

Abstract
While outcomes for children with T-cell acute lymphoblastic leukemia (T-ALL) and T-lymphoblastic lymphoma (T-LL) have improved significantly with contemporary therapy, outcomes for patients with relapsed or refractory (r/r) disease remain dismal. Improved risk stratification and the incorporation of novel therapeutics have the potential to improve outcomes further in T-ALL/T-LL by limiting relapse risk and improving salvage rates for those with r/r disease. In this review we will discuss the challenges and new opportunities for improved risk stratification in T-ALL and T-LL. We will further discuss the recent incorporation of the novel therapeutics nelarabine and bortezomib into front-line therapy for children with T-ALL and T-LL. Finally, we will address new classes of targeted small molecule inhibitors, immunotherapeutics, and chimeric antigen receptor T-cell therapies under investigation in r/r T-ALL and T-LL.
Keywords: acute lymphoblastic leukemia, chimeric antigen receptor T-cells, proteasome inhibitors, immunotherapy, small molecule inhibitors
Introduction
Acute lymphoblastic leukemia (ALL) is the most common pediatric malignancy, and T-cell ALL (T-ALL) accounts for approximately 15% of pediatric ALL cases. Lymphoblastic lymphoma (LL) is the second most common type of non-Hodgkin lymphoma (NHL) in children and adolescents, with 70–80% of cases arising from T-lymphoblasts (T-LL). Survival for children and young adults with T-ALL and T-LL has improved remarkably over the past 50 years. Five-year overall survival (OS) exceeds 85% for newly diagnosed patients treated on contemporary protocols.1–4 The improvement in outcomes has been achieved by carefully designed sequential randomized clinical trials performed by multiple cooperative groups.4–13
Treatment of T-LL has shifted from an NHL-based approach to an ALL-based approach, as studies have shown superior outcomes for patients with T-LL treated on ALL-like regimens.3,14–17 Many cooperative groups now treat T-ALL and T-LL patients on the same trial using slightly modified therapy, and therapeutic differences have narrowed with time.3 While there is debate on whether T-ALL and T-LL represent separate entities or a spectrum of the same disease, it is clear they share many similar molecular alterations.3,18,19 Furthermore, T-ALL patients can present with lymphomatous disease while T-LL patients often have low-level (5–25%) marrow involvement.20 T-LL patients can relapse into the marrow and meet the definition of T-ALL (>25% marrow blasts). T-LL is less likely to involve the CNS at diagnosis or relapse.3 The primary difference between T-ALL and T-LL is the propensity of T-ALL to “invade” extra-lymphatic spaces.
Despite improved outcomes for patients with de novo disease, outcomes for patients with relapsed/refractory (r/r) T-ALL and T-LL are dismal, with 3-year OS <25%.3,21 Thus, the primary goal of front-line treatment is to prevent relapse. Unfortunately, further intensification of cytotoxic therapy is not feasible because of treatment-related mortality. Moreover, it is unlikely that further intensification will add benefit as was shown with a number of recent trials in B-ALL.22–25 Two approaches that have potential to improve cure rates in T-ALL and T-LL are more accurate risk stratification and the incorporation of effective novel therapies.
Improved Risk Stratification in T-ALL
A cornerstone of B-ALL treatment is patient risk stratification, allocating patients into prognostic groups based on genetic alterations, clinical variables, and treatment response, including minimal residual disease (MRD) and treating poor risk patients with more intensive or alternative agents and lower risk patients with less intensive therapy. In T-ALL, MRD is the only known independent prognostic variable.2,26,27 No genetic alterations or clinical variables have been found that are reproducibly prognostic independent of MRD.28,29 Thus, risk stratification is limited to response (morphology and MRD) in most cooperative groups and the majority of patients who relapse are classified as favorable risk. Better understanding of disease biology has the potential to improve risk stratification. In 2017, the Children’s Oncology Group (COG) - St. Jude Children’s Research Hospital (SJCRH) - TARGET (Therapeutically Applicable Research to Generate Effective Treatments) initiative published detailed genomic analysis on 264 T-ALL patients, demonstrating T-ALL could be grouped based on biology (Figure 1).30
Figure 1. T-ALL Biology.
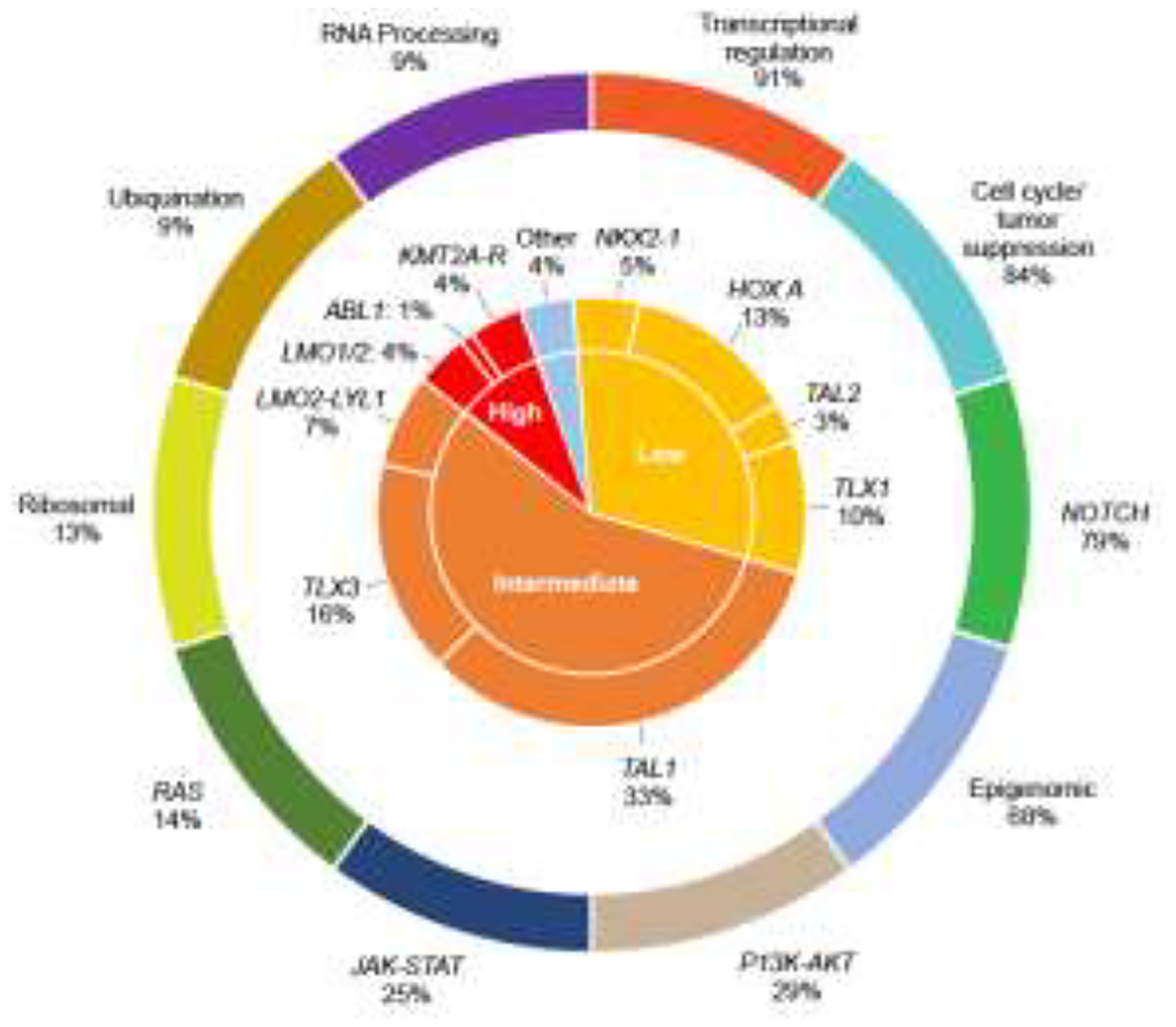
T-ALL can be divided into groups based on genetic alterations that lead to dysregulation of potentially targetable functional pathways (outer ring) or based on increased expression of different transcription factors (inner ring). Percents are frequencies in the TARGET T-ALL cohort.30 High risk (<70% 5-year event-free survival), Intermediate risk (70–90%), Low risk (>90%) – rates are not adjusted for MRD. Adapted from Teachey and Pui, Lancet Oncology 2019.2
Genomic profiling efforts by other groups have identified the same genetic alterations and confirmed the complexity and heterogeneity of T-ALL disease biology.28,29 A number of genomic classifiers have been investigated to determine prognostic significance in T-ALL. The absence of biallelic TCRγ (ABD) phenotype was associated with a poor response to induction chemotherapy in patients treated on COG P9404 (NCT01230983) and DFCI 00–01 (NCT00165178),31 but analysis of patients treated in an MRD-stratified approach on the MRC trial UKALL2003 (NCT00222612) showed equivalent outcomes independent of ABD status, suggesting this phenotype is not prognostic independent of MRD.32 The impact of alterations in NOTCH1, PTEN, RAS, and FBXW7 on outcomes in T-ALL have been investigated retrospectively by various groups, with discordant results.28,29,33–35 Our group recently performed comprehensive genomic profiling on ~1300 cases of T-ALL treated on the COG AALL0434 (NCT00408005) clinical trial through a NCI/Gabriella Miller Kids First award (1X01HD100702-01). Prior genomic studies of T-ALL have had three major limitations. First, most studies excluded high-risk and refractory patients because remission samples were used for germline DNA and these were unavailable for patients who had persistent disease after the first month of therapy. Thus, analyses were not powered to correlate genomics with outcome as many patients with events were excluded.30 Our study included all AALL0434 subjects with available samples and used a multi-pronged approach for isolating germline DNA including removing blasts by sorting and enriching normal mononuclear cells. Second, T-ALL disease biology is heterogenous and complex. Unlike B-ALL, T-ALL patients generally cannot be easily grouped by sentinel genetic alterations that drive outcome such as ETV6-RUNX1.2 Our novel unpublished data demonstrate alterations in T-ALL can be grouped by dysregulated pathways that strongly correlate with outcome. Finally, several studies demonstrated that many of the biologically relevant alterations in T-ALL occur in non-coding regions of the genome and the total number of published T-ALL genomes is <200.30,36–38 Many drivers of transcription factor deregulation cannot be identified without whole-genome sequencing (WGS). This often occurs because rearrangement breakpoints can be distant from the affected gene, do not result in expression of a chimeric transcript, or are missed by RNA-sequencing (RNA-seq). Our integrated RNA-seq and WGS analyses allows for prioritization of deregulated genes by expression analysis, then focused analysis of structural variants from the WGS. Preliminary results from our study were recently presented at the European Hematology Association (EHA) Annual meeting in 2022. We found >60% of driver lesions in T-ALL are non-coding.39 We anticipate this study will be powered to identify prognostic genomic alterations or groups of alterations that can be combined with MRD to improve risk stratification.
In 2009 a novel T-ALL subtype that had a distinctive gene expression profile and immunophenotype, similar to early T-cell precursors (ETPs) and subsequently termed ETP-ALL, was described in 17 patients.40 While early retrospective analyses found ETP ALL cases had markedly inferior outcomes in comparison to patients without ETP,31,40–43 more recent studies suggested that ETP and Non-ETP T-ALL have similar outcomes.43–46 However, small numbers of ETP patients have limited the conclusions of these studies. Despite similar eventfree survival (EFS) and OS rates for ETP and Non-ETP T-ALL on contemporary studies, the types of events differ. Non-ETP T-ALL patients are more likely to experience relapse, while patients with ETP-ALL are more likely to have refractory disease.27,43 Preliminary data from AALL0434 demonstrated significantly higher rates of induction failure (>25% blasts on end-induction marrow evaluation) and detectable MRD in patients with ETP-ALL as compared to non-ETP T-ALL.27. A complete analysis of ~1300 cases prospectively assessed for ETP status and treated on AALL0434 is ongoing and we anticipate the results of this study will define the prognostic importance of ETP status in a large, uniformly treated, centrally classified contemporary T-ALL cohort.
Risk Stratification Challenges in T-LL
Risk stratification in T-LL presents a particular challenge. Various clinical characteristics including age, sex, stage, presence of a mediastinal mass, and lactate dehydrogenase level, have not been consistently shown to have independent prognostic significance.47 Low-stage (I/II) disease is rare4,11 and is therefore not a suitable parameter to identify patients for therapy de-escalation. On COG A5971 (NCT00004228), patients with CNS disease had inferior outcome as compared to patients with disseminated disease but no CNS involvement.48 This finding could not be validated on AALL0434 because patients with CNS3 status were excluded due to low expected patient numbers.4
Similar to MRD in T-ALL, minimal disseminated disease (MDD), assessed using flow cytometry or PCR-based methods, has been evaluated for prognostic significance in T-LL.40,49,50 A retrospective analysis of samples from patients treated on COG A5971 demonstrated that over two-thirds of patients had detectable MDD at diagnosis. Further, MDD ≥1% at diagnosis was prognostic of inferior EFS.40 Similar findings were noted using an MDD cutoff of >3% in patients treated on two recent AIEOP protocols (LNH-97 and EuroLB-02).50 However, on AALL0434, patients with MDD ≥1% at diagnosis were prospectively stratified to high risk (HR) therapy, and no difference in disease free survival (DFS) was noted for HR patients (85.0% ± 3.4%) versus those treated with standard risk (SR) therapy (87.4% ± 4.0%). Further, MDD ≥1% at diagnosis was not prognostic of EFS.4 On AALL1231 (NCT02112916) T-LL patients treated with SR therapy had diagnostic MDD <1% and some patients treated with intermediate risk (IR) therapy had diagnostic MDD ≥1%. While detailed analysis of the prognostic effect of MDD in this cohort is ongoing, 4 year EFS and OS for the SR and IR groups were equivalent.51 These data from AALL0434 and AALL1231 suggest that diagnostic MDD may not have prognostic relevance in the context of contemporary T-LL therapy.
As with T-ALL, numerous studies have examined the role of genomic lesions in determining prognosis in T-LL. Loss of heterozygosity on chromosome 6q (LOH6q) was shown retrospectively to confer a higher risk of relapse in patients treated on NHL-BFM95.52 In a comparison with T-ALL samples treated on the same protocol, it was noted that the regions of LOH6q were unique between the two groups, and T-ALL patients with LOH6q did not carry an increased relapse risk.53 A subsequent retrospective analysis of a larger T-LL cohort treated on BFM protocols demonstrated a markedly inferior EFS and confirmed an increased risk of relapse for patients with LOH6q.18 Alterations in KMT2D have also been associated with an increased risk for relapse in T-LL patients treated on BFM studies.54 As in T-ALL, numerous studies have evaluated the prognostic significance of alterations in NOTCH1, FBXW7, and PTEN.18,35,54–56 In contrast to T-ALL, the available data consistently demonstrate a favorable prognosis for T-LL patients with NOTCH1 and/or FBXW7 mutations. A recently proposed genetic classifier utilized NOTCH1, RAS, PTEN, and PI3K mutation status along with LOH6q to define 3 risk groups for relapse56 and will be prospectively validated as part of an ongoing international clinical trial LBL-2018 (NCT04043494). Further, patients with NOTCH1/FBXW7 wild-type will be stratified to the HR group on LBL-2018, which should further validate the prognostic significance of these genomic alterations in T-LL.
Novel Therapies in De Novo T-ALL and T-LL
COG recently tested two novel agents, nelarabine and bortezomib, in sequential phase 3 international studies, based on promising results from trials in patients with r/r disease.57,58 AALL0434 included a 2 × 2 pseudo-factorial randomization comparing Capizzi-style escalating methotrexate plus pegaspargase (CMTX) vs. high-dose methotrexate (HDMTX), with/without six 5-day courses of nelarabine on an augmented Berlin-Frankfurt-Münster (aBFM) backbone.7,13 T-ALL patients were randomized to receive CMTX vs. HDMTX, and patients who were defined as intermediate- or high-risk were randomized to ± nelarabine (>90% of patients). The 4-year disease-free survival (DFS) for T-ALL patients on the nelarabine vs. no nelarabine arms were 88.9 ± 2.2% versus 83.3 ± 2.5%, respectively.7,13 The best outcome was on the CMTX plus nelarabine arm with a 4-year DFS of 92.2% ± 2.8%. In contrast, the 4-year DFS on the HD-MTX/no nelarabine arm, the standard of care throughout much of the world, was 78.0% ± 3.7%.7,13 All patients with T-LL received CMTX, and HR patients (64%) were randomized to ± nelarabine. The 4-year EFS and OS for T-LL patients were 84.7% ± 2.3% and 89.0% ± 2.0%, respectively.4 No difference was seen for HR patients treated with nelarabine (4-year DFS 85.1% ±4.8%) versus without nelarabine (85.0% ± 4.9%), though the study was not adequately powered to detect a difference.4
AALL1231 randomized children and young adults with T-ALL to an aBFM backbone with or without the proteasome inhibitor bortezomib during Induction and Delayed Intensification. Additional modifications were made to the AALL0434 backbone in order to eliminate cranial radiation (CRT) in >90% of patients on AALL1231: glucocorticoids were intensified using dexamethasone instead of prednisone and two extra doses of pegaspargase were given. T-LL patients have not historically received prophylactic CRT, based on low CNS relapse rates. Nevertheless, the same backbone modifications were made for patients with T-LL and T-ALL. On AALL0434, >90% of T-ALL patients received CRT. On AALL1231, <10% of patients received CRT. An evaluation of comparable T-ALL patients on AALL0434 who received CRT and AALL1231 patients who did not demonstrated similar EFS (p=0.4) and OS (p=0.6), suggesting that the backbone modifications in AALL1231 allowed for elimination of CRT without compromising outcomes.51 T-LL patients treated on the control arm of AALL1231 had inferior outcome compared to T-LL patients on AALL043451 and inferior outcome compared to T-ALL patients treated on the control arm of AALL1231 (Figure 2). In contrast, outcomes for T-LL patients treated on AALL0434 were superior to patients with T-ALL.4,13 From these data we we can infer that patients with T-ALL benefited from intensification of glucocorticoid therapy. Patients with T-LL, however, had increased treatment related mortality from the intensified backbone without a reduction in relapse.51 On AALL1231, 4-year EFS and OS were not statistically different for patients treated without bortezomb (Arm A) versus with bortezomib (Arm B) overall51 or in T-ALL (Figure 2A). In contrast, T-LL patients had improved EFS and OS with bortezomib: 4-year EFS (86.4 ± 4.0 % vs 76.5 ± 5.1%; p=0.041); 4-year OS (89.5 ± 3.6% vs 78.3 ± 4.9%, p=0.009; Figure 2B).51 This was the first study to demonstrate a benefit in OS for newly diagnosed T-LL utilizing a small molecule inhibitor.
Figure 2. T-LL patients had improved OS with bortezomib.
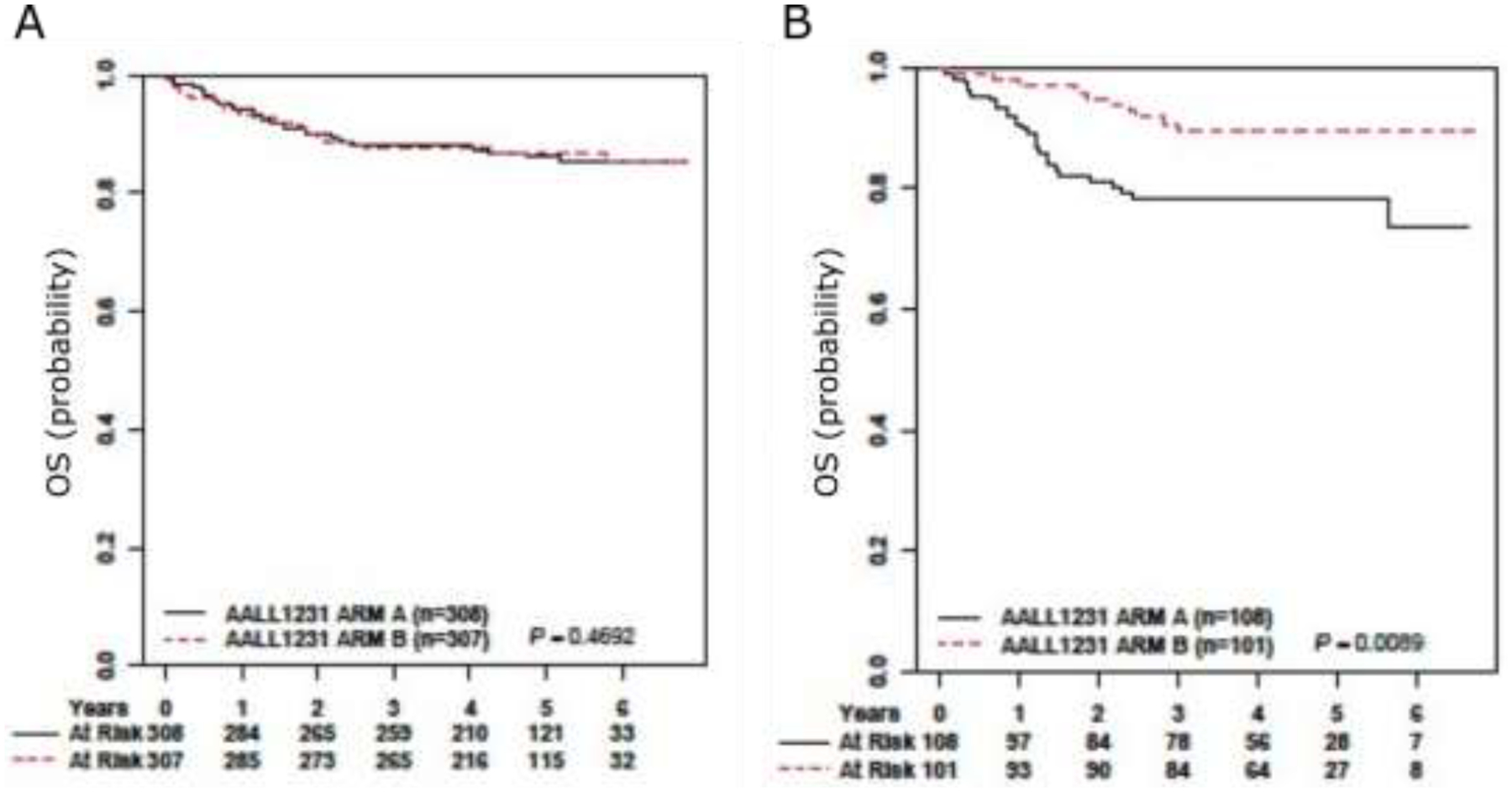
4-year OS for AALL1231 T-ALL (left panel) patients randomized to the bortezomib arm was 87.9%± 2.1% compared with 88.3%±2.1% without bortezomib (P=.469). 4-year OS for T-LL (right panel) was 89.5%±3.6% with bortezomib vs 78.3%±4.9% without bortezomib (P=.009). Adapted from Teachey et al. Journal of Clinical Oncology 2022.51
Over the past 30 years, therapy has been harmonized for T-ALL and T-LL, as they have been considered a spectrum of the same disease. The data from AALL0434 and AALL1231 clearly change that paradigm. T-ALL patients benefited from nelarabine and glucocorticoid intensification, whereas T-LL patients benefited from bortezomib. We hypothesize that there are intrinsic biologic differences or extrinsic factors such as the microenvironment separating T-ALL from T-LL that may lead to these differences in response to certain medications.
Novel Therapies for R/R T-ALL and T-LL
A number of promising targeted agents and immunotherapies are in different stages of development for r/r T-ALL. While beyond the scope to discuss these in detail, a few examples will be mentioned. A number of signaling pathways including Notch1, PI3K/Akt/mTOR, Jak/Stat, and MAPK are frequently dysregulated in T-ALL.1,2 The most commonly activated pathway in T-ALL is Notch1, leading to considerable enthusiasm targeting this pathway.21 Early clinical trials using gamma-secretase inhibitors (GSIs), which block the ability of Notch to activate transcription, were hampered by gastrointestinal (GI) toxicity.59 Preclinical studies demonstrated GI toxicity could be ameliorated either with alternative doses and schedules or with the use of corticosteroids; however, the lack of efficacy of GSIs in adult solid tumors and toxicity in early clinical trials has hindered further development.21,59 Jak/Stat signaling is frequently activated in T-ALL, especially in ETP-ALL.60 Aberrant Jak/Stat signaling is also a key driver of corticosteroid resistance and Jak/Stat inhibitors are particularly effective in preclinical models of ETP-ALL.61 Based on these data, some children with newly diagnosed ETP-ALL are eligible for treatment with the Jak/Stat inhibitor ruxolitinib in SJCRH Total Therapy XVII (NCT03117751). Deregulation of cell cycle machinery is common in T-ALL.62 Cyclin D3, in particular, has an essential role in T-ALL disease initiation and progression, which can be blocked with CDK4/6 inhibitors, including palbociclib and ribociclib.62 Multiple studies have shown CDK4/6 inhibitors are effective in T-ALL preclinical models, leading to the development of COG AINV18P1 (NCT03792256), a phase 1 trial combining palbociclib with cytotoxic chemotherapy in r/r ALL/LL.63,64 PI3K/Akt/mTOR signaling is also frequently activated in T-ALL. Moreover, inhibitors of this pathway are active as single agents and in combination with chemotherapy in preclinical T-ALL models, leading to trials combining the mTOR inhibitor everolimus with chemotherapy and with the CDK4/6 inhibitor ribociclib in r/r T-ALL (NCT03740334 and NCT01523977).64,65 Many of the therapeutic targets described above for T-ALL have relevance in T-LL as well, given the overlapping genomic landscape previously discussed, but limited preclinical work has focused specifically on T-LL. A recent publication from De Smedt and colleagues identified PIM1 as a potential therapeutic target in T-ALL and T-LL.66 The authors utilized a patient-derived xenograft model from a pediatric patient with T-LL and a TCRβ-PIM1 translocation and demonstrated that treatment with a second-generation PIM1 inhibitor synergized with dexamethasone and prolonged survival.66 This work provides an intriguing avenue for further study.
A number of preclinical studies have suggested targeting the apoptotic machinery including members of the B-cell lymphoma 2 (BCL2) protein family may be effective in T-ALL.67 BH3 profiling has demonstrated ETP-ALL is often dependent on BCL2, while non-ETP T-ALL is dependent on BCL-XL.67 Combining low-dose navitoclax and venetoclax can target both BCL-XL and BCL-2, while avoiding the excess toxicity seen with standard dosing of navitoclax in chronic lymphocytic leukemia trials.68,69 Recently, promising results from a phase 1 dose-escalation trial combining venetoclax and low-dose navitoclax with chemotherapy in r/r ALL/LL were published (NCT03181126). The combination was well-tolerated and 10 of 19 T-ALL patients obtained morphologic CR; six of those were MRD negative (<10−4).70
Independent studies from the Bourquin and Yang labs performed ex vivo drug response profiling in a large panel of primary T-ALL samples, finding a high percentage of pediatric T-ALL cases (30–44%) are sensitive to the tyrosine kinase inhibitor dasatinib.71,72 Both groups demonstrated the responses were not due to the presence of ABL-class fusions. Using phosphoproteomics, the Borquin lab identified aberrant activation of SRC in sensitive samples and also showed a SRC kinase inhibitor was effective in preclinical models.71 Using RNAseq and network-based systems pharmacology, the Yang lab identified pre-TCR-LCK activation as the driver of sensitivity to dasatinib. LCK is a SRC-family kinase.72 Both groups demonstrated T-ALL samples were resistant to other ABL-class inhibitors, such as imatinib, that do not target SRC family kinases.
The use of novel immunotherapies has transformed the therapeutic landscape in B-ALL over the past decade. Translating immunotherapies, including chimeric antigen receptor T-cells (CAR-T) has been difficult in T-ALL because of concerns over “on-target/off-tumor” toxicity, fratricide, and the possibility of contamination of T-ALL blasts during CAR-T manufacturing, leading to lentiviral transfection of leukemic blasts.73 Immunophenotypic profiling of T-ALL blasts suggests CD2, CD5, CD7, and CD38 are commonly expressed at both diagnosis and relapse in T-ALL.74 While all four of these antigens are expressed to varying degrees on normal hematopoietic cells, none of them are highly expressed on other non-hematopoietic cells or tissues, making them attractive immunotherapy targets.74 CD38-directed immunotherapies were among the first to translate into clinical trials in T-ALL, because there are a number of FDA-approved anti-CD38 monoclonal antibodies that have been shown to be safe and effective in multiple myeloma. The anti-CD38 monoclonal antibody daratumumab was found to be highly effective in preclinical models of T-ALL,75,76 leading to an ongoing trial combining daratumumab with cytotoxic chemotherapy in r/r T-ALL (NCT03384654). Preliminary data from this trial were recently presented at the 2022 American Society of Clinical Oncology (ASCO) and EHA annual meetings.77,78 An overall response rate (ORR; complete remission + complete remission with incomplete count recovery) of 83.3% was observed in pedatric patients with T-ALL and an ORR of 60% was noted in young adult patients with T-ALL. In T-LL patients an ORR of 40% was seen. Further, the toxicity profile was manageable.77,78 These preliminary data are extremely encouraging. CD47 represents another potential immunotherapeutic target in T-ALL and is overexpressed in T-ALL as compared to T-LL.79 Preclinical work has validated CD47 as a therapeutic target,80–82 and a recent study showed that combined therapy with daratumumab + an anti-CD47 monoclonal antibody had superior efficacy at clearing bone marrow disease burden when given post-chemotherapy than either antibody therapy alone in patient-derived xenograft models of T-ALL.83 These promising preclinical data support a role for clinical development of CD47-directed therapy in T-ALL.
Preclinical studies using allogenic and autologous CAR-T cells targeting CD2, CD5, and CD7 have also shown considerable promise, leading to a number of on-going clinical trials in r/r T-ALL (NCT04572308, NCT04840875, NCT03081910).84,85 Several case reports have noted efficacy of CD7-directed CAR-T cell products in patients with r/r T-ALL/T-LL.86–88 Further, a recently published phase I trial of a donor-derived CD7-directed CAR-T cell product in 20 patients with r/r T-ALL demonstrated efficient expansion, a favorable safety profile, and a 90% CR rate, though 60% of patients experienced grade 1–2 graft-versus-host disease.89 A limited follow-up study of 5 pediatric patients using an autologous CD7-directed product was presented at the 2022 ASCO annual meeting. The authors reported no cases of GVHD and an 80% CR rate.90 While challenges remain in the translation of CAR-T cell therapy in T-ALL/T-LL, these early data are very promising.
There are several challenges facing the incorporation of novel agents into treatment for both de novo and r/r T-ALL/T-LL. Toxicity associated with the up-front chemotherapy backbones used in T-ALL and T-LL limits the opportunities for incorporation of a novel therapeutic. However, adding novel agents into front-line therapy offers a critical opportunity to limit relapse, which is key to improving outcomes in T-ALL/T-LL. AALL0434 incorporated nelarabine into the Consolidation, Delayed Intensification, and Maintenance phases of therapy, while AALL1231 incorporated bortezomib during Induction and Delayed Intensification. Therefore, future front-line studies will need to identify novel agents with non-overlapping toxicity or focus on adding novel therapy to blocks such as Interim Maintenance. Further complications arise when considering the differential effects of nelarabine and bortezomib in T-ALL and T-LL. Based on the data from AALL0434 and AALL1231, some centers are now treating T-ALL and T-LL differently, with T-ALL patients received nelarabine and T-LL patients receiving bortezomib. T-ALL and T-LL may be treated differently on future clinical studies, raising the need for separate statistical consideration of these groups. Therapy for relapsed disease faces its own set of challenges. Improvement in patient outcomes has led to a concomitant decrease in patients with relapsed disease, making it difficult to enroll an adequate number of patients to complete a trial. Further, disease kinetics in r/r T-ALL/T-LL are rapid, resulting in the need for immediate treatment in most patients. As many early phase clinical trials are open only at select centers, access to novel therapies is limited for many patients. These considerations raise the need for novel, flexible clinical trial designs that expand access to as many patients as possible. Figure 3 depicts potential strategies for incorporation of novel therapies into the treatment of r/r T-ALL/T-LL, which currently consists of chemotherapy followed by hematopoietic stem cell transplantation.
Figure 3. Incorporation of novel therapeutics into therapy for relapsed/refractory T-ALL/T-LL.
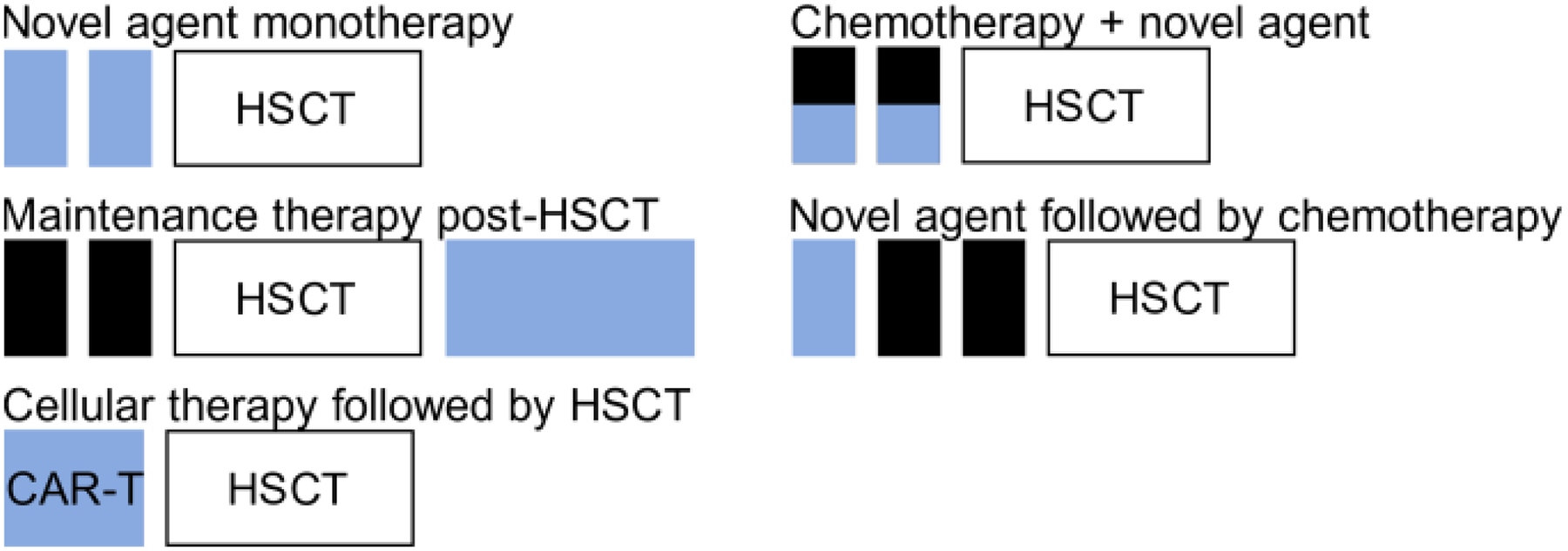
Blue boxes represent potential opportunities to incorporate novel therapeutics into therapy for relapsed/refractory T-ALL/T-LL. Abbreviations: CAR-T – chimeric antigen receptor T-cell; HSCT – hematopoietic stem cell transplant
Conclusion
In summary, the outcomes for children and young adults with T-ALL and T-LL have greatly improved over time, particularly with the incorporation of novel agents into established chemotherapy backbones. However, not all patients are cured, and new approaches are needed for these patients. Improved understanding of T-ALL and T-LL biology may lead to more accurate risk stratification, identifying patients who are likely to relapse. Novel precision medicine and immunotherapeutic approaches offer promise for patients with r/r T-lymphoblastic disease and may ultimately be incorporated into up-front therapy to reduce relapses.
Funding:
This work was funded in part by R01CA193776, R01CA264837, X01HD100702, and R03CA256550 and the Leukemia and Lymphoma Society (DTT)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest:
Summers RJ: none
Teachey DT: DTT has patents pending on CD38 CAR-T cells for hematologic malignancies and biomarkers for cytokine release syndrome. DTT receives research funding from BEAM Therapeutics and NeoImmune Tech. DTT serves on advisory boards for Sobi, Janssen, and BEAM Therapeutics.
References
- 1.Teachey DT, O’Connor D: How I treat newly diagnosed T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma in children. Blood 135:159–166, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teachey DT, Pui C-H: Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. The Lancet Oncology 20:e142–e154, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burkhardt B, Hermiston ML: Lymphoblastic lymphoma in children and adolescents: review of current challenges and future opportunities. British Journal of Haematology 185:1158–1170, 2019 [DOI] [PubMed] [Google Scholar]
- 4.Hayashi RJ, Winter SS, Dunsmore KP, et al. : Successful Outcomes of Newly Diagnosed T Lymphoblastic Lymphoma: Results From Children’s Oncology Group AALL0434. Journal of Clinical Oncology 38:3062–3070, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vora A, Goulden N, Mitchell C, et al. : Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol 15:809–18, 2014 [DOI] [PubMed] [Google Scholar]
- 6.Vrooman LM, Stevenson KE, Supko JG, et al. : Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31:1202–10, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winter SS, Dunsmore KP, Devidas M, et al. : Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children’s Oncology Group AALL0434 Methotrexate Randomization. J Clin Oncol 36:2926–2934, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moricke A, Zimmermann M, Valsecchi MG, et al. : Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127:2101–12, 2016 [DOI] [PubMed] [Google Scholar]
- 9.Pieters R, de Groot-Kruseman H, Van der Velden V, et al. : Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34:2591–601, 2016 [DOI] [PubMed] [Google Scholar]
- 10.Pui CH, Campana D, Pei D, et al. : Treating childhood acute lymphoblastic leukemia without cranial irradiation. The New England journal of medicine 360:2730–41, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landmann E, Burkhardt B, Zimmermann M, et al. : Results and conclusions of the European Intergroup EURO-LB02 trial in children and adolescents with lymphoblastic lymphoma. Haematologica 102:2086–2096, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Termuhlen AM, Smith LM, Perkins SL, et al. : Outcome of newly diagnosed children and adolescents with localized lymphoblastic lymphoma treated on Children’s Oncology Group trial A5971: A report from the Children’s Oncology Group. Pediatric Blood & Cancer 59:1229–1233, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunsmore KP, Winter SS, Devidas M, et al. : Children’s Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. Journal of Clinical Oncology 38:3282–3293, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiter A, Schrappe M, Ludwig WD, et al. : Intensive ALL-type therapy without local radiotherapy provides a 90% event-free survival for children with T-cell lymphoblastic lymphoma: a BFM group report. Blood 95:416–21, 2000 [PubMed] [Google Scholar]
- 15.Uyttebroeck A, Suciu S, Laureys G, et al. : Treatment of childhood T-cell lymphoblastic lymphoma according to the strategy for acute lymphoblastic leukaemia, without radiotherapy: long term results of the EORTC CLG 58881 trial. Eur J Cancer 44:840–6, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Sun XF, Xia ZJ, Zhen ZJ, et al. : Intensive chemotherapy improved treatment outcome for Chinese children and adolescents with lymphoblastic lymphoma. Int J Clin Oncol 13:436–41, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Sandlund JT, Pui CH, Zhou Y, et al. : Effective treatment of advanced-stage childhood lymphoblastic lymphoma without prophylactic cranial irradiation: results of St Jude NHL13 study. Leukemia 23:1127–30, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonn BR, Rohde M, Zimmermann M, et al. : Incidence and prognostic relevance of genetic variations in T-cell lymphoblastic lymphoma in childhood and adolescence. Blood 121:3153–3160, 2013 [DOI] [PubMed] [Google Scholar]
- 19.Basso K, Mussolin L, Lettieri A, et al. : T-cell lymphoblastic lymphoma shows differences and similarities with T-cell acute lymphoblastic leukemia by genomic and gene expression analyses. Genes, Chromosomes and Cancer 50:1063–1075, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Arber DA, Orazi A, Hasserjian R, et al. : The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391–2405, 2016 [DOI] [PubMed] [Google Scholar]
- 21.Raetz EA, Teachey DT: T-cell acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2016:580–588, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeha S, Pei D, Choi J, et al. : Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. Journal of Clinical Oncology 37:3377–3391, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burke MJ, Salzer WL, Devidas M, et al. : Replacing cyclophosphamide/cytarabine/mercaptopurine with cyclophosphamide/etoposide during consolidation/delayed intensification does not improve outcome for pediatric B-cell acute lymphoblastic leukemia: a report from the COG. Haematologica 104:986–992, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salzer WL, Burke MJ, Devidas M, et al. : Toxicity associated with intensive postinduction therapy incorporating clofarabine in the very high-risk stratum of patients with newly diagnosed high-risk B-lymphoblastic leukemia: A report from the Children’s Oncology Group study AALL1131. Cancer 124:1150–1159, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salzer WL, Burke MJ, Devidas M, et al. : Impact of Intrathecal Triple Therapy Versus Intrathecal Methotrexate on Disease-Free Survival for High-Risk B-Lymphoblastic Leukemia: Children’s Oncology Group Study AALL1131. J Clin Oncol 38:2628–2638, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrappe M, Valsecchi MG, Bartram CR, et al. : Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood 118:2077–84, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Wood BL, Winter SS, Dunsmore KP, et al. : T-Lymphoblastic Leukemia (T-ALL) Shows Excellent Outcome, Lack of Significance of the Early Thymic Precursor (ETP) Immunophenotype, and Validation of the Prognostic Value of End-Induction Minimal Residual Disease (MRD) in Children’s Oncology Group (COG) Study AALL0434. Blood 124:1–1, 2014. 24993873 [Google Scholar]
- 28.Petit A, Trinquand A, Chevret S, et al. : Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 131:289–300, 2018 [DOI] [PubMed] [Google Scholar]
- 29.Jenkinson S, Kirkwood AA, Goulden N, et al. : Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia 30:39–47, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Easton J, Shao Y, et al. : The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nature Genetics 49:1211–1218, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gutierrez A, Dahlberg SE, Neuberg DS, et al. : Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol 28:3816–23, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farah N, Kirkwood AA, Rahman S, et al. : Prognostic impact of the absence of biallelic deletion at the TRG locus for pediatric patients with T-cell acute lymphoblastic leukemia treated on the Medical Research Council UK Acute Lymphoblastic Leukemia 2003 trial. Haematologica 103:e288–e292, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bandapalli OR, Zimmermann M, Kox C, et al. : NOTCH1 activation clinically antagonizes the unfavorable effect of PTEN inactivation in BFM-treated children with precursor T-cell acute lymphoblastic leukemia. Haematologica 98:928–936, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tesio M, Trinquand A, Ballerini P, et al. : Age-related clinical and biological features of PTEN abnormalities in T-cell acute lymphoblastic leukaemia. Leukemia 31:2594–2600, 2017 [DOI] [PubMed] [Google Scholar]
- 35.Park M-J, Taki T, Oda M, et al. : FBXW7andNOTCH1mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. British Journal of Haematology 145:198–206, 2009 [DOI] [PubMed] [Google Scholar]
- 36.Mansour MR, Abraham BJ, Anders L, et al. : An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346:1373–1377, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rahman S, Magnussen M, León TE, et al. : Activation of the LMO2 oncogene through a somatically acquired neomorphic promoter in T-cell acute lymphoblastic leukemia. Blood 129:3221–3226, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu S, Qian M, Zhang H, et al. : Whole-genome noncoding sequence analysis in T-cell acute lymphoblastic leukemia identifies oncogene enhancer mutations. Blood 129:3264–3268, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pölönen P, Elsayed A, Montefiori L, et al. : Comprehensive genome characterization reveals new subtypes and mechanisms of oncongene deregulation in childhood T-ALL. HemaSphere 6, 2022 [Google Scholar]
- 40.Coustan-Smith E, Mullighan CG, Onciu M, et al. : Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 10:147–56, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inukai T, Kiyokawa N, Campana D, et al. : Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children’s Cancer Study Group Study L99–15. Br J Haematol 156:358–65, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Ma M, Wang X, Tang J, et al. : Early T-cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med 6:416–20, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Conter V, Valsecchi MG, Buldini B, et al. : Early T-cell precursor acute lymphoblastic leukaemia in children treated in AIEOP centres with AIEOP-BFM protocols: a retrospective analysis. The Lancet Haematology 3:e80–e86, 2016 [DOI] [PubMed] [Google Scholar]
- 44.Patrick K, Wade R, Goulden N, et al. : Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol 166:421–4, 2014 [DOI] [PubMed] [Google Scholar]
- 45.Zuurbier L, Gutierrez A, Mullighan CG, et al. : Immature MEF2C-dysregulated T-cell leukemia patients have an early T-cell precursor acute lymphoblastic leukemia gene signature and typically have non-rearranged T-cell receptors. Haematologica 99:94–102, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burns MA, Place AE, Stevenson KE, et al. : Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: Results from DFCI ALL Consortium Protocols 05–001 and 11–001. Pediatric Blood & Cancer 68, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burkhardt B, Mueller S, Khanam T, et al. : Current status and future directions of T-lymphoblastic lymphoma in children and adolescents. British Journal of Haematology 173:545–559, 2016 [DOI] [PubMed] [Google Scholar]
- 48.Termuhlen AM, Smith LM, Perkins SL, et al. : Disseminated lymphoblastic lymphoma in children and adolescents: results of the COG A5971 trial: a report from the Children’s Oncology Group. Br J Haematol 162:792–801, 2013 [DOI] [PubMed] [Google Scholar]
- 49.Stark B, Avigad S, Luria D, et al. : Bone marrow minimal disseminated disease (MDD) and minimal residual disease (MRD) in childhood T-cell lymphoblastic lymphoma stage III, detected by flow cytometry (FC) and real-time quantitative polymerase chain reaction (RQ-PCR). Pediatric Blood & Cancer 52:20–25, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Mussolin L, Buldini B, Lovisa F, et al. : Detection and role of minimal disseminated disease in children with lymphoblastic lymphoma: The AIEOP experience. Pediatr Blood Cancer 62:1906–13, 2015 [DOI] [PubMed] [Google Scholar]
- 51.Teachey DT, Devidas M, Wood BL, et al. : Children’s Oncology Group Trial AALL1231: A Phase III Clinical Trial Testing Bortezomib in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia and Lymphoma. Journal of Clinical Oncology, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burkhardt B, Bruch J, Zimmermann M, et al. : Loss of heterozygosity on chromosome 6q14–q24 is associated with poor outcome in children and adolescents with T-cell lymphoblastic lymphoma. Leukemia 20:1422–1429, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Burkhardt B, Moericke A, Klapper W, et al. : Pediatric precursor T lymphoblastic leukemia and lymphoblastic lymphoma: Differences in the common regions with loss of heterozygosity at chromosome 6q and their prognostic impact. Leukemia & Lymphoma 49:451–461, 2008 [DOI] [PubMed] [Google Scholar]
- 54.Khanam T, Sandmann S, Seggewiss J, et al. : Integrative genomic analysis of pediatric T-cell lymphoblastic lymphoma reveals candidates of clinical significance. Blood 137:2347–2359, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Callens C, Baleydier F, Lengline E, et al. : Clinical Impact of NOTCH1 and/or FBXW7 Mutations, FLASH Deletion, and TCR Status in Pediatric T-Cell Lymphoblastic Lymphoma. Journal of Clinical Oncology 30:1966–1973, 2012 [DOI] [PubMed] [Google Scholar]
- 56.Balbach ST, Makarova O, Bonn BR, et al. : Proposal of a genetic classifier for risk group stratification in pediatric T-cell lymphoblastic lymphoma reveals differences from adult T-cell lymphoblastic leukemia. Leukemia 30:970–973, 2016 [DOI] [PubMed] [Google Scholar]
- 57.Horton TM, Whitlock JA, Lu X, et al. : Bortezomib reinduction chemotherapy in high-risk ALL in first relapse: a report from the Children’s Oncology Group. Br J Haematol 186:274–285, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berg SL, Blaney SM, Devidas M, et al. : Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children’s Oncology Group. J Clin Oncol 23:3376–82, 2005 [DOI] [PubMed] [Google Scholar]
- 59.Real PJ, Tosello V, Palomero T, et al. : Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med 15:50–8, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maude SL, Dolai S, Delgado-Martin C, et al. : Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 125:1759–67, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meyer LK, Huang BJ, Delgado-Martin C, et al. : Glucocorticoids paradoxically facilitate steroid resistance in T cell acute lymphoblastic leukemias and thymocytes. J Clin Invest 130:863–876, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sawai CM, Freund J, Oh P, et al. : Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 22:452–65, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raetz E, Teachey D, Minard C, et al. : Safety of Palbociclib in Combination with Chemotherapy in Children and Young Adult Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia and Lymphoma: A Children’s Oncology Group Pilot Study. Blood, ASH Annual Meeting Abstracts, 2020, pp 20–21 [Google Scholar]
- 64.Pikman Y, Alexe G, Roti G, et al. : Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia. Clin Cancer Res 23:1012–1024, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Place AE, Pikman Y, Stevenson KE, et al. : Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 65:e27062, 2018 [DOI] [PubMed] [Google Scholar]
- 66.De Smedt R, Peirs S, Morscio J, et al. : Pre-clinical evaluation of second generation PIM inhibitors for the treatment of T-cell acute lymphoblastic leukemia and lymphoma. Haematologica 104:e17–e20, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chonghaile TN, Roderick JE, Glenfield C, et al. : Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov 4:1074–87, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roberts AW, Seymour JF, Brown JR, et al. : Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients With Relapsed or Refractory Disease. Journal of Clinical Oncology 30:488–496, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilson WH, O’Connor OA, Czuczman MS, et al. : Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. The Lancet Oncology 11:1149–1159, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pullarkat VA, Lacayo NJ, Jabbour E, et al. : Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov 11:1440–1453, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frismantas V, Dobay MP, Rinaldi A, et al. : Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood 129:e26–e37, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gocho Y, Liu J, Hu J, et al. : Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nature Cancer, 2021, pp 284–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Diorio C, Teachey DT: Harnessing immunotherapy for pediatric T-cell malignancies. Expert Rev Clin Immunol 16:361–371, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leong S, Inglott S, Papaleonidopoulou F, et al. : CD1a is rarely expressed in pediatric or adult relapsed/refractory T-ALL: implications for immunotherapy. Blood Adv 4:4665–4668, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bride KL, Vincent TL, Im SY, et al. : Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia (T-ALL). Blood, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vogiatzi F, Winterberg D, Lenk L, et al. : Daratumumab eradicates minimal residual disease in a preclinical model of pediatric T-cell acute lymphoblastic leukemia. Blood 134:713–716, 2019 [DOI] [PubMed] [Google Scholar]
- 77.Hogan LE, Bhatla T, Teachey DT, et al. : Efficacy and safety of daratumumab (DARA) in pediatric and young adult patients (pts) with relapsed/refractory T-cell acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LL): Results from the phase 2 DELPHINUS study. Journal of Clinical Oncology 40:10001–10001, 2022 [Google Scholar]
- 78.Vora A, Bhatla T, Teachey DT, et al. : Efficacy and safety of daratumumab in pediatric and young adult patients with relapsed/refractory T-cell acute lymphoblastic leukemia or lymphoblastic lymphoma: results from phase 2 Delphinus study. HemaSphere 6, 2022 [Google Scholar]
- 79.Raetz EA, Perkins SL, Bhojwani D, et al. : Gene expression profiling reveals intrinsic differences between T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Pediatric Blood & Cancer 47:130–140, 2006 [DOI] [PubMed] [Google Scholar]
- 80.Chao MP, Alizadeh AA, Tang C, et al. : Therapeutic Antibody Targeting of CD47 Eliminates Human Acute Lymphoblastic Leukemia. Cancer Research 71:1374–1384, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leclair P, Liu C-C, Monajemi M, et al. : CD47-ligation induced cell death in T-acute lymphoblastic leukemia. Cell Death & Disease 9, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang W, Wang W-T, Fang K, et al. : MIR-708 promotes phagocytosis to eradicate T-ALL cells by targeting CD47. Molecular Cancer 17, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Müller K, Vogiatzi F, Winterberg D, et al. : Combining daratumumab with CD47 blockade prolongs survival in preclinical models of pediatric T-ALL. Blood, 2022 [DOI] [PubMed] [Google Scholar]
- 84.Cooper ML, DiPersio JF: Chimeric antigen receptor T cells (CAR-T) for the treatment of T-cell malignancies. Best Pract Res Clin Haematol 32:101097, 2019 [DOI] [PubMed] [Google Scholar]
- 85.Fleischer LC, Spencer HT, Raikar SS: Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. Journal of Hematology & Oncology 12, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dai H-P, Cui W, Cui Q-Y, et al. : Haploidentical CD7 CAR T-cells induced remission in a patient with TP53 mutated relapsed and refractory early T-cell precursor lymphoblastic leukemia/lymphoma. Biomarker Research 10, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li S, Wang X, Yuan Z, et al. : Eradication of T-ALL Cells by CD7-targeted Universal CAR-T Cells and Initial Test of Ruxolitinib-based CRS Management. Clinical Cancer Research 27:1242–1246, 2021 [DOI] [PubMed] [Google Scholar]
- 88.Xie L, Ma L, Liu S, et al. : Chimeric antigen receptor T cells targeting CD7 in a child with high-risk T-cell acute lymphoblastic leukemia. Int Immunopharmacol 96:107731, 2021 [DOI] [PubMed] [Google Scholar]
- 89.Pan J, Tan Y, Wang G, et al. : Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-in-Human, Phase I Trial. Journal of Clinical Oncology 39:3340–3351, 2021 [DOI] [PubMed] [Google Scholar]
- 90.Zhao L, Pan J, Tang K, et al. : Autologous CD7-targeted CAR T-cell therapy for refractory or relapsed T-cell acute lymphoblastic leukemia/lymphoma. Journal of Clinical Oncology 40:7035–7035, 2022 [Google Scholar]