

SUMMARY
Patients with galactosemia who carry the S135L (c.404C>T) variant of GALT, documented to encode low-level residual GALT activity, have been under-represented in most prior studies of outcomes in Type 1 galactosemia. What is known about the acute and long-term outcomes of these patients, therefore, is based on very limited data. Here, we present a study comparing acute and long-term outcomes of 12 patients homozygous for S135L, 25 patients compound heterozygous for S135L, and 105 patients homozygous for two GALT-null (G) alleles. This is the largest cohort of S135L patients characterized to date. Acute disease following milk exposure in the newborn period was common among patients in all 3 comparison groups in our study, as were long-term complications in the domains of speech, cognition, and motor outcomes. In contrast, while at least 80% of both GALT-null and S135L compound heterozygous girls and women showed evidence of an adverse ovarian outcome, prevalence was only 25% among S135L homozygotes. Further, all young women in this study with even one copy of S135L achieved spontaneous menarche; this is true for only about 33% of women with classic galactosemia. Overall, we observed that while most long-term outcomes trended milder among groups of patients with even one copy of S135L, many individual patients, either homozygous or compound heterozygous for S135L, nonetheless experienced long-term outcomes that were not mild. This was true despite detection by newborn screening and both early and life-long dietary restriction of galactose. This information should empower more evidence-based counseling for galactosemia patients with S135L.
Keywords: galactosemia, S135L variant, acute symptoms, long-term outcomes, POI, speech delay, neurocognitive outcomes
INTRODUCTION
Type 1 galactosemia, resulting from a deficiency of galactose-1-P uridylyltransferase (GALT), the second enzyme in the Leloir pathway of galactose metabolism (Holden et al 2003), comprises a continuum of autosomal recessive conditions that are typically subdivided into 3 categories: classic galactosemia (CG), clinical variant galactosemia (CVG), and biochemical/ Duarte variant galactosemia (DG) (Berry 2020). CG results from profound deficiency of GALT. Patients with CG demonstrate less than 1% residual GALT activity in red blood cells (RBC), or other tissues, even when highly sensitive methods are applied (Demirbas et al 2019).
Neonates with CG may experience a rapid progression of acute symptoms following exposure to breast milk or cow’s milk-based formula, both of which contain abundant lactose (Jenness 1974) that metabolizes to glucose and galactose. Acute symptoms may include neonatal cataracts, failure to thrive, feeding problems, compromised liver function, coagulopathies, hypoglycemia, and E. coli sepsis, which can be lethal (Berry 2020). Immediate and life-long dietary restriction of galactose, often initiated in response to a positive newborn screen result, may prevent or resolve the acute symptoms of CG (Berry 2020). However, despite rigorous dietary intervention, most patients with CG grow to experience one or more long-term complications affecting the domains of speech, cognitive development, motor outcomes, and ovarian function, among others (Rubio-Gozalbo et al 2019). The mechanisms of pathophysiology underlying long-term complications in CG, other than cataracts (Ai et al 2000), remain unknown and may differ by tissue (Haskovic et al 2020; Fridovich-Keil and Berry 2022).
In contrast to patients with CG, patients with DG are compound heterozygotes who carry one profound (G) and one very mildly-affected (D2) allele of GALT; together these alleles confer about 25% residual GALT activity (Fridovich-Keil et al 1993). Newborns with DG may be identified by NBS on the basis of diminished red blood cell (RBC) GALT activity or elevated galactose metabolites (Pyhtila et al 2015). However, recent studies confirm that these patients demonstrate no increased prevalence of acute symptoms following milk exposure in infancy (Fridovich-Keil et al 2022), and also no increased prevalence of long-term complications later in childhood regardless of galactose exposure in the first year of life (Carlock et al 2019). A prior study reported normal markers of ovarian function in a cohort of girls with DG (Badik et al 2011).
Positioned between CG and DG is CVG, that results from the presence of hypomorphic GALT variants that leave 1–10% residual GALT activity, at least in some tissues (Berry 2020). The variant most associated with CVG is S135L, a missense substitution initially reported at high prevalence among galactosemia patients of African ancestry (Baker et al 1966; Segal 1969; Lai et al 1996). This genetic variant is also relatively common among patients of Brazilian (Garcia et al 2016) and Portuguese (Coelho et al 2014) ancestry. Patients either homozygous or compound heterozygous for S135L have been reported to be at risk for acute symptoms in infancy following exposure to milk, but unlikely to experience significant long-term complications as long as dietary restriction of galactose is initiated shortly after birth (Berry 2020).
Published reports of benign long-term outcomes among patients with S135L have relied on very limited data, however, as most large studies of patient long-term outcomes in galactosemia have included very few patients who self-identified as having African ancestry, and/or who carried the S135L genetic variant (Waggoner et al 1990; Schweitzer et al 1993; Spencer et al 2013; Frederick et al 2017; Frederick et al 2018; Rubio-Gozalbo et al 2019). The goal of the present study was to expand what is known about the acute and long-term outcomes of patients with S135L to facilitate more evidence-based prognostic counseling and decision-making by affected families and their healthcare providers.
Specifically, this study was designed to address two questions. First, how do the newborn and long-term outcomes of patients with one versus two copies of S135L compare with outcomes experienced by CG patients completely deficient in GALT? Second, because the patients in our cohort reported a range of durations in early high galactose (milk) dietary exposure, we also asked whether there was any apparent relationship between the extent of dietary galactose exposure early in life and the prevalence of complications later in childhood or adulthood. While still limited, our results substantially extend what is known about the outcomes of a subset of patients long under-represented in studies of patient outcomes in Type 1 galactosemia.
METHODS
Participant recruitment:
Most S135L patients included in this study were enrolled following recruitment efforts aimed at (a) galactosemia patients and families using social media sites maintained by the Galactosemia Foundation (www.galactosemia.org) or (b) metabolic specialists identified by recruitment efforts targeting the international professional listserv, metab-L, and the homepage of the Society for the Study of Inborn Errors of Metabolism (SSIEM). Recruitment occurred predominantly between September 2020 and October 2021. The GALT genotypes of all participants were confirmed from medical records before enrollment.
Relevant clinical and laboratory data for each participant were obtained in one of two ways. Either (a) the participant was invited to enroll directly in our ongoing IRB-approved research study of galactosemia (Emory IRB 00024933; PI: JL Fridovich-Keil) in which case data were gathered using direct online surveys and authorized access to medical records, or (b) if the patient was unavailable, the referring healthcare provider was invited to enroll and submit relevant non-protected health information (non-PHI) from available medical records for the patient using a custom questionnaire (Supplemental Data) approved by the Emory IRB and administered via REDCap. Collaborating healthcare providers were also advised to obtain any required local IRB/ethics approval prior to participation.
Data collection via a custom healthcare provider survey:
The 32-question healthcare provider survey used for this study was developed by 3 authors (Fridovich-Keil, Stepien, Katler) and administered online via REDCap (Harris et al 2009); a PDF version is presented here as Supplemental Data. Our survey consisted of yes/no questions, questions scoring the degree of dietary galactose restriction using a 4-part Likert scale response (Joshi et al 2015), and free-response queries. We also asked about participant demographics using racial and ethnic categories recommended by the US National Institutes of Health (NIH; https://grants.nih.gov/grants/guide/notice-files/not-od-15-089.html#:~:text=The%20revised%20standards%20contain%20five,%22Not%20Hispanic%20or%20Latino.%22), timing and method of galactosemia diagnosis, acute symptoms, if any, relevant lab results, and specific long-term outcomes (e.g., speech, cognition, bone health, motor, ovarian function, and general health including neuropsychiatric outcomes). To be clear, no patient PHI was requested or received using the healthcare provider survey. Healthcare provider surveys for 38 participants were received; however, one participant was later excluded from the study because they were discovered to have Duarte (DG) galactosemia (S135L/ D2). Our final data set therefore included information about 37 patients with S135L: 12 with a GALT genotype of S135L/S135L (Supplemental Table 6) and 25 with a GALT genotype of S135L/G (Supplemental Table 7).
Data analysis:
Data from this study were analyzed in aggregate and compared to results from appropriate subsets of 105 patients defined as GALT-null based on the predicted residual GALT activity associated with their GALT genotype (Riehman et al 2001; Spencer et al 2013). These GALT-null cases, selected from our ongoing study of outcomes in CG (Emory IRB 00024933; PI: JL Fridovich-Keil) served as an approximately age and sex-matched comparison group for prevalence of acute symptoms, long-term complications, and biochemical markers including RBC GALT activity and galactose-1-phosphate (Gal-1P).
For comparisons of Gal-1P values (Figure 1), we applied a linear mixed-effects model (repeated measures ANOVA) using GALT genotype (S135L/S135L versus S135L/G versus G/G) as a predictor and patient code as a random effect to account for repeated measures from some patients. To be clear, the value of “n” presented for each box and whisker plot in Figure 1 lists the number of patients, which may be less than the number of data points in that time frame. The p values presented above each triplet of box and whisker plots derive from the fixed effect of GALT genotype from the repeated measures ANOVA. Where this overall p-value was <0.05, we also show significant individual p-values obtained by pairwise comparisons of estimated marginal means, adjusted using the Tukey method (Tukey 1949) for comparing a family of 3 estimates.
Figure 1:
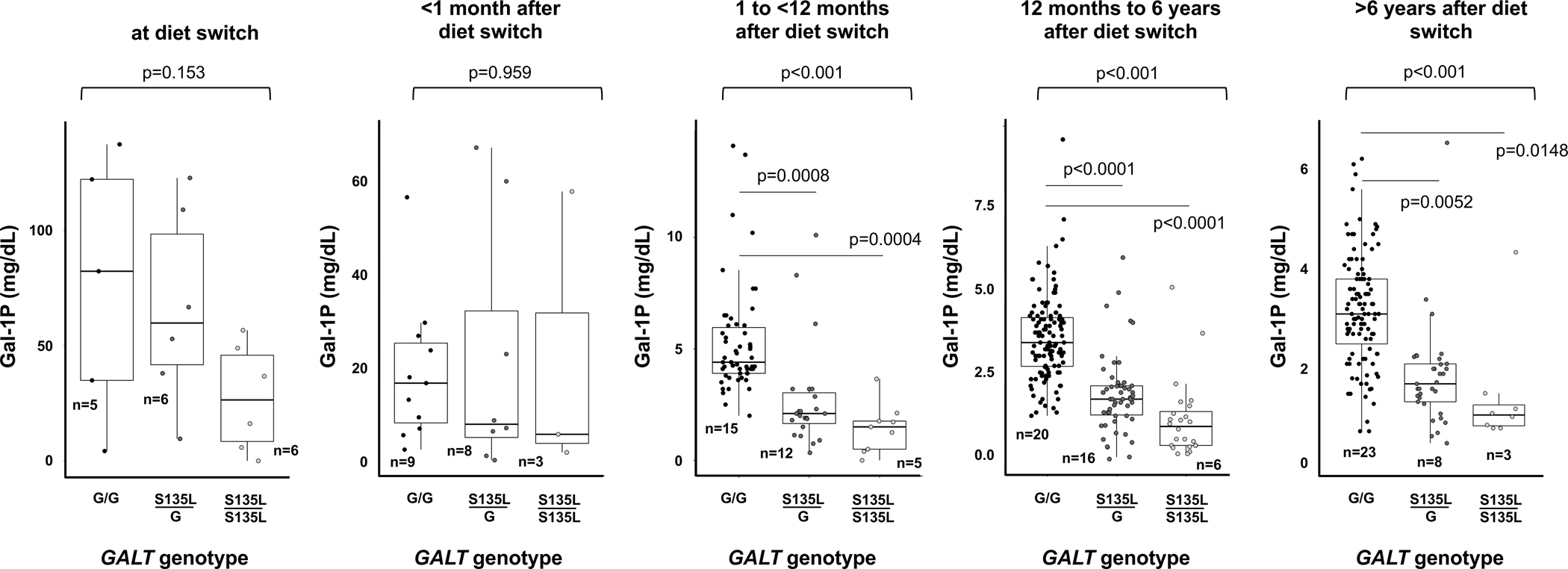
RBC Gal-1P values in each of 5 time periods following the initiation of dietary galactose restriction among patients either homozygous or compound heterozygous for S135L and GALT-null controls. The p values reported above each panel of box and whisker plots derive from the linear mixed-effects models (repeated measures ANOVA) (see Methods). Pairwise comparisons were only performed where the group p<0.05, and among pairwise comparisons only those with p<0.05 following multiple test correction are shown. The n listed by each box and whisker plot indicates the number of patients represented. In some cases, where some patients contributed more than one data point for a given time period, n is therefore less than the number of data points plotted. Relatedness of repeated measures from the same patient was accounted for by including a patient code as a random effect in the analysis (see Methods).
For acute and long-term outcomes (Figure 3), we first performed a Fisher’s exact test on the 2×3 contingency table for the data representing each outcome domain; these p values are presented above each corresponding triplet of bars. Where the group p-values were <0.05 we proceeded to pairwise Fisher’s exact test comparisons and applied the Benjamini-Hochberg method (Benjamini and Hochberg 1995) for multiple test correction. For simplicity, only pairwise p-values demonstrating significance are shown in Figure 3.
Figure 3:
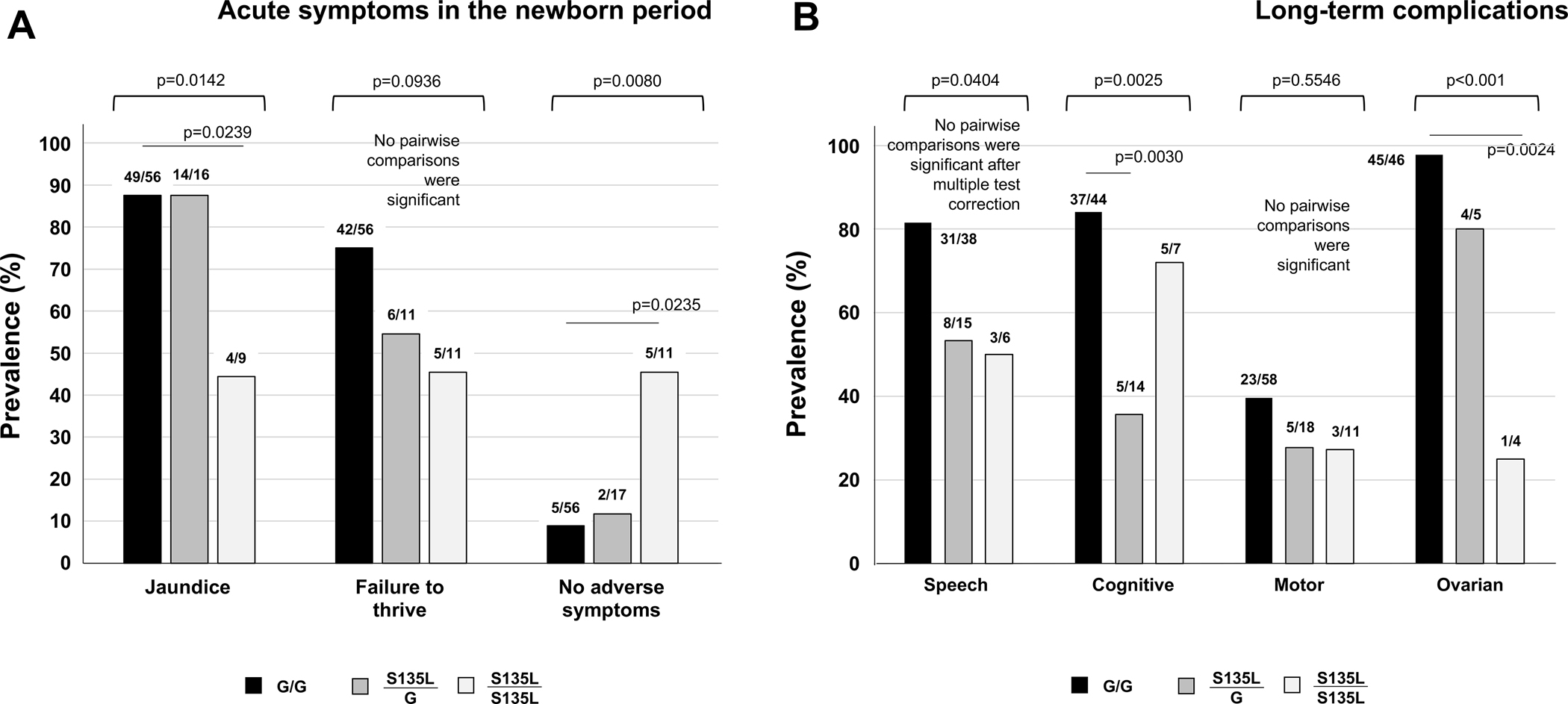
Prevalence of acute and long-term outcomes among galactosemia patients with S135L versus GALT-null controls. (A) Common symptoms in the newborn period, and (B) Long-term developmental and ovarian complications. The p values listed above each triplet of bars derive from 2×3 contingency tables performed as described in Methods. Where the overall p value for a triplet was <0.05, we also performed pairwise comparisons (see Methods), and these are presented only if significant following multiple test correction.
Due to the small sample sizes, we addressed only prevalence and not severity of each outcome. Further, due to the broad distribution of responses concerning degree of dietary galactose restriction collected using the Likert scale questions in our survey, specific comparison group sizes stratified by diet were too small to support meaningful statistical analyses, so these results were analyzed in a descriptive manner only.
RESULTS
Study participants with S135L
Table 1 summarizes the combined demographic characteristics of all participants in this study, listing only those racial categories that included at least one study participant. The 12 patients homozygous for S135L are described individually in Supplemental Table 6; the 25 patients compound heterozygous for S135L are described individually in Supplemental Table 7. To protect patient privacy, we only list specific G alleles in Supplemental Table 7 if they were common; for rare alleles that might uniquely identify a patient, we listed the non-S135L allele as “G.”
Table 1:
Demographic characteristics of participants with S135L/S135L (n=12), S135L/G (n=25), and GALT-null (n=105) galactosemia
| Category | S135L/S135L (n=12) | S135L/G (n=25) | GALT-null controls (n=105) |
|---|---|---|---|
|
| |||
| Sex | |||
| Female | 6 (50%) | 11 (44%) | 51 (48.6%) |
| Male | 6 (50%) | 14 (56%) | 54 (51.4%) |
| Other/unknown | 0 (0%) | 0 (0%) | 0 (0%) |
|
| |||
| Mean age (years) | 11.5 (2.5mo-26yr) | 8.8 (3mo-36yr) | 10.05 (1mo-71yr) |
|
| |||
| Race/Ethnicity | |||
| White Hispanic/Latino | 0 (0%) | 4 (16%) | 8 (8.4%) |
| White Not Hispanic/Latino | 1 (8.3%) | 3 (12%) | 95 (90.3%) |
| Black Hispanic/Latino | 0 (0%) | 0 (0%) | 0 (0%) |
| Black Not Hispanic/Latino | 10 (83.3%) | 8 (32%) | 0 (0%) |
| Mixed Race Hispanic/Latino | 0 (0%) | 3 (12%) | 0 (0%) |
| Mixed Race, Not Hispanic/Latino | 1 (8.3%) | 6 (24%) | 2 (1.3%) |
| Unknown | 0 (0%) | 1 (4%) | 0 (0%) |
|
| |||
| Continent of Residence | |||
| North America | 7 (58.3%) | 18 (72%) | 102/105 (97.1%) |
| South America | 0 (0%) | 3 (12%) | 1/105 (1%) |
| Europe / UK / Turkey | 5 (41.7%) | 4 (16%) | 1/105 (1%) |
| Unknown | 0 (0%) | 0 (0%) | 1/105 (1%) |
Of the 37 participants with S135L, 25 lived in the United States, 9 lived in Europe, Turkey, or the United Kingdom, and 3 lived in South America. All laboratory and outcome data for these patients were provided directly from medical records via a referring healthcare provider, with appropriate IRB approvals, using the survey presented in Supplemental Data.
Selection of a GALT-null comparison group:
Our GALT-null comparison group was selected from among volunteers enrolled in our ongoing longitudinal study of outcomes in CG (Emory IRB00024933; PI: JL Fridovich-Keil). Specifically, we restricted enrollment in this group to individuals for whom GALT genotype information was available with predicted GALT activity of <0.4% the normal level (Riehman et al 2001; Spencer et al 2013), and for whom we also had acute and/or long-term outcome information in the relevant domains. Applying these criteria, we identified a cohort of 105 GALT-null individuals, with an average age and sex distribution that was comparable to both the S135L homozygous and S135L/G compound heterozygous groups (Table 1).
Race and ethnicity:
In this study, 83% of S135L homozygotes and 32% of S135L compound heterozygotes identified as Black or African American; 8.3% and 28%, respectively, identified as White; and 8.3% and 36%, respectively, identified as Mixed Race (Table 1). In contrast, >98% of volunteers in the GALT-null comparison group identified as White, and <2% identified as Mixed Race. No participant in the any of the 3 groups identified as Asian, American Indian, or Pacific Islander, so these categories were not included in Table 1. With regard to ethnicity, no S135L homozygote identified as Hispanic/ Latino; however, 28% of S135L compound heterozygotes and about 8% of participants in the GALT-null comparison group identified as Hispanic/ Latino (Table 1).
Detection by NBS:
NBS for galactosemia is available in some countries or jurisdictions, but not in all. Combined, 67% of S135L homozygotes and 88% of S135L compound heterozygotes in this study were first identified by NBS, albeit sometimes delayed beyond the newborn period (Supplemental Tables 1, 6, 7). Of note, one infant homozygous for S135L (FKT487, Supplemental Table 6) was missed by newborn screening because their total galactose (Tgal; galactose + galactose-1-phosphate) result at the time of testing was normal; this patient was diagnosed by genetic testing at 18 months. A second patient homozygous for S135L (FKT525, Supplemental Table 6) received a “borderline” initial NBS result for galactosemia due to a normal Tgal with GALT activity that was only slightly below the laboratory reference cut-off. A repeat NBS screen at 30 days of life showed elevated Tgal and deficient GALT activity, at which point the baby was switched to soy formula while confirmatory testing was pursued. Finally, one infant compound heterozygous for S135L (FKT516) was reported to have a normal newborn screening Tgal level despite a low GALT enzyme activity (Supplemental Table 7).
GALT activity and metabolite levels detected in RBCs
Clinical GALT activity measurements from RBCs were available for 11/12 S135L homozygotes, 22/25 S135L compound heterozygotes (Supplemental Tables 6 and 7), and 48/105 GALT-null participants in this study; these values derived from multiple labs using different methods and units. Many participants in all 3 comparison groups (7/11 S135L/S135L, 9/22 S135L/G, and 28/48 GALT-null) reported undetectable (0.0 units) GALT activity; the remainder reported trace RBC GALT activity. Of note, prior studies (Demirbas et al 2019) document that some commonly used laboratory tests are unable to detect the cryptic GALT activity present in RBC samples from patients with S135L. That some patients with S135L were reported to have zero RBC GALT activity, while others were not, may therefore reflect technical rather than biological differences.
RBC Gal-1P levels were available for a subset of participants in all 3 comparison groups. Clustered by time period following the switch to a low galactose diet, these data showed two important patterns (Figure 1). First, in every time period, and even when differences were not statistically significant, the rank order of median Gal-1P values was the same: GALT-null (G/G) were the highest, S135L/G were intermediate, and S135/L/S135L were the lowest. Second, while the median Gal-1P values between genotype groups were clearly distinguishable in the later time periods (Figure 1), there was a great spread within each genotype group with substantial overlap of Gal-1P values among all genotypes at all time points.
Representative graphs of longitudinal RBC Gal-1P values from individual participants, arranged by GALT genotype, are presented in Figure 2. As expected, all 6 participants depicted experienced a precipitous drop in RBC Gal-1P following dietary restriction of galactose, which occurred immediately following the first time point. All 4 participants with even one S135L allele reached the anticipated baseline for Gal-1P (dashed line at 1 mg/dL). Of the 2 GALT-null participants in this figure, both showed Gal-1P values that plateaued above the baseline.
Figure 2:
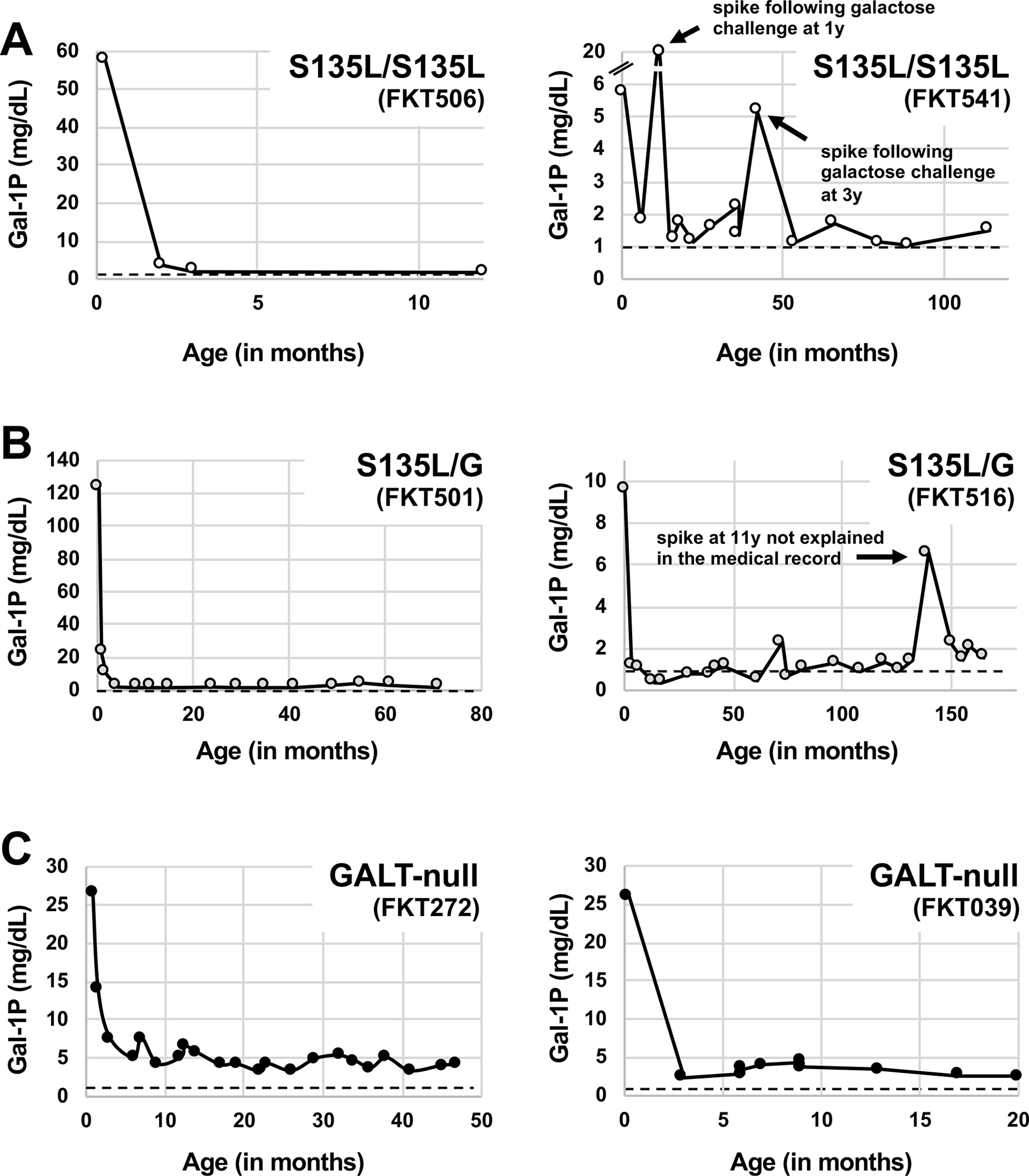
Longitudinal RBC Gal-1P values (all converted to units of mg/dL) reported for representative patients from each of the GALT genotype comparison groups in this study (A) S135L homozygotes (FKT506 and FKT541), (B) S135L/G compound heterozygotes (FKT501 and FKT516), and (C) GALT-null patients (FKT272 and FKT039). The dashed line in each graph represents a common upper limit for unaffected controls (1mg/dL Gal-1P). Arrows in individual graphs presented in panels A and B indicate Gal-1P “spikes” discussed in the text.
An unexpected but important observation that emerged from two of the graphs – one illustrating longitudinal Gal-1Ps from an S135L homozygote (FKT541) and the other illustrating longitudinal Gal-1Ps from an S135L compound heterozygote (FKT516) – was the appearance of one or more points in each graph well above the baseline (Figure 2). For FKT541, Gal-1P spikes occurred at both 1 and 3 years, both times following a galactose challenge noted contemporaneously in the medical record. For FKT516, a single Gal-1P spike occurred at age 11 years, but no dietary explanation was recorded in the medical record. As explained in the Discussion, these spikes imply that, unlike patients with DG, patients homozygous or compound heterozygous for S135L remain unable to efficiently metabolize galactose, at least in red blood cells, well beyond infancy.
Neonatal History and Acute Findings
Most participants in all 3 comparison groups experienced acute symptoms following exposure to milk in infancy, and the prevalence of acute symptoms was highest among GALT-null patients and lowest among S135L homozygotes (Figure 3A). For example, 87.5% of both S135L compound heterozygotes (14/16) and GALT-null patients (49/56) experienced jaundice, but among S135L homozygotes the prevalence was only 44.4% (4/9). Similarly, the prevalence of failure to thrive was 75% (42/56) among GALT-null patients, about 55% (6/11) among S135L/G compound heterozygotes, and about 45% (5/11) among S135L homozygotes.
Other than jaundice and failure to thrive, other acute symptoms reported for S135L homozygotes included elevated liver enzymes, coagulopathy, and vomiting. Other acute symptoms reported for S135L compound heterozygotes included elevated liver enzymes, hyperbilirubinemia, thrombocytopenia, vomiting, and cataracts. Finally, other acute symptoms reported for babies in the GALT-null comparison group included poor feeding, vomiting, diarrhea, and cataracts, among other problems. For all three genotype groups, given the small numbers of babies who experienced more than 1 week of galactose exposure, these lists should not be considered comprehensive. There were no cases of neonatal death in our study cohort. Combined, fewer than 9% (5/56) of GALT-null patients and about 12% (2/17) of S135L/G compound heterozygotes avoided acute symptoms altogether, whereas more than 45% (5/11) of S135L homozygotes did not report any acute symptoms.
Assessing the prevalence of acute symptoms with regard to duration of galactose exposure in infancy further distinguished the 3 GALT genotype groups. Specifically, among infants who were exposed to milk for 1–7 days and who reported outcome from the newborn period, 0/4 (0%) S135L homozygotes, but 6/7 (almost 86%) S135L compound heterozygotes reported adverse outcomes in infancy (Supplemental Tables 6–7). In contrast, among GALT-null patients reporting outcomes from the infant period, of 51 infants exposed to milk for even 1 day all reported at least some adverse acute symptoms.
Prevalence of long-term complications
A large fraction of participants in all 3 comparison groups experienced adverse long-term outcomes, and as was seen with acute symptoms, the prevalence of each complication was highest among GALT-null patients and lower among those with at least 1 copy of S135L (Figure 3B). Adverse speech outcomes scored among those 2 years and older were reported for 82% (31/38) of GALT-null patients, 53% (8/15) of S135L/G patients, and 50% (3/6) of S135L homozygotes (Supplemental Table 2, Figure 3B).
Adverse cognitive outcomes, including learning difficulties, intellectual deficits, or a requirement for non-speech-related special educational services in school, also scored among those 2 years or older, were reported for 84% (37/44) of GALT-null patients, 36% (5/14) of S135L/G patients, and 71% (5/7) of S135L homozygotes (Supplemental Table 3, Figure 3B).
Adverse motor outcomes were the least prevalent of the long-term complications scored for this study and were reported for 40% (23/58) of GALT-null patients, 28% (5/18) of S135L/G patients, and 27% (3/11) of S135L homozygotes (Supplemental Table 4, Figure 3B). Specific diagnoses included: early hypotonia, spasticity, ataxia, intention tremors, gait instability, problems with fine motor skills (holding pencil, buttoning, typing, tying shoelaces), and problems with gross motor skills (walking, throwing, catching).
Evidence of adverse ovarian outcomes, including either hormone levels outside the control range (e.g., low estradiol or Anti-Mullerian Hormone (AMH), elevated follicle stimulating hormone (FSH)), or clinical symptoms (e.g., primary or secondary amenorrhea, or irregular cycles) were reported for almost 98% (45/46) of GALT-null girls and women, 80% (4/5) of S135L/G girls and women, and 25% (1/4) of S135L homozygous young women (Supplemental Table 5, Figure 3B). Further, while close to 1/3 of all girls with classic galactosemia require exogenous hormones to achieve menarche (Waggoner et al 1990; Schweitzer et al 1993; Spencer et al 2013; Frederick et al 2017; Frederick et al 2018; Rubio-Gozalbo et al 2019), none of the nine young women in this study with either one or two copies of S135L required exogenous hormones to achieve menarche. However, four of the five young women in this study who are compound heterozygotes for S135L reported evidence of ovarian dysfunction which included severely diminished ovarian reserve (noted at ages 6, 10, 11 and 33 years). Of note, one woman who is a compound heterozygote for S135L reported two pregnancies achieved naturally despite irregular cycles. For all of the adverse long-term outcomes listed above, there was no clear relationship between prior experience of acute symptoms and later outcome complications, or between presence versus absence of detected RBC GALT activity and later outcome complications (Supplemental Tables 2–5).
Impact of prolonged milk exposure on prevalence of long-term complications among patients with S135L
One goal of this study was to explore whether extended exposure to milk might alter the prevalence of long-term complications among patients homozygous, or compound heterozygous, for S135L. In short, we saw no clear relationship between the duration of galactose exposure prior to diagnosis and adverse outcomes relating to speech, cognitive, motor, or ovarian outcomes among either S135L homozygotes or compound heterozygotes in our study (Supplemental Tables 2–5). Similarly, we saw no clear relationship between the rigor of dietary galactose restriction after diagnosis and any of the long-term adverse outcomes studied here (Supplemental Tables 2–5).
Of note, with regard to the impact of possible dairy re-introduction after infancy, two S135L compound heterozygotes in this study both reported that they initially had a dairy-restricted diet in infancy but then “relaxed” the diet, allowing some dairy after age 1 year. One of these children (FKT510) reported expressive speech delay at age 2 years 10 months, while the other (FKT499) reported no developmental problems at age 6 years.
DISCUSSION
Here we report on the acute and long-term outcomes of 37 patients with S135L: 12 homozygotes and 25 compound heterozygotes. This is the largest cohort of galactosemia patients with S135L yet reported in a study of outcomes. Some of our findings confirm observations noted in prior publications, but others are novel and highlight the range of outcomes experienced by these patients. Combined, our results substantially expand what is known about the biochemical and clinical outcomes of patients with S135L.
First, among patients in our cohort who reported relevant data, almost 55% (5/11) of infants homozygous for S135L, and more than 88% (15/17) of infants compound heterozygous for S135L, experienced at least one adverse outcome in infancy following exposure to milk. This result confirms the importance of newborn screening and immediate dietary intervention for infants with S135L, and the potential seriousness of a false negative result in this population (Crushell et al 2009).
Second, even among patients with S135L in our cohort who were switched to low galactose formula within the first week of life, adverse long-term outcomes still occurred. Specifically, among S135L homozygotes in this category for whom relevant data were available, one of two reported speech problems. Among S135L compound heterozygotes in this category, four of seven reported speech problems. Further, we saw no clear evidence of a relationship between RBC GALT activity level and presence or absence of acute symptoms in infancy or prevalence of adverse long-term outcomes later in childhood (Supplemental Tables 3–7). This result mirrors what has been reported previously for patients with classic galactosemia (Hughes et al 2009), leaving open the question of why some patients experience more severe long-term outcomes than others. The possibility of genetic modifiers outside the GALT locus is one strong possibility that remains to be explored. Of note, we also saw no clear relationship between rigor of dietary galactose restriction after diagnosis and presence or absence of any of the long-term complications scored for this study (Supplemental Tables 2–5). Our cohorts were not large enough, or characterized in sufficient detail, to support further stratification testing for differences in severity in addition to presence or absence of specific complications.
The single exceptional outcome observed among patients with S135L GALT in this study was ovarian function, scored by presence or absence of evidence of an adverse ovarian outcome, either hormonal (generally AMH below a laboratory-defined threshold or FSH above a laboratory-defined threshold) or clinical (generally primary or secondary amenorrhea, or irregular cycles). In short, S135L compound heterozygotes showed almost the same near-complete penetrance of evidence of an adverse ovarian outcome reported for patients with GALT-null CG (Waggoner et al 1990; Schweitzer et al 1993; Spencer et al 2013; Frederick et al 2017; Frederick et al 2018; Rubio-Gozalbo et al 2019). Of note, however, all five of the S135L compound heterozygous young women in this study achieved spontaneous menarche; this was not the case for at least 1/3 of other young women with CG reported from studies in the US and Europe (Waggoner et al 1990; Schweitzer et al 1993; Spencer et al 2013; Frederick et al 2017; Frederick et al 2018; Rubio-Gozalbo et al 2019). Similarly, among the four young women homozygous for S135L in this study, all four also achieved spontaneous menarche and only one (25%) showed later evidence of an adverse ovarian outcome (irregular cycles at age 21). This is an intriguing observation, but small cohort sizes limit conclusions. Further, given current uncertainty about the mechanism(s) underlying adverse ovarian outcomes in CG, hypothesized to range from direct ovo-toxicity of galactose or one or more galactose metabolites, to glycosylation defects, to epigenetic mechanisms (Forges et al 2006; Rubio-Gozalbo et al 2010; Fridovich-Keil et al 2011), why ovarian function might be preferentially spared, or at least adverse outcomes delayed, among young women with S135L, while other outcomes are less so, remains unclear.
Finally, our data help to address the question of whether patients with S135L remain compromised, beyond infancy, in their ability to metabolize galactose. That two patients in the study each demonstrated one or more transient spikes in RBC Gal-1P -- FKT541 at ages 1 and 3, and FKT516 at age 11 (Figure 2) -- suggests that galactose metabolism remains a long-term challenge for these children, at least in red blood cells. Of course, what this result reveals about galactose metabolism in other tissues is unclear. Regardless, the RBC result stands in stark contrast to DG, where affected children are typically able to consume normal levels of dietary galactose by age 1 year without experiencing elevation of RBC Gal-1P (Fridovich-Keil et al 1993).
Our study had both strengths and limitations. Perhaps the greatest strength was the relatively large cohort sizes of both S135L homozygotes and compound heterozygotes recruited from across the U.S., UK, Turkey and Europe, and South America. However, while 12 and 25 are large cohort sizes for homozygous and compound heterozygous patients with S135L, respectively, not all patients could be scored for all outcomes, and these numbers are not large enough to minimize the impact of individual differences. Therefore, whether our results reported here reflect outcomes in the greater community of galactosemia patients with S135L remains unknown.
Another limitation specific to our study of S135L compound heterozygotes derives from variability in the identities of the non-S135L “G” alleles in these patients. To be clear, some of these G alleles were well-characterized, e.g., Q188R, but others were not. In Supplemental Table 7 we list the specific G allele in each patient only if it was common; we code the allele as “G” if it was rare to protect the privacy of the study participant. Because many of these G alleles were either rare, poorly characterized, or both, grouping them into functionally meaningful categories for analysis was not possible.
Our reliance on medical records for biochemical and outcome data rather than direct assessment, and the lack of standardization across medical records, were additional limitations. For example, an “expressive speech delay” reported for one child might have been scored differently for another child at another clinic. Further, all data for patients with S135L were gathered from prospective medical records, whereas the control group data were collected using a combination of medical records review and both prospective and retrospective parent surveys. This difference could have introduced some element of bias. Finally, while our comparison groups of patients were approximately matched by age and distribution of biological sex, we did not have sufficient information for many patients to address other potential covariates, such as family socioeconomic circumstances, that might have impacted developmental outcomes.
Supplementary Material
1-sentence take-home message:
Patients with Type 1 galactosemia who carry one or two copies of the S135L (c.404C>T) genetic variant of GALT demonstrate generally milder long-term outcomes than their GALT-null counterparts; however, these patients may experience significant acute and long-term complications despite early and rigorous dietary restriction of galactose.
ACKNOWLEDGMENTS
We gratefully acknowledge the many patients and their families and healthcare providers who contributed to this study. We also specifically thank Dave Cutler for guidance on statistical analysis, Caitlin Manello for assistance with some of the data gathering, and Grace Carlock for assistance with REDCap early in the project. This work was supported in part by NIH Grant DK107900 (to JLFK).
Footnotes
Conflict of Interest
Quinton Katler, Karolina Stepien, Nathan Paull, Sneh Patel, Michael Adams, Mehmet Cihan Balci, Gerard Berry, Annet Bosch, Angela De La O, Didem Demirbas, Julianna Edman, Melanie Goff, Stephanie Hacker, Ina Knerr, Kristen Lancaster, Hong Li, Bryce Mendelsohn, Brandi Nichols, Wladimir Bocca Vieira de Rezende Pinto, Júlio César Rocha, Estela Rubio-Gozalbo, Michael Saad-Naguib, Sabine Scholl-Buergi, Sarah Searcy, Paulo Victor Sgobbi de Souza, Angela Wittenauer, and Judith Fridovich-Keil all declare that they have no conflict of interest with this work. Can Ficicioglu declares the following potential conflicts of interest: consulting fees from Horizon, Takeda, Travere, Swedish Orphan, and Sanofi, and honoraria from Sanofi and Horizon.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent for this study was obtained from all patients, or from their healthcare providers who shared non-protected health information, as appropriate.
Animal Rights:
This article does not contain any studies with animal subjects performed by the any of the authors.
Data Availability Statement:
All data for this manuscript are included in Supplemental Tables.
REFERENCES:
- Ai Y, Zheng Z, O’Brien-Jenkins A, et al. (2000) A mouse model of galactose-induced cataracts. Human molecular genetics 9: 1821–1827. [DOI] [PubMed] [Google Scholar]
- Badik JR, Castaneda U, Gleason TJ, et al. (2011) Ovarian function in Duarte galactosemia. Fertility and sterility 96: 469–473 e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker L, Mellman WJ, Tedesco TA, Segal S (1966) Galactosemia: Symptomatic and asymptomatic homozygotes in one Negro sibship. J Pediatrics 68: 551–558. [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 57: 289–300. [Google Scholar]
- Berry G (2020) Classic Galactosemia and Clinical Variant Galactosemia. In Editor ed.êds. Book Classic Galactosemia and Clinical Variant Galactosemia Seattle (WA): University of Washington. [PubMed] [Google Scholar]
- Carlock G, Fischer ST, Lynch ME, et al. (2019) Developmental Outcomes in Duarte Galactosemia. Pediatrics 143: e20182516. [DOI] [PubMed] [Google Scholar]
- Coelho AI, Ramos R, Gaspar A, et al. (2014) A frequent splicing mutation and novel missense mutations color the updated mutational spectrum of classic galactosemia in Portugal. J Inherit Metab Dis 37: 43–52. [DOI] [PubMed] [Google Scholar]
- Crushell E, Chukwu J, Mayne P, Blatny J, Treacy EP (2009) Negative screening tests in classical galactosaemia caused by S135L homozygosity. J Inherit Metab Dis 32: 412–415. [DOI] [PubMed] [Google Scholar]
- Demirbas D, Huang X, Daesety V, et al. (2019) The ability of an LC-MS/MS-based erythrocyte GALT enzyme assay to predict the phenotype in subjects with GALT deficiency. Mol Genet Metab 126: 368–376. [DOI] [PubMed] [Google Scholar]
- Forges T, Monnier-Barbarino P, Leheup B, Jouvet P (2006) Pathophysiology of impaired ovarian function in galactosaemia. Human reproduction update 12: 573–584. [DOI] [PubMed] [Google Scholar]
- Frederick AB, Cutler DJ, Fridovich-Keil JL (2017) Rigor of non-dairy galactose restriction in early childhood, measured by retrospective survey, does not associate with severity of five long-term outcomes quantified in 231 children and adults with classic galactosemia. J Inherit Metab Dis 40: 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick AB, Zinsli AM, Carlock G, Conneely K, Fridovich-Keil JL (2018) Presentation, progression, and predictors of ovarian insufficiency in classic galactosemia. J Inherit Metab Dis 41: 785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridovich-Keil JL, Berry GT (2022) Pathophysiology of long-term complications in classic galactosemia: What we do and do not know. Mol Genet Metab 137: 33–39. [DOI] [PubMed] [Google Scholar]
- Fridovich-Keil JL, Carlock G, Patel S, Potter NL, Coles CD (2022) Acute and early developmental outcomes of children with Duarte galactosemia. JIMD Rep 63: 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridovich-Keil JL, Gambello MJ, Singh RH, Sharer JD (1993) Duarte Variant Galactosemia. In Adam MP, Mirzaa GM, Pagon RA et al. eds. GeneReviews((R)) Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E (2011) Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis 34: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DF, Camelo JS Jr., Molfetta GA, et al. (2016) Clinical profile and molecular characterization of Galactosemia in Brazil: identification of seven novel mutations. BMC Med Genet 17: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG (2009) Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 42: 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskovic M, Coelho AI, Bierau J, et al. (2020) Pathophysiology and targets for treatment in hereditary galactosemia: A systematic review of animal and cellular models. J Inherit Metab Dis 43: 392–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden HM, Rayment I, Thoden JB (2003) Structure and function of enzymes of the Leloir pathway for galactose metabolism. The Journal of biological chemistry 278: 43885–43888. [DOI] [PubMed] [Google Scholar]
- Hughes J, Ryan S, Lambert D, et al. (2009) Outcomes of siblings with classical galactosemia. The Journal of pediatrics 154: 721–726. [DOI] [PubMed] [Google Scholar]
- Jenness R (1974) Proceedings: Biosynthesis and composition of milk. J Invest Dermatol 63: 109–118. [DOI] [PubMed] [Google Scholar]
- Joshi A, Kale S, Chandel S, Pal D (2015) Likert Scale: Explored and Explained. British Journal of Applied Science and Technology 7: 396–403. [Google Scholar]
- Lai K, Langley SD, Singh RH, Dembure PP, Hjelm LN, Elsas LJ (1996) A prevalent mutation for galactosemia among black Americans. Journal of Pediatrics 128: 89–95. [DOI] [PubMed] [Google Scholar]
- Pyhtila BM, Shaw KA, Neumann SE, Fridovich-Keil JL (2015) Newborn screening for galactosemia in the United States: looking back, looking around, and looking ahead. JIMD Rep 15: 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehman K, Crews C, Fridovich-Keil JL (2001) Relationship between genotype, activity, and galactose sensitivity in yeast expressing patient alleles of human galactose-1-phosphate uridylyltransferase. The Journal of biological chemistry 276: 10634–10640. [DOI] [PubMed] [Google Scholar]
- Rubio-Gozalbo ME, Gubbels CS, Bakker JA, Menheere PP, Wodzig WK, Land JA (2010) Gonadal function in male and female patients with classic galactosemia. Human reproduction update 16: 177–188. [DOI] [PubMed] [Google Scholar]
- Rubio-Gozalbo ME, Haskovic M, Bosch AM, et al. (2019) The natural history of classic galactosemia: lessons from the GalNet registry. Orphanet J Rare Dis 14: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer S, Shin Y, Jakobs C, Brodehl J (1993) Long-Term Outcome in 134 Patients with Galactosemia. European journal of pediatrics 152: 36–43. [DOI] [PubMed] [Google Scholar]
- Segal S (1969) The Negro variant of congenital galactosemia. In Hsia DY ed. Galactosemia Springfield: Charles C. Thomas, 176-. [Google Scholar]
- Spencer JB, Badik JR, Ryan EL, et al. (2013) Modifiers of ovarian function in girls and women with classic galactosemia. The Journal of clinical endocrinology and metabolism 98: E1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukey JW (1949) Comparing Individual Means in the Analysis of Variance. Biometrics 5: 99–114. [PubMed] [Google Scholar]
- Waggoner DD, Buist NR, Donnell GN (1990) Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis 13: 802–818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data for this manuscript are included in Supplemental Tables.