

Abstract
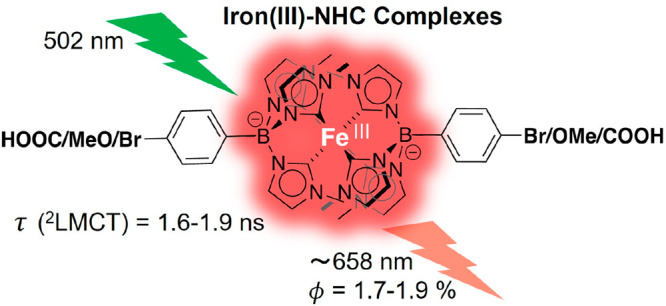
Fe(III) complexes with N-heterocyclic carbene (NHC) ligands belong to the rare examples of Earth-abundant transition metal complexes with long-lived luminescent charge-transfer excited states that enable applications as photosensitizers for charge separation reactions. We report three new hexa-NHC complexes of this class: [Fe(brphtmeimb)2]PF6 (brphtmeimb = [(4-bromophenyl)tris(3-methylimidazol-2-ylidene)borate]–, [Fe(meophtmeimb)2]PF6 (meophtmeimb = [(4-methoxyphenyl)tris(3-methylimidazol-2-ylidene)borate]–, and [Fe(coohphtmeimb)2]PF6 (coohphtmeimb = [(4-carboxyphenyl)tris(3-methylimidazol-2-ylidene)borate]–. These were derived from the parent complex [Fe(phtmeimb)2]PF6 (phtmeimb = [phenyltris(3-methylimidazol-2-ylidene)borate]– by modification with electron-withdrawing and electron-donating substituents, respectively, at the 4-phenyl position of the ligand framework. All three Fe(III) hexa-NHC complexes were characterized by NMR spectroscopy, high-resolution mass spectroscopy, elemental analysis, single crystal X-ray diffraction analysis, electrochemistry, Mößbauer spectroscopy, electronic spectroscopy, magnetic susceptibility measurements, and quantum chemical calculations. Their ligand-to-metal charge-transfer (2LMCT) excited states feature nanosecond lifetimes (1.6–1.7 ns) and sizable emission quantum yields (1.7–1.9%) through spin-allowed transition to the doublet ground state (2GS), completely in line with the parent complex [Fe(phtmeimb)2]PF6 (2.0 ns and 2.1%). The integrity of the favorable excited state characteristics upon substitution of the ligand framework demonstrates the robustness of the scorpionate motif that tolerates modifications in the 4-phenyl position for applications such as the attachment in molecular or hybrid assemblies.
Short abstract
A series of well-defined Fe(III) complexes through a hexa-NHC scorpionate framework with electron-donating and -withdrawing substituents were synthesized and fully characterized. All three Fe(III) complexes show the visible photoluminescence around 630 nm at room temperature through ligand-to-metal charge-transfer (2LMCT) excited states with nanosecond lifetimes (1.6−1.7 ns) and generous emission quantum yields (1.7−1.9%) through spin-allowed transition to the doublet ground state (2GS), exclusively similar to the parent complex [Fe(phtmeimb)2]PF6 (2.0 ns and 2.1%).
Introduction
The development of photosensitizers based on Earth-abundant, inexpensive, and nontoxic metals, with the goal of replacing the to-date widely used noble metals, has attracted a lot of interest in the field of coordination chemistry in recent years.1 Such research is motivated by the desire to use solar-energy conversion processes on a large scale. Until recently, the field of solar energy conversion based on coordination compounds has to a large degree focused on octahedral metal complexes of noble metals with low-spin 4d6 or 5d6 electronic configurations using different second and third row transition metals (TMs) including Ru(II), Re(I), Os(II), and Ir(III).2,3 The ligand field splitting in such transition metal complexes is inherently larger than that for the corresponding complexes containing first row TMs such as Cr, Mn, Fe, and Co. This shifts the metal-centered (MC) states to higher energies in the former case, which in turn results in slow deactivation of the photoactive charge-transfer (CT) states.4 Together with the employment of π-accepting ligands such as 2,2-bipyridyl (bpy), in metal complexes involving 4d6 or 5d6 metal cations, this has led to the development of many metal complexes that have metal-to-ligand charge-transfer (MLCT) states lower than MC states in energy.5,6 This implies that, for the MLCT state, a state with demonstrated importance for photofunctional applications for metal complexes based on 4d6 and 5d6 metal cations, the deactivation of the excited state via the MC states is a concern only at elevated temperatures.5 Efficient intersystem crossing from 1MLCT states usually populates 3MLCT states that exhibit slow radiative and nonradiative relaxation to the ground state. Additionally, 4d6 and 5d6 metal complexes with π accepting ligands display a relatively wide visible light absorption window and favorable redox properties of the GS and the 3MLCT state. As a result, they are heavily featured in photophysical applications.2 In parallel, four-coordinate 5d8 complexes involving Pt(II) and Au(III) have also been investigated and successfully used in photophysical applications thanks to the strong ligand field connected to these third-row transition metals.5,7
There have been some reports about photoactive Earth-abundant metal complexes, foremost from metal complexes containing TMs such as Cu(I), Cr(0), Mn(I/IV), and Co(III).5 The problem with first row transition metal complexes for photophysical applications in general is that the weak ligand field results in their MC states being relatively low in energy, providing a fast deactivation pathway and reducing the efficiency of the photofunctional MLCT states.4 Of the first row transition metals, iron is by far the most abundant.8 For Fe(II) polypyridyl complexes, the most widely studied direct base metal analogues of the successful Ru-, Os- and Ir-polypyridyl photosensitizers, the MLCT states are deactivated on the 100 fs time scale to the low-lying MC states.6 McCusker and Heinze reported attempts to increase the MLCT excited state lifetime by employing ligands with increased bite angle and introducing π-accepting and/or push–pull moieties.9,10 Recently, McCusker reported a cage compound involving the Fe(II)(bpy)3 motif, exhibiting a 2.6 ps MLCT lifetime, the longest recorded to date for an iron polypyridine complex.11 However, the introduction of strongly σ-donating N-heterocyclic carbene (NHC) ligands in the field of photoactive iron complexes6,12 has significantly increased the excited state lifetime of Fe(II) MLCT states, reaching up to 528 ps.13 By increasing the ligand field strength, the MC states increase in energy, thus slowing down the deactivation of the MLCT state.6,14,15 The photophysical properties of Fe–NHC metal complexes have been further improved using different approaches.16−20
By employing the facial tridentate scorpionate pre-NHC ligand [phtmeimbH3](PF6)2 (where phtmeimbH3 = [phenyltris(3-methyl-1H-imidazol-3-ium-1-yl)borate)]2+), reported by Smith,21 based on the corresponding pre-NHC ligand originally developed by Fehlhammer,22 [htmeimbH3](PF6)2 (where htmeimbH3 = [hydridotris(3-methyl-1H-imidazol-3-ium-1-yl)borate)]2+), we synthesized the corresponding Fe(III) complex [Fe(phtmeimb)2]PF6 (where phtmeimb = [phenyltris(3-methylimidazol-2-ylidene)borate]–).23 This complex showed an LMCT excited-state lifetime of 2 ns and an intense fluorescence with a 2.1% quantum yield,23 constituting the second example of room temperature photoluminescence from an iron complex.24 Further, the LMCT excited state was oxidatively and reductively quenched in bimolecular reactions using standard electron donors and acceptors, which was the first example of such quenching involving an iron charge-transfer state being demonstrated.23,25 Very recently, Therien synthesized an Fe–NHC–porphyrin conjugate that showed photoluminescence from a state with considerable MLCT contribution,26 and Bauer found both MLCT and LMCT photoluminescence from photoexcited states of an Fe(III)–NHC-cyclometalated complex, as communicated in a preliminary report.27 In fact, there are only three examples of iron complexes with a nanosecond excited CT state lifetime outside the class of NHC complexes, namely, the Fe(II) complexes reported by Herbert involving strongly electron-donating amide ligands28 and the cyclometalated Fe(II) complex with a phenylphenanthroline framework reported by Berkefeld.29
Given the few examples of complexes with iron-based photoluminescence and/or long-lived iron-based CT states, it is clearly a challenging task to generate new iron-based complexes possessing photophysical properties that allow for efficient applications. However, there is an interest in modifying existing, promising iron NHC complexes, as described by the increase in reported Fe–NHC complexes from 2013 to date.14,15 Most new complexes are based on structural variations of the ligand framework of the first iron complex having a 3MLCT lifetime above 1 ps, the Fe–NHC complex [Fe(pbmi)2](PF6)2 (where pbmi = 1,1′-(pyridine-2,6-diyl)bis(3-methylimidazol-2-ylidene)).12
Here, we report the first series of modifications of [Fe(phtmeimb)2]PF6 with the purpose of finding possible structure–(photo)functional relationships based on the presence of electron-withdrawing and -donating substituents. To this end, the 4-position of the phenyl group present in the framework of the NHC scorpionate ligand [phtmeimbH3]− was substituted with either bromo, carboxyl, or methoxy groups, respectively. The resulting Fe(III) complexes [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 are shown in Figure 1. The 4-phenyl position was chosen, as it constitutes an obvious point of further functionalization when considering possibilities for application, for instance, for immobilization of the [Fe(phtmeimb)2]PF6 framework on a surface, allowing for potential photofunctional applications based on the [Fe(phtmeimb)2]+ chromophore attached to semiconductors, such as dye-sensitized solar cells30,31 and photocatalysts.32,33
Figure 1.

Chemical structures of the Fe(III) complexes investigated in this study.
Results and Discussion
Synthesis and Spectroscopic Characterizations
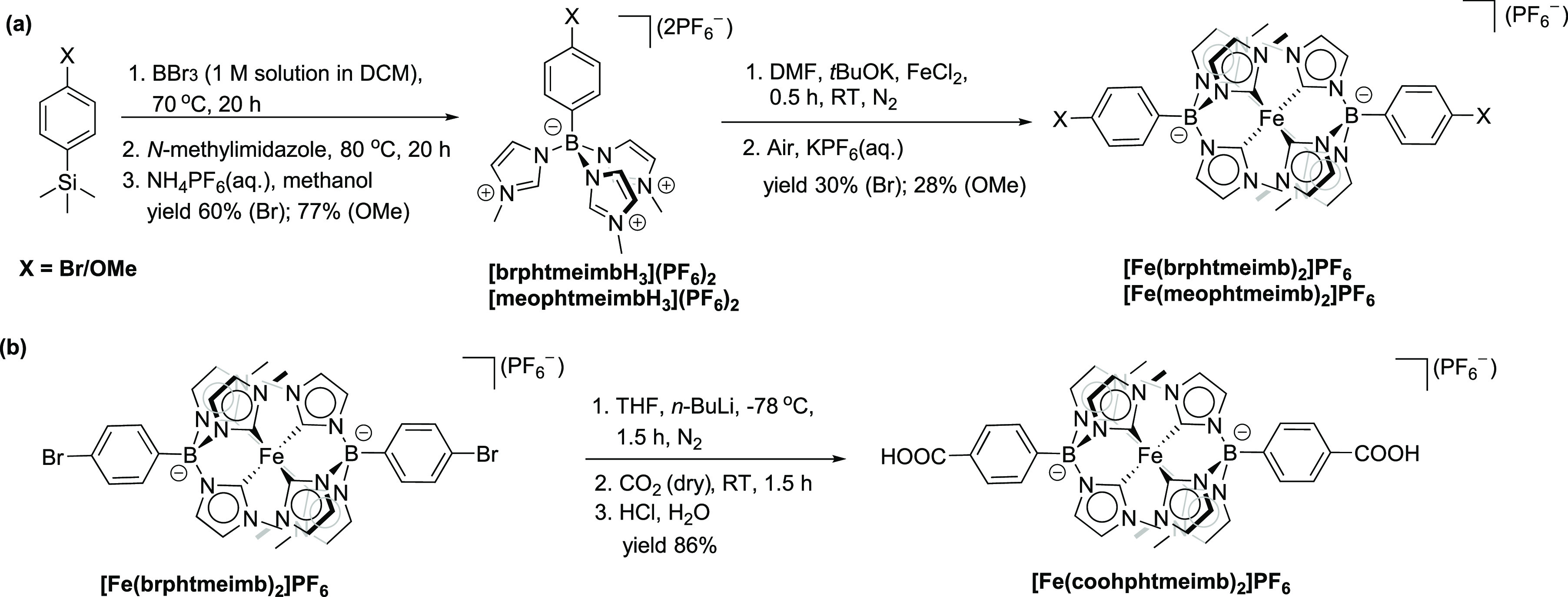
The tridentate facial pre-NHC ligands [brphtmeimbH3](PF6)2 (brphtmeimbH3 = [(4-bromophenyl)tris(3-methyl-1H-imidazol-3-ium-1-yl)borate]2+ and [meophtmeimbH3](PF6)2 (meophtmeimbH3 = [(4-methoxyphenyl)tris(3-methyl-1H-imidazol-3-ium-1-yl)borate]2+ were synthesized as shown in Scheme 1a, following the synthetic procedure of the parent pre-NHC ligand [phtmeimbH3](PF6)2,23 starting from commercially available 4-bromophenyltrimethylsilane and 4-methoxyphenyltrimethylsilane, respectively. The iron(III) complexes [Fe(brphtmeimb)2]PF6 and [Fe(meophtmeimb)2]PF6 were efficiently synthesized under a nitrogen atmosphere from FeIICl2 and the corresponding free carbene ligands, generated in situ from [brphtmeimbH3](PF6)2 and [meophtmeimbH3](PF6)2, respectively, followed by spontaneous oxidation of Fe(II) into Fe(III) during the workup procedure in air, as shown in Scheme 1a (for details, see Supporting Information section S1). The iron(III) complex [Fe(coohphtmeimb)2]PF6 (coohphtmeimb = [(4-carboxyphenyl)tris(3-methyl-1H-imidazol-3-ium-1- yl)borate]2+ was synthesized by carboxylation of [Fe(brphtmeimb)2]PF6 using n-BuLi and CO2, as shown in Scheme 1b (for details, see Supporting Information section S1). The identity and purity of [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 were established by 1H NMR and 13C NMR spectroscopy, high-resolution mass spectrometry, elemental analysis, single crystal X-ray diffraction (scXRD), cyclic voltammetry, 57Fe Mößbauer spectroscopy, magnetic susceptibility measurements, and electron paramagnetic resonance spectroscopy.
Scheme 1. (a) Synthesis of [brphtmeimbH3](PF6)2, [Fe(brphtmeimb)2]PF6, [meophtmeimbH3](PF6)2, and [Fe(meophtmeimb)2]PF6; (b) Synthesis of [Fe(coohphtmeimb)2]PF6.
The 1H NMR and 13C{1H} NMR spectra of pre-NHC ligands [brphtmeimbH3](PF6)2 and [meophtmeimbH3](PF6)2 together with the metal complexes [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 coincided well with their proposed chemical structures. All metal complexes showed well-resolved 1H NMR signals in the range from 15 to −13 ppm, despite being d5 complexes (for details, see Supporting Information section S2), as previously observed for the parent complex [Fe(phtmeimb)2]PF6.23 Dark red single crystals of [Fe(brphtmeimb)2]PF6 and [Fe(meophtmeimb)2]PF6 suitable for scXRD analysis were grown from a saturated acetonitrile solution of the respective complex by slow diffusion of diethyl ether at room temperature. Purple single crystals of [Fe(coohphtmeimb)2]PF6 suitable for scXRD analysis were grown from a saturated acetonitrile:methanol (3:2) solution of the complex by slow diffusion of diethyl ether at room temperature (see Supporting Information section S4 for details). The molecular structure (Figure 2) and the +III oxidation state of iron in all complexes were unambiguously confirmed by scXRD analysis. The coordination of the iron center to the bis-tridentate NHC ligand resulted in the complexes [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6, which resulted in a near-perfect octahedral geometry. The average Fe–C bond lengths and C–Fe–C bite angles are 2.008–1.976 Å and 87.28–86.70°, respectively, in all complexes. The observed bond lengths and angles are very similar (1.996 Å and 86.9°, respectively) to the reported parent [Fe(phtmeimb)2]PF6 complex (for details, see Supporting Information section S5).23 Notably, there are no significant deviations in the observed Fe–C bond length and C–Fe–C angles between the complexes substituted with −Br, −OMe, and −COOH groups at the 4-phenyl position in the respective ligand.
Figure 2.

scXRD Molecular structures of [Fe(phtmeimb)2]PF623 (a), [Fe(brphtmeimb)2]PF6 (b), [Fe(meophtmeimb)2]PF6 (c), and [Fe(coohphtmeimb)2]PF6 (d). Thermal ellipsoids are shown at 50% probability. Hydrogen atoms, counterions, and solvent molecules are omitted for clarity. Fe = orange; B = purple; N = blue; C = gray; Br = brown; O = red.
Magnetism, EPR, and Mößbauer Study
The magnetization study for [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 is shown in Supporting Information Figure S14, together with data for the parent [Fe(phtmeimb)2]PF6 complex. The magnetization (M) for all three systems shows the expected variation with the reduced field (BT–1) indicating a saturation not much above 1 Bohr magneton (μβ). This agrees with the common S = 1/2 ground state for the expected low-spin electronic configuration (t2g5) imposed by the strong-field carbene ligands. For all three systems, the isothermally measured magnetization curves are superimposable in the (BT–1) plot, demonstrating the absence of zero field splitting as required for the low-spin (LS) t2g5 ground state. The temperature variations of the magnetic susceptibilities of the three systems are slightly different. In all three cases, the χT product shows a temperature dependence, reflecting an incompletely quenched orbital angular momentum. The near orbital degeneracy of the t2g orbitals expected for these close-to-octahedral structures will also make the electronic g-factor very sensitive to vibronic couplings.34 This can contribute to the minor differences observed in the susceptibilities but, more importantly, will also provide a mechanism for very system dependent broadening of EPR signals. For temperature-coded data, see Supporting Information section S5 and Figures S15–S17.
The EPR spectra of [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 do not show any EPR signal at X-band frequencies, in either perpendicular or parallel mode (for details, see Supporting Information section S6), similar to their parent complex [Fe(phtmeimb)2]PF6.23
The low-temperature (80 K) Mößbauer spectra of [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 reveal a quadrupole split doublet with almost the same center shift (CS) and magnitude of the electric quadrupole splitting (QS) indicating low-spin Fe(III), just as the parent [Fe(phtmeimb)2]PF6 complex at 80 K (Supporting Information Figure S18).23 Furthermore, the 295 K spectrum of [Fe(meophtmeimb)2]PF6 also shows an extra doublet (spectral intensity 25(5)%) with CS and QS representative of low-spin Fe(II) (Supporting Information Figure S19). The Lorentzian line width Γ for the main Fe(III) component in the three spectra was only about 0.27(1) mm/s at 295 K, unveiling a narrow distribution in crystal environments in all samples. The spectra at low temperature show an asymmetric doublet structure with broad lines (Figure S18). The fitting results are presented in Table S6 which also includes the result from [Fe(phtmeimb)2]PF6.23 The Fe(II) doublet seen in the room temperature spectrum for [Fe(meophtmeimb)2]PF6 is hardly detected in the low-temperature (80 K) spectrum (Figure S18). An analysis shows a maximum spectral intensity of less than 3% for this Fe(II) component at around 80 K in the [Fe(meophtmeimb)2]PF6 sample. This could be due to different Mößbauer recoil free factors (f) for Fe(III) and Fe(II) in this complex. The f-factors become more equal at lower temperature, which is why a maximum limit of the ratio Fe(II)/Fe(III) to less than 3% can be determined. One explanation to the origin of the Fe(II) impurity(ies) is given in the caption of Supporting Information Figure S19. The center shift and electric quadrupole splitting for all of the doublets above fall in the range of reported values for low-spin S = 1/2, Fe(III) ions.23,24 In Supporting Information Figure S20, areas of different Fe valencies are presented in a |QS| vs CS (at 80 K) diagram for other Fe-carbenes. The asymmetries of the Fe(III) doublet found at 80 K can furthermore be explained on the basis of magnetic relaxation effects35 and a negative sign of QS. The relaxation time of the magnetic moment (in fact, the Mößbauer effect detects the magnetic hyperfine field acting at the Fe nucleus) of the Fe(III) ion at 80 K is comparable to the observation time τ of the Mößbauer effect. The observation time τ corresponds to the mean lifetime of the nuclear excited level, in the case of 57Fe spectroscopy to ∼70 ns. Magnetization, EPR, and Mößbauer results used to assign the spin state of the investigated compounds are summarized in Table 1.
Table 1. Magnetization, EPR, and Mößbauer Results Used to Assign the Spin State of the Investigated Compounds.
| Complex | Magnetization | EPR | Mößbauer (CS and QS), mm/s |
|---|---|---|---|
| [Fe(phtmeimb)2]+ 23 | S = 1/2; g ≈ 2.00 | No Signal | (−0.090 and 1.539);low spin S = 1/2 |
| [Fe(brphtmeimb)2]+ | S = 1/2; g ≈ 2.00 | No Signal | (−0.081 and 1.666);low spin S = 1/2 |
| [Fe(meophtmeimb)2]+ | S = 1/2; g ≈ 2.00 | No Signal | (−0.089 and 1.620);low spin S = 1/2 |
| [Fe(coohphtmeimb)2]+ | S = 1/2; g ≈ 2.00 | No Signal | (−0.056 and 1.595);low spin S = 1/2 |
Steady State Spectroscopy
The steady state absorption spectra of [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 are very similar to the parent complex [Fe(phtmeimb)2]PF6 (Figure 3). No significant trend in the absorption maxima shift can be discerned with the given experimental accuracy. These only minor differences suggest that the para-substitution of the phenyl moiety has little impact on the excited state of [Fe(phtmeimb)2]PF6. In fact, one can suspect that phenyl rings are not involved in the transitions in the lower energy manifold. This notion is corroborated by the electrochemical data (see below). Therefore, the transition peaking at around 500 nm is assigned to an 2LMCT-band for all complexes, as already elucidated for the parent compound.23 The steady state absorption data for all complexes is collected in Table 2.
Figure 3.
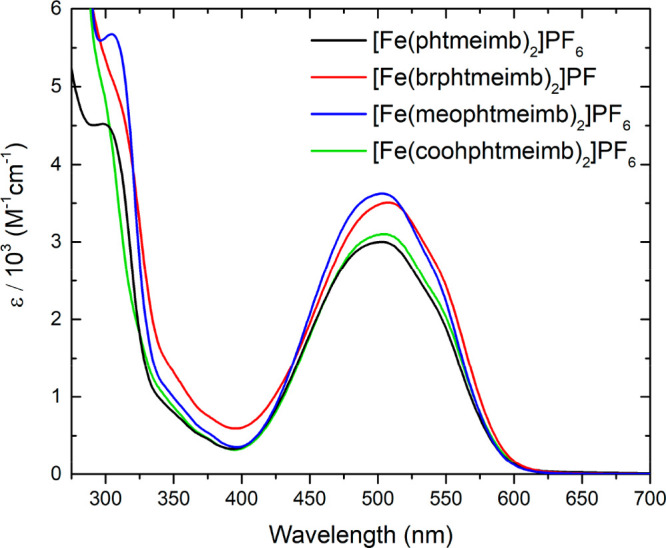
UV–vis absorption spectra of [Fe(phtmeimb)2]PF6,23 [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6. All complexes were measured in acetonitrile except for [Fe(coohphtmeimb)2]PF6 which was measured in methanol (to enable higher concentration).
Table 2. Steady State Photophysical Properties Including the Absorption Maximum (Abs max), Its Full Width at Half-Maximum (FWHM), the Peak Extinction Coefficient (ε), the Emission Maximum (Em max), Its FWHM, the Emission Quantum Yield (ϕ), and Finally the 0–0 Energy (E00) of All Complexes Discussed in This Report.
| Substituent | Abs maxa(nm/eV) | Abs fwhm (eV) | Ext coeffε (M–1 cm–1) | Em maxa(nm/eV) | Em fwhm (eV) | ϕ (%) | Ε00 (eV) |
|---|---|---|---|---|---|---|---|
| [Fe(phtmeimb)2]PF6b | 502/2.47 | 0.58 | 3000 | 655/1.89 | 0.43 | 2.1 | 2.14 |
| [Fe(brphtmeimb)2]PF6 | 508/2.44 | 0.60 | 3500 | 658/1.88 | 0.43 | 1.8 | 2.13 |
| [Fe(meophtmeimb)2]PF6 | 503/2.74 | 0.59 | 3600 | 658/1.88 | 0.43 | 1.7 | 2.13 |
| [Fe(coohphtmeimb)2]PF6 | 505/2.46 | 0.58 | 3100 | 661/1.88 | 0.43 | 1.9 | 2.14 |
Defined as where the derivative of the spectrum with respect to wavelength is zero.
Data from ref (23).
The steady state emission spectra for [Fe(phtmeimb)2]PF6, [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and Fe(coohphtmeimb)2]PF6 are shown in Figure 4. After excitation at 502 nm, all complexes show a broad emission centered around 658 nm, nearly identical to what was observed for the parent complex, [Fe(phtmeimb)2]PF6.23 To further investigate the nature of the emission, excitation spectra were recorded for [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6. The emitted intensity probed at 625 nm as a function of excitation wavelength agrees for all three complexes reasonably with their absorption spectra (see Figures S21–S23), which is an indication that the emission indeed originates from the excited 2LMCT state of the complex. The para-substituted complexes tend to have a slightly lower emission quantum yield compared to the parent complex. Based on comparative measurements to the 2.1% quantum yield reference that was reported for [Fe(phtmeimb)2]PF6,23 the quantum yield of [Fe(brphtmeimb)2]PF6 is 1.8%, that of [Fe(meophtmeimb)2]PF6 is 1.7%, and that of [Fe(coohphtmeimb)2]PF6 is 1.9%. The data for all complexes is collected in Table 2.
Figure 4.

Emission spectra of [Fe(phtmeimb)2]PF623 compared to [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6. All complexes are dissolved in acetonitrile. The emission spectrum of [Fe(phtmeimb)2]PF6 has been normalized at its peak wavelength; others are scaled by their relative emission quantum yields.
Cyclic Voltammetry and Spectroelectrochemistry
The voltammetric characterization of [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 (Figure 5 and Table 3) revealed two reversible one-electron waves that can attributed to the Fe(III/II) and Fe(IV/III) couples in analogy to the parent complex [Fe(phtmeimb)2]PF6.23 In comparison to the latter, the potentials of the metal-centered couples show only very moderate shifts of about 30 mV toward higher potentials for the electron-withdrawing bromide and carboxylic acid substituents and similar shifts in the opposite direction for the electron-donating methoxy substituent. Analogous but more pronounced substituent effects were found for the potential of the first ligand oxidation that is shifted relative to the parent complex by +80 and −280 mV in [Fe(brphtmeimb)2]PF6 and [Fe(meophtmeimb)2]PF6, respectively. Further reduction of the Fe(II) state was observed for [Fe(brphtmeimb)2]PF6 and [Fe(coohphtmeimb)2]PF6. The peaks at −2.3 and −2.7 V, respectively, can be tentatively attributed to the reduction of the functionalized aryl moieties rather than the actual carbene ligands. The latter are not reduced within the available potential window in the case of the parent complex and [Fe(meophtmeimb)2]PF6, and spectroscopic data confirms that the same situation also applies to [Fe(brphtmeimb)2]PF6 and [Fe(coohphtmeimb)2]PF6.
Figure 5.

Differential pulse and cyclic voltammograms of [Fe(brphtmeimb)2]PF6 (1 mM, 0.1 V s–1, red line) (top), [Fe(meophtmeimb)2]PF6 (1.2 mM, 0.1 V s–1, blue line) (middle), and [Fe(coohphtmeimb)2]PF6 (0.6 mM, 0.05 V s–1, green line, * postpeak due to adsorption of [Fe(coohphtmeimb)2] on the electrode) (bottom) in acetonitrile (0.1 M N(n-butyl)4PF6, black line).
Table 3. Electrochemical and UV–vis Spectroscopic Dataa.
|
E°/V |
λmax/nm(ε/103 M–1 cm–1) |
|||||
|---|---|---|---|---|---|---|
| Complex | Fe(III/II)b | Fe(IV/III)b | Loxc | n = 0(Fe(II), MLCT) | n = 1(Fe(III), LMCT) | n = 2(Fe(IV), LMCT) |
| [Fe(phtmeimb)2]n+ | –1.16 | 0.26 | 1.67 | 348 (10.8) | 502 (3.0) | 715 (6.8) |
| [Fe(brphtmeimb)2]n+ | –1.14 | 0.28 | 1.75 | 352 (12.8) | 504 (3.3) | 715 (8.9) |
| [Fe(meophtmeimb)2]n+ | –1.19 | 0.24 | 1.39 | 363 (13.5) | 505 (3.3) | 716 (10.1) |
| [Fe(coohphtmeimb)2]n+ | –1.13 | 0.29 | >1.9 | 361 (13.9) | 505 (3.1) | 717 (8.1) |
In acetonitrile with 0.1 M N(n-butyl)4PF6 vs Fc.
Half-wave potential (CV).
Peak potential (DPV).
Spectroelectrochemistry data featuring the Fe(III) ground state together with the corresponding Fe(II) and Fe(IV) states obtained from controlled potential bulk electrolysis is shown in Figures 6–8 for [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6. The data is summarized in Table 3. For the Fe(III) and Fe(IV) complexes, the energies of their lowest energy LMCT bands are within error margins indistinguishable from those of the parent complex (Table 3).23 At a first glance, this appears to be at odds with the trends in electrochemical potentials, in particular the significantly lowered potential for ligand oxidation of [Fe(meophtmeimb)2]PF6 in combination with the marginal effects on the metal couples. Also, the MLCT bands of the Fe(II) complexes are rather similar in energy. This places the potentials for ligand reduction involved in the electronic excitation in all cases well below the lower limit of the potential window, while electrochemical reduction of [Fe(brphtmeimb)2]n+ can be observed already at −2.3 V. These results support the notion that the observed electrochemical ligand oxidation and reduction processes are not involved in the spectroscopic transitions if they are essentially localized on the phenyl rings as one might anticipate in particular for ligand oxidation of [Fe(meophtmeimb)2]n+ and ligand reduction of [Fe(brphtmeimb)2]n+.
Figure 6.

UV–vis spectroelectrochemistry of [Fe(brphtmeimb)2]PF6 (red line) in acetonitrile (0.1-M N(n-butyl)4PF6). Left: Reduction at −1.44 V generating [Fe(brphtmeimb)2] (blue line). Right: Oxidation at 0.76 V generating [Fe(brphtmeimb)2]2+ (blue line).
Figure 8.

UV–vis spectroelectrochemistry of [Fe(coohphtmeimb)2]PF6 (red line) in acetonitrile (0.1 M N(n-butyl)4PF6). Left: Reduction at −1.44 V generating [Fe(coohphtmeimb)2] (blue line). Right: Oxidation at 0.76 V generating [Fe(coohphtmeimb)2]2+ (blue line).
Figure 7.

UV–vis spectroelectrochemistry of [Fe(meophtmeimb)2]PF6 (red line) in acetonitrile (0.1 M N(n-butyl)4PF6). Left: Reduction at −1.44 V generating [Fe(meophtmeimb)2] (blue line). Right: Oxidation at 0.76 V generating [Fe(meophtmeimb)2]2+ (blue line).
Transient Absorption (TA) Spectroscopy
The transient absorption (TA) spectra of [Fe(phtmeimb)2]PF6, [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 in acetonitrile recorded 100 ps after excitation in the 2LMCT band at ∼500 nm are shown in Figure 9a. The TA spectra of all complexes share the same spectral features, and the selected time delay is representative for showing the fully developed spectra that later only decay. At 500 nm, the ground state bleach (GSB) region is overwhelmed by excited state absorption (ESA). The pronounced ESA below 450 nm is in line with the LMCT assignment and the corresponding absorption of the Fe(II) ground state (see Figures 6–8). Additional ESA with a peak around 580 nm and the broad absorption in the red and near-infrared region can be attributed to the NHC ligand radical with the superimposed stimulated emission signal peaking around 700 nm. The stimulated emission dynamics unambiguously reports on the evolution of the emissive excited state population. For [Fe(brphtmeimb)2]PF6, the selected kinetics at the before mentioned wavelengths are shown in Figure 9b; all kinetics follow the same exponential decay. The kinetics for [Fe(meophtmeimb)2]PF6 and [Fe(coohphtmeimb)2]PF6 are similar (see Supporting Information Figures S24 and S25). The excited state decay can be accurately described by a single exponential model and a global fit36 to the data resulting in a universal lifetime of ∼2 ns at all observed features and for all [Fe(phtmeimb)2]PF6 derivatives; see Table 4. These results are in good agreement with emission lifetimes (of 1.9 ns) determined by time-correlated single photon counting (TC-SPC, in Supporting Information section S10, Figure S27, and Table S7). The excited state lifetimes of all three derivatives are thus very similar to the 2 ns previously reported for the parent complex [Fe(phtmeimb)2]PF6.23
Figure 9.

(a) Measured TA spectra at 100 ps of [Fe(phtmeimb)2]PF6, [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 in acetonitrile compared. The data has been chirp and background corrected and cut to remove excitation (∼500 nm) scatter. (b) Kinetics at selected wavelengths of [Fe(brphtmeimb)2]PF6, also including the single exponential fit from global analysis (measured data is shown as symbols and the fit as solid lines). The kinetics for the other complexes are shown in Supporting Information Figures S25 and S26.
Table 4. Collected Photophysical Properties Including the Emission Quantum Yield (φ), Excited State Lifetime (τ), Radiative Decay Rate (kr), and Non-Radiative Decay Rate (knr) of All Complexes Discussed in This Report.
| Complex | ϕ (%) | τ (ns) | kr(107 s–1) | knr(108 s–1) |
|---|---|---|---|---|
| [Fe(phtmeimb)2]PF6 23 | 2.1 | 2.0 | 1.1 | 5.0 |
| [Fe(brphtmeimb)2]PF6 | 1.8 | 1.7 | 1.1 | 5.8 |
| [Fe(meophtmeimb)2]PF6 | 1.7 | 1.7 | 1.0 | 5.8 |
| [Fe(coohphtmeimb)2]PF6 | 1.9 | 1.6 | 1.2 | 6.1 |
Based on the quantum yield and the lifetime of the excited state, the substituents of [Fe(phtmeimb)2]PF6 have very minor effects on both radiative and nonradiative decay pathways (Table 4). (The nonradiative decay rate is increased from 5.0 × 108 s–1 ([Fe(phtmeimb)2]PF6) to ∼6 × 108 s–1 for the substituted complexes.) Since the energetic positions of the MC states are very similar for all complexes (based on quantum chemical calculations, see Supporting Information section S11), the accelerated nonradiative decay of the substituted complexes could be due to faster internal conversion directly from the 2LMCT excited state to the ground state. The change in photophysical properties, however, is only minor, which means that introducing substituents to the [Fe(phtmeimb)2]PF6 framework still preserves strong photoluminescence from the 2LMCT state with an ∼2 ns lifetime.
Quantum Chemical Calculations
Key points on the potential energy surfaces of [Fe(phtmeimb)2]+ as well as [Fe(brphtmeimb)2]+, [Fe(meophtmeimb)2]+, and [Fe(coohphtmeimb)2]+ congeners were calculated by using unrestricted density functional theory (DFT). For brevity and due to the close similarity between the different studied molecules, only the [Fe(brphtmeimb)2]+ energy profile has been plotted in Figure 10. The quantum chemical results reveal a doublet ground state (2GS) and quartet (4MC) and hextet states (6MC) stable under phenyl group functionalization following the same energy trend as previously reported for [Fe(phtmeimb)2]+.23 The calculated spin density for the doublet ground state of all the investigated complexes is found to be mainly located on the metal and carbene lone pairs, as shown for [Fe(brphtmeimb)2]+ in Figure 10. Overall, the results from the quantum chemical calculations highlight the similarity of the electronic structure properties across the full series of complexes, including the lack of involvement of the phenyl-based moieties, which is consistent with the overall observed lack of electronic communication between the metal center and the side groups. The spin density on the metal in the relaxed quartet and hextet states indicates the same metal center nature of these states for the three iron carbene derivatives. All spin densities for [Fe(phtmeimb)2]+ and congeners are displayed in Supporting Information Table S8. The relaxed ground state geometries of the three iron complexes are in good agreement with the reported X-ray structures. The average iron–carbene distances are also reported in Supporting Information Tables S8–S12 for all complexes and suggest unremarkable structural changes due to addition to bromide, methoxy, or carboxylic groups in the 4-position of the phenyl.
Figure 10.
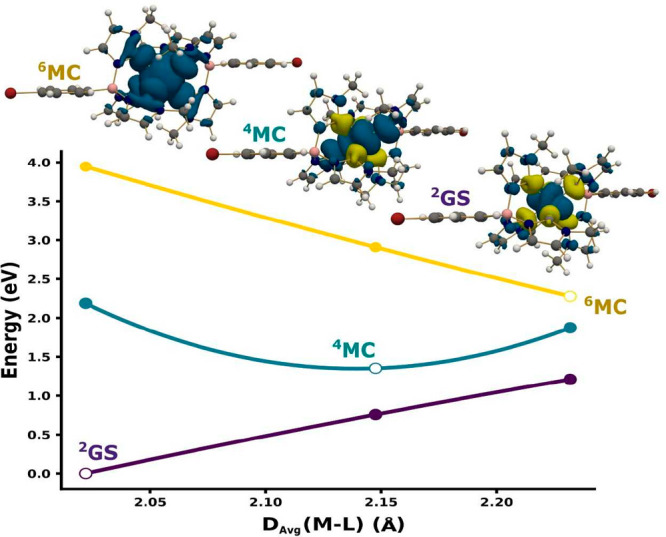
Energy graphical representation of the [Fe(brphtmeimb)2]PF6 doublet ground state (2GS), metal center quartet state (4MC), and metal center hextet state (6MC) along the internal coordinate metal–ligand averaged bond distance (Davg(M–L)). The energies of the relaxed geometries in the doublet, quartet, and hextet multiplicities are represented by empty points, while filled points correspond to the point energies calculated at the corresponding structures. The calculated spin densities for 2[Fe(brphtmeimb)2]+, 4[Fe(brphtmeimb)2]+, and 6[Fe(brphtmeimb)2]+ are also displayed. All calculated values are given in Supporting Information Tables S8–S12.
Conclusion
In conclusion, for the triad [Fe(brphtmeimb)2]PF6, [Fe(meophtmeimb)2]PF6, and [Fe(coohphtmeimb)2]PF6 that are building on the parent compound [Fe(phtmeimb)2]PF6 containing the scorpionate ligand [phtmeimb]−, the substitution of the latter in the 4-phenyl position with either −Br, −OMe, or −COOH substituents did not result in any significant changes of the ground state properties such as geometry and magnetic properties, however adding three new iron complexes with ns lifetime and visible photoluminescence to the existing very small library of such complexes. Electrochemistry and quantum chemistry calculations indicate weak electronic communication between the phenyl moiety of the scorpionate ligand and the iron center, leading to only marginal electrochemical shifts between the complexes. The essentially identical charge-transfer absorption bands of the three complexes in their Fe(II), Fe(III), and Fe(IV) states, further suggest that the spectroscopically relevant ligand orbitals do not extend over the phenyl moieties. Importantly, the 2LMCT excited state of the substituted Fe(III) complexes not only retains the excited state energy but also shows only modestly reduced emission quantum yields and excited state lifetimes relative to the parent complex. This demonstrates that the favorable photophysical properties, characteristic of the parent complex, could be exploited in prospective photoactive assemblies with the 4-phenyl position as an attachment point. Our results reveal remarkably small effects of both electron-withdrawing and -donating substituents on the ground and excited state properties, thereby demonstrating that the [Fe(phtmeimb)2]PF6 motif should tolerate a wide range of modification for the above purposes without loss of the favorable photofunctionalities.
Acknowledgments
The Swedish Strategic Research Foundation (SSF, EM16-0067) and the Knut and Alice Wallenberg (KAW, 2018.0074) Foundation are gratefully acknowledged for support. O.P. thanks the Carl Trygger Foundation for Postdoc Fellowship. R.L. acknowledges financial support by the Swedish Research Council (VR, 2020-05058). P.P. acknowledges the Swedish Research Council (VR, 2021-05313) for financial support as well as the NSC and LUNARC supercomputing centres for providing computing resources. K.W. acknowledges support from the Swedish Research Council (VR, 2020-03207), the Swedish Energy Agency (Energimyndigheten, P48747-1), the LMK Foundation, and the Sten K Johnson Foundation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c02410.
Synthesis, 1H and 13C NMR spectra, HR-MS spectra, single crystal X-ray diffraction, magnetic susceptibility and magnetization measurements, Mößbauer spectroscopy, electron paramagnetic resonance measurements, steady state spectroscopy, steady state absorption, steady state emission, transient absorption spectroscopy, TCSPC data, and quantum chemistry, including figures and tables (PDF)
The authors declare no competing financial interest.
This Article was published ASAP on October 24, 2022 with production errors. Chemical names have been updated in the Abstract, and Scheme 1 and the Supporting Information file have been replaced. The corrected version was reposted on October 26, 2022.
Supplementary Material
References
- Bozic-Weber B.; Constable E. C.; Housecroft C. E. Light harvesting with Earth abundant d-block metals: Development of sensitizers in dye-sensitized solar cells (DSCs). Coord. Chem. Rev. 2013, 257, 3089–3106. 10.1016/j.ccr.2013.05.019. [DOI] [Google Scholar]
- Balzani V.; Credi A.; Venturi M. Photochemistry and photophysics of coordination compounds: An extended view. Coord. Chem. Rev. 1998, 171, 3–16. 10.1016/S0010-8545(98)90005-4. [DOI] [Google Scholar]
- Ford P. C. From curiosity to applications. A personal perspective on inorganic photochemistry. Chem. Sci. 2016, 7, 2964–2986. 10.1039/C6SC00188B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker J. K. Electronic structure in the transition metal block and its implications for light harvesting. Science 2019, 363, 484–488. 10.1126/science.aav9104. [DOI] [PubMed] [Google Scholar]
- a Wenger O. S. Photoactive Complexes with Earth-Abundant Metals. J. Am. Chem. Soc. 2018, 140 (42), 13522–13533. 10.1021/jacs.8b08822. [DOI] [PubMed] [Google Scholar]; b Wenger O. S. A bright future for photosensitizers. Nat. Chem. 2020, 12, 323–324. 10.1038/s41557-020-0448-x. [DOI] [PubMed] [Google Scholar]; c Zhang Y.; Schulz M.; Wächtler M.; Karnahl M.; Dietzek B. Cu(i) vs. Ru(ii) photosensitizers: elucidation of electron transfer processes within a series of structurally related complexes containing an extended π-system. Coord. Chem. Rev. 2018, 356, 127–146. 10.1016/j.ccr.2017.10.016. [DOI] [PubMed] [Google Scholar]; d Kaufhold S.; Rosemann N. W.; Chábera P.; Lindh L.; Losada I. B.; Uhlig J.; Pascher T.; Strand D.; Wärnmark K.; Yartsev A.; Persson P. Microsecond Photoluminescence and Photoreactivity of a Metal-Centered Excited State in a Hexacarbene–Co(III) Complex. J. Am. Chem. Soc. 2021, 143 (3), 1307–1312. 10.1021/jacs.0c12151. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hockin B. M.; Li C.; Robertson N.; Zysman-Colman E. Photoredox catalysts based on earth-abundant metal complexes. Catal. Sci. Technol. 2019, 9, 889–915. 10.1039/C8CY02336K. [DOI] [Google Scholar]; f Cheung K. P. S.; Sarkar S.; Gevorgyan V. Visible Light-Induced Transition Metal Catalysis. Chem. Rev. 2022, 122 (2), 1543–1625. 10.1021/acs.chemrev.1c00403. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Wegeberg C.; Häussinger D.; Wenger O. S. Pyrene-Decoration of a Chromium(0) Tris(diisocyanide) Enhances Excited State Delocalization: A Strategy to Improve the Photoluminescence of 3d6Metal Complexes. J. Am. Chem. Soc. 2021, 143 (38), 15800–15811. 10.1021/jacs.1c07345. [DOI] [PubMed] [Google Scholar]; i Büldt L. A.; Guo X. W.; Vogel R.; Prescimone A.; Wenger O. S. A Tris(diisocyanide)chromium(0) Complex Is a Luminescent Analog of Fe(2,2′-Bipyridine)32+. J. Am. Chem. Soc. 2017, 139, 985–992. 10.1021/jacs.6b11803. [DOI] [PubMed] [Google Scholar]; j Herr P.; Kerzig C.; Larsen C. B.; Haussinger D.; Wenger O. S. Manganese(i) complexes with metal-to-ligand charge transfer luminescence and photoreactivity. Nat. Chem. 2021, 13, 956–962. 10.1038/s41557-021-00744-9. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Persson P.; Sundström V.; Wärnmark K. Fe N-Heterocyclic Carbene Complexes as Promising Photosensitizers. Acc. Chem. 2016, 49, 1477–1485. 10.1021/acs.accounts.6b00186. [DOI] [PubMed] [Google Scholar]
- Yam V. W. W.; Wong K. M. C. Luminescent metal complexes of d6, d8 and d10 transition metal centres. Chem. Commun. 2011, 47, 11579–11592. 10.1039/c1cc13767k. [DOI] [PubMed] [Google Scholar]
- Greenwood N. N.; Earnshaw A.. Chemistry of the Elements; Pergamon Press: Oxford, U.K., 1986. [Google Scholar]
- Britz A.; Gawelda W.; Assefa T. A.; Jamula L. L.; Yarranton J. T.; Galler A.; Khakhulin D.; Diez M.; Harder M.; Doumy G.; March A. M.; Bajnóczi Eĭ.; Németh Z.; Pápai M.; Rozsályi E.; Szemes D. S.; Cho H.; Mukherjee S.; Liu C.; Kim T. K.; Schoenlein R. W.; Southworth S. H.; Young L.; Jakubikova E.; Huse N.; Vankó G.; Bressler C.; McCusker J. K. Using Ultrafast X-ray Spectroscopy To Address Questions in Ligand-Field Theory: The Excited State Spin and Structure of [Fe(dcpp)2]2+. Inorg. Chem. 2019, 58 (14), 9341–9350. 10.1021/acs.inorgchem.9b01063. [DOI] [PubMed] [Google Scholar]
- Reuter T.; Kruse A.; Schoch R.; Bauer M.; Lochbrunner S.; Heinze K. Higher MLCT lifetime of carbene iron(II) complexes by chelate ring expansion. Chem. Commun. 2021, 57, 7541–7544. 10.1039/D1CC02173G. [DOI] [PubMed] [Google Scholar]
- Paulus B. C.; Adelman S. L.; Jamula L. L.; McCusker J. K. Leveraging excited-state coherence for synthetic control of ultrafast dynamics. Nature 2020, 582, 214–219. 10.1038/s41586-020-2353-2. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Harlang T.; Canton S. E.; Chábera P.; Suárez-Alcántara K.; Fleckhaus A.; Vithanage D. A.; Göransson E.; Corani A.; Lomoth R.; Sundström V.; Wärnmark K. Towards longer-lived metal-to-ligand charge transfer states of iron(ii) complexes: an N-heterocyclic carbene approach. Chem. Commun. 2013, 49, 6412–6414. 10.1039/c3cc43833c. [DOI] [PubMed] [Google Scholar]
- Chábera P.; Kjaer K. S.; Prakash O.; Honarfar A.; Liu Y.; Fredin L. A.; Harlang T. C. B.; Lidin S.; Uhlig J.; Sundström V.; Lomoth R.; Persson P.; Wärnmark K. FeII Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-to-Ligand Charge-Transfer Excited-State Lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. 10.1021/acs.jpclett.7b02962. [DOI] [PubMed] [Google Scholar]
- Kaufhold S.; Wärnmark K. Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes. Catalysts 2020, 10 (1), 132. 10.3390/catal10010132. [DOI] [Google Scholar]
- Lindh L.; Chábera P.; Rosemann N. W.; Uhlig J.; Wärnmark K.; Yartsev A.; Sundström V.; Persson P. Photophysics and Photochemistry of Iron Carbene Complexes for Solar Energy Conversion and Photocatalysis. Catalysts 2020, 10 (3), 315. 10.3390/catal10030315. [DOI] [Google Scholar]
- a Liu L.; Duchanois T.; Etienne T.; Monari A.; Beley M.; Assfeld X.; Haacke S.; Gros P. C. A new record excited state 3MLCT lifetime for metalorganic iron(ii) complexes. Phys. Chem. Chem. Phys. 2016, 18, 12550–12556. 10.1039/C6CP01418F. [DOI] [PubMed] [Google Scholar]; b Francés-Monerris A.; Magra K.; Darari M.; Cebrián C.; Beley M.; Domenichini E.; Haacke S.; Pastore M.; Assfeld X.; Gros P. C.; Monari A. Synthesis and Computational Study of a Pyridylcarbene Fe(II) Complex: Unexpected Effects of fac/mer Isomerism in Metal-to-Ligand Triplet Potential Energy Surfaces. Inorg. Chem. 2018, 57 (16), 10431–10441. 10.1021/acs.inorgchem.8b01695. [DOI] [PubMed] [Google Scholar]; c Magra K.; Domenichini E.; Francés-Monerris A.; Cebrián C.; Beley M.; Darari M.; Pastore M.; Monari A.; Assfeld X.; Haacke S.; Philippe C. G. Impact of the fac/mer Isomerism on the Excited-State Dynamics of Pyridyl-carbene Fe(II) Complexes. Inorg. Chem. 2019, 58 (8), 5069–5081. 10.1021/acs.inorgchem.9b00138. [DOI] [PubMed] [Google Scholar]; d Duchanois T.; Etienne T.; Beley M.; Assfeld X.; Perpète E. A.; Monari A.; Gros P. C. Heteroleptic Pyridyl-Carbene Iron Complexes with Tuneable Electronic Properties. Eur. J. Inorg. Chem. 2014, 2014, 3747–3753. 10.1002/ejic.201402356. [DOI] [Google Scholar]; e Magra K.; Francés-Monerris A.; Cebrian C.; Monari A.; Haacke S.; Gros P. C. Bidentate Pyridyl-NHC Ligands: Synthesis, Ground and Excited State Properties of Their Iron(II) Complexes and the Role of the fac/mer Isomerism. Eur. J. Inorg. Chem. 2021, 10.1002/ejic.202100818. [DOI] [Google Scholar]
- Reuter T.; Kruse A.; Schoch R.; Lochbrunner S.; Bauer M.; Heinze K. Higher MLCT lifetime of carbene iron(II) complexes by chelate ring expansion. Chem. Commun. 2021, 57, 7541–7544. 10.1039/D1CC02173G. [DOI] [PubMed] [Google Scholar]
- a Zimmer P.; Müller P.; Burkhardt L.; Schepper R.; Neuba A.; Steube J.; Dietrich F.; Flörke U.; Mangold S.; Gerhards M.; Bauer M. N-Heterocyclic Carbene Complexes of Iron as Photosensitizers for Light-Induced Water Reduction. Eur. J. Inorg. Chem. 2017, 2017, 1504–1509. 10.1002/ejic.201700064. [DOI] [Google Scholar]; b Dierks P.; Vukadinovica Y.; Bauer M. Photoactive iron complexes: more sustainable, but still a challenge. Inorg. Chem. Front. 2022, 9, 206–220. 10.1039/D1QI01112J. [DOI] [Google Scholar]
- Nair S. S.; Bysewski O. A.; Kupfer S.; Wächtler M.; Winter A.; Schubert U. S.; Dietzek B. Excitation Energy-Dependent Branching Dynamics Determines Photostability of Iron(II)–Mesoionic Carbene Complexes. Inorg. Chem. 2021, 60 (12), 9157–9173. 10.1021/acs.inorgchem.1c01166. [DOI] [PubMed] [Google Scholar]
- a Becker M.; Wyss V.; Housecroft C. E.; Constable E. C. The influence of alkyl chains on the performance of DSCs employing iron(II) N-heterocyclic carbene sensitizers. Dalton Trans. 2021, 50, 16961–16969. 10.1039/D1DT03252F. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Karpacheva M.; Wyss V.; Housecroft C. E.; Constable E. C. There Is a Future for N-Heterocyclic Carbene Iron(II) Dyes in Dye-Sensitized Solar Cells: Improving Performance through Changes in the Electrolyte. Materials 2019, 12 (24), 4181. 10.3390/ma12244181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshaw A. P.; Bontchev R. P.; Smith J. M. Oxidation of the Tris(carbene)borate Complex PhB(MeIm)3MnI(CO)3 to MnIV[PhB(MeIm)3]2(OTf)2. Inorg. Chem. 2007, 46 (10), 3792–3794. 10.1021/ic070187w. [DOI] [PubMed] [Google Scholar]
- Kernbach U.; Ramm M.; Luger P.; Fehlhammer W. P. A Chelating Triscarbene Ligand and Its Hexacarbene Iron Complex. Angew. Chem., Int. Ed. Engl. 1996, 35, 310–312. 10.1002/anie.199603101. [DOI] [Google Scholar]; Angew. Chem. 1996, 108, 333 - 335.10.1002/ange.19961080313
- Kjær K. S.; Kaul N.; Prakash O.; Chábera P.; Rosemann N. W.; Honarfar A.; Gordivska O.; Fredin L. A.; Bergquist K.; Häggström L.; Ericsson T.; Lindh L.; Yartsev A.; Styring S.; Huang P.; Uhlig J.; Bendix J.; Strand D.; Sundström V.; Persson P.; Lomoth R.; Wärnmark K. Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 2019, 363, 249–253. 10.1126/science.aau7160. [DOI] [PubMed] [Google Scholar]
- Chábera P.; Kjaer K. S.; Prakash O.; Honarfar A.; Liu Y.; Fredin L. A.; Harlang T. C. B.; Lidin S.; Uhlig J.; Sundström V.; Lomoth R.; Persson P.; Wärnmark K. FeII Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-to-Ligand Charge-Transfer Excited-State Lifetime. J. Phys. Chem. Lett. 2018, 9 (3), 459–463. 10.1021/acs.jpclett.7b02962. [DOI] [PubMed] [Google Scholar]; b Chábera P.; Liu Y.; Prakash O.; Thyrhaug E.; Nahhas A.; Honarfar A.; Essén S.; Fredin L. A.; Harlang T. C. B.; Kjær K. S.; Handrup K.; Ericson F.; Tatsuno H.; Morgan K.; Schnadt J.; Häggström L.; Ericsson T.; Sobkowiak A.; Lidin S.; Huang P.; Styring S.; Uhlig J.; Bendix J.; Lomoth R.; Sundström V.; Persson P.; Wärnmark K. A low-spin Fe(iii) complex with 100-ps ligand-to-metal charge transfer photoluminescence. Nature 2017, 543, 695–699. 10.1038/nature21430. [DOI] [PubMed] [Google Scholar]
- a Rosemann N. W.; Chábera P.; Prakash O.; Kaufhold S.; Wärnmark K.; Yartsev A.; Persson P. Tracing the Full Bimolecular Photocycle of Iron(III)–Carbene Light Harvesters in Electron-Donating Solvents. J. Am. Chem. Soc. 2020, 142 (19), 8565–8569. 10.1021/jacs.0c00755. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Aydogan A.; Bangle R. E.; Cadranel A.; Turlington M. D.; Conroy D. T.; Cauët E.; Singleton M. L.; Meyer G. J.; Sampaio R. N.; Elias B.; Troian-Gautier L. Accessing Photoredox Transformations with an Iron(III) Photosensitizer and Green Light. J. Am. Chem. Soc. 2021, 143 (38), 15661–15673. 10.1021/jacs.1c06081. [DOI] [PubMed] [Google Scholar]
- Jiang T.; Bai Y.; Zhang P.; Han Q.; Mitzi D. B.; Therien M. J. Electronic structure and photophysics of a supermolecular iron complex having a long MLCT-state lifetime and panchromatic absorption. Proc. Natl. Acad. Sci.U.S.A. 2020, 117 (34), 20430–20437. 10.1073/pnas.2009996117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M.; Steube J.; Päpcke A.; Bokareva O.; Reuter T.; Demeshko S.; Schoch R.; Hohloch S.; Meyer F.; Heinze K.; Kühn O.; Lochbrunner S.. Janus-type dual emission of a Cyclometalated Iron(III) complex. 10.21203/rs.3.rs-64316/v1. [DOI] [PMC free article] [PubMed]
- a Braun J. D.; Lozada I. B.; Kolodziej C.; Burda C.; Newman K. M. E.; Lierop J. v.; Davis R. L.; Herbert D. E. Iron(II) coordination complexes with panchromatic absorption and nanosecond charge-transfer excited state lifetimes. Nat. Chem. 2019, 11, 1144–1150. 10.1038/s41557-019-0357-z. [DOI] [PubMed] [Google Scholar]; b Larsen C. B.; Braun J. D.; Lozada I. B.; Kunnus K.; Biasin E.; Kolodziej C.; Burda C.; Cordones A. A.; Gaffney K. J.; Herbert D. E. Reduction of Electron Repulsion in Highly Covalent Fe-Amido Complexes Counteracts the Impact of a Weak Ligand Field on Excited-State Ordering. J. Am. Chem. Soc. 2021, 143 (49), 20645–20656. 10.1021/jacs.1c06429. [DOI] [PubMed] [Google Scholar]
- Leis W.; Argüello Cordero M. A.; Lochbrunner S.; Schubert H.; Berkefeld A. A Photoreactive Iron(II) Complex Luminophore. J. Am. Chem. Soc. 2022, 144 (3), 1169–1173. 10.1021/jacs.1c13083. [DOI] [PubMed] [Google Scholar]
- O'Regan B.; Gratzel M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. 10.1038/353737a0. [DOI] [Google Scholar]
- Sharma K.; Sharma V.; Sahrama S. S. Dye-Sensitzed Solar Cells: fundamentals and Current Statues. Nanoscale Res. Lett. 2018, 13, 381. 10.1186/s11671-018-2760-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Niu F.; Mortelliti M. J.; Sheridan M. V.; Sherman B. D.; Zhu Y.; McBride J. R.; Dempsey J. L.; She S.; Dares C. J.; Li F.; Meyer T. J. A stable dye-sensitized photoelectrosynthesis cell mediated by a NiO overlayer for water oxidation. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 12564–12571. 10.1073/pnas.1821687116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M.; Leung M. K. H.; Leung D. Y. C.; Sumathy K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renewable and Sustainable Energy Reviews 2007, 11, 401–425. 10.1016/j.rser.2005.01.009. [DOI] [Google Scholar]
- a Griffith J. S.The Theory of Transition-Metal Ions; Cambridge Univ. Press: 1964; Chapter 12. [Google Scholar]; b Duelund L.; Toftlund H. Electron paramagnetic resonance characteristics of some non-heme low-spin iron(III) complexes. Spectrochim. Acta, Part A 2000, 56, 331–340. 10.1016/S1386-1425(99)00243-7. [DOI] [PubMed] [Google Scholar]
- Gutlich P.; Bill E.; Trautwein A. X.. Mössbauer Spectroscopy and Transition Metal Chemistry; Springer-Verlag: Berlin, Heidelberg, 2011. [Google Scholar]
- Müller C.; Pascher T.; Eriksson A.; Chábera P.; Uhlig J. KiMoPack: A python Package for Kinetic Modeling of the Chemical Mechanism. J. Phys. Chem. J. Phys. Chem. A 2022, 126 (25), 4087–4099. 10.1021/acs.jpca.2c00907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.