

Abstract
Background
Porcine epidemic diarrhea virus (PEDV), an enteric coronavirus, has become the major causative agent of acute gastroenteritis in piglets since 2010 in China.
Results
In the current study, 91 complete spike (S) gene sequences were obtained from PEDV positive samples collected from 17 provinces in China from March 2020 to March 2021. A phylogenetic analysis showed that 92.3% (84 out of 91) of the identified strains belonged to GII subtype, while 7.7% (7 out of 91) were categorized as S-INDEL like strains and grouped within GI-c clade. Based on a recombination analysis, six of S-INDEL like strains were recombinant strains originated from S-INDEL strain FR/001/2014 and virulent strain AJ1102. In addition, PEDV variant strains (CH/GDMM/202012, CH/GXDX/202010 et al) carrying novel insertions (360QGRKS364 and 1278VDVF1281) in the S protein were observed. Furthermore, the deduced amino acid sequences for the S protein showed that multiple amino acid substitutions in the antigenic epitopes in comparison with the vaccine strains.
Conclusions
In conclusion, these data provide novel molecular evidence on the epidemiology and molecular diversity of PEDV in 2020–2021. This information may help design a strategy for controlling and preventing the prevalence of PEDV variant strains in China.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12917-022-03481-4.
Keywords: PEDV, Spike gene, Phylogenetic analysis, S-INDEL like strain, Recombination
Background
Coronaviruses (CoVs) can infect a wide variety of animals, and cause respiratory, enteric, and other diseases [1]. So far, the most relevant enteric coronaviruses in pigs include porcine epidemic diarrhea virus (PEDV), transmissible gastroenteritis virus (TGEV), and recently identified viruses, such as porcine deltacoronavirus (PDCoV) and swine acute diarrhea syndrome coronavirus (SADS-CoV) [2, 3]. Among these CoVs, PEDV has the highest detection rate and up to 100% mortality in neonatal piglets. The acute diarrhea caused by PEDV was characterized by severe vomiting, dehydration, and watery diarrhea [4]. Outbreaks of PED brought huge economic losses to pig production worldwide [5].
PEDV is an enveloped virus, whose genome is composed of a positive sense, single-stranded, and non-segmented RNA with a size of approximately 28 kb. PEDV genome encodes open reading frame (ORF) 1ab, spike (S), ORF3, envelope (E), membrane (M), and nucleoprotein (N) from 5′ to 3′ untranslated region (UTR) [6]. S protein is a glycoprotein peplomer located on the viral surface, and contains 1383–1386 amino acids (aa) in most strains. Despite the S protein of PEDV cannot be demarcated by a protease cleavage site, it is divided into S1 and S2 subunits based on the homology with other coronaviruses [7, 8]. The receptor for PEDV is still unknown. However, S1 protein has been shown to bind to sialic acid glycans and the porcine aminopeptidase N (pAPN) is a receptor binding domain for facilitating viral invasion. S2 subunit is responsible for membrane fusion [9, 10]. S protein is also the main target for inducing neutralizing antibodies. The neutralizing epitope region COE (499–638 aa), four neutralizing B cell epitopes S1A (435–485 aa), SS2 (748–755 aa), SS6 (764–771 aa), and 2C10 (1368–1374 aa) have been identified on the S protein [11–14]. In addition, a specific linear B-cell epitope SE16 (722-731aa) had been identified to be required for a reactivity with the mAb 2E10. The epitope SE16 was localized on the surface of PEDV S protein [15].
PEDV was first reported in the United Kingdom in 1971 [16]. In China, PEDV was first identified and isolated in 1984. Since then, PEDV infection occurred sporadically and regionally. In 2010, a high virulent strain of PEDV emerged on pig farms in southern China, and caused up to 100% mortality in newborn piglets, and immediately swept throughout the country [17]. The presence of variant strain in China was identified by the detection of a field CH/FJND-3/2011 strain [18]. The AJ1102 strain isolated from a PEDV positive farm had become the prevalent variant for the time [19].
The epidemiological survey proceeded from February 2011 to March 2014 in 29 provinces of China showed that the PEDV positive rates for samples and pig farms were 61.10–78.49% and 71.43–83.47%, respectively. Genetic drift could be confirmed mainly by the genetic variation of S protein as compared with the Chinese commercialized vaccine strain CV777 [20, 21]. PEDV is mainly classified into two genotypes GI (classical) and GII (variant) on the basis of S gene [18]. In 2013, new PEDV variants containing new insertion and deletion in S gene versus prototype strain were reported in USA [22]. Subsequently, these variants, named S-INDEL-variant, were also detected and isolated in China [4]. S-INDEL strains were associated with milder clinical signs and lower mortality in suckling pigs.
Currently, PEDV is still the main pathogen that leads to the death of piglets on pig farms in China. High morbidity, variation, and recombination of viral genomes make it hard to prevent and control the prevalence of PEDV. To better understand the prevalence and molecular characteristics of PEDV in different regions of China, 91 PEDV positive samples were collected from 17 provinces of China. The full-length S genes were sequenced and analyzed with a focus on the variation of the neutralizing epitopes, the emergence of S-INDEL strains, and potential recombinant between different strains. These data systematically described the genetic and evolutionary characteristics of PEDV field strains in China from 2020 to 2021, and might promote the development of novel effective vaccines.
Results
Prevalence analysis and genome characteristics of PEDV based on S gene in China
In the PEDV positive farms, 115 clinical samples were found to be positive for PEDV based on the RT-PCR detection (Table 1). Among them, the S genes of 91 PEDV positive samples obtained in 17 provinces in China from 2020 to 2021 were successfully amplified and sequenced. This epidemiological survey covered the major pig-raising provinces in China (Table 2).
Table 1.
Detection rates of PEDV from Mar 2020 to Mar 2021 in China
| Month | 2020 | 2021 | Sum | ||
|---|---|---|---|---|---|
| Mar ~ Jun | Jul ~ Sep | Oct ~ Dec | Jan ~ Mar | ||
| Total samples | 260 | 130 | 203 | 88 | 681 |
| Samples sequenced | 21 | 14 | 46 | 10 | 91 |
| Positive samples | 28 | 21 | 51 | 15 | 115 |
| Positive rate (%) | 10.8 | 16.2 | 25.1 | 17.0 | 16.9 |
Table 2.
PEDV distribution in 17 provinces in China from March 2020 to March 2021 (The abbreviation of sampling provinces were given in supplementary Table 1)
| PEDV negative province | PEDV positive province reported | PEDV positive province in this study |
|---|---|---|
| Tibet | Xinjiang, Qinghai, Gansu, Ningxia, Chongqing, Shanxi, Zhejiang, Beijing, Tianjin, Heilongjiang, Jilin, Hainan, Taiwan, Shanghai | Inner Mongoria, Liaoning, Hebei, Shandong, Shaanxi, Henan, Anhui, Jiangsu, Sichuan, Hubei, Guizhou, Yunnan, Guangxi, Hunan, Jiangxi, Fujian, Guangdong |
Note: Bold letters stand for case of S-INDEL like strain
The length of S genes of 91 field strains was 4149–4176 nucleotides (nt), which encoded proteins of 1383–1392 amino acids. A phylogenetic analysis based on the sequenced S genes and 24 reference strains indicated that PEDV can be divided into two genotypes (GI and GII) and further classified into five subgroups, including GI-a, GI-b, GI-c, GII-a, and GII-b (Fig. 1). The GI-a clade comprised classical strains, such as CV777, CH/S, and DR13.The classical vaccine strains attenuated from CV777 and DR13 belonged to GI-b subgroup. Notably, 92.3% (84 out of 91) of the strains belonged to genotype GII. CH/HBTS/202010, CH/SCMY/202012, CH/GXLB/202005, and CH/GXLP/202007 were categorized as GII-a and other 80 belonged to GII-b subgroup. Meanwhile, CH/JSXZ/202012, CH/SCYB/202012, CH/SCGA/202103, CH/SCST/202004, CH/HNBR/202101, CH/JXXG/202101, and CH/HNSQ/202102 were located in the GI-c clade and closely related to PEDV S-INDEL strains found in the US (OH851), Korea (KNU-1406-1), and China (ZL29). They distributed in four provinces of China (Table 2).
Fig. 1.
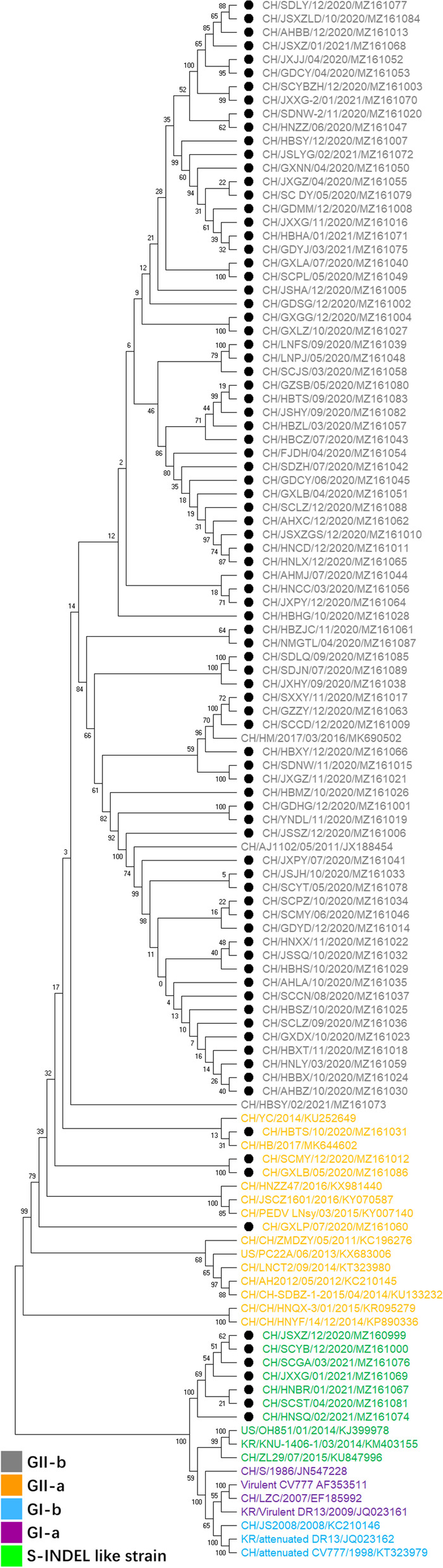
Geographical distribution and phylogenetic analysis of PEDV positive samples in China. Phylogenetic analysis of PEDV strains based on 91 S gene sequences identified in this study and 24 reference S gene sequences in Genbank. Multiple nucleotide sequence alignments are performed with ClustalW algorithm using MEGA 6.0 software. A neighbor-joining method based phylogenetic tree is automatically constructed with 1000 bootstrap replicates using MEGA 6.0 software. Labels at branch tips refer to the strain name and GenBank accession number. The black solid dots mark 91 PEDV S sequences detected in this study. Sequences from different genotypes are marked with different color. GI-a, GI-b, S-INDEL like strain, GII-a, and GII-b are labelled with purple, blue, green, orange, and grey, respectively
Sequence homology analysis
The nucleotide sequences of S genes of 91 samples had a homology of 94.2–100%. Four strains in GII-a subgroup shared homology of 97.3–99.7%, while eighty strains in GII-b subgroup shared homology of 96.8–100%. The newly detected seven S-INDEL like strains shared homology of 95.1–95.6% and 94.2–96.8% with non-S-INDEL like strains in subgroups GII-a and GII-b, respectively. The sequence identity among 7 strains in S-INDEL groups was 98.6–99.3% between each other. The strains in GII group shared homology of 93.2–93.8% and 96.7–99.1% with Chinese vaccine strain CV777 (GI-b) and AJ1102 (GII-b), respectively. In addition, the S-INDEL like strains had homology of 94.8–95.2% with Chinese vaccine strains (Table 3).
Table 3.
Nucleotide acid sequence homology of S genes of 91 PEDV strains and reference strains
| Genotype and reference strains | Genotypes | |||
|---|---|---|---|---|
| GIIa | GII-b | S-INDEL | ||
|
Percentage Identity (%) |
GII-a | 97.3–99.7 | 95.2–97.9 | 95.1–95.6 |
| GII-b | 95.2–97.9 | 96.8–100.0 | 94.2–96.5 | |
| S-INDEL | 95.1–95.6 | 98.6–99.3 | 98.6–99.3 | |
| CV777 | 93.2–93.8 | 92.7–93.7 | 94.8–95.2 | |
| AJ1102 | 96.7–97.0 | 96.9–99.1 | 94.9–95.1 | |
Deduced amino acids analysis of PEDV variants in China
In this study, the S-INDEL like strains were identified in seven samples. The deduced amino acid sequences of S proteins in the identified S-INDEL like strains were used to compare with the reference S-INDEL strains. A sequence alignment showed that four amino acid deletions (58QGVN61) were observed in the attenuated CV777 strain (GI-b) and S-INDEL like strains as compared with the virulent strain AJ1102 (GII-b). In addition, three mutations and one deletion of the deduced S protein were found in the identified S-INDEL like strains as compared with the reference S-INDEL strains (Fig. 2a).
Fig. 2.

Molecular characterization of the emergent PEDV strains. a Alignment of partial S protein sequences of 7 S-INDEL like strains in this study and the prototype S-INDEL strains in the United States (OH851 and Minnesota58), South Korea (KNU-1406-1), and China (ZL29). b Locations of unique amino acid (aa) insertions identified in the 91 detected sequences. The symbol “-” indicates an aa deletion. Unique variations of aa are labelled with yellow
A new amino acid insertion relative to the prototype in the S protein were found in five samples. The CH/GDMM/202012 had continuous 5 amino acid insertions (360QGRKS364) in the S1 domain. CH/GXDX/202010, CH/AHBZ/202010, CH/AHLA/202010, and CH/HNLY/202003 had 3 or 4 amino acid insertions (1279DVF1281 or 1278VDVF1281) in the S2 domain (Fig. 2b).
Recombination analysis of S-INDEL like strains
A recombination analysis of the 91 strains detected in this study and 178 reference strains were performed by using RDP4 software. The results derived from seven recombination methods indicated that six S-INDEL like strains (CH/HNBR/01/2021, CH/JSXZ/12/2020, CH/SCYB/12/2020, CH/SCGA/202103, CH/SCST/04/2020, and CH/HNSQ/02/2021) were recombinant strains originated from FR/001/2014 strain and virulent strain AJ1102 (Fig. 3). Further similarity comparisons were made using SimPlot among six S-INDEL like strains and their parental strains, the recombination breakpoints were found to be located within the nucleotides 720–1220, 743–1204 or 743–1201 of S genes.
Fig. 3.

Recombination analysis of six S-INDEL like strains. The recombinant events are identified by using a Simplot analysis. The query sequences from a CH/HNBR/01/2021, b CH/JSXZ/12/2020, c CH/SCYB/12/2020, d CH/SCGA/03/2021, e CH/SCST/04/2020, and f CH/HNSQ/02/2021 were used to compare with the parental sequences FR/001/2014 and AJ1102. The x-axis indicates the S gene sequences, and the y-axis represents the similarity value. The regions of recombinant breakpoint are shown within two red lines
Antigenic epitopes of PEDV S protein in Chinese strains
Five major neutralizing epitopes S1A (435–485aa), COE domain (499–638 aa), SS2 (748–755 aa), SS6 (764–771 aa), 2C10 (1368–1374 aa), and one linear B-cell epitope (SE16, 722–731 aa) have been identified on the surface of S protein. Among these epitopes, SS2, 2C10 and SE16 (data not shown) in all field PEDVs were highly conserved, with only one mutation (1374Y-C) in the 2C10 as compared to the two vaccine strains. The aa difference was compared among field strains and vaccine strain CV777 and AJ1102 strains. All of the field strains had two aa substitutions (764L-S/P or 766D-S/F). One mutation (765Q-H) in the epitope SS6 of CH/GXLZ/202010 was observed. In the S1A epitope, the field strains except for CH/GXLZ/202010 and CH/HNSQ/202102 had one mutation (479S-A). In the COE domain, all field strains except for CH/HNSQ/202102 had three substitutions (552T-S, 597G-S, and 636Q-E/R). Meanwhile, a serine substitution (520A-S) was observed in all field strains except for CH/HNXX/202011, CH/JSSQ/202010, and CH/SCCN/202008. In addition, multiple mutations on S protein were detected in the COE domain (499I-T, 502V-I, 524H-S/L/Y, 539F-L, 566K-N, 569D-A/N, 606Y-H, 612G-V/S, and 634P-S) (Fig. 4).
Fig. 4.

Comparison of the antigen epitopes of S proteins of field strains and the vaccine strains. Dots indicate the amino acids which are identical to those of reference strains. The colored amino acids indicate the mutations in the neutralizing epitopes S1A, COE, SS2, SS6, and 2C10. The red box shows the S-INDEL like strain sequence
Discussion
China is the largest country for pig farming in the world, with an annual pig slaughter of approximately 600 million in 2020. The re-emergence of PEDV, especially the variant strains, since 2010 has brought a great threat to Chinese pig industry. PEDV was the major causative agent that led to the diarrheal disease and death in piglets in comparison with other coronaviruses, such as TGEV, PDCoV, and SADS-CoV [23]. Although a variety of commercial vaccines based on classical GI strain CV777 and the variant GII strain AJ1102 have been employed to control the outbreak of PEDV, their efficacies have been always poor. The immunity failure was associated with the constant variation of virus and the insufficient mucosal immunity induced by the vaccines [24]. Therefore, understanding the prevalence and variation characterization of PEDV genome is of importance in preventing PEDV prevalence. In a recent study, 49 complete S genes were sequenced, and novel insertions, deletions, and multiple S gene recombination events were found in PEDV variant strains [25]. Lei et al. collected 184 specimens from pig farms in China in 2017–2018, and detected an average PEDV-positive rate of 38.04% [26]. Zhang et al. made a survey of molecular characteristics of PEDV in Henan province, China in 2015–2019 and found PEDV existed widely in both PEDV-vaccine immunized (25.00%) and non-immunized swine herds (62.29%). Sixteen of the sequenced PEDV Henan strains were located in the GII clade [27]. However, there is no nationwide epidemiological survey of PEDV in the past 2 years. Furthermore, African swine fever (ASF) outbreaks were declared in China since 2018 and quickly swept through the whole country [28]. Management and prevention measures of epidemic diseases have been altered in pig farms. It is still unknown about if the prevalence characterization of PEDV has changed under the background of ASF. In this study, 115 PEDV positive samples were collected in 17 provinces of China, including large swine-raising provinces, such as Sichuan, Henan, Hunan, and Shandong province. Our data showed that the positive rate of PEDV was from 10.8–25.1% from March 2020 to April 2021, thus indicating that PEDV was still one of the major pathogenic agents in Chinese swine herds. Our data also showed that the positive rate of PEDV was the highest in October and December. Thus, we should pay more attention to the prevention of PEDV when the weather turns cold, such as the turn of autumn and winter and the winter.
The full-length nucleotide sequences of S genes of 91 field strains were successfully sequenced. Based on the phylogenetic analysis for the S gene, all of the PEDV strains could be classified into two main genotypes and five subgroups, including GI-a (classical strain), GI-b (attenuated classical strain), GI-c (S-INDEL strain), GII-a, and GII-b (variant strain). All strains detected in this study shared 94.2–100% homology between each other. A majority of them belonged to GII genotype (84 out of 91) which represented the pandemic strains of PEDV in recent years. GII genotype strains shared 93.2–93.8% and 96.7–99.1% homology with attenuated Chinese vaccine strains CV777 and AJ1102, respectively. Vaccines based on CV777 strain was widely used and contributed to a good control of PEDV in China before 2010 [29]. However, the variant strains that were clustered into different subgroup (GII) shared lower homology with CV777 (GI). The different genotypes between CV777 and PEDV epidemic strains may lead to an incomplete protection provided by the vaccines in China since 2010.
The detected strains CH/JSXZ/202012, CH/SCYB/202012, CH/SCGA/202103, CH/SCST/202004, CH/HNBR/202101, CH/JXXG/202101, and CH/HNSQ/202102 were more related to S-INDEL strains. They were obtained from four provinces of China and classified into GI-c subgroup, thus suggesting that S-INDEL like strains have spread and circulated in China. Seven of S-INDEL like strains shared a high homology of 98.6–99.3% between each other. Although the presence of S-INDEL strains of PEDV has been described to induce less severe symptoms and low fatality rate as compared with non-INDEL strains [30, 31], the pathogenicity of this subgroup is still controversial. Severe diarrhea and vomiting, along with high mortality rates were associated with the S-INDEL PEDV strains in some European countries [32]. Therefore, the pathogenicity and infection rate of S-INDEL like strains in China should be constantly monitored.
To date, S-INDEL strains have been detected widely in the world, including America, Asia, and Europe. The S-INDEL strains could be separated into two distant clades (INDEL 1 and INDEL 2) based on the characteristics of the S-INDEL genotype [33]. INDEL 2 contained prototype American (OH851) and European strains (FR/001/2014), as well as the first identified Chinese S-INDEL strain ZL29. Some other Asia S-INDEL strains belonged to the INDEL 2 clade. A further phylogenetic analysis using the seven S-INDEL like strains and reference S-INDEL strains retrieved from the NCBI nucleotide database showed that all of the detected S-INDEL like strains were categorized into INDEL 2. However, they were clearly clustered within a new cluster in INDEL 2 clade (Fig. 5). Although the identified seven S-INDEL like strains had the common four aa deletion (58QGVN61) which was in line with the reference S-INDEL strains, they had additional novel three amino acid mutations and one deletion located at the positions 156–160 aa. As previously described, INDEL 2 clade might originate from virulent strains DR13, Italy/7239/2009 or other field NON-INDEL strains, whereas INDEL 1 showed a common ancestor, including CV777 or other PEDV strains detected in China before 2010 [24, 34]. According to our results, the novel S-INDEL like strains detected in this study were originated from FR/001/2014 in INDEL 2 clade and virulent strain AJ1102. This indicated that natural recombinant events might exist among variant strains and vaccine strains in China. Therefore, the use of live vaccine strains should be carefully considered especially in PEDV positive pig farms.
Fig. 5.
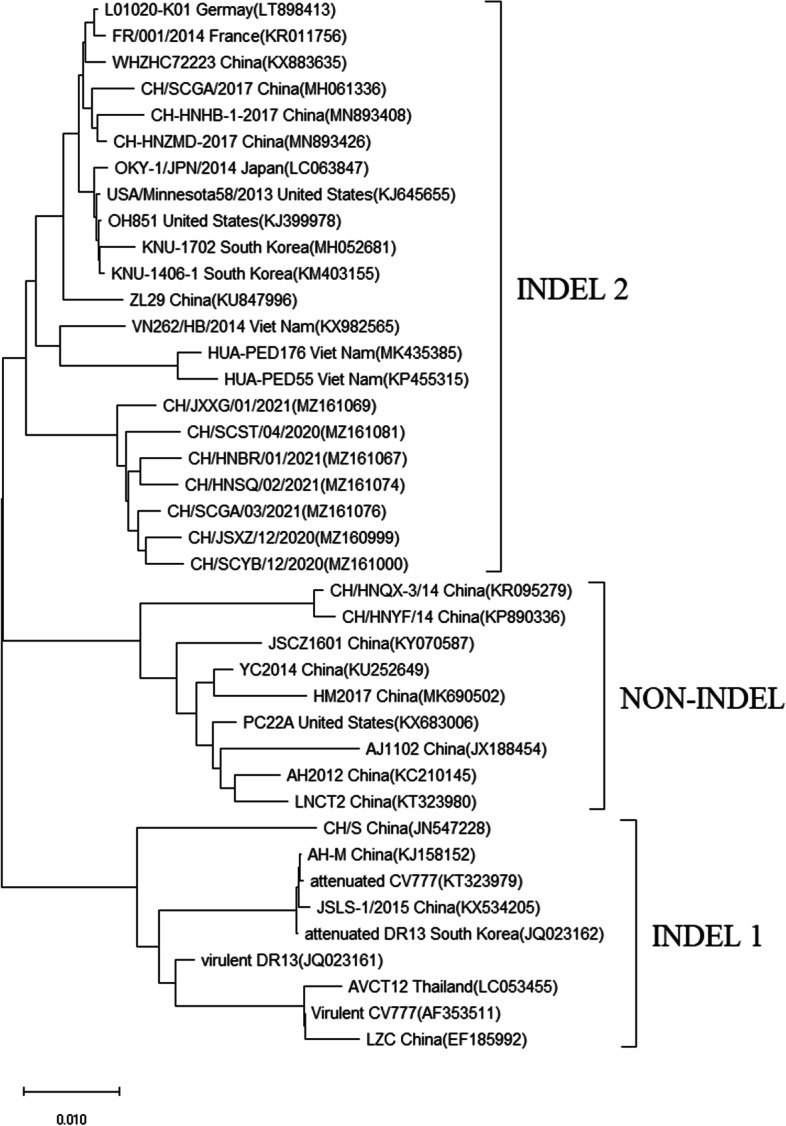
Phylogenetic analysis based on the S genes of S-INDEL like strains and the reference S-INDEL strains around the world. The evolutionary tree was constructed by the neighbor-joining method using MEGA6 software. Bootstrap values are indicated for each node, based on 1000 replicates. The positions of seven S-INDEL like strain are annotated by solid black circles
Spike protein is the most variable protein of PEDV. The variation of S protein is considered to be responsible for the changes of viral antigenicity, determination of the genetic diversity for PEDV and affected the viral virulence [35, 36]. To evaluate whether the antigenicity of PEDV field strains has changed as compared with vaccine strains used in China, the neutralizing antigenic epitopes on S protein were analyzed. Several single nucleotide polymorphisms (SNPs) were observed on the S1A,SS6,and 2C10 epitopes, and COE domain. Five main serine substitution (520A-S, 552T-S, 597G-S, 764L-S, and 766D-S) and one Glutamate substitution (636Q-E) were found in the field strains as compared with the vaccine strain CV777. One common substitution in S1A (479S-A) and multiple mutations (499I-T, 502V-I, 524H-S/L/Y, 539F-L, 566K-N, 569D-A/N, 606Y-H, 612G-V/S, and 634P-S) in individual sequences were detected in the field stains as compared with the virulent strain AJ1102. These mutations might involve in the immune escape and the antigenicity of virus, resulting in a less effective immune protection provided by the commercial vaccines CV777 and AJ1102. Notably, five PEDV samples with new insertion in the S protein sequences were detected. CH/GDMM/202012 had a continuous 5 aa insertion (360QGRKS364) located in S1 domain while the other four (CH/GXDX/202010, CH/AHBZ/202010, CH/AHLA/202010, and CH/HNLY/202003) had three or four amino acid insertions (1279DVF1281 or 1278VDVF1281) located in S2 domain. To the best of our knowledge, this is the first report for these novel S-insertion variants. The S-INDEL-variants were proved to have decreased virulence in host, whereas one strain with a novel four-amino-acid insertion in the COE domain was highly pathogenic to the neonatal pigs [37]. Although we did not obtain the isolated novel S-insertion strains, it could be predicted that the conformational structure of S protein might have changed, probably resulting in a change for the pathogenicity of these strains.
Conclusions
Collectively, this study described the nationwide investigation of the PEDV prevalence in China in recent years. The major causative virus strains were GII genotype variants with the ratio of 92.3%. Notably, seven S-INDEL like strains were detected in four provinces in China. Alignment of S deduced aa sequences revealed novel mutations and deletion compared with prototype S-INDEL strain. Based on recombination analyses, the novel S-INDEL like strains were originated from FR/001/2014 in INDEL 2 clade and virulent strain AJ1102. Moreover, variant PEDV strains with novel insertions (360QGRKS364 and 1278VDVF1281) in S protein sequences were detected and needed to be addressed on the specific function of insertions in the future work. In addition, we also identified multiple mutations in the aa sequences of S proteins of the variant strains compared to those of the vaccine strains. These PEDV mutants derived from genetic mutations, deletions, insertions, and recombination of the S genes might be the major cause of antigenic drift and immune failure. The molecular characterization of S protein should be investigated continuously and would work in the control and prevention of PED in China.
Materials and methods
Sample collection
A total of 115 field PEDV positive samples (intestine, intestinal content, feces, and anal swab) were collected from various pig farms located in 17 provinces of China from March 2020 to March 2021. Animal samples were collected from the farms of New Hope Liuhe Co., Ltd. and Jiangxi Zhengbang Technology Co., Ltd., respectively. We have acquired a consent from the farm owners. All samples were stored at − 80 °C before RNA extraction.
PCR detection of PEDV
The samples were diluted with phosphate-buffered saline and centrifuged at 8,000×g for 10 min at 4 °C and the supernatants were transferred into a 1.5 mL RNase-free tube. Viral RNA was extracted using the TIANamp Virus DNA/RNA Kit (TIANGEN) according to the manufacturer’s instructions. Reverse transcription was carried out using PrimeScript™ IV 1st strand cDNA Synthesis Mix (TaKara). The full-length S gene of PEDV was amplified using EmeraldAmp® PCR Master Mix (TaKara). The S1 was amplified by two pairs of primers (S1-1U, 5′- ATCGTCAGAGGCATTTTTAA-3′; S1-1L, 5′-ATCCATCACCATTAAACGAA-3′; S1-2U, 5′-ATGTTGTGTTAGGCTTGTTG-3′; S1-2L, 5′-CACTAACAGGCGTGTTGTAA-3′), and the S2 was amplified by three primers (S2-U, 5′-CTGATTCTGGACAGTTGTTA-3′; S2-1L, 5′-TTGGACAGCATCCAAAGACA-3′; S2-2L, 5′-CTTCGAGACATCTTTGACAA-3′) [38]. PCR products were purified and recovered using AxyPrep™ DNA Gel Extraction Kit (Axygen), and then subjected to sequencing by Sangon Biotech company (Shanghai, China).
Genetic and phylogenetic analysis
The 91 complete S gene sequences of PEDV obtained in this study have been uploaded to GenBank with accession numbers from MZ160999 to MZ161089. Twenty-four reference sequences retrieved from the NCBI nucleotide database and the 91 identified sequences were used for the phylogenetic analysis. Multiple nucleotide sequence alignments were performed with Clustal W algorithm using MEGA 6.0 software. A phylogenetic tree based on S gene sequence was constructed by the neighbor-joining method using MEGA 6.0 software, with bootstrap values calculated for each node from 1000 replicates [39, 40].
Deduced amino acid analysis of PEDV variants
To elucidate the genetic characteristics of S-INDEL like strains detected in this study, partial S gene sequences were aligned with reference S-INDEL strains from US (OH851), South Korea (KNU-1406-1), and China (ZL29) using MEGA 6.0 software. The novel mutation, insertion, and deletion in S proteins were also detected in this study.
Recombination analysis
In order to investigate the recombination events occurred in the detected variant strains, especially the S-INDEL, all of the detected strains and 178 reference strains obtained from Genbank were analyzed using program RDP4 [41]. Potential recombinant strains, parental strains, and possible recombination breakpoints were identified by a series of methods including RDP, GeneConv, SiScan, 3Seq, BootScan, MaxChi, and Chimaera. Recombination events were examined by at least three of the methods mentioned above with a cutoff of p < 0.05. Based on the fragment size of genome that contributed to the recombinants, the original sequences were defined as major parents (contributing the larger fragment) and the minor parents (contributing the small fragment). The recombinant strain was further analyzed by Simplot v3.5.1 software [42].
Analysis of the antigenic epitopes on the S proteins
In order to study the antigenic variation of PEDV strains, all of the ninety-one S protein sequences were used for a sequence alignment. Twenty-eight representative sequences were selected and compared with Chinese PEDV vaccine strain (attenuated CV777 and AJ1102). A focus was on the neutralizing epitope regions including S1A, COE domain, SS2, SS6, 2C10, and the linear B-cell epitope SE16 [43, 44].
Additional Files
Additional file 1: Table S1. Information of PEDV strains obtained in this study. The information including strain name, collection date, collection region, S gene length, virus isolation and accession number.
Acknowledgements
The authors thank Dr. Chuang Lyu from Qingdao Jiazhi Biotechnology Co., Ltd. in Qingdao, Shandong province for proofreading the manuscript.
Abbreviations
- PEDV
Porcine epidemic diarrhea virus
- S
Spike
- CoVs
Coronaviruses
- TGEV
Transmissible gastroenteritis virus
- PDCoV
Porcine deltacoronavirus
- SADS-CoV
Swine acute diarrhea syndrome coronavirus
- ORF
Open reading frame
- E
Envelope
- M
Membrane
- N
Nucleoprotein
- UTR
Untranslated region
- aa
Amino acids
- pAPN
Porcine aminopeptidase N
- nt
Nucleotides
- ASF
African swine fever
- SNPs
Single nucleotide polymorphisms
Authors’ contributions
Conceptualization, HZ, CW2 (Chuang Wang), LQ, CW1 (Chengbao Wang); Methodology, XW; Formal analysis and investigation, LS, MX, LZ, HW, HY and FL Writing—original draft preparation, HZ, LS XW and MX; Writing—review and editing, CW2 (Chuang Wang), LQ and CW1 (Chengbao Wang) Funding Acquisition, CW1 (Chengbao Wang) Resources: CW1 (Chengbao Wang) Supervision, CW1 (Chengbao Wang). All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Grant numbers: SKLVEB2016KFKT014), China Postdoctoral Science Foundation (Grant numbers: 2017 M610659 and 2018 T111113), Science and Technology Department of Jiangxi Province (Grant numbers: 20194ABC28008), and the Fundamental Research Funds for the Central Universities (Grant numbers: 2452019053). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The nucleotide sequence data obtained in this study are available in GenBank, and their accession numbers are MZ160999-MZ161089. The nucleic acid sequences referenced in this paper were obtained from National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/nuccore).
Declarations
Ethics approval and consent to participate
This study was performed according to the animal welfare guidelines of the World Organization for Animal Health, and animal sampling was strictly carried out in accordance with guidelines established by the Ethics of Animal Experiments of Northwest A&F University, Yangling, China. We got the verbal consent to collect the samples like rectal swab, and intestinal tissues from the farm owners. The animal experiments were carried out in strict accordance with guidelines established by the Ethics of Animal Experiments of Northwest A&F University, Yangling, China. All the protocols were approved by this committee (Permit Number: 2014BAD23B11). The field study did not include endangered or protected species. No specific permissions were required for the collection of samples because the samples were collected from public or non-protected areas.
Consent for publication
Not applicable.
Competing interests
Author LS, XW and LQ are employed by Shandong New Hope Liuhe Group Co., Ltd.; Author XW and LQ are employed by Qingdao Jiazhi Biotechnology Co., Ltd.; Author MX, LZ, HY, FL and CW2 (Chuang Wang) are employed by Zhengbang Group Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The remaining authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hong Zhuang, Leilei Sun, Xiaobo Wang and Min Xiao contributed equally to this work and share first authorship.
Contributor Information
Chuang Wang, Email: wangchuang@zhengbang.com.
Liting Qin, Email: qinlt@newhope.cn.
Chengbao Wang, Email: wangchengbao@nwafu.edu.cn.
References
- 1.Woo PC, Lau SK, Yip CC, Huang Y, Tsoi HW, Chan KH, et al. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol. 2006;80(14):7136–7145. doi: 10.1128/JVI.00509-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Byrum B, Zhang Y. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg Infect Dis. 2014;20(7):1227–1230. doi: 10.3201/eid2007.140296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gong L, Li J, Zhou Q, Xu Z, Chen L, Zhang Y, et al. A New Bat-HKU2-like Coronavirus in Swine, China, 2017. Emerg Infect Dis. 2017;23(9). [DOI] [PMC free article] [PubMed]
- 4.Wang D, Fang L, Xiao S. Porcine epidemic diarrhea in China. Virus Res. 2016;226:7–13. doi: 10.1016/j.virusres.2016.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song D, Park B. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes. 2012;44(2):167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kocherhans R, Bridgen A, Ackermann M, Tobler K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes. 2001;23(2):137–144. doi: 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duarte M, Tobler K, Bridgen A, Rasschaert D, Ackermann M, Laude H. Sequence analysis of the porcine epidemic diarrhea virus genome between the nucleocapsid and spike protein genes reveals a polymorphic ORF. Virology. 1994;198(2):466–476. doi: 10.1006/viro.1994.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W, van Kuppeveld FJM, He Q, Rottier PJM, Bosch BJ. Cellular entry of the porcine epidemic diarrhea virus. Virus Res. 2016;226:117–127. doi: 10.1016/j.virusres.2016.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li BX, Ge JW, Li YJ. Porcine aminopeptidase N is a functional receptor for the PEDV coronavirus. Virology. 2007;365(1):166–172. doi: 10.1016/j.virol.2007.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang SH, Bae JL, Kang TJ, Kim J, Chung GH, Lim CW, et al. Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol Cells. 2002;14(2):295–299. [PubMed] [Google Scholar]
- 12.Sun D, Feng L, Shi H, Chen J, Cui X, Chen H, et al. Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Vet Microbiol. 2008;131(1–2):73–81. doi: 10.1016/j.vetmic.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okda FA, Lawson S, Singrey A, Nelson J, Hain KS, Joshi LR, et al. The S2 glycoprotein subunit of porcine epidemic diarrhea virus contains immunodominant neutralizing epitopes. Virology. 2017;509:185–194. doi: 10.1016/j.virol.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang CY, Cheng IC, Chang YC, Tsai PS, Lai SY, Huang YL, et al. Identification of neutralizing monoclonal antibodies targeting novel conformational epitopes of the porcine epidemic Diarrhoea virus spike protein. Sci Rep. 2019;9(1):2529. doi: 10.1038/s41598-019-39844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong N, Meng Q, Jiao Y, Wu Y, Zuo Y, Wang H, et al. Identification of a novel B-cell epitope in the spike protein of porcine epidemic diarrhea virus. Virol J. 2020;17(1):46. doi: 10.1186/s12985-020-01305-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood EN. An apparently new syndrome of porcine epidemic diarrhoea. Vet Record. 1977;100(12):243–244. doi: 10.1136/vr.100.12.243. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Vlasova AN, Kenney SP, Saif LJ. Emerging and re-emerging coronaviruses in pigs. Curr Opin Virol. 2019;34:39–49. doi: 10.1016/j.coviro.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Liu X, Shi D, Shi H, Zhang X, Feng L. Complete genome sequence of a porcine epidemic diarrhea virus variant. J Virol. 2012;86(6):3408. doi: 10.1128/JVI.07150-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bi J, Zeng S, Xiao S, Chen H, Fang L. Complete genome sequence of porcine epidemic diarrhea virus strain AJ1102 isolated from a suckling piglet with acute diarrhea in China. J Virol. 2012;86(19):10910–10911. doi: 10.1128/JVI.01919-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen N, Li S, Zhou R, Zhu M, He S, Ye M, et al. Two novel porcine epidemic diarrhea virus (PEDV) recombinants from a natural recombinant and distinct subtypes of PEDV variants. Virus Res. 2017;242:90–95. doi: 10.1016/j.virusres.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Chen J, Shi D, Shi H, Zhang X, Yuan J, et al. Immunogenicity and antigenic relationships among spike proteins of porcine epidemic diarrhea virus subtypes G1 and G2. Arch Virol. 2016;161(3):537–547. doi: 10.1007/s00705-015-2694-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Byrum B, Zhang Y. New variant of porcine epidemic diarrhea virus, United States, 2014. Emerg Infect Dis. 2014;20(5):917–919. doi: 10.3201/eid2005.140195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su M, Li C, Qi S, Yang D, Jiang N, Yin B, et al. A molecular epidemiological investigation of PEDV in China: characterization of co-infection and genetic diversity of S1-based genes. Transbound Emerg Dis. 2020;67(3):1129–1140. doi: 10.1111/tbed.13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo J, Fang L, Ye X, Chen J, Xu S, Zhu X, et al. Evolutionary and genotypic analyses of global porcine epidemic diarrhea virus strains. Transbound Emerg Dis. 2019;66(1):111–118. doi: 10.1111/tbed.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan B, Jiao D, Zhang R, Zhou J, Guo R, Yu Z, et al. Origin and epidemic status of porcine epidemic diarrhea virus variants in China. Transbound Emerg Dis. 2020;67(3):1364–1370. doi: 10.1111/tbed.13444. [DOI] [PubMed] [Google Scholar]
- 26.Tan L, Li Y, He J, Hu Y, Cai X, Liu W, et al. Epidemic and genetic characterization of porcine epidemic diarrhea virus strains circulating in the regions around Hunan, China, during 2017-2018. Arch Virol. 2020;165(4):877–889. doi: 10.1007/s00705-020-04532-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Han F, Yan X, Liu L, Shu X, Hu H. Prevalence and phylogenetic analysis of spike gene of porcine epidemic diarrhea virus in Henan province, China in 2015-2019. Infect Genet Evol. 2021;88:104709. doi: 10.1016/j.meegid.2021.104709. [DOI] [PubMed] [Google Scholar]
- 28.Bao YJ, Qiu J, Luo Y, Rodriguez F, Qiu HJ. The genetic variation landscape of African swine fever virus reveals frequent positive selection and adaptive flexibility. Transbound Emerg Dis. 2021. [DOI] [PubMed]
- 29.Yang X, Huo JY, Chen L, Zheng FM, Chang HT, Zhao J, et al. Genetic variation analysis of reemerging porcine epidemic diarrhea virus prevailing in Central China from 2010 to 2011. Virus Genes. 2013;46(2):337–344. doi: 10.1007/s11262-012-0867-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin CM, Annamalai T, Liu X, Gao X, Lu Z, El-Tholoth M, et al. Experimental infection of a US spike-insertion deletion porcine epidemic diarrhea virus in conventional nursing piglets and cross-protection to the original US PEDV infection. Vet Res. 2015;46:134. doi: 10.1186/s13567-015-0278-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vlasova AN, Marthaler D, Wang Q, Culhane MR, Rossow KD, Rovira A, et al. Distinct characteristics and complex evolution of PEDV strains, North America, may 2013-February 2014. Emerg Infect Dis. 2014;20(10):1620–1628. doi: 10.3201/eid2010.140491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stadler J, Zoels S, Fux R, Hanke D, Pohlmann A, Blome S, et al. Emergence of porcine epidemic diarrhea virus in southern Germany. BMC Vet Res. 2015;11:142. doi: 10.1186/s12917-015-0454-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Nova PJG, Cortey M, Diaz I, Puente H, Rubio P, Martin M, et al. A retrospective study of porcine epidemic diarrhoea virus (PEDV) reveals the presence of swine enteric coronavirus (SeCoV) since 1993 and the recent introduction of a recombinant PEDV-SeCoV in Spain. Transbound Emerg Dis. 2020;67(6):2911–2922. doi: 10.1111/tbed.13666. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Yim-Im W, Chen Q, Zheng Y, Schumacher L, Huang H, et al. Identification of porcine epidemic diarrhea virus variant with a large spike gene deletion from a clinical swine sample in the United States. Virus Genes. 2018;54(2):323–327. doi: 10.1007/s11262-018-1542-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Diep N, Choijookhuu N, Fuke N, Myint O, Izzati UZ, Suwanruengsri M, et al. New tropisms of porcine epidemic diarrhoea virus (PEDV) in pigs naturally coinfected by variants bearing large deletions in the spike (S) protein and PEDVs possessing an intact S protein. Transbound Emerg Dis. 2020;67(6):2589–2601. doi: 10.1111/tbed.13607. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki T, Terada Y, Enjuanes L, Ohashi S, Kamitani W. S1 subunit of spike protein from a current highly virulent porcine epidemic diarrhea virus is an important determinant of virulence in piglets. Viruses. 2018;10(9). [DOI] [PMC free article] [PubMed]
- 37.Ji Z, Shi D, Shi H, Wang X, Chen J, Liu J, et al. A porcine epidemic diarrhea virus strain with distinct characteristics of four amino acid insertion in the COE region of spike protein. Vet Microbiol. 2021;253:108955. doi: 10.1016/j.vetmic.2020.108955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Liu X, Shi D, Shi H, Zhang X, Li C, et al. Detection and molecular diversity of spike gene of porcine epidemic diarrhea virus in China. Viruses. 2013;5(10):2601–2613. doi: 10.3390/v5102601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu H, Jung K, Vlasova AN, Chepngeno J, Lu Z, Wang Q, et al. Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J Clin Microbiol. 2015;53(5):1537–1548. doi: 10.1128/JCM.00031-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26(19):2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lole Kavita S, Bollinger Robert C, Paranjape Ramesh S, Gadkari D, Kulkarni Smita S, Novak Nicole G, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected Seroconverters in India, with evidence of Intersubtype recombination. J Virol. 1999;73(1):152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Q, Liu X, Fang Y, Zhou P, Wang Y, Zhang Y. Detection and phylogenetic analyses of spike genes in porcine epidemic diarrhea virus strains circulating in China in 2016-2017. Virol J. 2017;14(1):194. doi: 10.1186/s12985-017-0860-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng Q, Fang L, Ding Z, Wang D, Peng G, Xiao S. Rapid manipulation of the porcine epidemic diarrhea virus genome by CRISPR/Cas9 technology. J Virol Methods. 2020;276:113772. doi: 10.1016/j.jviromet.2019.113772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Information of PEDV strains obtained in this study. The information including strain name, collection date, collection region, S gene length, virus isolation and accession number.
Data Availability Statement
The nucleotide sequence data obtained in this study are available in GenBank, and their accession numbers are MZ160999-MZ161089. The nucleic acid sequences referenced in this paper were obtained from National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/nuccore).