

Abstract
Background:
Proliferation of vascular smooth muscle cells (VSMCs) is a hallmark of arterial diseases, especially in arterial restenosis following angioplasty or stent placement. VSMCs reprogram their metabolism to meet the increased requirements of lipids, proteins and nucleotides for their proliferation. De novo purine synthesis is one of critical pathways for nucleotide synthesis. However, its role in proliferation of VSMCs in these arterial diseases has not been defined.
Methods:
De novo purine synthesis in proliferative VSMCs was evaluated by LC-MS/MS. The expression of ATIC (5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase), the critical bifunctional enzyme in the last two steps of the de novo purine synthesis pathway, was assessed in VSMCs of proliferative arterial neointima. Global and VSMC-specific knockout of Atic mice were generated and used for examining the role of ATIC-associated purine metabolism in the formation of arterial neointima and atherosclerotic lesions.
Results:
In this study, we found that de novo purine synthesis was increased in proliferative VSMCs. Upregulated purine synthesis genes, including ATIC, were observed in the neointima of the injured vessels and atherosclerotic lesions both in mice and humans. Global or specific knockout of Atic in VSMCs inhibited cell proliferation, attenuating the arterial neointima in models of mouse atherosclerosis and arterial restenosis.
Conclusions:
These results reveal that de novo purine synthesis plays an important role in VSMC proliferation in arterial disease. These findings suggest that targeting ATIC is a promising therapeutic approach to combat arterial diseases.
Keywords: ATIC, de novo purine synthesis, vascular smooth muscle cells, proliferation, atherosclerosis, arterial diseases
Introduction
Vascular smooth muscle cells (VSMCs) are the major cells in the medial layers of arteries and normally exhibit a contractile phenotype that contributes to the regulation of blood vessel tone, blood flow distribution and blood pressure in normal mature blood vessels. In response to vascular injury during disease progression, VSMCs switch to an activated, synthetic and proliferative phenotype1 and contribute to the development of a variety of arterial diseases, including atherosclerosis, in-stent restenosis, bypass graft occlusion and hypertension2, 3. Thus, targeting VSMC proliferation is critical in the treatment of arterial disorders.
Nucleotides (e.g., ATP, GTP, dATP, dGTP, TTP, CTP, dTTP and dCTP) are essential for a large number of biological processes in cells. Proliferative cells require increased nucleotide synthesis for DNA replication and RNA production to meet the enhanced demand for protein synthesis during cell proliferation4. To obtain adequate nucleotide pools, increased expression of genes associated with nucleotide synthesis, including genes involved in purine and pyrimidine synthesis, is required. Nucleotides can be acquired through salvage pathways via the recycling of existing nucleosides and nucleobases and exogenous nucleoside uptake5, or through de novo synthesis from small molecule precursors such as glutamine, glycine, aspartate and N10-formyl-tetrahydrofolate (THF)6. While the salvage pathways are the predominant pathways supplying nucleotides to quiescent terminally differentiated cells, a few studies have indicated that de novo synthesis of nucleotides can provide nucleotides to highly growing and proliferating cells7–10.
De novo purine synthesis (DNPS) produces purines through 10 sequential steps that convert 5-phosphoribose-1-pyrophosphate (PRPP) to inosine monophosphate (IMP) using six enzymes11. These six enzymes include one trifunctional enzyme (GART), two bifunctional enzymes (PAICS and ATIC), and three monofunctional enzymes (GPAT, FGAMS, and ADSL)11. The synthesized IMP is converted into guanosine monophosphate (GMP) or adenosine monophosphate (AMP), which can be utilized for RNA and DNA synthesis7 (Figure 1A). Emerging evidence has implicated that enhanced DNPS is tightly associated with the cell proliferation of cancers10, 12–14. However, the role and regulation of DNPS in vascular cells have not been examined.
Figure 1. De novo purine metabolism is increased in VSMCs of proliferative arterial diseases.

(A) Schematic showing the purine biosynthetic pathway and associated enzymes and metabolites. (B) Heat map showing the gene expression of venous bypass grafts from rabbit at indicated time points after transplantation by re-analysis of reported microarray dataset. (C) Heat map displaying the fold change of mRNA levels of indicated genes in the carotid arteries from mice with or without ligation injury for 5 days (n = 4–7). (D) Heat map showing fold change of mRNA levels of indicated genes in human saphenous vein grafts after ex vivo culture for 7 days (n = 3–6). (E) Representative Western blots and their quantification showing indicated protein expression in carotid arteries from mice after sham or ligation injury for 7 days (n = 6). (F) Representative Western blots and their quantification showing indicated protein expression in human saphenous vein grafts after ex vivo culture for 7 days (n = 6). (G) Schematic of the incorporation of nitrogen and carbon from glutamine, glycine and formate into the purine ring. (H) Normalized peak areas of 15N-labeled purine intermediates, as measured by targeted LC-MS/MS, in resting and proliferative HCSMCs labeled with 4 mM 15N-amide-glutamine for 4 h (n = 3). (I) Normalized peak areas of 13C-labeled purine intermediates, as measured by targeted LC-MS/MS, in resting and proliferative HCSMCs labeled with 1 mM 13C2-glycine for 4 h (n = 3). (J) Relative incorporation of 14C from glycine and formate and relative incorporation of 3H from hypoxanthine into DNA and RNA in resting and proliferative HCSMCs labeled with 2 μCi U-14C-glycine, 14C-formate and 3H-hypoxanthine for 12 hours, respectively (n = 3). The data are represented as mean ± SEM, *P < 0.05, **P < 0.01 and ***P < 0.001 for indicated comparisons.
The bifunctional enzyme, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase (ATIC), catalyzes the last two essential steps of the DNPS pathway, converting 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) to IMP. It has been reported that ATIC expression is increased in many cancer cells and could be a promising target of DNPS in the development of cancer therapies. In this study, we showed that ATIC-mediated DNPS is the dominant source of DNA/RNA building blocks for proliferative VSMCs, and global or specific deletion of ATIC in VSMCs markedly attenuated neointima formation and atherosclerotic lesions in mice. This study reveals an important causal role of DNPS in the proliferation of VSMCs and subsequent arterial disorders and identifies ATIC as a promising target for the treatment of arterial diseases.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. The sources of reagents and detailed methods are provided in the Supplemental Material.
Animal Experiments
Mice were housed in temperature-controlled cages on a 12:12-hour light:dark cycle and given free access to water and normal chow. All animal experiments and care were approved by the IACUC (Institutional Animal Care & Use Committee) of Augusta University in accordance with the National Institutes of Health guidelines. Researchers were blinded to the assignment of diet and evaluation of the experimental outcomes.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism (La Jolla, CA, USA). Data are represented as mean ± SEM. Data normality was assessed via the Shapiro-Wilk test. Brown-Forsythe test or F test were used to evaluate the homogeneity of variance. Two-group comparisons were performed with unpaired/paired two-tailed t-test or unpaired two-tailed t-test with Welch’s correction (for the cases where the variances were unequal) when normal distribution was satisfied. Otherwise, nonparametric Mann-Whitney test or Wilcoxon matched-pairs signed rank test was used for data sets with non-normal distribution. Multiple group comparisons were performed with one-way ANOVA followed by a Bonferroni post hoc analysis, two-way ANOVA followed by Tukey post hoc test (for experiments with two factors) or Brown-Forsythe ANOVA and Welch’s ANOVA test with Dunnett’s T3 multiple comparison (for the cases where the variances were unequal) when normal distribution was satisfied; otherwise, non-parametric Kruskal-Wallis test followed by Dunn’s post hoc analysis was used. Mixed-effect ANOVA followed by a Bonferroni post hoc test was used for growth curve statistical analysis. Tests used for statistical analysis for each experiment are specified in Table S3. Statistical significance was defined as follows: *p < 0.05, **p < 0.01, ***p < 0.001; ns indicates no significant difference. All biological experiments were repeated at least three times using independent cell cultures or individual animals (biological replications).
Results
De novo purine synthesis is upregulated in proliferative VSMCs
To explore the relevance of de novo purine synthesis and arterial diseases, published microarray data of rabbit vein grafts (GEO accession: GSE110398) were re-analyzed. Heat map analysis revealed that genes responsible for DNPS, including PRPS1, PPAT, GART and ATIC, were upregulated at 1 and 7 days post bypass grafting in rabbit (Figure 1B), and this was accompanied by increased proliferative markers and decreased contractile VSMC markers (Figure 1B). The ligated carotid artery mouse model is an animal model that mimics proliferative arterial disorder in humans15. We examined the mRNA expression of purine synthesis genes in ligated mouse carotid arteries with qPCR analysis. The mRNA levels of DNPS-associated genes, including the key enzymes Ppat, Gart, Pfas, Paics as well as Atic, were much higher in the left ligated carotid arteries at day 5 post ligation than those in the right control carotid arteries. These dynamic changes of DNPS-associated genes in sham and ligated arteries paralleled with those of genes associated with proliferation (Figure 1C, Figure S1A). The mRNA levels of Hprt, which is responsible for purine salvage synthesis, was increased in the left ligated carotid arteries, whereas the mRNA levels of other genes of purine salvage synthesis, including Aprt, were comparable between ligated and control arteries (Figure S1A). Additionally, we cultured human saphenous veins for 7 days to induce neointima formation of vein grafts. These samples were assayed with qPCR, and mRNA levels of DNPS-associated genes were also increased, which is consistent with the proliferative marker expression pattern (Figure 1D, Figure S1B–C). Increased expression of some of the above DNPS genes, such as ATIC and GART, in the ligated carotid arteries and human vein grafts with neointima was validated at the protein level with Western blots. Notably, the enhanced ATIC, GART, and PFAS expression was in line with upregulation of the proliferative marker PCNA and downregulation of the contractile smooth muscle cell (SMC) markers Calponin 1 and SMMHC at the protein level (Figure 1E–F), supporting the association of DNPS with proliferation.
Platelet-derived growth factor (PDGF), which promotes VSMC proliferation (Figure S1D), significantly upregulated the mRNA levels of DNPS genes in human coronary smooth muscle cells (HCSMCs) (Figure S1E). To determine the effect of upregulated DNPS genes on de novo purine metabolism, we measured the relative DNPS flux in PDGF-induced HCSMCs. PDGF-treated HCSMCs cultured in medium with 5% dialyzed FBS were incubated with stable isotope-labeled 15N-amide-glutamine and 13C2-glycine, both of which contribute to purine ring synthesis (Figure 1G). The cell extracts were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The normalized peak area of 15N-glutamine was lower in proliferative HCSMCs compared to resting HCSMCs, which may reflect the increased utilization of 15N-glutamine in proliferative VSMCs. Moreover, the normalized peak area of 15N labeled purine intermediates, including purine nucleotides (IMP, AMP, ADP, ATP, GMP, GDP) and purine deoxynucleotides (dAMP, dATP, dGDP, dGTP), were enhanced in response to proliferative signal stimulation (Figure 1H and S1F). Similarly, isotopic tracing with 13C2-glycine, which provides two carbon atoms to the purine ring (Figure 1G), also revealed that de novo purine synthesis in proliferative VSMCs was significantly increased (Figure 1I and S1G). Furthermore, we used the radiotracers U-14C-glycine and 14C-formate to examine the contribution of DNPS to newly assembled nucleic acid synthesis (Figure 1G), and used G-3H-hypoxanthine to determine the purine salvage pathway to nucleic acid synthesis in proliferative VSMCs (Figure S1H). As shown in Figure 1J, the incorporation of 14C from glycine and formate, but not 3H from hypoxanthine, into DNA/RNA was dramatically increased in proliferative VSMCs, indicating that de novo purine synthesis is highly associated with de novo RNA/DNA synthesis and subsequent proliferation of VSMCs.
ATIC is upregulated in VSMCs of proliferative arterial diseases
To study the role of de novo purine synthesis in proliferative arterial disease, we firstly examined the expression of ATIC, the bifunctional enzyme of the last two steps in DNPS, in VSMCs of proliferative arterial diseases. Immunofluorescence staining of ATIC and the smooth muscle marker ACTA2 as well as proliferative marker Ki67 revealed a much greater level of ATIC expression in proliferative VSMCs in the intima area of human atherosclerotic lesions than that in the media layer (Figure S2A). Immunostaining of ATIC, ACTA2 and EdU in mouse carotid arteries showed that ATIC was expressed at a higher level by proliferative VSMCs in the neointima area of ligated carotid arteries than VSMCs in the media layer of ligated carotid arteries or uninjured mouse carotid arteries (Figure 2A–B). ATIC expression was also increased in proliferative VSMCs of atherosclerotic plaques from Apoe−/− mice compared to that of C57BL/6 wild-type (WT) mice (Figure 2C–D).
Figure 2. ATIC expression is upregulated in proliferative VSMCs in arterial disease.

(A) Representative and quantification immunofluorescent staining of ATIC and EdU in the carotid arteries from mice after ligation injury for 4 weeks. The area highlighted with the white line is the arterial intima (I) and media (M) (n = 8–9). (B) Representative immunohistochemical (IHC) staining of ATIC in the carotid arteries from mice after ligation injury for 4 weeks. (C-D) Representative (C) and quantification (D) immunofluorescent staining of ATIC and EdU on the aortic sinus from Apoe−/− or wild-type (WT) mice fed with Western diet for 3 months (n = 6–9). The data are represented as mean ± SEM, *P < 0.05, **P < 0.01 and ***P < 0.001 for indicated comparisons.
We examined ATIC expression in proliferative VSMCs in vitro. As shown in Figure S2B, PDGF significantly upregulated ATIC protein expression in HCSMCs. Additionally, targeted re-analysis of published RNA-seq data (GEO accession: GSE69637) showed that ATIC was upregulated in human saphenous vein SMCs following stimulation with PDGF and IL-1α (Figure S2C). Re-analysis of RNA-seq data of rat VSMCs (GEO accession: GSE111714) showed a marked increase of Atic in rat VSMCs treated with PDGF for 24 hours (Figure S2D). Collectively, these data reveal that ATIC expression is increased in proliferative VSMCs in human and rodent diseased arteries, and in cultured activated VSMCs, suggesting that ATIC-mediated DNPS induction is highly associated with proliferative VSMCs.
ATIC knockdown decreased de novo purine synthesis-mediated nucleic acid assembling in proliferative VSMCs
We determined the role of ATIC in de novo purine synthesis in proliferative VSMCs using ATIC siRNA (Figure S3A–B). The peak areas of 15N-glutamine in ATIC knockdown and control HCSMCs were comparable. However, a significant accumulation of 15N labeled AICAR was seen in ATIC knockdown cells, indicating that ATIC knockdown impedes de novo purine metabolic flux (Figure 3A). Subsequently, lower levels of 15N labeled de novo purine products, including AMP, IMP, GTP, ATP, GMP and dAMP, were seen in ATIC-knockdown proliferative HCSMCs than in control cells (Figure 3A and S3C). Furthermore, U-14C-glycine radiolabeled DNA and RNA was decreased in ATIC-knockdown HCSMCs compared with that of controls (Figure 3B), demonstrating that ATIC-mediated de novo purine synthesis is critical for de novo nucleic acid assembling in proliferative VSMCs. We applied EdU and EU incorporation assays to evaluate the overall DNA and RNA synthesis rates. The percentage of EdU-positive cells was lower in proliferative ATIC-knockdown HCSMCs than that of control cells (Figure 3C), and similar results were obtained using alternative ATIC siRNA pools that targeted a different site of ATIC mRNA (Figure S3D–F). In addition, the level of EU incorporation into ATIC knockdown cells was reduced compared with that in control cells in vehicle- and PDGF-activated HCMSCs, respectively (Figure 3D). ATIC knockdown had a slight effect on de novo protein synthesis in PDGF-activated HCSMCs (Figure S3G). These results indicate ATIC knockdown inhibits do novo nucleotide synthesis and further impedes overall nucleic acid assembling in proliferative VSMCs.
Figure 3. ATIC knockdown decreases de novo purine metabolism-mediated nucleotide synthesis in proliferative VSMCs.

(A) Normalized peak areas of 15N-labeled purine intermediates, as measured by targeted LC-MS/MS, in proliferative HCSMCs transfected with control or ATIC siRNA and labeled with 4 mM 15N-amide-glutamine for 4 h (n = 3). (B) Relative incorporation of 14C from glycine into DNA and RNA in resting and proliferative HCSMCs transfected with control or ATIC siRNA and labeled with 2 μCi U-14C-glycine for 12 hours (n = 4). (C) Representative images and quantification of flow cytometry analysis showing EdU incorporation in HCSMCs transfected with control or ATIC siRNA for 48 hours and treated with PDGF (20 ng/ml) for 18 hours (n = 3). (D) Representative images and quantification of 5-EU staining showing newly synthesized RNA synthesis in HCSMCs transfected with control or ATIC siRNA for 48 hours and treated with vehicle or PDGF (20 ng/ml) for 12 hours (n = 5). (E) Representative images and quantification of flow cytometry analysis showing EdU incorporation in HCSMCs transfected with control or ATIC siRNA for 48 hours and treated with PDGF (20 ng/ml) and supplemented with adenine (20 μM) or hypoxanthine (100 μM) for 18 hours (n = 8). (F) Representative images and quantification of 5-EU staining showing new RNA synthesis in HCSMCs transfected with control or ATIC siRNA for 48 hours and treated with vehicle or PDGF (20 ng/ml) and supplemented with adenine (20 μM) or hypoxanthine (100 μM) for 12 hours (n = 5). The data are represented as mean ± SEM, *P < 0.05, **P < 0.01 and ***P < 0.001 for indicated comparisons.
Adenine and hypoxanthine are purine bases that can be converted into purine nucleotides to supply nucleotide pools through salvage pathways (Figure 1A). We investigated whether the inhibitory effect of ATIC knockdown on DNA/RNA synthesis was rescued by the addition of exogenous purine bases that bypassed the requirement for ATIC to maintain adequate nucleotide pools. DNA and RNA synthesis assays showed that, in proliferative HCSMCs activated by PDGF, either adenine (20μM) or hypoxanthine (100μM) was sufficient to fully rescue ATIC knockdown-induced diminished DNA (Figure 3E) and RNA synthesis (Figure 3F). These results indicate that ATIC knockdown in proliferative cells results in a major change in nucleic acid synthesis.
ATIC is required to promote VSMC proliferation and cell cycle phase progression in vitro
We next determined the effect of ATIC knockdown on VSMC proliferation. Cell counting and WST-1 proliferation assays showed that the growth of ATIC-knockdown HCSMCs was remarkably decreased versus control HCSMCs (Figure 4A–4B). Consistent with the above decreased proliferation phenotype, Western blots showed that PDGF-induced Cyclin D1 and PCNA were markedly diminished in ATIC knockdown HCSMCs (Figure 4C). To investigate the role of ATIC in mice, we generated floxed Atic (AticF/F) mice (Figure S4A). AticF/F mice were then crossed with Rosa26Cre/ERT2 mice to obtain AticF/F;Rosa26Cre/ERT2 mice. We isolated and cultured mouse aortic smooth muscle cells (MASMCs) from Rosa26Cre/ERT2 and AticF/F;Rosa26Cre/ERT2 mice, and treated the cells with 4-hydroxytamoxifen to obtain Atic control and knockout (AticWT and AticiKO) MASMCs (Figure S4B–C). AticWT and AticiKO MASMCs were analyzed with proliferation assays, with results consistent with that of ATIC-knockdown HCSMCs (Figure 4D–4H). Additionally, an exogenous purine supplement of adenine (20μM) or hypoxanthine (100μM) partially rescued the ATIC knockdown-induced inhibitory effect of VSMC proliferation (Figure 4I). Together, these data demonstrate that ATIC-mediated de novo purine synthesis is required to promote VSMC proliferation.
Figure 4. ATIC is required for VSMC proliferation in vitro.

(A) Growth curves of HCSMCs transfected with control or ATIC siRNA for the indicated times (n = 6). (B) WST-1 cell proliferation assay of HCSMCs transfected with control or ATIC siRNA for 72 hours at indicated culture condition (n = 6–7). (C) Representative Western blot and quantification of the indicated proteins normalized to β-actin in HCSMCs transfected with control or ATIC siRNA for 48 hours and treated with PDGF (20 ng/ml) for 24 hours (n = 4). (D) Representative images and quantification of flow cytometry analysis of EdU staining in MASMCs isolated from AticWT or AticiKO mice (n = 7). (E) Representative EdU staining (green) and the percentage of EdU-positive MASMCs isolated from AticWT or AticiKO mice (n = 6–7). (F) Growth curves of MASMCs isolated from AticWT or AticiKO mice and cultured for the indicated times (n = 6). (G) WST-1 cell proliferation assay of MASMCs isolated from AticWT or AticiKO mice (n = 6–7). (H) Representative Western blot and quantification of the indicated proteins normalized to β-actin in MASMCs isolated from AticWT or AticiKO mice (n = 4–6). (I) Growth curves of HCSMCs transfected with control or ATIC siRNA and treated with adenine (20 μM) or hypoxanthine (100 μM) for the indicated times (n = 6). Black * for siATIC vs siCTRL, blue * for siATIC vs siATIC+Ade, green * for siATIC vs siATIC+Hyp. (J) Heat map showing the expression of cell cycle-associated genes in AticWT or AticiKO MASMCs as revealed by RNA sequencing. (K-L) Flow cytometry analysis of cell cycle for MASMCs isolated from AticWT or AticiKO mice (K) and for HCSMCs transfected with control or ATIC siRNA (L) (n = 6). The data are represented as mean ± SEM, *P < 0.05, **P < 0.01 and ***P < 0.001 for indicated comparisons.
To further gain insight on the effect of Atic deficiency on VSMCs, we performed whole-transcriptome RNA-seq analysis with AticWT and AticiKO MASMCs. We observed significant alterations in gene expression, with 716 upregulated and 563 downregulated genes in AticiKO MASMCs compared with AticWT MASMCs (Figure S4D). The functional annotation of these differentially expressed genes (DEGs) was performed by Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. GO analysis showed that downregulated DEGs in AticiKO MASMCs were mainly enriched in vasculature development, cell proliferation and differentiation genes, and upregulated DEGs were enriched in cell stress and defense responses genes (Figure S4E). KEGG pathway analysis displayed that Atic knockout (KO) significantly decreased the expression of genes involved in cancer-related pathways, Rap1 signaling, and TGF-β signaling as well as cell cycle-associated pathways. Moreover, Atic KO significantly increased the expression of genes involved in RNA degradation and vascular smooth muscle contraction (Figure S4F). Proper regulation of the cell cycle is critical for cell proliferation in eukaryotes. Previous studies have uncovered that classes of periodic genes oscillate during the cell cycle and regulate cell cycle progression16, 17. Upon further analysis of the DEGs between AticiKO and AticWT groups, we also found that a series of known cell cycle positive regulators were decreased in AticiKO MASMCs, including Ccne1, Ccne2 (Cyclin E), Pcna, Ccna2 (Cyclin A2) and Ccnb2 (Cyclin B2), which are required for the G1/M transition, S phase progression, and G2/M transition, respectively (Figure 4J). Additionally, increased mRNA levels of negative cell cycle regulators such as Cdkn2b, Cdkn1a, and Cdkn1b were also observed in AticiKO MASMCs compared to AticWT MASMCs (Figure 4J). The effect of ATIC-knockdown or Atic-KO on the cell cycle of VSMCs was determined with flow cytometry. ATIC-knockdown or Atic-KO resulted in an accumulation of cell populations in S phase, indicating S phase arrest in VSMCs (Figure 4K–4L). The above data indicate that ATIC-mediated de novo purine synthesis is crucial for cell cycle progression, and subsequent cell proliferation in VSMCs in vitro.
We also investigated whether ATIC is involved in VSMC migration and phenotypic changes. A scratch wound healing assay showed that the percentage of wound closure was comparable between control and ATIC knockdown HCSMCs after 8 hours (Figure S4G). In the Boyden chamber migration assay, there is no statistical difference in the number of migrated cells through the filter membrane between ATIC knockdown and control groups (Figure S4H). ATIC knockdown upregulated SM22α expression but had no effect on ACTA2 expression under PDGF-stimulated conditions in vitro (Figure S4I), indicating a modest role of ATIC in VSMC phenotypic change in vitro.
VSMC-specific deletion of Atic attenuates formation of arterial neointima in mice
To investigate the role of ATIC in VSMCs in vivo, AticF/F mice were crossed with Myh11Cre/ERT2 mice to generate AticF/F;Myh11Cre/ERT2 mice, which were referred to as AticiΔVSMC mice, following tamoxifen treatment (Figure 5A). Western blots with mouse medial layers of aortas showed a significant decrease in the level of ATIC in aortas of AticiΔVSMC mice versus control mice, which were Myh11Cre/ERT2 mice treated with tamoxifen (Figure 5B). To examine the effect of Atic deficiency in the VSMCs in arterial neointima formation, 9-week-old AticiΔVSMC and control mice were subjected to left common carotid artery ligation. The ligated arteries were collected 21 days later (Figure 5C). Hematoxylin-eosin (HE) staining of serial cross-sections of carotid arteries with relative distances to the ligation site showed that the neointima area and neointima/media ratio were decreased in AticiΔVSMC mice compared to those of control mice (Figure 5D–E). The percentage of EdU-positive VSMCs in the cross-sections of arterial neointima was significantly reduced in AticiΔVSMC mice compared to control mice (Figure 5F). In contrast, external elastic lamina (EEL) circumference of ligated carotid arteries in AticiΔVSMC mice did not differ from that in control mice, indicating that VSMC-specific Atic deficiency does not affect constrictive remodeling but does affect neointima hyperplasia (Figure S5A). Furthermore, compared to control Myh11Cre/ERT2 mice, Atic knockout in VSMCs significantly attenuated expression of the proliferative marker PCNA in ligated arteries 7 days post-injury, but had no effect on the reduction of the VSMC differentiation markers SMMHC and ACTA2 and had only a modest effect on SM22α (Figure 5G). Collectively, these data indicate that VSMC-specific deletion of Atic attenuates neointima formation through suppressing VSMC proliferation.
Figure 5. VSMC-specific deletion of Atic attenuates neointima formation in a model of vascular injury.

(A) Strategy for generating AticiΔVSMC mice by crossing AticF/F mice with Myh11Cre/ERT2 mice and tamoxifen treatment of the consequent mice. (B) Representative Western blot and quantification showing ATIC expression in media of aortas from Myh11Cre/ERT2 and AticiΔVSMC mice. (C) Schematic timeline of tamoxifen treatment and carotid artery ligation model. (D) Representative HE-stained cross-sections of carotid arteries from mice with or without left common carotid artery ligation for 21 days. Sections are 200 to 600μm from the site of ligation. Yellow lines indicate the internal elastic lamina. (E) Quantification of the arterial neointima area and the ratio of neointima area to medial area of injured carotid arteries. (n = 5–6). (F) Representative EdU-positive stained cells in the neointima of carotid arteries from Myh11Cre/ERT2 and AticiΔVSMC mice and their quantification(n=7). (G) Representative Western blot and quantification of the indicated proteins normalized to β-actin in the sham and ligation-injured carotid arteries of Myh11Cre/ERT2 and AticiΔVSMC mice 7 days post ligation injury (n = 4). Data are represented as mean ± SEM, *P < 0.05, **P < 0.01 and ***P < 0.001 for indicated comparisons.
Global Atic deficiency reduces formation of arterial neointima in mice
In order to evaluate ATIC as a potential therapeutic target, we assessed the effect of global Atic deficiency in neointima formation. We crossed AticF/F mice with Rosa26Cre/ERT2 mice and generated AticF/F;Rosa26Cre/ERT2 mice. These mice were referred to as AticiKO mice following tamoxifen treatment (Figure 6A). Western blots showed a marked decrease of ATIC in carotid arteries, aortas and liver, and a modest decrease in kidney, spleen and lung in AticiKO mice (Figure S6A). Immunostaining showed a significant decrease in the level of ATIC in the aortas of AticiKO mice versus AticWT mice (Figure S6B). Additionally, we examined histological changes in these tissues/organs of AticiKO mice but did not find any apparent abnormalities in tissue/organs even with markedly decreased ATIC such as in aorta and liver (Figure S6C).
Figure 6. Global Atic deficiency attenuates neointima formation in a model of vascular injury.
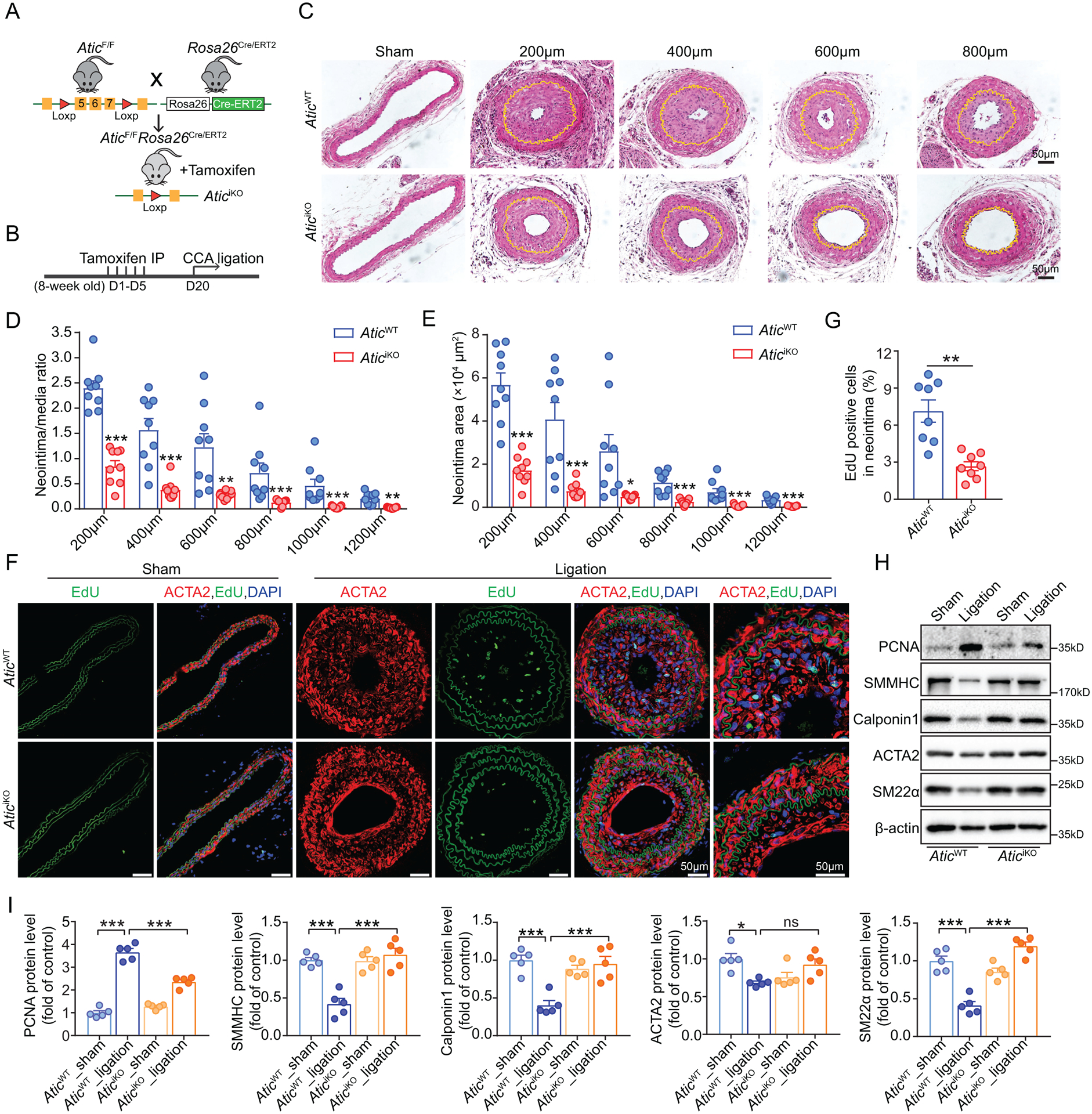
(A) Strategy for generating AticiKO mice by crossing AticF/F mice with Rosa26Cre/ERT2 mice and tamoxifen treatment of the consequent mice. (B) Schematic timeline of tamoxifen treatment and carotid artery ligation model. (C) Representative HE-stained cross-sections of carotid arteries from the mice with or without left common carotid artery ligation for 28 days. Sections are 200 to 800 μm from the site of ligation. Yellow lines indicate the internal elastic lamina. (D-E) Quantification of the arterial neointima area and the ratio of neointima area to medial area of injured carotid arteries. (n = 9). (F-G) Representative EdU-positive stained cells in the neointima of carotid arteries from AticWT and AticiKO mice and their quantification (n = 8). (H-I) Representative Western blot (H) and quantification (I) of the indicated proteins normalized to β-actin in the sham and ligation-injured carotid arteries of AticWT and AticiKO mice 7 days post ligation injury (n = 5). Data are represented as mean ± SEM, *P < 0.05, **P < 0.01 and ***P < 0.001 for indicated comparisons.
Following the experimental design indicated in Figure 6B, we performed left common carotid artery ligation injury in AticiKO and AticWT mice. HE staining of serial cross-sections of carotid arteries with relative distances to the ligation site showed that the neointima area and neointima/media ratio were dramatically decreased in AticiKO mice compared to AticWT mice (Figure 6C–6E). Furthermore, the percentage of EdU-positive VSMCs in the cross sections of arterial neointima was significantly decreased in AticiKO mice compared to AticWT mice (Figure 6F–6G). Nonetheless, the EEL circumference of ligated common carotid artery in AticiKO mice did not differ from that in AticWT mice, indicating that Atic global deficiency had no effect on constrictive remodeling after ligation injury (Figure S5B). Additionally, we also found that, compared to control AticWT mice, Atic knockout significantly attenuated induction of the proliferative marker PCNA in ligated arteries 7 days post-injury, which was accompanied with upregulation of decreased VSMC differentiation markers SMMHC, Calponin 1, ACTA2 and SM22α in the injured carotid arteries of AticiKO mice (Figure 6H–6I). Collectively, these data imply that ATIC is required to promote a differentiated-to-proliferative phenotypic switch of VSMCs in vivo. We compared the intima/media ratio and intima area of AticiKO (Figure 6D–6E) and AticiΔVSMC (Figure 5E) mice to the corresponding perimeters of their controls and found that the inhibitory effect on neointima formation was much greater in AticiKO mice than AticiΔVSMC mice at sites of 600, 800 and 1000 μm (Figure S7).
VSMC-specific or global deletion of Atic alleviates atherosclerosis in Apoe−/− mice
VSMC proliferation plays a vital role in the development of atherosclerotic lesions2, 3. To investigate the involvement of VSMC Atic in formation of atherosclerotic lesions, we bred the AticF/F;Myh11Cre/ERT2 mice with Apoe−/− mice to generate Apoe−/−;AticF/F;Myh11Cre/ERT2 mice. These mice and their controls were treated with tamoxifen, and the consequent Apoe−/−;AticiΔVSMC mice and controls were fed with Western diet for 12 weeks. No significant differences in the level of plasma cholesterol, triglycerides, or glucose were observed in these mice (Figure S8A). Oil Red O staining of whole aortas collected from these mice showed a significant decrease in lesion size in Apoe−/−;AticiΔVSMC mice compared to control mice (Figure 7A). HE staining and Oil Red O staining also showed smaller lesion areas and less lipid deposition in the aortic sinuses of Apoe−/−;AticiΔVSMC mice than control mice (Figure 7B). To further characterize the VSMC composition in atherosclerotic lesions, we performed ACTA2 staining to identify smooth muscle cells in the aortic sinus. The result showed that the ACTA2-positive area of the lesions was significantly decreased in the aortic sinus of Apoe−/−;AticiΔVSMC mice compared with that of Apoe−/−;Myh11Cre/ERT2 mice (Figure 7C). Thus, VSMC-specific deletion of Atic significantly decreased the VSMC content in the atherosclerotic lesions and reduced atherosclerotic lesion size in Apoe−/− mice fed with Western diet.
Figure 7. VSMC-specific and global Atic deficiency alleviates atherosclerosis in Apoe−/− mice fed with Western diet for 12 weeks.

(A) Representative images and quantification of Oil Red O staining of en face aortas from Apoe−/−;Myh11Cre/ERT2 and Apoe−/−;AticiΔVSMC mice fed with Western diet for 12 weeks (n = 12–14). (B) Representative HE (upper) and Oil Red O (lower) staining of aortic sinuses from Apoe−/−;Myh11Cre/ERT2 and Apoe−/−;AticiΔVSMC mice fed with Western diet for 12 weeks (n = 9). (C) Representative ACTA2 staining of smooth muscle cells in aortic sinuses from Apoe−/−;Myh11Cre/ERT2 and Apoe−/−;AticiΔVSMC mice fed with Western diet for 12 weeks and their quantification (n = 8–9). (D) Representative images and quantification of Oil Red O staining of en face aortas from Apoe−/−;AticWT and Apoe−/−;AticiKO mice fed with Western diet for 12 weeks (n = 11). (E) Representative HE (upper) and Oil Red O (lower) staining of aortic sinuses from Apoe−/−;AticWT and Apoe−/−;AticiKO male mice fed with Western diet for 12 weeks (n = 7–11). (F) Representative ACTA2 staining of smooth muscle cells in aortic sinuses from Apoe−/−;AticWT and Apoe−/−;AticiKO male mice fed with Western diet for 12 weeks and their quantification (n = 8–9). Data are represented as mean ± SEM, **P < 0.01 and ***P < 0.001 for indicated comparisons.
To investigate the effect of global Atic deficiency in atherosclerosis development, we bred the AticF/F;Rosa26Cre/ERT2 mice with Apoe−/− mice to generate Apoe−/−;AticF/F;Rosa26Cre/ERT2 mice. These mice and their controls were treated first with tamoxifen, and the consequent Apoe−/−;AticiKO mice and controls were fed with Western diet for 12 weeks to induce atherosclerosis. These mice exhibited no significant differences in the level of plasma glucose in both males and females, and plasma triglycerides in males as well as cholesterol in females, whereas it was noted that male Apoe−/−;AticiKO mice showed a modest decrease in plasma cholesterol and female Apoe−/−;AticiKO mice showed an increase in plasma triglycerides compared to their controls (Figure S8B–C). Oil Red O staining of whole aortas revealed that the size of atherosclerotic lesions was significantly reduced in Apoe−/−;AticiKO mice compared to that in Apoe−/−;AticWT mice in both male (Figure 7D) and female (Figure S8D) mice, respectively. In addition, the smaller lesion size and less lipid deposition were also observed in the aortic sinuses of Apoe−/−;AticiKO mice of both genders (Figure 7E and S8E–F). ACTA2 staining of aortic sinuses also showed a decreased VSMC content in atherosclerotic lesions of Apoe−/−;AticiKO mice compared with that of Apoe−/−;AticWT mice of both genders (Figure 7F and S8G). Thus, these data suggest that ATIC participates in proliferation of VSMCs in atherosclerotic lesions and facilitates the development of atherosclerosis in Apoe−/− mice fed with a Western diet. We compared the inhibitory effect on the lesion size (lesion of both en face aorta and aortic sinus) as well as ACTA2 area in lesions between Apoe−/−;AticiKO (Figure 7D–F) and Apoe−/−;AticiΔVSMC (Figure 7A–C) mice. There was a trend of a greater inhibitory effect on lesion size and SMC content in Apoe−/−;AticiKO mice compared to those of Apoe−/−;AticiΔVSMC mice, although the difference did not reach statistical significance (Figure S8H).
ATIC homodimerization inhibitor can improve metabolic syndrome in a mouse model through modulation of de novo purine synthesis18. We fed AticiKO and AticWT mice with high fat diet (HFD) to examine the role of ATIC in HFD-induced metabolic syndrome. AticiKO and AticWT mice displayed no significant differences in HFD-induced body-weight gain of both genders (Figure S9A–B), fat content or leanness of the body mass (Figure S9C–D), or the fasting blood glucose level (Figure S9E), resulting in no improvement in glucose clearance and insulin sensitivity as determined by glucose tolerance test (GTT) and insulin tolerance test (ITT), respectively (Figure S9F–I).
Discussion
In this study, we demonstrate that ATIC-mediated DNPS is enhanced to supply purine nucleotides for incorporation into DNA/RNA synthesis in proliferative VSMCs, which contributes to the formation of arterial neointima and atherosclerotic lesions (Figure 8). DNPS suppression by Atic VSMC-specific or global deletion reduces formation of arterial neointima and atherosclerotic lesions in mice, indicating that ATIC is a promising target for treatment of proliferative arterial diseases.
Figure 8. Role of ATIC-mediated de novo purine synthesis in VSMC proliferation in arterial proliferative diseases.
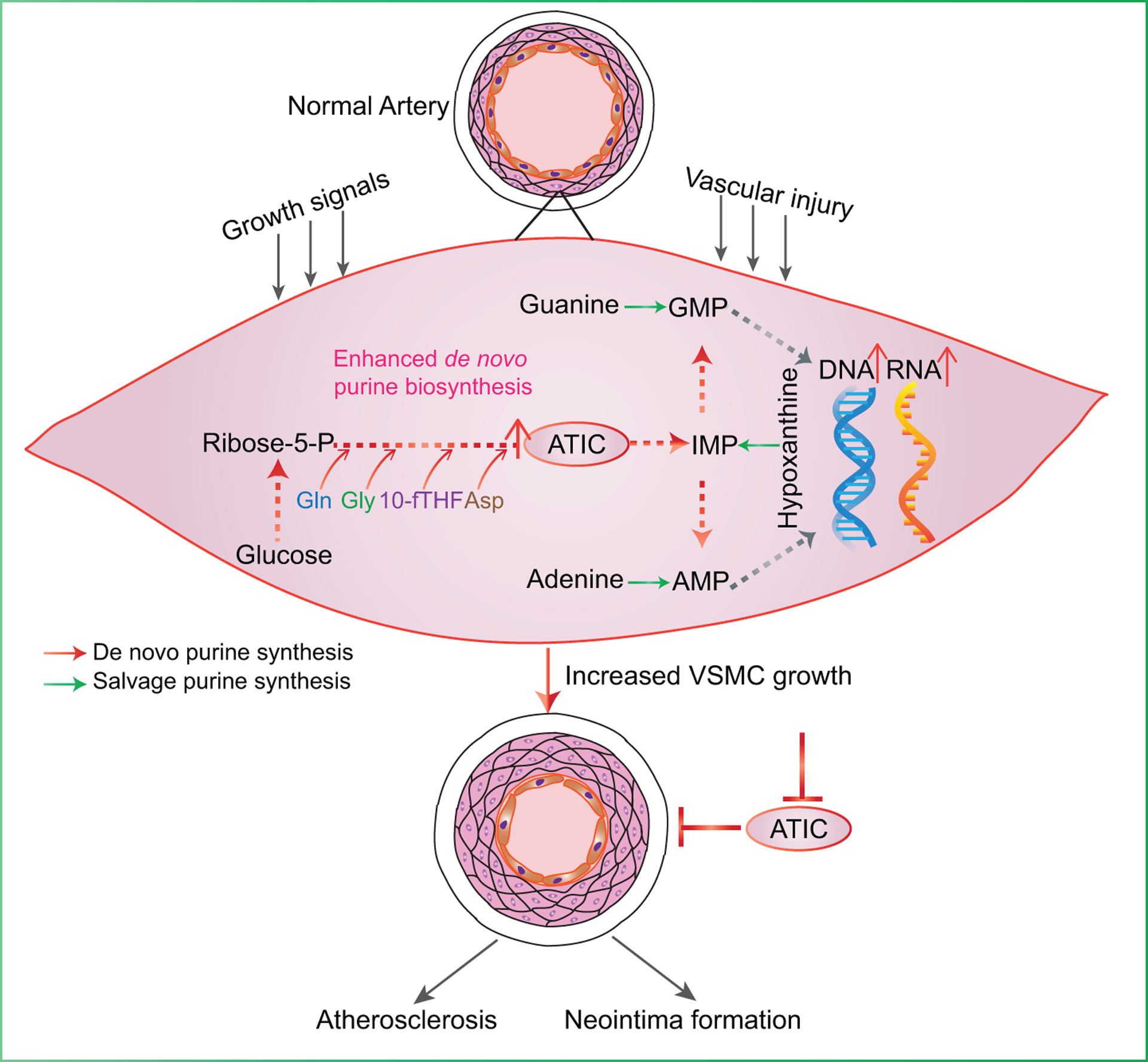
ATIC-mediated de novo purine synthesis is enhanced when cells are exposed to the stimulation of injury or growth signals in VSMCs, which can supply purine nucleotides for incorporation into DNA/RNA synthesis to meet the demand for VSMC proliferation. Proliferative VSMCs ultimately contribute to the formation of arterial neointima and atherosclerotic lesions.
DNPS is an important metabolic feature for proliferative VSMCs and proliferative arterial diseases. In the development of many proliferative arterial diseases, VSMCs switch to a proliferative phenotype. This is driven or accompanied by cellular metabolic adaptations. Past studies have emphasized involvement of increased glucose metabolism19, glutamine uptake20 and long-chain fatty acid metabolism21 in VSMC proliferation and development of proliferative arterial diseases. However, the role of purine metabolism and its regulation in VSMC proliferation and related arterial disorders remains largely unknown. A prominent finding in this study is that proliferative VSMCs stimulate purine synthesis. This was first evidenced with increased gene expression for most of the major enzymes of de novo purine synthesis and some enzymes of the salvage pathway of purine synthesis in both proliferative vessel walls and in vitro cultured proliferative VSMCs. A high rate of DNPS shifting is attributed to many signaling pathways including phosphatidylinositol 3-kinase-Akt (PI3K/Akt)22, MYC10, and mechanistic target of rapamycin complex 1 (mTORC1)23. Bioinformatics analysis of the human ATIC promoter reveals a conserved DNA-binding motif for MYC in the promoter region of the ATIC gene, suggesting a possible role of MYC in transcriptional regulation of ATIC expression. Very likely, c-MYC-mediated upregulation of purine metabolism genes, including ATIC, is one of the major pathways for increased purine metabolism, although this mechanism needs further investigation in a future study.
DNPS is associated with increased synthesis of nucleotides and nucleic acids in proliferative cells. Proliferative VSMCs with increased expression of DNPS display high levels of nucleotides and nucleic acids including RNA and DNA, which are evidenced by higher levels of these traced metabolites in proliferative VSMCs than resting cells. It has been reported that de novo purine synthesis highly correlates with cell cycle progression. In proliferating cells, purine synthesis is upregulated during the G1 and S phases of the cell cycle to meet the nucleotide demand24. In T cells, purine nucleotides regulate both the G1 to S phase transition and progression of S phase25. Another study illustrates that de novo synthesis of purine nucleotides increases the cell growth rate through promotion of the G1 to S transition26. ATIC deficiency directly constrained the DNPS flux and inhibited DNA/RNA synthesis, further leading to arrest of the cell cycle of proliferative VSMCs. Therefore, it is conceivable that the nucleotide demand for cell cycle progression may be attributed to ATIC-mediated purine nucleotide synthesis in proliferative VSMCs.
Our data also showed that some of the genes associated with the purine salvage pathway, such as hypoxanthine guanine phosphoribosyltransferase (HPRT), were increased in proliferative vessel walls and PDGF-treated VSMCs (Figure 1B, S1A, S1E). However, a tracing assay indicates a modest contribution of purine salvage synthesis to DNA/RNA synthesis in cultured proliferative VSMCs (Figure 1J). Recent studies have demonstrated a crucial role of purine salvage synthesis in cell proliferation27, 28. Thus, the role of the salvage purine pathway in VSMCs, especially in the formation of arterial proliferation in vivo, requires further study.
ATIC deletion does not modulate activity of AMP-activated protein kinase (AMPK) in VSMCs. AICAR is the substrate of ATIC and accumulates as a result of ATIC inactivation29. AICAR, as an analogue of AMP30, binds to the AMPKγ subunit and activates AMPK31 and its downstream signaling pathways18, 32, 33. Past studies with methotrexate (MTX), a drug with an inhibitory effect on ATIC, showed AMPK activation and consequent antitumor34, 35 and anti-diabetic effects36. Recently, studies with a novel specific ATIC inhibitor also shows AMPK activation in its inhibitory effect on tumor growth37 and improvement on metabolic disorder18. In our study, ATIC-knockdown VSMCs showed an increased level of 15N labeled AICAR (Figure 1I); however, no AMPK activation was observed in ATIC-knockdown VSMCs (Figure S10A), indicating, when compared with cells used in other studies, that AICAR accumulation in VSMCs may not reach to the level that activates AMPK or that the AMPK subunit of VSMCs is not sensitive to accumulated AICAR.
ATIC deletion-associated DNPS inhibition does not reduce mTORC1 signaling in VSMCs. mTORC1 is a nutrient sensor and its activation state is influenced by nutrient availability such as amino acids, glucose and cholesterol38–40. Recent studies have demonstrated that mTORC1 can also sense intracellular purine nucleotide availability through Rheb GTPase, and thereafter purine synthesis inhibition lowers the mTORC1 activity, leading to suppressed proliferation of tumor cells41, 42. However, our study did not show decreased P-S6K1 and P-S6, the downstream signaling of mTORC1 in ATIC-knockdown VSMCs compared with control cells (Figure S10B), indicating that ATIC knockdown-mediated inhibition of VSMC proliferation is independent of the mTORC1/S6K1 pathway.
ATIC inhibition is a novel approach to target VSMC proliferation and reduce neointimal formation. VSMC proliferation is a hallmark of arterial proliferative diseases. In the past decades, many strategies to target VSMC proliferation, including cyclin-dependent kinase (CDK) inhibitors43, vascular brachytherapy44, antisense molecules45 and paclitaxel coating on stent46 have either not been developed further, not been widely adopted or are being currently questioned by recent systematic review and meta-analysis, implying the necessity to develop other approaches or molecules in the treatment of proliferative artery diseases. VSMC-specific Atic deletion reduces arterial neointima formation and formation of atherosclerotic lesions, and a similar effect was seen in global Atic-inducible deletion mice. With evidence of increased ATIC gene expression in proliferative vessel walls of humans, it will be interesting to further test the effect of ATIC inhibition in large animals and humans.
ATIC has long been regarded as a druggable target. The beneficial effect of MTX in the treatment of tumors, autoimmune diseases and rheumatological disorders has been attributed to its inhibition of ATIC47–49. A few medicinal small molecules that inhibit ATIC have been recently generated. These ATIC inhibitors showed either an anti-proliferative effect on cancer cells or improved diet-induced metabolic syndrome in mice18, 37, 50. In our study these inhibitors showed a modest suppression on VSMC proliferation in vitro (Figure S11). With the continuous effort on medicinal development of ATIC inhibitors in the field, ATIC compounds with greater inhibitory effect on VSMC proliferation is expected to be available soon and will be a promising approach in the treatment of proliferative arterial disease. ATIC mutation causes human fetal abnormality29. Inducible Atic deletion mice are viable with normal growth and histological abnormality for the major organs even these organs are with marked loss of ATIC are not observed. Thus, ATIC inhibition may affect growth or development, but is a safe target in adults for disease treatment. Further study on the side effects of ATIC inhibition will indicate whether ATIC inhibition is suitable for systemic therapeutic application or a local application such as coat of stent.
Supplementary Material
Clinical Perspective.
What is new?
ATIC-mediated de novo purine metabolism was enhanced to support nucleotide synthesis in proliferative VSMCs.
Global or VSMC-specific deletion of Atic inhibited cell proliferation and attenuated the arterial neointima formation in models of mouse atherosclerosis and arterial restenosis.
What are the clinical implications?
Our findings provide novel insights into the reprogramming of purine metabolism underlying VSMC proliferation in the development of arterial diseases.
Our study reveals that targeting ATIC is a promising therapeutic approach to combat arterial diseases.
Acknowledgments
QM, JMA, IBS, CZ and YH designed the study; QM, QY, JX, and GW provided the methods, conducted the experiments and analyzed the data; XZ, DK, ZL, XM, YZ, YC, QD, HWK and MH conducted the experiments and analyzed the data; ZD, LG, WS, NLW, DF and JM provided critical suggestions and insight during the study, and QM and YH wrote the manuscript. All authors reviewed the report and approved the final version.
Funding Sources
This work was supported in part by grants from National Science Foundation of China Grant 82070461; Shenzhen Science and Technology Innovation Committee Grants GXWD20201231165807007-20200818123312001; Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions 2019SHIBS0004; American Heart Association Grant 19TPA34910043; National Institutes of Health Grants R35CA197459, P01 CA120964, R01HL142097, R01EY030500, R01EY033369, R01EY033737 and R01GM024129.
Non-standard Abbreviations and Acronyms
- VSMC
vascular smooth muscle cell
- ATIC
5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/inosine monophosphate cyclohydrolase
- DNPS
de novo purine synthesis
- PRPP
5-phosphoribose-1-pyrophosphate
- IMP
inosine monophosphate
- GMP
guanosine monophosphate
- AMP
adenosine monophosphate
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- PDGF
platelet-derived growth factor
- HCSMC
human coronary smooth muscle cell
- HPRT
hypoxanthine guanine phosphoribosyltransferase
- AMPK
AMP-activated protein kinase
- MTX
methotrexate
Footnotes
Conflict of Interest Disclosures
None.
References
- 1.Owens GK, Kumar MS and Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz SM, deBlois D and O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–65. [DOI] [PubMed] [Google Scholar]
- 3.Dzau VJ, Braun-Dullaeus RC and Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–56. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J and Thompson CB. Metabolic regulation of cell growth and proliferation. Nature reviews Molecular cell biology. 2019;20:436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murray AW. The biological significance of purine salvage. Annu Rev Biochem. 1971;40:811–26. [DOI] [PubMed] [Google Scholar]
- 6.Hartman SC and Buchanan JM. Nucleic acids, purines, pyrimidines (nucleotide synthesis). Annu Rev Biochem. 1959;28:365–410. [DOI] [PubMed] [Google Scholar]
- 7.Lane AN and Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong X, Zhao F and Thompson CB. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr Opin Genet Dev. 2009;19:32–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villa E, Ali ES, Sahu U and Ben-Sahra I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel). 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Yang K, Xie Q, Wu Q, Mack SC, Shi Y, Kim LJY, Prager BC, Flavahan WA, Liu X, Singer M, Hubert CG, Miller TE, Zhou W, Huang Z, Fang X, Regev A, Suva ML, Hwang TH, Locasale JW, Bao S and Rich JN. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat Neurosci. 2017;20:661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pareek V, Pedley AM and Benkovic SJ. Human de novo purine biosynthesis. Crit Rev Biochem Mol Biol. 2021;56:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barfeld SJ, Fazli L, Persson M, Marjavaara L, Urbanucci A, Kaukoniemi KM, Rennie PS, Ceder Y, Chabes A, Visakorpi T and Mills IG. Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer. Oncotarget. 2015;6:12587–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goswami MT, Chen G, Chakravarthi BV, Pathi SS, Anand SK, Carskadon SL, Giordano TJ, Chinnaiyan AM, Thomas DG, Palanisamy N, Beer DG and Varambally S. Role and regulation of coordinately expressed de novo purine biosynthetic enzymes PPAT and PAICS in lung cancer. Oncotarget. 2015;6:23445–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Q, Lin M, Feng X, Ma F, Zhu Y, Liu X, Qu C, Sui H, Sun B, Zhu A, Zhang H, Huang H, Gao Z, Zhao Y, Sun J, Bai Y, Jin J, Hong X, Zou C and Zhang Z. Targeting CLK3 inhibits the progression of cholangiocarcinoma by reprogramming nucleotide metabolism. J Exp Med. 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A and Lindner V. Remodeling With Neointima Formation in the Mouse Carotid Artery After Cessation of Blood Flow. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17:2238–2244. [DOI] [PubMed] [Google Scholar]
- 16.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO and Botstein D. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bar-Joseph Z, Siegfried Z, Brandeis M, Brors B, Lu Y, Eils R, Dynlacht BD and Simon I. Genome-wide transcriptional analysis of the human cell cycle identifies genes differentially regulated in normal and cancer cells. Proc Natl Acad Sci U S A. 2008;105:955–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asby DJ, Cuda F, Beyaert M, Houghton FD, Cagampang FR and Tavassoli A. AMPK Activation via Modulation of De Novo Purine Biosynthesis with an Inhibitor of ATIC Homodimerization. Chem Biol. 2015;22:838–48. [DOI] [PubMed] [Google Scholar]
- 19.Hall JL, Chatham JC, Eldar-Finkelman H and Gibbons GH. Upregulation of glucose metabolism during intimal lesion formation is coupled to the inhibition of vascular smooth muscle cell apoptosis. Role of GSK3beta. Diabetes. 2001;50:1171–9. [DOI] [PubMed] [Google Scholar]
- 20.Osman I, He X, Liu J, Dong K, Wen T, Zhang F, Yu L, Hu G, Xin H, Zhang W and Zhou J. TEAD1 (TEA Domain Transcription Factor 1) Promotes Smooth Muscle Cell Proliferation Through Upregulating SLC1A5 (Solute Carrier Family 1 Member 5)-Mediated Glutamine Uptake. Circ Res. 2019;124:1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunaga H, Matsui H, Anjo S, Syamsunarno MR, Koitabashi N, Iso T, Matsuzaka T, Shimano H, Yokoyama T and Kurabayashi M. Elongation of Long-Chain Fatty Acid Family Member 6 (Elovl6)-Driven Fatty Acid Metabolism Regulates Vascular Smooth Muscle Cell Phenotype Through AMP-Activated Protein Kinase/Kruppel-Like Factor 4 (AMPK/KLF4) Signaling. J Am Heart Assoc. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha A, Connelly S, Jiang J, Zhuang S, Amador DT, Phan T, Pilz RB and Boss GR. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol Cell. 2014;55:264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM and Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science (New York, NY). 2016;351:728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pedley AM and Benkovic SJ. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem Sci. 2017;42:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quemeneur L, Gerland LM, Flacher M, Ffrench M, Revillard JP and Genestier L. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J Immunol. 2003;170:4986–95. [DOI] [PubMed] [Google Scholar]
- 26.Kondo M, Yamaoka T, Honda S, Miwa Y, Katashima R, Moritani M, Yoshimoto K, Hayashi Y and Itakura M. The rate of cell growth is regulated by purine biosynthesis via ATP production and G(1) to S phase transition. J Biochem. 2000;128:57–64. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Wang Y, Han N, Wang X and Ruan M. HPRT promotes proliferation and metastasis in head and neck squamous cell carcinoma through direct interaction with STAT3. Exp Cell Res. 2021;399:112424. [DOI] [PubMed] [Google Scholar]
- 28.Young GH, Lin JT, Cheng YF, Ho CF, Kuok QY, Hsu RC, Liao WR, Chen CC and Chen HM. Modulation of adenine phosphoribosyltransferase-mediated salvage pathway to accelerate diabetic wound healing. FASEB J. 2021;35:e21296. [DOI] [PubMed] [Google Scholar]
- 29.Marie S, Heron B, Bitoun P, Timmerman T, Van Den Berghe G and Vincent MF. AICA-ribosiduria: a novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am J Hum Genet. 2004;74:1276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabina RL, Holmes EW and Becker MA. The enzymatic synthesis of 5-amino-4-imidazolecarboxamide riboside triphosphate (ZTP). Science. 1984;223:1193–5. [DOI] [PubMed] [Google Scholar]
- 31.Henin N, Vincent MF and Van den Berghe G. Stimulation of rat liver AMP-activated protein kinase by AMP analogues. Biochim Biophys Acta. 1996;1290:197–203. [DOI] [PubMed] [Google Scholar]
- 32.Brooks HB, Meier TI, Geeganage S, Fales KR, Thrasher KJ, Konicek SA, Spencer CD, Thibodeaux S, Foreman RT, Hui YH, Roth KD, Qian YW, Wang T, Luo S, Torrado A, Si C, Toth JL, Mc Cowan JR, Frimpong K, Lee MR, Dally RD, Shepherd TA, Durham TB, Wang Y, Wu Z, Iversen PW and Njoroge FG. Characterization of a novel AICARFT inhibitor which potently elevates ZMP and has anti-tumor activity in murine models. Sci Rep. 2018;8:15458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, Jin C, Xu M, Zhou L, Li D and Yin Y. Bifunctional enzyme ATIC promotes propagation of hepatocellular carcinoma by regulating AMPK-mTOR-S6 K1 signaling. Cell Commun Signal. 2017;15:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Racanelli AC, Rothbart SB, Heyer CL and Moran RG. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009;69:5467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothbart SB, Racanelli AC and Moran RG. Pemetrexed indirectly activates the metabolic kinase AMPK in human carcinomas. Cancer Res. 2010;70:10299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pirkmajer S, Kulkarni SS, Tom RZ, Ross FA, Hawley SA, Hardie DG, Zierath JR and Chibalin AV. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes. 2015;64:360–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spurr IB, Birts CN, Cuda F, Benkovic SJ, Blaydes JP and Tavassoli A. Targeting tumour proliferation with a small-molecule inhibitor of AICAR transformylase homodimerization. Chembiochem. 2012;13:1628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C and Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–94. [DOI] [PubMed] [Google Scholar]
- 39.Patel J, Wang X and Proud CG. Glucose exerts a permissive effect on the regulation of the initiation factor 4E binding protein 4E-BP1. Biochem J. 2001;358:497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, Jiang X, van Eijkeren RJ, Davis OB, Louie SM, Perera RM, Covey DF, Nomura DK, Ory DS and Zoncu R. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emmanuel N, Ragunathan S, Shan Q, Wang F, Giannakou A, Huser N, Jin G, Myers J, Abraham RT and Unsal-Kacmaz K. Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep. 2017;19:2665–2680. [DOI] [PubMed] [Google Scholar]
- 42.Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, Ben-Sahra I and Manning BD. The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep. 2017;21:1331–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andres V Control of vascular cell proliferation and migration by cyclin-dependent kinase signalling: new perspectives and therapeutic potential. Cardiovasc Res. 2004;63:11–21. [DOI] [PubMed] [Google Scholar]
- 44.Negi SI, Torguson R, Gai J, Kiramijyan S, Koifman E, Chan R, Randolph P, Pichard A, Satler LF and Waksman R. Intracoronary Brachytherapy for Recurrent Drug-Eluting Stent Failure. JACC Cardiovasc Interv. 2016;9:1259–1265. [DOI] [PubMed] [Google Scholar]
- 45.Wang D and Atanasov AG. The microRNAs Regulating Vascular Smooth Muscle Cell Proliferation: A Minireview. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Royce S, Chakraborty A and Zhao Y. US Food and Drug Administration Perspective on “Mortality and Paclitaxel-Coated Devices: An Individual Patient Data Meta-Analysis”. Circulation. 2020;141:1870–1871. [DOI] [PubMed] [Google Scholar]
- 47.Allegra CJ, Hoang K, Yeh GC, Drake JC and Baram J. Evidence for direct inhibition of de novo purine synthesis in human MCF-7 breast cells as a principal mode of metabolic inhibition by methotrexate. J Biol Chem. 1987;262:13520–6. [PubMed] [Google Scholar]
- 48.Friedman B and Cronstein B. Methotrexate mechanism in treatment of rheumatoid arthritis. Joint Bone Spine. 2019;86:301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan ES and Cronstein BN. Molecular action of methotrexate in inflammatory diseases. Arthritis Res. 2002;4:266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fales KR, Njoroge FG, Brooks HB, Thibodeaux S, Torrado A, Si C, Toth JL, Mc Cowan JR, Roth KD, Thrasher KJ, Frimpong K, Lee MR, Dally RD, Shepherd TA, Durham TB, Margolis BJ, Wu Z, Wang Y, Atwell S, Wang J, Hui YH, Meier TI, Konicek SA and Geeganage S. Discovery of N-(6-Fluoro-1-oxo-1,2-dihydroisoquinolin-7-yl)-5-[(3R)-3-hydroxypyrrolidin-1-yl]t hiophene-2-sulfonamide (LSN 3213128), a Potent and Selective Nonclassical Antifolate Aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase (AICARFT) Inhibitor Effective at Tumor Suppression in a Cancer Xenograft Model. J Med Chem. 2017;60:9599–9616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.