

Abstract
Alzheimer’s disease (AD) has multiple clinically and pathologically defined subtypes where the underlying causes of such heterogeneity are not well established. Rare TREM2 variants confer significantly increased risk for clinical AD in addition to other neurodegenerative disease clinical phenotypes. Whether TREM2 variants are associated with atypical clinical or pathologically defined subtypes of AD is not known. We studied here the clinical and pathological features associated with TREM2 risk variants in an autopsy-confirmed cohort. TREM2 variant cases were more frequently associated with non-amnestic clinical syndromes. Pathologically, TREM2 variant cases were associated with an atypical distribution of neurofibrillary tangle density with significantly lower hippocampal NFT burden relative to neocortical NFT accumulation. In addition, NFT density but not amyloid burden was associated with an increase of dystrophic microglia. TREM2 variant cases were not associated with an increased prevalence, extent, or severity of co-pathologies. These clinicopathological features suggest that TREM2 variants contribute to clinical and pathologic AD heterogeneity by altering the distribution of neurofibrillary degeneration and tau-dependent microglial dystrophy, resulting in hippocampal sparing and non-amnestic AD phenotypes.
Keywords: TREM2, comorbidity, tau, dementia, behavioral variant, primary progressive aphasia
Introduction
Alzheimer’s disease (AD), the most common form of dementia, is typically clinically characterized by episodic memory deficits followed by progressive impairment in executive, visual, language and neuropsychiatric domains [10]. This clinical course strongly correlates with a stereotypical progression of neurofibrillary tangle (NFT) degeneration, which affects memory-related medial temporal lobe structures prior to neocortical involvement [2, 3]. AD can also manifest as non-amnestic syndromes including logopenic variant primary progressive aphasia (lvPPA), behavioral variant of AD (bvAD) with behavioral/dysexecutive deficits, posterior cortical atrophy (PCA), and corticobasal syndrome (CBS) [10, 16, 36]. This phenotypic variability has been associated with an atypical regional distribution of tau [19, 32, 39], but the mechanisms, including potential genetic modifiers, that contribute to this non-stereotypic distribution are poorly understood.
TREM2 encodes a transmembrane protein that is preferentially expressed in microglia and modulates the innate immune response in the brain [17, 46]. Genetic studies have identified that a rare heterozygous loss-of-function variant p.R47H in TREM2 is associated with an increased risk of AD with an odds ratio comparable to the strongest non-Mendelian genetic risk factor APOE ε4 [14, 21]. Additional coding variants p.R62H, p.H157Y and p.D87N in TREM2 have also been identified as being associated with AD risk [14, 20, 49, 52]. The R47H TREM2 variant is also associated with other neurodegenerative diseases such as Parkinson’s disease (PD) [23], frontotemporal dementia (FTD) [5], and sporadic amyotrophic lateral sclerosis [6]. Homozygous loss of function variants ofTREM2 also cause Nasu-Hakola disease, characterized as a sclerosing leukodystrophy with or without multiple bone cysts [11, 47]. These reports indicate that TREM2 variants have pleiotropic effects which may manifest with variable clinical and pathological phenotypes in the context of neurodegeneration.
Although TREM2 variants have been extensively validated to confer AD risk [27], relatively less is known about the impact of TREM2 variants on clinical and pathological features of AD. Likely due to the low frequency of TREM2 variants, most retrospective studies have conducted in small patient series with inconsistent findings with regard to disease duration [24, 42, 51, 57], age at onset [24, 44, 51], and presenting symptoms [29, 44, 51]. Moreover, most series were based on a clinical diagnosis of probable AD without postmortem confirmation. Neuropathologic studies of TREM2 variant cases with ADNC have described increased tau and amyloid-β (Aβ) burden in multiple brain regions [41, 45], a relative loss of amyloid-associated microglia [35, 41], larger amyloid plaque size [22] and increased severity of α-synucleinopathy [24].
There is a need to better understand the clinicopathologic profiles of AD patients with TREM2 variants in order to better understand the factors that contribute to disease heterogeneity which may improve clinical diagnostic accuracy and eventually to inform the development of disease-modifying therapies. Thus, we investigated the clinical and pathological features associated with autopsy confirmed TREM2 variant AD cases. We report here the largest series of autopsy-confirmed TREM2 variant AD cases to date including comprehensive analysis of clinical phenotypes, regional patterns of NFT and their associations with amyloid and microglial pathologies, and co-morbid neurodegenerative disease pathologies.
Materials and methods
Participants
Genetic, demographic, and diagnostic information on autopsy cases is presented in Table 1. 54 cases with 14 TREM2 variants were identified from 1509 autopsy cases from the Center for Neurodegenerative Disease Research (CNDR) brain bank at the University of Pennsylvania [54]. 18 cases had low or no AD neuropathologic change (ADNC) where the primary neuropathological diagnosis included progressive supranuclear palsy (n=1), multiple system atrophy (n=2), frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) (n=3), tauopathy (n=1), amyotrophic lateral sclerosis (n=8), low ADNC (n=1), unremarkable adult brain (n=1), and primary age-related tauopathy (PART) (n=1). Of the remaining 36 cases with intermediate or high ADNC, 5 cases were associated with TREM2 variants of unknown significance, resulting in 31 cases with 4 TREM2 variants that have been demonstrated as definite (R47H and R62H) or possible (D87N and H127Y) AD risk modifiers (high ADNC, n=30; intermediate ADNC, n=1). In this cohort, 2 cases were found to also harbor a C9orf72 expansion mutation associated with FTLD-TDP. A comparison cohort of 119 cases without rare TREM2 variants but with intermediate or high ADNC and clinical documentation to define typical amnestic versus atypical non-amnestic AD was identified from cases referred to the CNDR brain bank from the Penn Memory Center.
Table 1.
Clinical characteristics and neuropathologic diagnosis of cases with TREM2 variants
| Case No. | Genetic variation | Sex | Age at death (yrs) | PMI (hrs) | Brain weight (g) | Age at onset (yrs) | Disease duration (yrs) | ABC score | Neuropathological diagnosis | Clinical diagnosis | Typical, amnestic AD |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Intermediate or high ADNC with TREM2 variants (AD TREM2 variants) | |||||||||||
| 1 | TREM2 R47H | F | 76 | 23 | 1053 | 68 | 8 | A3B3C3 | High ADNC LBD | Probable AD | Yes |
| 2 | TREM2 R47H APOE3/3 | M | 60 | 12 | 1136 | 56 | 4 | A3B3C3 | High ADNC | lvPPA | No |
| 3 | TREM2 R47H APOE2/3 | M | 62 | 8 | 871 | 53 | 9 | A3B3C3 | High ADNC | Probable AD | Yes |
| 4 | TREM2 R47H APOE3/4 | F | 93 | 7 | 999 | 75 | 18 | A3B3C2 | High ADNC LATE | Probable AD | Yes |
| 5 | TREM2 R47H | F | 87 | 13 | 970 | 76 | 11 | A3B3C3 | High ADNC LATE | Probable AD | Yes |
| 6 | TREM2 R62H C9orf72 expansion | F | 72 | 5 | 1200 | 66 | 6 | A3B3C3 | High ADNC FTLD-TDP | svPPA | No |
| 7 | TREM2 H157Y APOE3/4 | F | 79 | 4 | 909 | 69 | 10 | A3B3C3 | High ADNC LBD | Probable AD | Yes |
| 8 | TREM2 R47H APOE3/3 | F | 70 | 18 | 1010 | 59 | 11 | A3B3C3 | High ADNC LBD | Probable AD, language impairment-predominant | No |
| 9 | TREM2 R62H | M | 77 | 4 | 1100 | 71 | 6 | A3B3C3 | High ADNC | bvFTD | No |
| 10 | TREM2 R47H APOE3/4 | M | 82 | 9 | 1285 | 70 | 12 | A3B3C3 | High ADNC LATE | CVD/AD | No |
| 11 | TREM2 R62H APOE3/3 | F | 61 | 18 | 997 | 52 | 9 | A3B3C3 | High ADNC | Probable AD | Yes |
| 12 | TREM2 R47H APOE3/3 | F | 83 | 20 | 1130 | 76 | 7 | A3B3C3 | High ADNC HS | DLB | No |
| 13 | TREM2 D87N APOE3/4 | M | 74 | 19 | 1470 | 68 | 6 | A3B3C3 | High ADNC LBD | DLB | No |
| 14 | TREM2 R62H | M | 69 | 5 | 1187 | 60 | 9 | A3B3C3 | High ADNC | DLB/AD | No |
| 15 | TREM2 R47H APOE3/4 | F | 71 | 4.5 | 1087 | 62 | 9 | A3B3C3 | High ADNC LBD | PPA, mixed | No |
| 16 | TREM2 R47H APOE4/4 | F | 87 | 4 | 957 | 63 | 24 | A3B3C3 | ALS High ADNC | MND | No |
| 17 | TREM2 R47H APOE3/3 | M | 64 | 16.5 | 1320 | 55 | 9 | A3B3C3 | High ADNC LBD | Probable AD | Yes |
| 18 | TREM2 R47H APOE3/4 | M | 78 | 18 | 1162 | 67 | 11 | A3B3C3 | High ADNC LATE | FTD-NOS | No |
| 19 | TREM2 R62H | M | 75 | 11 | 1221 | 62 | 13 | A3B3C3 | High ADNC MSA | svPPA | No |
| 20 | TREM2 R47H | M | 61 | 11 | 1471 | 53 | 8 | A3B3C3 | High ADNC LBD | Probable AD | Yes |
| 21 | TREM2 R47H APOE3/3 | F | 80 | 7 | 1000 | 59 | 21 | A3B3C3 | High ADNC LATE | Behavioral/dysexecutive variant of AD | No |
| 22 | TREM2 R62H APOE3/4 | F | 65 | 13.5 | 1060 | 51 | 14 | A3B3C3 | High ADNC LATE | Probable AD | Yes |
| 23 | TREM2 R47H APOE3/4 | F | 79 | 43.5 | 1214 | 68 | 11 | A3B3C3 | High ADNC LATE | Probable AD | Yes |
| 24 | TREM2 R47H APOE3/3 | F | 68 | 9 | 1000 | 60 | 8 | A3B3C3 | High ADNC LBD | Probable AD | Yes |
| 25 | TREM2 R62H APOE2/3 | M | 86 | 12 | 1243 | 74 | 12 | A3B3C3 | High ADNC | Probable AD | Yes |
| 26 | TREM2 R47H APOE3/4 | M | 71 | 20 | 1164 | 63 | 8 | A3B3C3 | High ADNC LBD | PCA | No |
| 27 | TREM2 R47H APOE3/4 | M | 77 | 9 | 1280 | 74 | 3 | A3B3C3 | High ADNC LATE | Probable AD | Yes |
| 28 | TREM2 R47H APOE3/4 | F | 74 | 16 | 1199 | 60 | 14 | A3B3C3 | High ADNC LBD | Probable AD | Yes |
| 29 | TREM2 R47H APOE3/4 | M | 77 | 14 | 1307 | 68 | 9 | A3B3C3 | High ADNC LBD | Probable AD | Yes |
| 30 | TREM2 R47H C9orf72 expansion | F | 65 | 3.5 | 910 | 55 | 10 | A3B3C2 | FTLD-TDP High ADNC | bvFTD | No |
| 31 | TREM2 R47H APOE3/3 | M | 59 | 14 | 1352 | NA | NA | A2B2C2 | Intermediate ADNC | Normal | Yes |
| 32 | TREM2 F170L APOE2/3 | F | 88 | 5 | 1236 | 82 | 6 | A3B3C3 | High ADNC LBD | Probable AD | Yes |
| 33 | TREM2 L121R | M | 49 | 5 | 1002 | 34 | 15 | A2B3C3 | Intermediate ADNC | FTD-NOS | No |
| 34 | TREM2 L221I APOE4/4 | F | 64 | 14 | 1099 | 53 | 11 | A3B3C3 | High ADNC | Probable AD | Yes |
| 35 | TREM2 R47C APOE3/3 | M | 79 | 20 | 1147 | 72 | 7 | A3B3C3 | High ADNC | Probable AD | Yes |
| 36 | TREM2 S16F APOE3/4 | M | 76 | 14.5 | 1317 | 69 | 7 | A3B3C2 | High ADNC LBD | Probable AD, language impairment-predominant | No |
|
| |||||||||||
| No or low ADNC with TREM2 variants (Non-AD TREM2 variants) | |||||||||||
| 37 | TREM2 c.677–6T>C | M | 51 | 17 | 1405 | - | - | A0B0C0 | ALS | PLS | - |
| 38 | TREM2 D87N | F | 43 | 14 | 1079 | - | - | A0B0C0 | ALS, other (No TDP-43) | MND | - |
| 39 | TREM2 L133L | M | 91 | 20 | 1489 | - | - | A0B2C0 | Tauopathy, unclassifiable | DLB/FTD-NOS | - |
| 40 | TREM2 R47H | M | 69 | 5 | 1200 | - | - | A1B1C0 | MSA, CVD | MSA-p | - |
| 41 | TREM2 S149G | M | 77 | NA | 1040 | - | - | A1B1C2 | FTLD-TDP | svPPA | - |
| 42 | TREM2 A130V | F | 56 | 6 | 1353 | - | - | A0B0C0 | MSA | MSA-p | - |
| 43 | TREM2 R62H | M | 63 | 9 | 1430 | - | - | A0B0C0 | ALS | MND | - |
| 44 | TREM2 D87N | M | 48 | 3 | 1380 | - | - | A0B0C0 | ALS | MND | - |
| 45 | TREM2 R62H | F | 49 | 6 | 591 | - | - | A1B0C0 | FTLD-TDP | FTD-NOS | - |
| 46 | TREM2 R47H | F | 66 | 6 | 951 | - | - | A1B1C0 | Low ADNC | Schizophrenia | - |
| 47 | TREM2 R47H | M | 43 | 30.5 | 1545 | - | - | A0B0C0 | Unremarkable adult brain | Depression | - |
| 48 | TREM2 R62H SOD1 variant | F | 63 | 6.5 | 1392 | - | - | A0B1C0 | ALS, other (No TDP-43) | MND | - |
| 49 | TREM2 R47H | F | 80 | 16 | 1400 | - | - | A0B2C0 | PART | Schizophrenia | - |
| 50 | TREM2 R47H | M | 63 | NA | 120 | - | - | A0B0C0 | PSP | PSP | - |
| 51 | TREM2 R62H | F | 61 | 18 | 1425 | - | - | A0B0C0 | FTLD-TDP ALS | bvFTD | - |
| 52 | TREM2 R62H | F | 56 | 16 | 1146 | - | - | A1B1C2 | ALS HS | MND | - |
| 53 | TREM2 R136Q | M | 76 | 4 | 1213 | - | - | A1B1C0 | ALS Low ADNC | MND | - |
| 54 | TREM2 R47H C9orf72 expansion | F | 68 | 8 | 1099 | - | - | A0B1C0 | ALS FTLD-TDP | FTD-NOS/ALS | - |
F Female, M Male, PMI post-mortem interval, A Amyloid score, B Braak score, C, CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) score, F female, M male, PMI post-mortem interval, A amyloid score, B Braak score, C, CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) score, ADNC Alzheimer’s disease neuropathologic change, LBD Lewy body disease, FTD-NOS frontotemporal dementia, not otherwise specified, bvFTD behavioral variant of frontotemporal dementia, PPA primary progressive aphasia, lvPPA logopenic variant of primary progressive aphasia, svPPA semantic variant of primary progressive aphasia, MND motor neuron disease, CVD cerebrovascular disease, DLB dementia with Lewy bodies, FTLD-TDP frontotemporal lobar degeneration with TDP-43 (transactive response DNA binding protein 43 kDa) inclusions, LATE limbic-predominant age-related TDP-43 encephalopathy, MSA-p multiple system atrophy-parkinsonian type, ALS amyotrophic lateral sclerosis, PLS primary lateral sclerosis, PCA posterior cortical atrophy, HS hippocampal sclerosis, PART primary age-related tauopathy, PSP progressive supranuclear palsy
Clinical assessment
Clinical records were extracted from the CNDR Integrated Neurodegenerative Disease Database [54]. These include sex, age at death, age at onset, disease duration, presenting symptoms and clinical diagnosis at the time of death by the clinician who assessed the patient, and MMSE scores. Based on the clinical diagnosis and presenting symptoms, clinical phenotype was determined according to the consensus and accepted criteria for typical amnestic or atypical non-amnestic syndromes including logopenic variant of primary progressive aphasia (lvPPA), posterior cortical atrophy (PCA), behavioral variant of frontotemporal dementia (bvFTD), corticobasal syndrome (CBS), and behavioral/dysexecutive variant of AD [1, 8, 12, 36, 43].
Neuropathologic assessment
Human brain tissues were obtained at autopsy and fixed using either 10% neutral buffered formalin or 70% ethanol as described [54]. Tissues were then embedded in paraffin blocks and cut into 6 μm thick sections for histological staining with hematoxylin and eosin (H&E) and well-characterized primary antibodies to detect tau (PHF1), Aβ (NAB228), TDP-43 (1D3), and α-synuclein (SYN303) [54]. Up to sixteen regions are routinely examined in the CNDR neuropathology evaluations as described [54]. Each brain region was semi-quantitatively scored for the severity of neuropathological lesions (0, absent; 0.5, rare; 1, mild; 2, moderate; 3, severe). According to consensus guidelines, proteinopathies and vascular pathology were evaluated as follows. NAB228 and PHF1 positive Aβ, neurofibrillary tangles, and neuritic plaques were evaluated to determine the level of Alzheimer’s disease neuropathologic change (ADNC) [31]. α-synuclein positive Lewy body pathology was assigned into amygdala-predominant, brainstem, limbic or neocortical Lewy body disease (LBD) [30]. TDP-43 proteinopathy was classified into ALS-TDP [4], FTLD-TDP with types A-E [25], and limbic-predominant age-related TDP-43 encephalopathy neuropathological change (LATE-NC) with three stages (stage 1, amygdala; stage 2, hippocampus; stage 3, middle frontal cortex) [34]. The presence of large cerebral infarcts and semi-quantitatively scored cerebral amyloid angiopathy and arteriosclerosis lesions were further evaluated for a low, intermediate, or high likelihood that vascular pathology contributed to cognitive impairment [50].
Quantitative image analysis of regional NFT densities
For quantitative analysis of regional NFT densities, five brain regions were selected as representative association cortices (middle frontal cortex, superior temporal cortex, and angular cortex) and hippocampal subfields (CA1 and subiculum) as previously described [32]. For each case, PHF1 stained regions were scanned at 20x magnification using a Leica Aperio AT2 scanner and analyzed using QuPath software. Using an unbiased method to reduce sampling bias, tiles (0.125 mm2 per tile) were systematically selected at regularly spaced intervals across each region and then exported to ImageJ software (NIH, Bethesda, MD) for manual NFT counting. NFT density was expressed as the number of PHF1 positive NFT averaged across the selected tiles in each region. All counts were performed in a blinded manner.
Classification of neuropathological subtypes of AD
On the basis of regional NFT counts, cases with Braak stage V or VI were classified into hippocampal-sparing (HpSp), limbic-predominant (LP), or typical neuropathologic subtypes of AD using the threshold-based mathematical algorithm as previously described by Murray et al [32]. Cases were defined as HpSp AD if they met the following three criteria: 1) The ratio of the average hippocampal NFT to the average cortical NFT was less than 1.1 corresponding to less than the 25th percentile of AD cases. 2) All three of the hippocampal NFT densities [CA1 (median = 12), subiculum (median = 20), CA1-subiculum average (median = 17)] had to be less than the median values. 3) At least three of the cortical NFT measures [middle frontal (median = 5), superior temporal (median = 10), angular (median = 8), and cortical average (median = 8)] had to be greater than or equal to the median values. Cases were defined as limbic-predominant AD if they met the reverse criteria as follows: 1) The ratio of the average hippocampal NFT to the average cortical NFT was greater than 3.6 corresponding to greater than the 75th percentile of AD cases. 2) All three of the hippocampal NFT densities had to be greater than the median values. 3) At least three of the cortical NFT measures had to be less than or equal to the median values. Cases who did not meet criteria for either HpSp or LP AD were considered typical AD.
Statistical analysis
R-4.1.5 was used for statistical analyses. Continuous variables between AD TREM2 variant and AD TREM2 wild-type groups were compared by unpaired t-test with results expressed as mean ± standard deviation, Mann-Whitney U test with results expressed as median ± SD, or linear mixed effects models with TREM2 genotype and interaction term of TREM2 genotype and brain region as main fixed effects and brain region as a fixed effect covariate. Associations between categorical variables were determined by Fisher’s exact test. Associations between binary or ordinal dependent variables and either of continuous or binary independent variables were evaluated by binomial or ordinal logistic regression. Correlations between continuous variables with a single measurement were determined by Pearson’s or Spearman’s correlation coefficient and those with repeated measurements were evaluated by linear mixed effects models. For longitudinal cognitive analysis, a linear mixed effect model was used with fixed effects including TREM2 genotype and interval from MMSE test to death, and the main effect, the interaction of interval from MMSE test to death and study groups. TREM2 wild-type and middle frontal cortex were set as reference factors for TREM2 genotype and brain region in linear mixed effects models if appropriate. Shapiro-Wilk normality test was conducted if appropriate. A p value < 0.05 was considered statistically significant.
Results
AD TREM2 variants are associated with non-amnestic clinical syndromes
Clinical information is provided for each AD case in Table 1. Clinical features were compared between AD TREM2 variant cases (n = 31, mean age at death ± SD = 73.61 ± 8.91 years, and female = 51.61 %) and AD TREM2 wild-type controls (n = 119, mean age at death ± SD = 75.17 ± 10.14 years, female = 52.94 %; age at death, p = 0.405; sex, p = 1) as summarized in Table 2. With regards to clinical presentation, the AD TREM2 variant group had a high proportion of non-amnestic clinical syndromes compared to the AD TREM2 wild-type group (p=0.002, Table 2). Indeed, of the 31 AD patients with TREM2 variants, only 16 cases (51.61 %, Table 2 and Fig. 1a) met criteria for a typical amnestic syndrome with initial episodic memory deficits and progressive dementia. The remaining 15 cases (48.39 %, Table 2 and Fig. 1a), including 2 cases carrying the C9orf72 expansion that were diagnosed with semantic variant of primary progressive aphasia (svPPA) and bvFTD, had non-amnestic syndromes (behavioral/dysexecutive variant of AD, n=1, 3.23%; bvFTD, n=2, 6.45%; lvPPA, n=1, 3.23%; svPPA, n=2, 6.45%; mixed PPA showing both non-fluent and fluent forms of PPA (n=1), n=1, 3.23%; PCA, n=1, 3.23%; dementia with Lewy bodies (DLB), n=2, 6.45%; frontotemporal dementia, not otherwise specified (FTD-NOS), n=1, 3.23%; motor neuron disease (MND), n=1, 3.23%; probable AD, language impairment-predominant, n=1, 3.23%; mixed cerebrovascular disease/AD, n=1, 3.23%; mixed DLB/AD, n=1, 3.23%).
Table 2.
Clinical features of AD patients with TREM2 variants and TREM2 wild-type
| AD TREM2 variants | AD TREM2 wild-type | p value | |
|---|---|---|---|
|
| |||
| N | 31 | 119 | |
| Female, N (%) | 16 (51.61 %) | 63 (52.94 %) | †1 |
| Age at death, yrs | 73.61 ± 8.91 | 75.17 ± 10.14 | ‡0.405 |
| Clinical phenotype | †0.002* | ||
| Typical, amnestic AD, n (%) | 16 (51.61 %) | 96 (80.67 %) | |
| (Clinical diagnosis) | |||
| Probable AD | 15 (48.39 %) | 87 (73.11 %) | |
| Possible AD | 0 | 6 (5.04 %) | |
| Normal | 1 (3.23 %) | 3 (2.52 %) | |
| Atypical, non-amnestic AD, n (%) | 15 (48.39 %) | 23 (19.33 %) | |
| (Clinical diagnosis) | |||
| Behavioral/dysexecutive variant of AD | 1 (3.23 %) | 0 | |
| bvFTD | 2 (6.45 %) | 1 (0.84 %) | |
| CBS | 0 | 2 (1.68 %) | |
| lvPPA | 1 (3.23 %) | 0 | |
| svPPA | 2 (6.45 %) | 1 (0.84 %) | |
| PPA, mixed | 1 (3.23 %) | 0 | |
| PPA, non-specified | 0 | 1 (0.84 %) | |
| PCA | 1 (3.23 %) | 1 (0.84 %) | |
| DLB | 2 (6.45 %) | 3 (2.52 %) | |
| FTD-NOS | 1 (3.23 %) | 11 (9.24 %) | |
| MND | 1 (3.23 %) | 0 | |
| VaD | 0 | 1 (0.84 %) | |
| Probable AD, frontal features-predominant | 0 | 1 (0.84 %) | |
| Probable AD, hallucination and confusion-predominant | 0 | 1 (0.84 %) | |
| Probable AD, language impairment-predominant | 1 (3.23 %) | 0 | |
| CVD/AD | 1 (3.23 %) | 0 | |
| DLB/AD | 1 (3.23 %) | 0 | |
|
| |||
| N | 30 | 117 | |
| Age at onset, yrs | 63 ± 7.62 | 63 ± 10.46 | #0.684 |
| Disease Duration, yrs | 9 ± 4.53 | 9 ± 3.92 | #0.789 |
| Early-onset AD, n (%) | 16 (53.33 %) | 61 (52.14 %) | †1 |
|
| |||
| N | 18 | 86 | |
| Last MMSE score | 8.5 ± 6.54 | 7 ± 7.61 | #0.513 |
Values are mean ± standard deviation
Fisher’s exact test
T-test
Mann-Whitney U test
p<0.05 is statistically significant.
bvFTD behavioral variant of frontotemporal dementia, CBS corticobasal syndrome, PPA primary progressive aphasia, lvPPA logopenic variant of primary progressive aphasia, svPPA semantic variant of primary progressive aphasia, PCA posterior cortical atrophy, DLB dementia with Lewy bodies, FTD-NOS frontotemporal dementia, not otherwise specified, MND motor neuron disease, VaD vascular dementia, CVD cerebrovascular disease
Fig. 1. Clinical and pathological features of AD patients with TREM2 risk variants.
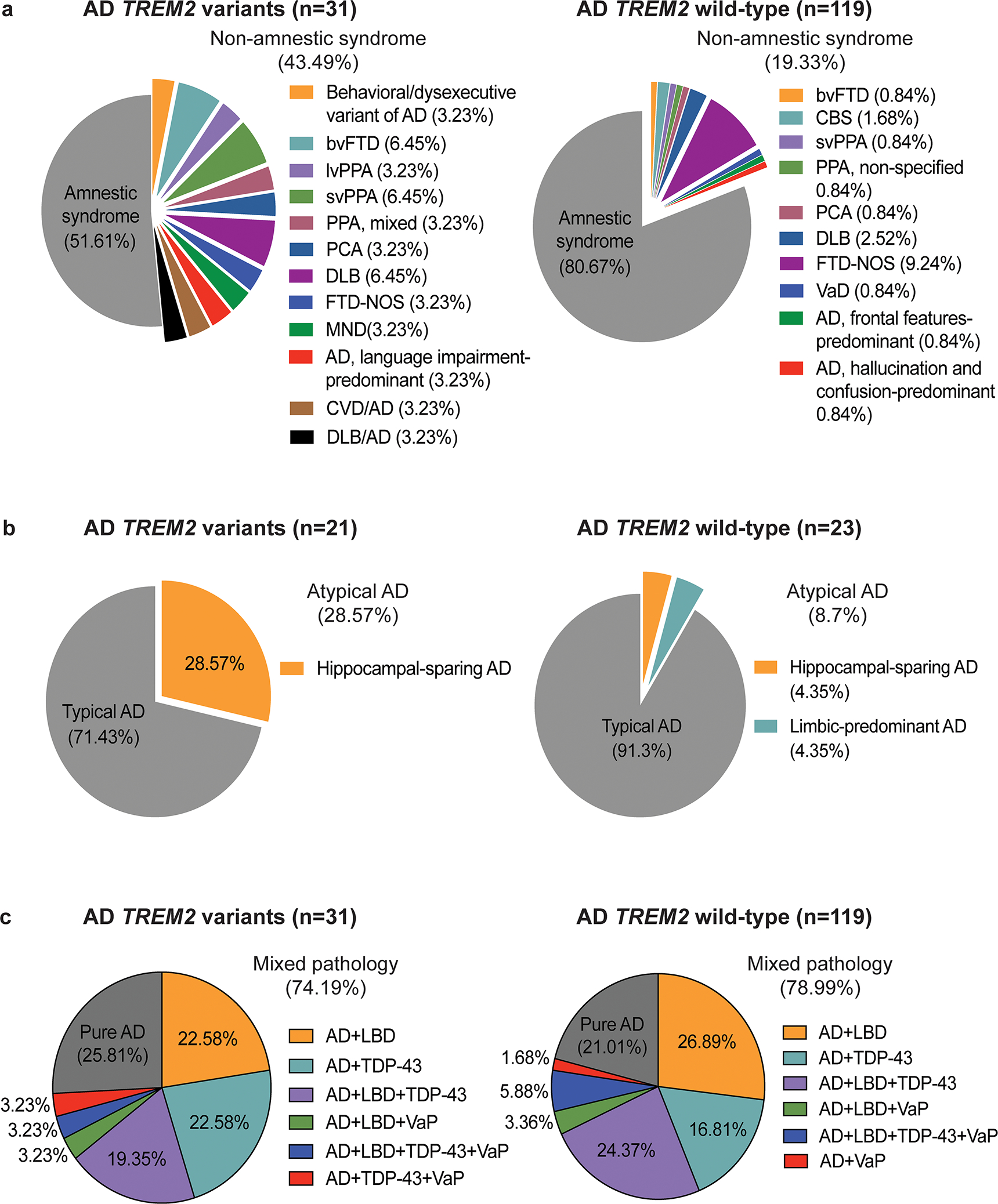
Pie charts depicts the proportion of (a) clinical phenotypes, (b) neuropathological subtype of AD, and (c) comorbid pathologies observed in AD cases with TREM2 variants versus TREM2 wild-type, showing the relatively high proportion of (a) non-amnestic syndromes and (b) hippocampal-sparing AD, but a similar proportion of (c) mixed pathology in AD TREM2 variant cases compared to AD TREM2 wild-type cases. Abbreviations: bvFTD= behavioral variant of frontotemporal dementia, lvPPA= logopenic variant of primary progressive aphasia, svPPA= semantic variant of primary progressive aphasia, PCA= posterior cortical atrophy, DLB= dementia with Lewy bodies, FTD-NOS= frontotemporal dementia, not otherwise specified, MND= motor neuron disease, CVD= cerebrovascular disease, VaD= vascular dementia, LBD= Lewy body disease, VaP = vascular pathology.
In contrast, the cohort of AD TREM2 wild-type cases had 96 of 119 cases (80.67 %, Table 2 and Fig. 1a) with a typical amnestic syndrome and 23 cases with an atypical non-amnestic syndrome (19.33%, Table 2 and Fig. 1a) including bvFTD (n= 1, 0.84%), CBS (n=2, 1.68 %), svPPA (n=1, 0.84%), PPA, non-specified (n=1, 0.84%), PCA (n=1, 0.84%), DLB (n=3, 2.52 %), FTD-NOS (n=11, 9.24 %), vascular disease [49] (n=1, 0.84%), probable AD, frontal features-predominant (n=1, 0.84%), and probable AD, hallucination and confusion-predominant (n=1, 0.84%). Despite the difference in clinical phenotype, no significant differences were detected between AD TREM2 variant cases and wild-type cases in terms of age at onset (TREM2 variants, n = 30, 63 ± 7.62; TREM wild-type, n = 117, 63 ± 10.46; p = 0.684, Table 2), disease duration (TREM2 variants, n = 30, 9 ± 4.53; TREM2 wild-type, n = 117, 9 ± 3.92; p = 0.789, Table 2), and the proportion of early-onset AD cases (TREM2 variants, n = 16 of 30, 53.33 %; TREM wild-type, n = 61 of 117, 52.14 %; p = 1, Table 2).
For the subset of cases where MMSE scores were available within 5 years of death, there was no difference in MMSE between AD with TREM2 variant cases (n = 18, 8.5 ± 6.54) and AD with TREM2 wild-type cases (n = 86, 7 ± 7.61, p = 0.513, Table 2). However, for the subset of cases with more than two MMSE scores (TREM2 variants, n = 12; TREM wild-type, n =87), we further evaluated the effect of TREM2 variants on longitudinal cognitive decline using a linear mixed effects model. There was a significant interaction effect between genotype and the interval from MMSE to death where TREM2 variant cases exhibited a faster decline in MMSE (β = −0.885, SE = 0.414; p = 0.033, Supplemental Fig. 1) compared to TREM2 wild-type cases. Together, these findings suggest TREM2 variants are associated with non-amnestic clinical syndromes as well as accelerated cognitive decline.
TREM2 variants and Hippocampal NFT density
21 of 31 randomly selected TREM2 variant and sex- and age-matched but otherwise randomly selected 23 of 119 randomly selected TREM2 wild-type cases were included for quantitative image analysis. To explore whether TREM2 variants are associated with an altered distribution of NFT pathology, PHF1-stained sections from three association cortices (middle frontal, superior temporal, and angular cortices) and two hippocampal subfields (CA1 and subiculum) were examined for NFT density (Fig.2, Supplemental table 1). There was no difference in NFT density between TREM2 variant versus wild-type groups in three association cortices including middle frontal cortex (AD TREM2 variants, n = 21, mean ± SD = 7.56 ± 3.80; AD TREM2 wild-type, n = 23, mean ± SD = 6.38 ± 2.68; p = 0.246, Fig.2b), superior temporal cortex (AD TREM2 variants, n = 21, mean ± SD = 10.05 ± 4.28; AD TREM2 wild-type, n = 23, mean ± SD = 9.59 ± 3.98; p = 0.717, Fig. 2b), and angular cortex (AD TREM2 variants, n = 21, mean ± SD = 9.13 ± 4.00; AD TREM2 wild-type, n = 23, mean ± SD = 8.28 ± 2.77; p = 0.419, Fig. 2b). NFT density averaged across these three association cortices also did not differ between the two groups (AD TREM2 variants, n=21, median ± SD = 10.19 ± 3.49; AD TREM2 wild-type, n = 23, median ± SD = 7.71 ± 2.66; p = 0.182, Fig. 2b). Similarly, no difference was detected in NFT density in the hippocampal CA1 subfield between AD TREM2 variants (n = 21, median ± SD = 12.43 ± 5.44) and AD TREM2 wild-type (n = 23, median ± SD = 14.93 ± 8.69; p = 0.256, Fig. 2b). However, NFT density in the subiculum (AD TREM2 variants, n = 21, median ± SD = 9.82 ± 13.16; AD TREM2 wild-type, n = 23, median ± SD = 15.86 ± 9.32; p = 0.002, Fig. 2b) and average hippocampal NFT density (AD TREM2 variants, n = 21, median ± SD = 10.66 ± 8.73); AD TREM2 wild-type, n = 23, median ± SD = 16.29 ± 8.62; p=0.023, Fig. 2b) were significantly lower in AD TREM2 variant cases compared to AD TREM2 wild-type control cases. The ratio of average hippocampal to average cortical NFT density was also significantly lower in AD TREM2 variant cases (n = 21, median ± SD=1.26 ± 0.68) compared to AD TREM2 wild-type controls (n = 23, median ± SD =1.84 ± 1.22; p=0.005, Fig. 2b).
Fig. 2. Regional NFT pathology in AD patients with TREM2 risk variants.
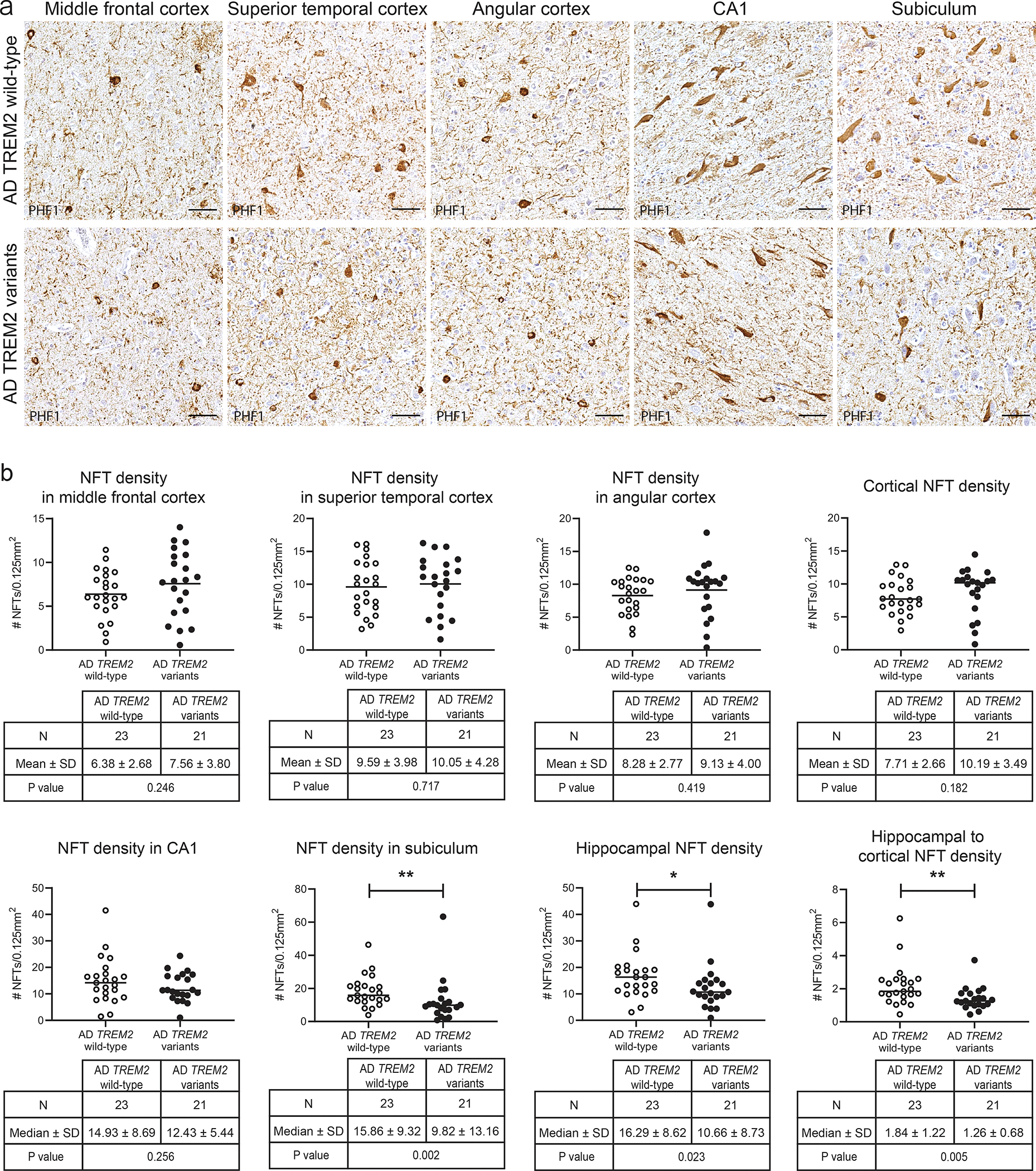
(a) PHF immunohistochemistry shows similar NFT burden in the middle frontal cortex, superior temporal cortex, angular cortex, and CA1 between AD TREM2 variant and AD TREM2 wild-type cases. PHF1 immunostaining of the subiculum shows less numerous NFT pathology in AD TREM2 variant cases than in AD TREM2 wild-type cases. Scale bars = 50 μm. (b) Graphs represent the quantification of NFT density in the middle frontal cortex, superior temporal cortex, angular cortex, cortical average, CA1, subiculum, hippocampal average, and the ratio of the average hippocampal NFT to the average cortical NFT in AD with TREM2 variants versus AD with TREM2 wild-type. NFT density was expressed as the number of PHF1-positive NFT pathology averaged across at least 12 sampling image fields (per 0.125 mm2). Mean (NFT density in middle frontal cortex, superior temporal cortex and angular cortex) and median values (cortical NFT density, NFT density in CA1 and subiculum, hippocampal NFT density, and hippocampal to cortical NFT density) are indicated. *p < 0.05 and **p < 0.01, as determined by unpaired t-test (NFT density in middle frontal cortex, superior temporal cortex and angular cortex) and Mann-Whitney U test (cortical NFT density, NFT density in CA1 and subiculum, hippocampal NFT density, and hippocampal to cortical NFT density).
As analyses were done on both ethanol and formalin fixed tissues, we verified that neurofibrillary tangle counts did not differ between these two types of fixatives based on quantification of a subset of cases for which both ethanol and formalin fixed tissues were available (n=10, r = 0.978; p < 0.001 by Pearson’s correlation coefficient, Supplemental Fig. 2). Moreover, to validate that these NFT quantifications were clinically relevant, we found that NFT density correlated well with MMSE scores obtained within five years of death (n=23, r = −0.478; p = 0.021 by Pearson’s correlation coefficient, Supplemental Fig. 3a). However, Aβ burden (n=15, ρ = −0.081; p = 0.775 by Spearman’s correlation coefficient, Supplemental Fig. 3b) and neuritic plaque density (n=15, ρ = 0.068; p = 0.809 by Spearman’s correlation coefficient, Supplemental Fig. 3c) did not correlate with MMSE. These results indicate our method for determining NFT density are technically sound and clinically relevant, thereby supporting the above finding that TREM2 variants in AD are associated with a decrease in the severity of hippocampal NFT burden relative to the neocortex.
TREM2 variants and Hippocampal-sparing AD
Using criteria defined by Murray et al. [32] with the notable difference that NFT counts were based on PHF1 stained sections as opposed to Thioflavin-S stained sections, cases were neuropathologically assigned to HpSp, typical, or limbic-predominant ADNC. Of the AD cases with TREM2 variants, 15 of 21 cases (71.43 %) including 2 cases carrying the C9orf72 expansion were defined as typical ADNC. The remaining 6 cases (28.57 %, Table 3 and Fig. 1b) were HpSp ADNC. Of the 6 cases with HpSp ADNC, 4 had non-amnestic syndromes including lvPPA (n = 1), mixed PPA (n=1), MND (n = 1), and PCA (n = 1). Of the 23 AD cases with TREM2 wild-type genotypes, 21 cases (91.3 %) were typical ADNC, while one had HpSp ADNC (4.35 %) with FTD-NOS and one with limbic-predominant AD with an amnestic syndrome (4.35 %, Table 3 and Fig. 1b). Fisher’s exact test revealed HpSp ADNC to be more common in AD TREM2 variant cases compared to AD TREM2 wild-type cases (p = 0.046, Table 3). These results suggest that TREM2 variants are associated with HpSp ADNC.
Table 3.
Neuropathologic subtypes of AD TREM2 variants and AD TREM2 wild-type
| AD TREM2 variants | AD TREM2 wild-type | p value (Fisher’s exact test) | |
|---|---|---|---|
|
| |||
| N | 21 | 23 | |
|
| |||
| Typical AD, n (%) | 15 (71.43 %) | 21 (91.3 %) | 0.046* (Typical AD vs Hippocampal-sparing AD) |
| Amnestic syndrome, n | 8 | 18 | |
| (Clinical diagnosis) | |||
| Probable AD | 8 | 18 | |
| Non-amnestic syndrome, n | 7 | 3 | |
| (Clinical diagnosis) | |||
| svPPA | 1 | 0 | |
| Probable AD, language impairment-predominant | 1 | 0 | |
| CVD/AD | 1 | 0 | |
| DLB | 1 | 1 | |
| FTD-NOS | 1 | 0 | |
| Behavioral/dysexecutive variant of AD | 1 | 0 | |
| bvFTD | 1 | 1 | |
| CBS | 0 | 1 | |
|
| |||
| Hippocampal-sparing AD, n (%) | 6 (28.57 %) | 1 (4.35 %) | |
| Amnestic syndrome, n | 2 | 0 | |
| (Clinical diagnosis) | |||
| Probable AD | 2 | 0 | |
| Non-amnestic syndrome, n | 4 | 1 | |
| (Clinical diagnosis) | |||
| lvPPA | 1 | 0 | |
| PPA, mixed | 1 | 0 | |
| MND | 1 | 0 | |
| PCA | 1 | 0 | |
| FTD-NOS | 0 | 1 | |
|
| |||
| Limbic-predominant AD, n (%) | 0 | 1 (4.35 %) | |
| Typical, amnestic AD, n | 0 | 1 | |
| (Clinical diagnosis) | |||
| Probable AD | 0 | 1 | |
p<0.05 is statistically significant.
ADNC Alzheimer’s disease neuropathologic change, svPPA semantic variant of primary progressive aphasia, CVD cerebrovascular disease, DLB dementia with Lewy bodies, FTD-NOS frontotemporal dementia, not otherwise specified, bvFTD behavioral variant of frontotemporal dementia, CBS corticobasal syndrome, PPA primary progressive aphasia, lvPPA logopenic variant of primary progressive aphasia, MND motor neuron disease, PCA posterior cortical atrophy
Using TREM2 variant cases in the current cohort, we previously reported that TREM2 variants with high ADNC did not exhibit altered regional Aβ burden, but did exhibit decreased Aβ plaque-associated microglia and increased neuritic plaque and tau accumulation, the latter determined by measuring the percent area occupied by PHF1 immunoreactivity [41]. This tau burden data was re-analyzed here using linear mixed effect models to corroborate whether there was evidence of altered regional distribution of tau. This model confirmed that overall tau burden was higher in high AD with TREM2 variants compared to wild-type cases (β = 5.197, SE = 2.319; p = 0.032, Supplemental table 3). However, relative to middle frontal cortex, tau burden in CA1 (β = −7.455, SE = 2.075; p = 0.001, Supplemental table 3) and subiculum (β = −4.796, SE = 2.117; p = 0.026, Supplemental table 3) was significantly lower in high AD TREM2 variant compared to wild-type cases. These findings suggest that TREM2 variants are associated with a relative increase in the severity of overall tau accumulation and a relative sparing of hippocampal tau burden characteristic of HpSp ADNC.
TREM2 variants and Aβ pathology
Based on our observations so far, we hypothesized that the distinct regional patterns of tau accumulation in association of TREM2 variants drive non-amnestic AD. To better understand the relationship between Aβ deposition and neurofibrillary tangle formation in TREM2 variant cases, the relationship between NFT density and Aβ burden or neuritic plaque density were examined across middle frontal cortex, CA1, subiculum, and hippocampus. Neither Aβ burden (β = −0.039, SE = 0.337; p = 0.910, Supplemental table 4) nor neuritic plaque density (β = −0.017, SE = 0.034; p = 0.624, Supplemental table 4) were related to NFT density across middle frontal, CA1, subiculum, and hippocampus in AD with TREM2 variant cases. These suggest that NFT accumulation does not correlate well with Aβ or neuritic plaque deposition in the context of high ADNC with TREM2 risk variants.
To determine whether TREM2 variants are associated with atypical regional patterns of Aβ deposition, we analyzed the ratios of hippocampal to middle frontal cortical Aβ burden and neuritic plaque density [41]. There were no differences in the ratios of hippocampal to middle frontal cortical Aβ burden (AD TREM2 variants, n=9, median ± SD = 0.28 ± 0.57; AD TREM2 wild-type, n = 13, median ± SD = 0.27 ± 0.15; p = 0.744, Supplemental Fig. 4a) or neuritic plaque density (AD TREM2 variants, n=14, median ± SD = 1.12 ± 1.77; AD TREM2 wild-type, n = 12, median ± SD = 1.15 ± 1.56; p = 0.705, Supplemental Fig. 4b) between AD TREM2 variant and wild type cases. Thus, the atypical clinical phenotypes associated with TREM2 variants did not appear to be driven by altered regional distributions of Aβ amyloid or neuritic plaques.[41].
TREM2 variants and microglial response to NFT pathology
We previously described that TREM2 variant cases exhibit decreased numbers of microglia per amyloid plaque and an apparent increase in the proportion of microglia with a dystrophic morphology [41]. To better understand how these altered microglial profiles, in particular dystrophic microglia, are involved in AD pathogenesis in the context of TREM2 variants, the relationships between the proportions of microglial subtypes and the accumulations of specific AD neuropathologies were evaluated across middle frontal cortex, CA1, subiculum, and hippocampus (Supplemental table 5). NFT density (β = −1.254, SE = 0.468; p = 0.014) but not Aβ burden (β = 1.082, SE = 0.779; p = 0.179) or neuritic plaque density (β = 0.053, SE = 0.055; p = 0.345) was negatively correlated with the proportion of homeostatic microglia. None of these AD pathologies were significantly correlated with the proportion of activated microglia (NFT density, β = 0.894, SE = 0.463; p = 0.066; Aβ burden, β = −0.974, SE = 0.775; p = 0.221; neuritic plaque density, β = −0.040, SE = 0.054; p = 0.462). Interestingly, NFT density was positively correlated with the proportion of dystrophic microglia (β = 0.324, SE = 0.130; p = 0.021), while Aβ burden (β = −0.099, SE = 0.216; p = 0.651) or neuritic plaque density (β = −0.011, SE = 0.015; p = 0.496) was not correlated with the proportion of dystrophic microglia. These findings suggest that in addition to the decreased number of amyloid-associated microglia we reported previously [35, 41], the overall altered TREM2-mediated microglia response may be linked to NFT pathology.
TREM2 variants and Concomitant pathologies
To assess whether atypical clinical phenotypes associated with TREM2 variants were associated with other co-existent neurodegenerative disease pathologies in AD, we evaluated frequencies of mixed pathology including vascular pathology, Lewy body disease (LBD), and TDP-43 proteinopathy in AD TREM2 variant compared to AD TREM2 wild-type cases (Table 4 and Fig. 1c). Of the 31 AD TREM2 variant cases, 8 cases (25.81%) had pure AD pathology defined as ADNC only or ADNC together with low probability of cerebrovascular pathology, while the majority of cases had mixed pathology (n = 23, 74.19%, Table 4 and Fig. 1c). Specifically, 7 cases exhibited LBD (22.58%, Fig.1c) and 7 cases exhibited TDP-43 proteinopathy (22.58%, Fig. 1c). 6 cases had both LBD and TDP-43 proteinopathy (19.35%, Fig.1c). One case had both TDP-43 proteinopathy and vascular pathology, one case exhibited both TDP-43 proteinopathy and vascular pathology, and one case exhibited LBD, TDP-43 proteinopathy and a coexistent moderate probability of vascular pathology (3.23% each, Fig.1c).
Table 4.
Concomitant pathologies in AD patients with TREM2 variants and TREM2 wild-type
| AD TREM2 variants | AD TREM2 wild-type | p value (Fisher’s exact) | ||
|---|---|---|---|---|
|
| ||||
| N | 31 | 119 | ||
|
| ||||
| Mixed pathology, n (%) | 0.628 | |||
| Yes | 23 (74.19 %) | 94 (78.99 %) | ||
| AD + LBD | 7 | 32 | ||
| AD + TDP-43 (LATE) | 5 | 18 | ||
| AD + TDP-43 (FTLD-TDP) | 1 | 1 | ||
| AD + TDP-43 (ALS-TDP) | 1 | 1 | ||
| AD + VaP | 0 | 2 | ||
| AD + LBD + TDP-43 (LATE) | 6 | 28 | ||
| AD + LBD + TDP-43 (FTLD-TDP) | 0 | 1 | ||
| AD + LBD + VaP | 1 | 4 | ||
| AD + TDP-43 (FTLD-TDP) + VaP | 1 | 0 | ||
| AD + LBD + TDP-43 (LATE) + VaP | 1 | 7 | ||
| No | 8 (25.81 %) | 25 (21.01 %) | ||
| AD only | 4 | 8 | ||
| AD + Low probability of VaP | 4 | 17 | ||
|
| ||||
| Vascular pathology (%) | 0.332 | |||
| Yes | 22 (70.97 %) | 95 (79.83 %) | ||
| Low probability | 19 | 82 | ||
| Moderate probability | 2 | 7 | ||
| High probability | 1 | 6 | ||
| No | 9 (29.03 %) | 95 (20.17 %) | ||
|
| ||||
| Lewy body disease, n (%) | 0.229 | |||
| Yes | 15 (48.39 %) | 72 (60.5 %) | ||
| Brain stem | 1 | 11 | ||
| Transitional or limbic | 1 | 22 | ||
| Diffuse or neocortical | 3 | 8 | ||
| Amygdala | 10 | 31 | ||
| No | 16 (51.61 %) | 47 (39.5 %) | ||
|
| ||||
| TDP-43 proteinopathy, n (%) | 1 | |||
| Yes | 15 (48.39 %) | 56 (47.06 %) | ||
| LATE-NC | 12 | 53 | ||
| Stage1 (Amygdala only) | 2 | 16 | ||
| Stage2 (+ Hippocampus) | 10 | 33 | ||
| Stage3 (+ Middle frontal gyrus) | 0 | 4 | ||
| FTLD-TDP | 2 | 2 | ||
| Type A | 2 | 1 | ||
| Type B | 0 | 1 | ||
| ALS-TDP | 1 | 1 | ||
| No | 16 (51.61 %) | 63 (52.94 %) | ||
LBD Lewy body disease, TDP-43 transactive response DNA binding protein 43 kDa, FTLD-TDP frontotemporal lobar degeneration with TDP-43 inclusions, ALS amyotrophic lateral sclerosis, VaP vascular pathology, LATE-NC limbic-predominant age-related TDP-43 encephalopathy
Likewise, in the cohort of 119 cases with AD TREM2 wild-type genotype, 25 cases (21.01%, Table 4 and Fig.1c) had pure AD and the remaining 94 cases (78.99%, Table 4 and Fig.1c) had co-morbid pathology including LBD (n= 32, 26.89%, Fig.1c), TDP-43 proteinopathy (n=20, 16.81%, Fig.1c), and vascular pathology (n=2, 1.68%, Fig.1c). 29 cases were found to exhibit both LBD and TDP-43 proteinopathy (24.37%, Fig.1c), while 4 cases had both LBD and vascular pathology (3.36%, Fig.1c). The remaining 7 cases had LBD, TDP-43 proteinopathy and vascular pathology (5.88%, Fig.1c). Statistically, the prevalence of mixed pathology did not differ between AD with TREM2 variants (n=23 of 31, 74.19 %) and AD with TREM2 wild-type genotypes (n=94 of 119, 78.99 %; p=0.628, Table 4).
Upon analyzing each type of co-morbid neuropathologic change, the frequencies of vascular pathology (TREM2 variants, n=22 of 31, 70.97 %; AD TREM2 wild-type, n=95 of 119, 79.83 %; p=0.332, Table 4), Lewy body disease (AD TREM2 variants, n=15 of 31, 48.39 %; AD TREM2 wild-type, n=72 of 119, 60.5 %; p=0.229, Table 4), and TDP-43 proteinopathy (AD TREM2 variants, n=15 of 31, 48.39 %; AD TREM2 wild-type, n=56, 47.06 %; p=1, Table 4) did not differ between the two groups. Thus, TREM2 variants did not appear to affect the prevalence of concomitant pathologies in AD.
We also evaluated the extent and severity of co-morbid neuropathologies in cases with or without TREM2 risk variants (Supplemental table 6). TREM2 variants were not associated changes in LBD stage (p = 0.120) or α-synuclein scores in amygdala (p = 0.177), hippocampus (p = 0.226), or middle frontal cortex (p = 0.434). Similarly, TREM2 variants were not associated with changes in LATE-NC stage (p = 0.738) or TDP-43 proteinopathy scores in amygdala (p = 0.766), hippocampus (p = 0.136), or middle frontal cortex (p = 0.704). There were no associations of TREM2 variants with cerebrovascular levels using VCING criteria (p = 0.825), the presence of large infarcts (p = 0.905), cerebral angiopathy scores in occipital lobe (p = 0.087) or arteriolosclerosis scores in occipital white matter (p = 0.391). In addition, TREM2 variants were not associated with the number of non-AD pathologies co-existing with AD (p = 0.496). These findings suggest that TREM2 variants did not appear to have an impact on the extent and severity of concomitant pathologies in AD.
Finally, we examined whether accumulations of specific AD neuropathologies were associated with the extent of non-AD neuropathologies (Supplemental table 7) in cases with TREM2 risk variants. Aβ load in middle frontal cortex was not associated with LBD stage (p = 0.571), LATE-NC stage (p = 0.285), or VCING levels (p = 0.360). Neuritic plaque density (LBD stages, p = 0.638; LATE-NC stages, p = 0.431; VCING levels, p = 0.851) and NFT density (LBD stages, p = 0.744; LATE-NC stages, p = 0.199; VCING levels, p = 0.991) in middle frontal cortex were also not associated with the extent of non-AD neuropathologic change. Likewise, Aβ burden (LBD stages, p = 0.418; LATE-NC stages, p = 0.198; VCING levels, p = 0.561), neuritic plaque density (LBD stages, p = 0.873; LATE-NC stages, p = 0.207; VCING levels, p = 0.958), and NFT density (LBD stages, p = 0.544; LATE-NC stages, p = 0.100; VCING levels, p = 0.233) in hippocampus were not associated the amount of comorbid neuropathologies. Overall, there was no evidence that TREM2 variants or specific AD neuropathologies were associated with increased prevalence or severity of non-AD comorbid neuropathologies.
Discussion
We report here the clinical and pathological phenotypes observed in autopsy-proven AD cases with TREM2 disease risk variants. Clinically, TREM2 variants were associated with non-amnestic clinical syndromes. These non-amnestic clinical phenotypes were not associated with amyloid pathology but rather an atypical, HpSp distribution of neurofibrillary degeneration. While TREM2 variants were associated with accelerated cognitive decline, TREM2 variants in AD were not associated with an increased frequency, extent, or severity of co-morbid neurodegenerative disease pathologies. Finally, the overall proportion of dystrophic microglia correlated with NFT density but not amyloid burden or neurtic plaque density. Thus, TREM2 variants appear to be associated with distinct clinicopathologic features including non-amnestic AD, an atypical distribution of NFT pathology, and more rapid cognitive decline, and these appear to be associated with the accumulation of dystrophic microglia independent of amyloid pathology.
Since the identification of rare variants in the coding sequence of TREM2 in association with risk for AD [14, 21], several groups have explored the effects of the variants on clinical features of the disease in AD patients. However, associations have been heterogeneous thus far, in part due to relatively small sample sizes due to the low allele frequency of TREM2 risk variants. Indeed, the R47H variant has been associated with altered disease duration in some [24, 42, 57], but not all studies [51]. One group showed that the variant decreased the age at onset of AD [51], in contrast with other cohorts [24, 44]. Additional clinical features, including neuropsychiatric symptoms, apraxia, and parkinsonian signs, have been previously associated with TREM2 variants in AD [29], while these atypical phenotypes were not noted in other cohorts [44, 51]. The majority of cases in these studies were categorized based on an antemortem diagnosis of AD without postmortem confirmation.
In this largest autopsy-confirmed cohort of TREM2 variants to date, we found that TREM2 variants were associated with non-amnestic clinical syndromes. The clinical phenotypes included behavioral/dysexecutive variant of AD, bvFTD, PPA, PCA, DLB, FTD-NOS, and MND, as well as mixed AD. Therefore, TREM2 variants may be associated with atypical AD clinical phenotypes, raising the prospect that some of previous associations with non-AD neurodegenerative disease clinical phenotypes may actually be due to underlying ADNC. Moreover, TREM2 variants were associated with accelerated global cognitive function. This result is consistent with the finding that atypical variants of AD often exhibit a more rapid cognitive decline [19].
Regional tau burden appears to correlate with various clinical manifestations in AD. For example, in vivo measurements of tau burden are higher in medial temporal lobe in patients with an amnestic presentation and in the clinically affected neocortical regions in those with non-amnestic presentations [7, 33, 37, 40], and they correlate with impairment in cognitive domains in a region-specific manner [37]. Likewise, postmortem studies identified relatively low hippocampal to cortical NFT burden in atypical variants of AD compared to typical amnestic AD [32, 39]. In the present study, our quantitative analysis revealed low NFT density in the hippocampus and a low ratio of hippocampal to cortical NFT density in AD TREM2 variant cases compared to TREM2 wild-type cases. This contrasts with the typical distribution profile of NFT pathology described by the Braak-staging scheme with relatively higher NFT burden in the medial temporal lobe compared to neocortex [2, 3]. Therefore, TREM2 variants appear to alter the distribution of NFT pathology resulting in a higher proportion of non-amnestic clinical presentations. Although our observation of the regional NFT burden corresponding to clinical syndromes is not surprising, it is notable that we, for the first time, detected atypical patterns of NFT accumulation associated with TREM2 variants.
Murray et al. formalized the definition of atypical neurofibrillary neurodegeneration based on the distribution of NFT pathology which defines three ADNC subtypes consisting of typical ADNC, HpSp ADNC, and limbic predominant ADNC [32]. Adopting these criteria, even though we employed PHF1 immunohistochemistry instead of Thioflavin staining, [32], we found that TREM2 variants were associated with HpSp AD. Given that we observed that TREM2 variants were associated with non-amnestic clinical syndromes, the overabundance of HpSp ADNC is consistent with previous studies demonstrating that HpSp AD is associated with non-amnestic clinical phenotypes [32, 56]. Although NFTs were detected by PHF1 immunohistochemistry instead of thioflavin-S fluorescence used by Murray et al., NFT counts measured by the two methods are strongly correlated [28] and analyses using thiofiavin-S and phospho-tau antibody staining have yielded similar results in terms of the proportion of ADNC subtypes [32, 56]. Moreover, the distinction between atypical and typical ADNC subtypes is statistical in nature, and we have applied here the same statistical criteria to an age- and sex-matched but otherwise random selection of AD cases which revealed significant differences between TREM2 variant versus TREM2 wild-type cases.
TREM2 is expressed on microglia and plays an important role in anti-inflammation and phagocytosis of cellular debris, both of which attenuate neurodegeneration [9]. TREM2 is differentially expressed across human brain regions, with higher levels in hippocampus and white matter and lower levels in cortical regions in healthy individuals [38, 53]. However, the expression pattern is inversed in AD brains showing lower hippocampal TREM2 compared to frontal TREM2 levels [38]. In addition, TREM2 expression in hippocampus remains stable across AD disease severity [38, 53]. These findings suggest that TREM2 may exhibit region-specific effects on AD progression.
Prokop et al. described that TREM2 variants were associated with an increase in overall tau burden including tau-positive dystrophic neurites associated with neuritic plaques, but not with Aβ burden [41]. We incorporated these findings into the current study to further evaluate relationship between NFT and Aβ burden in TREM2 risk variant cases. We found that NFT density was not correlated with Aβ burden or neuritic plaque density. These findings suggest that the increased overall tau burden observed in cases with TREM2 risk variants is not due to an increase in amyloid, raising the possibility that other mechanisms such as the loss of amyloid associated microglia may be responsible for the downstream formation of tauopathy.
Interactions between microglia and Aβ deposition play a pivotal role in inflammatory responses involving AD. Indeed, in vivo studies of TREM2 deficiency have focused on the upstream role of Aβ in the pathogenesis of AD, which includes disruption in microglial ability to Aβ phagocytosis and thereby an increase in Aβ accumulation, eventually facilitating tau pathology and contributing to neurodegeneration. [15, 26]. In support of this, we and others have reported a decrease in amyloid-associated microglia in TREM2 risk variant cases [35, 41]. Furthermore, Prokop et al. reported an altered proportion of dystrophic microglia but not homeostatic or activated microglia in TREM2 risk variants compared to wild-type cases [41]. In the current study, we further examined how the altered microglial profiles interact with AD pathology in the setting of TREM2 variants. We observed that overall dystrophic microglia load was related to NFT density and not Aβ or neuritic plaque accumulation, suggesting that TREM2 variants may alter microglial responses to tauopathy. This raises the potential that altered TREM2-mediated microglia response may be linked to Aβ as well as NFT pathology. Indeed, experimental model studies have suggested that TREM2 risk variants attenuate microglial reactivity in response to tauopathy, with the notable caveat that murine models do not typically exhibit dystrophic microglia [13].
Clinically-defined cohorts of AD have demonstrated considerable heterogeneity including the concomitant pathologies including Lewy body disease, TDP-43 proteinopathy, and vascular lesions [18, 34, 50]. These contribute to dementia clinical phenotypes and influence clinical presentations of AD [18, 34, 48, 50], although NFT pathology is a strong determinant of AD clinical profiles [55]. Thus, we hypothesized that different patterns of co-pathologies may be identified between AD patients with TREM2 variants versus TREM2 wild-type. However, there was no difference in the prevalence, extent, and severity of mixed pathology between AD with TREM2 variant cases and AD with TREM2 wild-type cases. In addition, none of AD pathologies associated with specific types of non-AD pathologic change. These findings are inconsistent with the previous report of increased density of α-synuclein burden observed in a study of one kindred with relatively few autopsied brains with or without the TREM2 R47H variant [24]. Overall, our comprehensive assessments of co-pathologies suggest that the atypical distribution of NFT pathology associated with TREM2 variants appears to be the main driver of the atypical, non-amnestic clinical phenotypes in AD. We have also identified two cases with TREM2 risk variants and the C9orf72 repeat expansion mutation. Both cases exhibited a high level of ADNC together with FTLD-TDP indicating that TREM2 risk variants appear to promote ADNC even in the setting of autosomal dominant FTLD-TDP.
A weakness of our study is the relatively small number of cases due to the relative rarity of TREM2 variants, although this cohort represents the largest series of autopsy-confirmed cases to date. Moreover, a subset of the clinical classifications was based on retrospective analysis of clinical reports. Although further replication studies need to confirm our findings in larger autopsy cohorts, our results suggest that TREM2 variants may be associated with non-amnestic clinical syndromes and an atypical distribution of NFT accumulation, which correlates with dystrophic microglia load. Thus, we speculate that clinical and pathological AD heterogeneity is driven at least in part by genetic variation, and that altered TREM2-dependent microglial reactivity appears to modify downstream patterns of neurofibrillary degeneration.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants P01AG066597, P30AG072979, U19AG062418, R01AG054519, RF1AG065341, K01AG061277, and R01NS109260. BK and EBL conceptualized the study, contributed to the methodology and design of the study, and drafted the manuscript. BK, ES, ATN, SP, BM, OO, JLR, MG, JP, DJI, DM-H, DW, JQT, CTM, VMVD, EBL were involved in data acquisition. BK and EBL performed the statistical analysis. All authors except JQT were involved in manuscript review and editing and have approved the final draft. EBL is and JQT was an editorial board member but had no role in the editorial handling of this manuscript. We especially thank Melissa E. Murray, Ph.D. for sharing background data and providing useful insights.
References
- 1.Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B et al. (2013) Criteria for the diagnosis of corticobasal degeneration. Neurology 80: 496–503 Doi 10.1212/WNL.0b013e31827f0fd1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: 239–259 Doi 10.1007/bf00308809 [DOI] [PubMed] [Google Scholar]
- 3.Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16: 271–278; discussion 278–284 Doi 10.1016/0197-4580(95)00021-6 [DOI] [PubMed] [Google Scholar]
- 4.Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M et al. (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74: 20–38 Doi 10.1002/ana.23937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broce I, Karch CM, Wen N, Fan CC, Wang Y, Tan CH et al. (2018) Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med 15: e1002487 Doi 10.1371/journal.pmed.1002487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P et al. (2014) TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol 71: 449–453 Doi 10.1001/jamaneurol.2013.6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho H, Kim HJ, Choi JY, Ryu YH, Lee MS, Na DL et al. (2019) (18)F-flortaucipir uptake patterns in clinical subtypes of primary progressive aphasia. Neurobiol Aging 75: 187–197 Doi 10.1016/j.neurobiolaging.2018.11.017 [DOI] [PubMed] [Google Scholar]
- 8.Crutch SJ, Schott JM, Rabinovici GD, Murray M, Snowden JS, van der Flier WM et al. (2017) Consensus classification of posterior cortical atrophy. Alzheimers Dement 13: 870–884 Doi 10.1016/j.jalz.2017.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deczkowska A, Weiner A, Amit I (2020) The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 181: 1207–1217 Doi 10.1016/j.cell.2020.05.003 [DOI] [PubMed] [Google Scholar]
- 10.Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K et al. (2014) Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 13: 614–629 Doi 10.1016/S1474-4422(14)70090-0 [DOI] [PubMed] [Google Scholar]
- 11.Ghezzi L, Carandini T, Arighi A, Fenoglio C, Arcaro M, De Riz M et al. (2017) Evidence of CNS β-amyloid deposition in Nasu-Hakola disease due to the TREM2 Q33X mutation. Neurology 89: 2503–2505 Doi 10.1212/wnl.0000000000004747 [DOI] [PubMed] [Google Scholar]
- 12.Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF et al. (2011) Classification of primary progressive aphasia and its variants. Neurology 76: 1006–1014 Doi 10.1212/WNL.0b013e31821103e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gratuze M, Leyns CE, Sauerbeck AD, St-Pierre MK, Xiong M, Kim N et al. (2020) Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J Clin Invest 130: 4954–4968 Doi 10.1172/jci138179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E et al. (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368: 117–127 Doi 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haass C (2021) Loss of TREM2 facilitates tau accumulation, spreading, and brain atrophy, but only in the presence of amyloid pathology. Neuron 109: 1243–1245 Doi 10.1016/j.neuron.2021.03.029 [DOI] [PubMed] [Google Scholar]
- 16.Hassan A, Whitwell JL, Josephs KA (2011) The corticobasal syndrome-Alzheimer’s disease conundrum. Expert Rev Neurother 11: 1569–1578 Doi 10.1586/ern.11.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hickman SE, El Khoury J (2014) TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem Pharmacol 88: 495–498 Doi 10.1016/j.bcp.2013.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K et al. (2007) Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184: 284–294 Doi 10.1016/j.brainres.2007.09.048 [DOI] [PubMed] [Google Scholar]
- 19.Jellinger KA (2020) Pathobiological Subtypes of Alzheimer Disease. Dement Geriatr Cogn Disord 49: 321–333 Doi 10.1159/000508625 [DOI] [PubMed] [Google Scholar]
- 20.Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D et al. (2014) Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet 23: 5838–5846 Doi 10.1093/hmg/ddu277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J et al. (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368: 107–116 Doi 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joshi P, Riffel F, Kumar S, Villacampa N, Theil S, Parhizkar S et al. (2021) TREM2 modulates differential deposition of modified and non-modified Abeta species in extracellular plaques and intraneuronal deposits. Acta Neuropathol Commun 9: 168 Doi 10.1186/s40478-021-01263-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kannarkat GT, Cook DA, Lee JK, Chang J, Chung J, Sandy E et al. (2015) Common Genetic Variant Association with Altered HLA Expression, Synergy with Pyrethroid Exposure, and Risk for Parkinson’s Disease: An Observational and Case-Control Study. NPJ Parkinsons Dis 1: Doi 10.1038/npjparkd.2015.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korvatska O, Leverenz JB, Jayadev S, McMillan P, Kurtz I, Guo X et al. (2015) R47H Variant of TREM2 Associated With Alzheimer Disease in a Large Late-Onset Family: Clinical, Genetic, and Neuropathological Study. JAMA Neurol 72: 920–927 Doi 10.1001/jamaneurol.2015.0979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK et al. (2017) Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 134: 65–78 Doi 10.1007/s00401-017-1679-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH et al. (2021) Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron 109: 1283–1301.e1286 Doi 10.1016/j.neuron.2021.02.010 [DOI] [PubMed] [Google Scholar]
- 27.Li R, Wang X, He P (2021) The most prevalent rare coding variants of TREM2 conferring risk of Alzheimer’s disease: A systematic review and meta-analysis. Exp Ther Med 21: 347 Doi 10.3892/etm.2021.9778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liesinger AM, Graff-Radford NR, Duara R, Carter RE, Hanna Al-Shaikh FS, Koga S et al. (2018) Sex and age interact to determine clinicopathologic differences in Alzheimer’s disease. Acta Neuropathol 136: 873–885 Doi 10.1007/s00401-018-1908-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luis EO, Ortega-Cubero S, Lamet I, Razquin C, Cruchaga C, Benitez BA et al. (2014) Frontobasal gray matter loss is associated with the TREM2 p.R47H variant. Neurobiol Aging 35: 2681–2690 Doi 10.1016/j.neurobiolaging.2014.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D et al. (2017) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: 88–100 Doi 10.1212/WNL.0000000000004058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al. (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123: 1–11 Doi 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW (2011) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 10: 785–796 Doi 10.1016/S1474-4422(11)70156-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nasrallah IM, Chen YJ, Hsieh M-K, Phillips JS, Ternes K, Stockbower GE et al. (2018) (18)F-Flortaucipir PET/MRI Correlations in Nonamnestic and Amnestic Variants of Alzheimer Disease. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 59: 299–306 Doi 10.2967/jnumed.117.194282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K et al. (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142: 1503–1527 Doi 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen AT, Wang K, Hu G, Wang X, Miao Z, Azevedo JA et al. (2020) APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol 140: 477–493 Doi 10.1007/s00401-020-02200-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ossenkoppele R, Pijnenburg YA, Perry DC, Cohn-Sheehy BI, Scheltens NM, Vogel JW et al. (2015) The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain 138: 2732–2749 Doi 10.1093/brain/awv191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ossenkoppele R, Schonhaut DR, Scholl M, Lockhart SN, Ayakta N, Baker SL et al. (2016) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139: 1551–1567 Doi 10.1093/brain/aww027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez SE, Nadeem M, He B, Miguel JC, Malek-Ahmadi MH, Chen K et al. (2017) Neocortical and hippocampal TREM2 protein levels during the progression of Alzheimer’s disease. Neurobiol Aging 54: 133–143 Doi 10.1016/j.neurobiolaging.2017.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petersen C, Nolan AL, de Paula França Resende E, Miller Z, Ehrenberg AJ, Gorno-Tempini ML et al. (2019) Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol 138: 597–612 Doi 10.1007/s00401-019-02036-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phillips JS, Das SR, McMillan CT, Irwin DJ, Roll EE, Da Re F et al. (2018) Tau PET imaging predicts cognition in atypical variants of Alzheimer’s disease. Hum Brain Mapp 39: 691–708 Doi 10.1002/hbm.23874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prokop S, Miller KR, Labra SR, Pitkin RM, Hoxha K, Narasimhan S et al. (2019) Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples. Acta Neuropathol 138: 613–630 Doi 10.1007/s00401-019-02048-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajagopalan P, Hibar DP, Thompson PM (2013) TREM2 and neurodegenerative disease. N Engl J Med 369: 1565–1567 Doi 10.1056/NEJMc1306509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J et al. (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134: 2456–2477 Doi 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenthal SL, Bamne MN, Wang X, Berman S, Snitz BE, Klunk WE et al. (2015) More evidence for association of a rare TREM2 mutation (R47H) with Alzheimer’s disease risk. Neurobiol Aging 36: 2443.e2421–2446 Doi 10.1016/j.neurobiolaging.2015.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roussos P, Katsel P, Fam P, Tan W, Purohit DP, Haroutunian V (2015) The triggering receptor expressed on myeloid cells 2 (TREM2) is associated with enhanced inflammation, neuropathological lesions and increased risk for Alzheimer’s dementia. Alzheimers Dement 11: 1163–1170 Doi 10.1016/j.jalz.2014.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med 23: 1018–1027 Doi 10.1038/nm.4397 [DOI] [PubMed] [Google Scholar]
- 47.Satoh JI, Kino Y, Yanaizu M, Saito Y (2018) Alzheimer’s disease pathology in Nasu-Hakola disease brains. Intractable Rare Dis Res 7: 32–36 Doi 10.5582/irdr.2017.01088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sennik S, Schweizer TA, Fischer CE, Munoz DG (2017) Risk Factors and Pathological Substrates Associated with Agitation/Aggression in Alzheimer’s Disease: A Preliminary Study using NACC Data. J Alzheimers Dis 55: 1519–1528 Doi 10.3233/JAD-160780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J et al. (2017) Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49: 1373–1384 Doi 10.1038/ng.3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skrobot OA, Attems J, Esiri M, Hortobagyi T, Ironside JW, Kalaria RN et al. (2016) Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain 139: 2957–2969 Doi 10.1093/brain/aww214 [DOI] [PubMed] [Google Scholar]
- 51.Slattery CF, Beck JA, Harper L, Adamson G, Abdi Z, Uphill J et al. (2014) R47H TREM2 variant increases risk of typical early-onset Alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimers Dement 10: 602–608.e604 Doi 10.1016/j.jalz.2014.05.1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK et al. (2017) Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement 13: 381–387 Doi 10.1016/j.jalz.2016.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strobel S, Grunblatt E, Riederer P, Heinsen H, Arzberger T, Al-Sarraj S et al. (2015) Changes in the expression of genes related to neuroinflammation over the course of sporadic Alzheimer’s disease progression: CX3CL1, TREM2, and PPARgamma. J Neural Transm (Vienna) 122: 1069–1076 Doi 10.1007/s00702-015-1369-5 [DOI] [PubMed] [Google Scholar]
- 54.Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL et al. (2014) A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 10: 477–484 e471 Doi 10.1016/j.jalz.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uemura MT, Robinson JL, Cousins KAQ, Tropea TF, Kargilis DC, McBride JD et al. (2022) Distinct characteristics of limbic-predominant age-related TDP-43 encephalopathy in Lewy body disease. Acta Neuropathol 143: 15–31 Doi 10.1007/s00401-021-02383-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uretsky M, Gibbons LE, Mukherjee S, Trittschuh EH, Fardo DW, Boyle PA et al. (2021) Longitudinal cognitive performance of Alzheimer’s disease neuropathological subtypes. Alzheimers Dement (N Y) 7: e12201 Doi 10.1002/trc2.12201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Lopez OL, Sweet RA, Becker JT, DeKosky ST, Barmada MM et al. (2015) Genetic determinants of disease progression in Alzheimer’s disease. J Alzheimers Dis 43: 649–655 Doi 10.3233/jad-140729 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.