

CASE REPORT
Pseudo hypoaldosteronism (PHA) is a type of channelopathy leading to life-threatening hyperkalemia, hyponatremia and metabolic acidosis in neonates. Type I PHA (PHAI) is characterized by either mutation in NR3C2 (MLR) gene or genes related to subunit of ENaC channel, whereas Type II (A to E) PHA is due to mutations in other genes. Type I PHA is further divided into systemic and renal forms based on the gene and organ involved. Systemic PHAI is a rare, multisystem disease presenting as severe salt wasting in neonates. In this article, we report a case of systemic pseudohypoaldosteronism type 1 in a 2 days old neonate with a novel mutation involving SCNN1B gene. Our patient appears to be the first reported case of systemic PHAI due to SCNN1B mutation from India.
Key words: systemic pseudohypoaldosteronism type 1, SCNN1B, hyponatremia, hyperkalemia, metabolic acidosis
INTRODUCTION
Pseudo hypoaldosteronism (PHA) is a rare genetic disorder characterized by severe hyperkalemia associated with hyponatremia and metabolic acidosis. It is of two types: Type I PHA (PHAI), which can either be autosomal recessive or autosomal dominant and Type II (PHAII), further subclassified in A to E based on genetic etiology, all have an autosomal dominant mode of inheritance with type IID being inherited in autosomal recessive manner also.
Autosomal dominant PHAI results from mutation in mineralocorticoid receptor (NR3C2 gene or MLR gene), whereas autosomal recessive PHAI results from mutation in any of the three subunits (alpha, beta, gamma) of epithelial sodium channels (ENaC)(1,2). Autosomal dominant subtype involves only the kidneys and is a milder form without systemic involvement, also known as renal PHAI. Autosomal recessive PHAI is a severe form, associated with multisystem involvement including kidneys, salivary glands, sweat glands, digestive glands and is called systemic PHAI. As a result of salt wasting from multiple systems, the patients develop renal manifestations like hyperkalemia, hyponatremia, pulmonary manifestations like wheezing, recurrent pulmonary infections, skin manifestations like miliaria, etc. (1,2). This disorder was first reported by Cheek and Perry in 1958 (3). Systemic PHAI is a very rare disease and only a few cases are reported to date. The estimated incidence of pseudohypoaldosteronism type 1 is around 1 in 80,000 individuals and that of systemic PHAI is 1 in 166,000 newborns (4,5).
Here we report a case of systemic pseudohypoaldosteronism type 1 in a neonate due to a novel variant detected in the gene SCNN1B.
CLINICAL CASE
A preterm (34 weeks) female neonate, full term, appropriate for gestational age with low birth weight, born of non-consanguineous marriage presented with thread like discharge from eyes and rashes all over the body on day 2 of life. The neonate had a history of sibling death due to sepsis and severe hyperkalemia at 28 days of life. Physical examination showed ropy discharge with crystallization of discharge from bilateral meibomian glands, miliaria all over the body with normal female genitalia and normal respiratory examination. (Figure 1)
Figure 1.
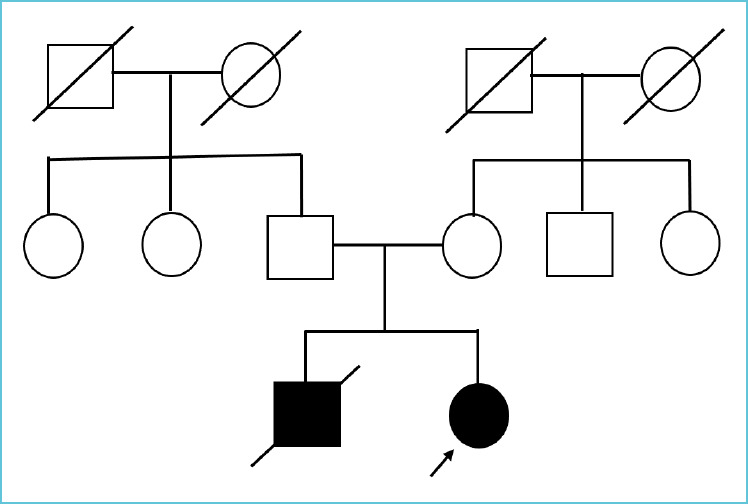
Pedigree chart
Investigations revealed severe hyperkalemia (K+ 6.7 mmol/L) and hyponatremia (Na+ 131 mmol/L). Sepsis parameters were negative. A salt wasting disorder was suspected and further investigations were done. As history of a sibling death was present, congenital adrenal hyperplasia was suspected and 17 alpha hydroxyprogesterone levels were determined, which was normal. On day 4 of life, the patient became lethargic with increased secretions from eyes and increased skin rashes. Investigations revealed worsening hyperkalemia (K+ 7.3 mmol/L), hyponatremia (Na+ 129mmol/L), metabolic acidosis. A possibility of systemic pseudohypoaldosteronism type 1 was kept and investigations were done, revealing elevated renin and aldosterone levels.
Genetic testing was done on day 5 of life in view of systemic PHAI, which revealed a homozygous NM_000336.2:c.585+1G>A mutation in intron 3 of SCNN1B gene. This variant is not reported in 1000 genomes and has an allele frequency of 0.0004% in gnomAD database. The gene SCNN1B has a low rate of benign loss of function variants as indicated by a high LoF variants Z-Score of 2.68. The c.585+1G>A variant is a loss of function variant in the gene SCNN1B, which is intolerant loss of function variant, as indicated by the presence of existing pathogenic loss of function variant NM_000336.2:c.1542+1G>A and 4 others. There are 6 downstream pathogenic loss of function variants, with the farthest being 402 residues downstream of the variant c.585+1G>A. Based on the above evidence, this variant has been classified as likely pathogenic according to ACMG guidelines (6) (Figure 2).
Figure 2.
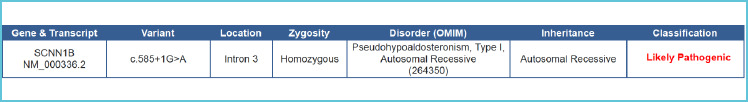
Next generation sequence findings of the case
Sequencing of the protein coding regions in genes associated was performed using Illumina next generation sequencing (NGS) systems at a mean coverage of 80-100x in the target region. GATK best practice framework was followed for variant identification. BWA-mem aligner was used to align the obtain sequences to human reference genome (GRCh37/hg19). Duplicate reads identification and removal, base quality recalibration and re-alignment of reads based on indels were done. Sention’s haplotypecaller module has been used to identify the variants which are relevant to the clinical indications (7). Along with this, deep variant analysis pipeline on Google cloud platform was used as a secondary pipeline to call genetic variants using inbuilt Sentieon modules (8). A total of 1425 genes related to the neonate’s phenotype were covered. Quality checks (QC) were performed on all to exclude variants where sequencing is of poor quality. Additional QC metrics included total homozygous and heterozygous calls (SNVs and indels), proportion of variant calls that were common, number of variants falling into different annotated consequence categories, number of extreme heterozygotes (alternate allele proportion 0.8).
As the patient had worsening hyperkalemia and metabolic acidosis, insulin, NaHCO3 and calcium polystyrene sulphonate were started. On day 24 of life, the patient had tachycardia, tachypnea, retractions and increased secretions, which was managed with chest physiotherapy, nebulization, O2 therapy and antibiotics. Respiratory distress settled on day 29 of life. Oxygen requirement decreased and the patient was discharged on day 34 on sodium supplements, sodium bicarbonate, calcium polystyrene sulphonate, nebulization with N-acetyl cysteine. The patient was on regular follow up. Eye discharge was persisting with persistent miliaria but no signs of respiratory distress. On day 70 of life, the patient went into sudden cardiac arrest at home. The cause was attributed to severe hyperkalemia but no laboratory investigations could be done to confirm the cause of death (Table 1).
Table 1.
Laboratory investigations of the case
| Laboratory parameter | Day of life 2 (admission) | Day of life 5 | Day of life 17 | Day of life 34 (discharge) |
|---|---|---|---|---|
| Sodium (mmol/L) | 131 | 129 | 141 | 140 |
| Potassium (mmol/L) | 6.7 | 7.3 | 5.5 | 4.2 |
| Urinary sodium (mmol/L) | 472 | |||
| Urinary potassium(mmol/L) | 9 | |||
| 17-alpha hydroxyprogesterone (ng/ml) | 3.9 | |||
| Plasma Renin (μIU/ml) | 267.2 | |||
| Aldosterone (ng/dL) | 1065 | |||
| Clinical exome (Next generation sequencing) | SCNN1B mutation |
DISCUSSION
Pseudo hypoaldosteronism type 1 was described first by Cheek and Perry in 1958. It is further of two types, namely renal PHAI and systemic PHAI. Renal PHAI occurs due to mutation in mineralocorticoid receptor (MLR). It is a milder variant as only kidneys are involved. Systemic PHAI is a more severe type characterized by a defect in any of the three subunits (alpha, beta, gamma) of ENaC sodium channels. Aldosterone, the principal mineralocorticoid of human body acts through mineralocorticoid receptors (MLR) and ENaC channels. ENaC is expressed in multiple tissues: sweat glands, meibomian glands, colon, salivary glands, etc. Hence, its mutation leads to multisystem involvement. Due to the impaired action of aldosterone, the renal regulation of sodium and potassium is disturbed leading to a triad of hyperkalemia, hyponatremia, and metabolic acidosis.
Systemic PHA type 1 is an autosomal recessive disease caused due to mutation in any of the three genes, namely SCNN1A (chromosome 12p13.31), SCNN1B (chromosome 16p12.1) or SCNN1G (chromosome 16p12.1). The risk increases in neonates born as a result of consanguinity (inbreeding). However, there was no inbreeding in our case. But there was an history of similar illness in sibling leading to death. Further genetic analysis of parents is required to know the carrier status.
To date, only about 35 cases of systemic pseudohypoaldosteronism have been reported around the world and only 2 cases in India. Out of which, the majority are due to mutation in SCNN1A gene and less than 10 cases are due to SCNN1B gene mutation. No case of SCNN1B mutation has been reported from India.
Most of the cases of systemic pseudohypoaldosteronism were reported in neonatal age groups, mostly in first two weeks of life. Neonates present with severe salt wasting manifestations, growth failure, letharginess and is associated with high mortality rates. As the age increases, salt wasting episodes tends to decrease and the patient might be asymptomatic and may lead a healthy life.
Investigations should include serum electrolytes, urinary electrolytes and a blood gas. Further investigation shows increased levels of serum aldosterone and renin activity. Genetic analysis can be done for confirmation of the disease and for any novel mutations.
Management includes sodium supplementation, combating hyperkalemia with albuterol nebulization, insulin, calcium gluconate infusion (with ECG monitoring), potassium binders and sodium bicarbonate for metabolic acidosis. Patients generally require lifelong treatment. As the age increases, the salt wasting manifestations decreases and patient may lead a normal healthy life.
LEARNING POINTS
When a neonate presents with severe life-threatening hyperkalemia, a possibility of systemic pseudohypoaldosteronism should be kept.
Systemic PHAI has high mortality rate when presented during infancy and mortality, morbidity decreases as age increases.
Treatment includes management of hyperkalemia, hyponatremia and metabolic acidosis. Prompt treatment might lead to the survival of patient and may lead a healthy life.
Footnotes
Contributions
Kamal Joshi: Data collection and primary manuscript.
Prashant Kumar Verma: Finalising manuscript.
Manidipa Barman: Preparing primary manuscript
REFERENCES
- 1.Furgeson SB, Linas S. Mechanisms of Type I and Type II Pseudohypoaldosteronism. J Am Soc Nephrol. 2010. Nov 1;21(11):1842–1845. [DOI] [PubMed] [Google Scholar]
- 2.Hanukoglu A. Type I pseudohypoaldosteronism includes two clinically and genetically distinct entities with either renal or multiple target organ defects. J Clin Endocrinol Metab. 1991. Nov;73(5):936–944. [DOI] [PubMed] [Google Scholar]
- 3.Cheek DB, Perry JW. A Salt Wasting Syndrome in Infancy. Arch Dis Child. 1958. Jun;33(169):252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amin N, Alvi NS, Barth JH, Field HP, Finlay E, Tyerman K, et al. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep. 2013;2013:130010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pseudohypoaldosteronism type 1: MedlinePlus Genetics [Internet]. [cited 2022 Apr 19]. Available from: https://medlineplus.gov/genetics/condition/pseudo-hypoaldosteronism-type-1/.
- 6.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. 2015. May;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freed D, Aldana R, Weber JA, Edwards JS. The Sentieon Genomics Tools - A fast and accurate solution to variant calling from next-generation sequence data [Internet]. bioRxiv; 2017. [cited 2022 Aug 16]. p. 115717. Available from:https://www.biorxiv.org/content/10.1101/115717v2.
- 8.Run DeepVariant | Cloud Life Sciences Documentation [Internet]. Google Cloud. [cited 2022 Aug 16]. Available from: https://cloud.google.com/life-sciences/docs/tutorials/deepvariant.