- [28].Cegla J, Neely RDG, France M et al. HEART UK consensus statement on Lipoprotein(a): A call to action. Atherosclerosis 2019; 291:62–70. [DOI] [PubMed] [Google Scholar]
- [29].Mach F, Baigent C, Catapano AL et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur. Heart J 2020; 41:111–188. [DOI] [PubMed] [Google Scholar]
- [30].Pearson GJ, Thanassoulis G, Anderson TJ et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol 2021; 37:1129–1150. [DOI] [PubMed] [Google Scholar]
- [31].Willeit P, Kiechl S, Kronenberg F et al. Discrimination and Net Reclassification of Cardiovascular Risk with Lipoprotein (a): Prospective 15-Year Outcomes in the Bruneck Study. J. Am. Coll. Cardiol 2014; 64:851–860. [DOI] [PubMed] [Google Scholar]
- [32].Nurmohamed NS, Kaiser Y, Schuitema PCE et al. Finding very high lipoprotein(a): the need for routine assessment. Eur. J. Prev. Cardiol 2022; 29:769–776. [DOI] [PubMed] [Google Scholar]; * The paper demonstrated that patients from a tertiary hospital with Lp(a) concentrations above the 99th percentile had nearly three-fold higher prevalence for ASCVD. In individuals with very high Lp(a), addition of Lp(a) concentrations to a risk prediction algorithm resulted in one-third of patients to be reclassified in risk in a primary prevention setting, and almost two thirds to be reclassified in secondary prevention setting.
- [33].Perrot N, Verbeek R, Sandhu M et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: The EPIC-Norfolk prospective population study. Atherosclerosis 2017; 256:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kronenberg F. Measuring lipoprotein(a): do it without ifs and buts. Eur J Prev Cardiol 2022; 29:766–768. [DOI] [PubMed] [Google Scholar]
- [35].Trinder M, Paruchuri K, Haidermota S et al. Repeat Measures of Lipoprotein(a) Molar Concentration and Cardiovascular Risk. J Am Coll Cardiol 2022; 79:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wilson DP, Jacobson TA, Jones PH et al. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol 2022. [DOI] [PubMed] [Google Scholar]
- [37].Reyes-Soffer G, Ginsberg HN, Berglund L et al. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol 2022; 42:e48–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cobbaert CM, Althaus H, Begcevic Brkovic I et al. Towards an SI-Traceable Reference Measurement System for Seven Serum Apolipoproteins Using Bottom-Up Quantitative Proteomics: Conceptual Approach Enabled by Cross-Disciplinary/Cross-Sector Collaboration. Clin Chem 2021; 67:478–489. [DOI] [PubMed] [Google Scholar]
- [39].Marcovina SM, Clouet-Foraison N, Koschinsky ML et al. Development of an LC-MS/MS Proposed Candidate Reference Method for the Standardization of Analytical Methods to Measure Lipoprotein(a). Clin Chem 2021; 67:490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kronenberg F Lipoprotein(a) measurement issues: Are we making a mountain out of a molehill? Atherosclerosis 2022; 349:123–135. [DOI] [PubMed] [Google Scholar]; * This review discusses many aspects of the difficulties in measuring Lp(a). It tries to distinguish between academic and practical concerns and warns to make a mountain out of a molehill.
- [41].Scharnagl H, Stojakovic T, Dieplinger B et al. Comparison of lipoprotein(a) serum concentrations measured by six commercially available immunoassays. Atherosclerosis 2019; 289:206–213. [DOI] [PubMed] [Google Scholar]
- [42].Arnett DK, Blumenthal RS, Albert MA et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019; 140:e563–e595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chinese Guideline on the Primary Prevention of Cardiovascular Diseases. Cardiology Discovery 2021; 1:70–104. [Google Scholar]