

Abstract
Accumulation of lipid-laden foam cells in the arterial wall plays a central role in atherosclerotic lesion development, plaque progression and late-stage complications of atherosclerosis. Yet, there are still fundamental gaps in our knowledge of the underlying mechanisms leading to foam cell formation in atherosclerotic arteries. The goal of this study was to investigate the role of receptor-independent macropinocytosis in arterial lipid accumulation and pathogenesis of atherosclerosis. Here, we showed that genetic inhibition of fluid-phase macropinocytosis in myeloid cells (LysMCre+ NHE1fl/fl) and repurposing of an FDA-approved drug that inhibits macrophage macropinocytosis, substantially decrease atherosclerotic lesion development in LDL receptor-deficient and ApoE−/− mice. Stimulation of macropinocytosis using genetic (H-RASG12V) and physiologically relevant approaches promotes internalization of unmodified native (nLDL) and modified (oxLDL and acLDL) lipoproteins in both wild-type and scavenger receptor (SR) knockout (CD36−/−/SR-A−/−) macrophages. Pharmacological inhibition of macropinocytosis in hypercholesterolemic wild-type and CD36−/−/SR-A−/− mice identifies an important role of macropinocytosis in LDL uptake by lesional macrophages in vivo and development of atherosclerosis. Furthermore, serial section high-resolution imaging, LDL immunolabeling and 3D reconstruction of subendothelial foam cells provide the first visual evidence of lipid macropinocytosis in both human and murine atherosclerotic arteries. In summary, our findings complement the SR paradigm of atherosclerosis and identify an entirely new therapeutic strategy to counter the development of atherosclerosis and reduce morbidity and mortality in patients with cardiovascular disease.
Keywords: Macropinocytosis, scavenger receptors, atherosclerosis, LDL, drug repurposing
One Sentence Summary:
Macrophage macropinocytosis: an important player and key therapeutic target in atherosclerosis.
Introduction
Atherosclerotic vascular disease is the underlying cause of myocardial infarction, ischemic stroke, stable and unstable angina, and peripheral artery disease. The development of atherosclerosis is initiated by subendothelial retention of plasma-derived apolipoprotein B (apoB)–containing lipoproteins (e.g., LDL) in focal areas of arteries, particularly regions in which laminar flow is disturbed by bends and at bifurcations (1). Subendothelial macrophages internalize LDL lipoproteins in the arterial wall, become lipid-laden foam cells that may undergo apoptosis and, if not coupled with efferocytic clearance, contribute to the formation and expansion of atherosclerotic lesions. Advanced atherosclerotic lesions may undergo destabilization, leading to rupture, thrombosis and compromised oxygen supply of affected organs (2). The most effective therapy to date that reduces atherosclerotic cardiovascular disease works by decreasing plasma LDL concentration, thereby reducing the likelihood that these cholesterol-rich particles will enter the arterial wall and become internalized by subendothelial macrophages (3). Despite the currently available drug-based therapies and advanced surgical interventions, atherosclerotic cardiovascular disease accounts for the majority of deaths in the Western world and its therapeutic management remains one of the most serious challenges in cardiovascular medicine (4).
Internalization of modified LDL [e.g., acetylated (ac) and oxidized (ox) LDL] by scavenger receptors (SR) has become the most widely accepted mechanism for macrophage foam cell formation during the last three decades (5–8). Molecular cloning identified SR family members CD36 and SR-A as high-affinity receptors for acLDL and oxLDL (9) and subsequent binding studies confirmed that deletion of CD36 and SR-A in macrophages (CD36−/−/SR-A−/−) inhibits 80% and 90% uptake of acLDL and oxLDL, respectively (7). Despite the dominance of this paradigm, several studies have demonstrated a partial (8, 10) or lack of inhibition (6, 11) of the atherosclerotic lesion area in hypercholesterolemic CD36−/−, SR-A−/− and CD36−/−/SR-A−/− mice. Unexpectedly, clinical studies reported an increased mortality rate from atherosclerotic coronary artery disease (CAD) in CD36-deficient patients (12), which may be due to the ubiquitous expression of CD36 and its pleiotropic functions that go beyond LDL uptake, lipid absorption and metabolism (13). Previous studies demonstrated that LDL undergoes oxidation in atherosclerotic lesions and amount of oxLDL correlate with plaque vulnerability (14). Despite this information, most clinical trials failed to support the benefits of antioxidant therapies in patients with atherosclerotic vascular disease (15). Although there are many potential and speculative explanations why antioxidants failed to improve clinical outcomes in patients with atherosclerosis (16), they also support the existence of a yet uncharacterized, LDL oxidation- and SR-independent cellular mechanism of lipid uptake in atherosclerotic vessels. Supporting this statement, in vitro studies by Kruth et al. have offered alternative LDL receptor (LDLR)- and SR-independent mechanisms of macrophage lipoprotein uptake via fluid-phase macropinocytosis (17–19). These studies demonstrated that stimulation of macrophage macropinocytosis in the presence of exogenous unmodified, native LDL (nLDL) promotes foam cell formation in vitro. Growth factors (PDGF, EGF, MCSF) and inflammatory cytokines (IFN-γ and TNF-α) stimulate macropinocytosis in vitro (20–24). Mechanistically, it was found that growth factors and inflammatory cytokines promote activation of their downstream effectors, Ras and PI3 kinase (PI3K), leading to dynamic phosphorylation of phosphatidylinositols, submembranous actin polymerization, membrane ruffling, macropinosome formation and macropinocytosis (25). Importantly, protein expression of these physiologically relevant macropinocytosis stimulators are elevated in both human and murine atherosclerotic arteries (26–28). In addition, cholesterol concentration found in atherosclerotic arteries saturate SR-mediated macrophage LDL uptake (29, 30), thus further supporting the role of receptor-independent uptake mechanisms of lipids in atherosclerotic vessels. To our knowledge, no previous studies have inhibited macropinocytosis selectively in myeloid cells in animal models of atherosclerosis, investigated the role of macropinocytosis-mediated LDL uptake in atherosclerosis development, quantified LDL macropinocytosis in atherosclerotic arteries, or analyzed human and murine atherosclerotic vessels for evidence of macropinocytosis-associated plasma membrane activities. Further, the present study repurposed a newly identified FDA-approved macropinocytosis inhibitor in a preclinical murine model to investigate its efficacy as a pharmacological treatment of atherosclerosis.
Results
Pharmacological inhibition of macropinocytosis decreases atherosclerotic lesion development in both wild-type and CD36−/−/SR-A−/− mice
Macropinocytosis (a.k.a. fluid-phase endocytosis) is an endocytic mechanism mediating internalization of extracellular fluid and non-specific bulk uptake of pericellular solutes. Pharmacological blockade of Na+-H+ exchanger 1 (NHE1) using 5-(N-ethyl-N-isopropyl)amiloride (EIPA) is currently regarded as the most effective and selective approach to inhibit macropinocytosis both in vitro and in vivo (31, 32). To investigate the relative contribution of macropinocytosis versus SR-mediated pathways to atherosclerosis development, we treated hypercholesterolemic C57BL/6 wild-type (WT) and CD36−/−/SR-A−/− mice with vehicle or EIPA using subcutaneously implanted osmotic pumps. Hypercholesterolemia was induced by adeno-associated viral overexpression of a PCSK9 gain-of-function mutant in combination with a Western-type diet as reported previously (33). A series of control experiments involving quantitation of nLDL internalization using Nile Red staining (Fig. S1, A–C) and direct Dil-nLDL uptake (Fig. S1, D & E) confirmed that EIPA inhibits macrophage macropinocytosis. EIPA treatment did not inhibit SR-mediated oxLDL (Fig. S1F), caveolin-dependent albumin (Fig. S1G), and clathrin-mediated transferrin endocytosis (Fig. S1H). Further, EIPA treatment did not induce cell death as determined using the LIVE/DEAD Fixable Cell Stain (Fig. S1I). Continuous infusion of EIPA substantially inhibited atherosclerotic lesion area in both WT (86.9%) and CD36−/−/SR-A−/−(88.6%) mice (Fig. 1, A–C, Fig. S2, A–D). EIPA treatment led to 83.4%, 88.2% and 89.3% reduction in atherosclerotic lesion area in the proximal, middle and distal LCA regions of WT mice in comparison to vehicle-treated control mice, respectively (Fig. 1, B & C). Vehicle-treated CD36−/−/SR-A−/− mice developed ~32% smaller lesion area compared to WT controls (Fig. 1C), consistent with previous reports (8, 10). Control experiments showing functional inhibition of oxLDL uptake in CD36−/−/SR-A−/− macrophages are shown in Fig. S2E. Histological characterization of WT and CD36−/−/SR-A−/− atherosclerotic arteries using H&E, Masson’s trichrome staining and CD68 immunostaining are shown in Fig. 1, D–G. Collagen content, atherosclerotic lesion area and CD68+ macrophage staining were significantly decreased in EIPA-treated mice compared with vehicle-treated WT and CD36−/−/SR-A−/− controls. Plasma cholesterol, body weight and blood pressure were not different between experimental groups (Fig. 1, H–J). To confirm the results obtained in male PCSK9-AAV-induced LDLR-downregulated mice (Fig. 1, A–C), we used male ApoE−/− and female LDLR-deficient mice. As shown in Fig. S3, A–G and Fig. S4, A–G, pharmacological inhibition of macropinocytosis using EIPA inhibited atherosclerosis development in Western diet-fed male ApoE−/− mice and in female LDLR-deficient animals.
Figure 1. The macropinocytosis inhibitor EIPA reduces atherosclerosis development in wild-type and SR knockout mice.

A-E, Male WT and CD36−−/SR-A−− mice treated ± EIPA following PCSK9-AAV + partial LCA ligation and fed a Western diet for 4 weeks. A, Representative images of isolated LCA (yellow arrows), scale bar: 1 mm. B, Oil Red O (ORO) staining for proximal, middle and distal LCA segments, scale bar: 100 μm. C, Quantification of ORO positive area (n = 8 - 13). D, Representative images of H&E, Masson’s trichrome and CD68 staining of LCA. E-G, Quantification of collagen deposition (E), lesion area (F) and CD68+ macrophages (G) in the LCA (n = 5). H, Total plasma cholesterol (n = 9 13). I, body weight prior to sacrifice (n = 9). J, Systolic blood pressure (n = 5). Data represent mean ± SEM. *p < 0.05 vs. vehicle.
Genetic stimulation of macropinocytosis promotes cholesterol accumulation in WT and CD36−/−/SR-A−/− macrophages, leading to foam cell formation
Macropinocytosis is stimulated by submembranous reorganization of the actin cytoskeleton, leading to formation of membrane ruffles that may circularize and close, subsequent cup formation and receptor-independent internalization of pericellular solutes via membrane-derived vesicles known as macropinosomes (Fig. 2A). To our knowledge, the relative contribution of SR-mediated lipid uptake vs. macropinocytosis to macrophage foam cell formation in vitro or in atherosclerotic lesions in vivo has not been investigated. We overexpressed constitutively active Ras (H-RASG12V) in WT and CD36−/−/SR-A−/− macrophages to stimulate membrane ruffle formation (Fig. 2, B & C) and determined macropinocytosis of nLDL, oxLDL and acLDL using ORO staining and Nile Red fluorescence. As shown in Fig. 2, D & E, uptake of nLDL (50 μg/ml) by both WT and CD36−/−/SR-A−/− macrophages was significantly increased following H-RASG12V overexpression-mediated macropinocytosis stimulation. Importantly, overexpression of H-RASG12V in CD36−/−/SR-A−/− macrophages stimulated ox- and acLDL (50 μg/ml) internalization, leading to foam cell formation (Fig. S5A). Preincubation of CD36−/−/SR-A−/− macrophages with EIPA inhibited Nile Red fluorescence confirming that ox- and acLDL internalization is mediated by macropinocytosis (Fig. S5A).
Figure 2. Characterization of lipid macropinocytosis by WT and CD36−/−/SR-A−/− macrophages in vitro.

A, A schematic diagram illustrating morphological plasma membrane changes during macropinocytosis. B-E, Wild-type and CD36−/−/SR-A−/− BMDM overexpressing HrasG12V or GFP control, ± EIPA (25 μM, 1 hr) treatment. B, Representative SEM images. Green arrows - linear ruffles; red arrows - curved ruffles or macropinocytic cups, scale bar 1 μm. C, Quantification of membrane ruffles (n = 3). D, HrasG12V overexpressing and GFP control macrophages were incubated with 50 μg/ml nLDL for 24 hrs. Representative images of ORO staining (n = 3), scale bar 50 μm. E, Cells were treated as described in (D) and Nile Red fluorescence was quantified by flow cytometry (n = 4). F, THP-1 macrophages were pretreated with vehicle or EIPA and incubated with nLDL or oxLDL (50 μg/ml) for 24 hrs in the presence or absence of macropinocytosis stimulator PMA. Macrophage LDL accumulation was quantified using Nile Red staining (n = 6 -10). Data are mean ± SEM. *p < 0.05 vs. vehicle, #p < 0.05 vs. AD HrasG12V.
Macrophage internalization of nLDL by macropinocytosis is linearly related to extracellular lipid concentration (34). On the contrary, ox- and acLDL saturate SR at concentrations between 25 - 50 μg/ml (35). Therefore, the next experiments were designed to investigate the relative contribution of macropinocytosis vs. SR to macrophage LDL internalization in vitro using the same experimental conditions. Macropinocytosis stimulator PMA increased uptake of both nLDL (50 μg/ml) and oxLDL (50 μg/ml) in the absence, but not in the presence, of EIPA (1.84-fold and 1.47-fold for nLDL and oxLDL, respectively) (Fig. 2F). In addition, macropinocytosis of nLDL (nLDL + PMA) and SR-mediated oxLDL uptake (oxLDL + vehicle) (1.84-fold and 2.43-fold for nLDL macropinocytosis and oxLDL uptake, respectively) are also shown in Fig. 2F. We next investigated macropinocytosis-dependent LDL uptake in lesional macrophages in vivo. Atherosclerotic AAV-PCSK9-injected WT and CD36−/−/SR-A−/− mice (14 weeks Western diet) were treated with vehicle or EIPA (25 mg/kg/day, i.p. for 2 days) and intravenously (tail vein) injected with Dil-nLDL (150 μg/mouse). The atherosclerotic aortas were isolated 24 hrs after LDL injection and Dil fluorescence determined in isolated lesional CD11b+ cells. As shown in Fig. S5B, CD11b+ cells internalize exogenously administered Dil-nLDL in both WT and CD36−/−/SR-A−/− mice and EIPA treatment suppressed internalization. Control experiments were performed to investigate whether EIPA regulates cholesterol efflux in macrophages. EIPA modestly (7.46%), but significantly, decreased HDL-mediated cholesterol efflux in lipid-laden macrophages (Fig. S5C). Taken together, these results suggest an important role for macropinocytosis in macrophage lipid uptake in both WT and CD36−/−/SR-A−/− mice.
Characterization of macropinocytosis in human macrophages using physiologically relevant stimulators
We investigated lipid internalization by human macrophages in response to physiologically relevant stimulators of macropinocytosis that are upregulated in atherosclerotic arteries, including platelet-derived growth factor (PDGF) (36) and macrophage colony-stimulating factor (MCSF) (37). Analysis of publicly available patient cohorts (Gene Expression Omnibus) confirmed increased expression of PDGF and MCSF in human atherosclerotic arteries compared to non-atherosclerotic control tissue (n = 32; Fig. 3, A & B). As shown in Fig. 3C, incubation of human THP-1 macrophages with PDGF and MCSF stimulated macropinocytosis of nLDL as shown by EIPA-inhibitable increase in Nile Red fluorescence. Further, physiological stimulation of macropinocytosis resulted in increased nLDL uptake by CD36−/−/SR-A−/− macrophages (Fig. 3D). Next, we investigated whether macropinocytosis stimulation regulates SR-A and CD36 expression in the plasma membrane and promotes changes in oxLDL uptake in human macrophages. Specificity of SR-A and CD36 antibodies was confirmed using CD36−/−/SR-A−/− macrophages (Fig. S2D). MCSF treatment (100 ng/ml, 10 min to 48 hrs) did not change CD36 and SR-A expression in the plasma membrane (Fig. S6, A & B). Confirming these results, pretreatment of macrophages with MCSF (60 min) did not change uptake of DiO-oxLDL (50 μg/ml, 4 hrs) (Fig. S6C). Finally, we examined whether MCSF and PDGF oxidize nLDL in the media of cultured macrophages using agarose gel electrophoresis. The relative electrophoretic mobility (REM) of LDL from conditioned media of vehicle-, MCSF- and PDGF-treated macrophages was not different from negative control nLDL, indicating no oxidative modification of LDL (i.e., negative charge) in response to MCSF and PDGF treatments (Fig. 3, E & F). Consistent with these results, non-pegylated (e.g., cell-impermeant) superoxide dismutase (SOD) and catalase did not inhibit MCSF- and PDGF-induced nLDL internalization (Fig. 3, G & H). Taken together, these results demonstrate that physiologically relevant stimulators of macropinocytosis promote nLDL uptake by human macrophages and may explain why some antioxidant therapies failed to attenuate progression of atherosclerotic vascular disease.
Figure 3. Physiologically relevant stimulation of macropinocytosis promotes lipid accumulation in wild-type and SR knockout macrophages.
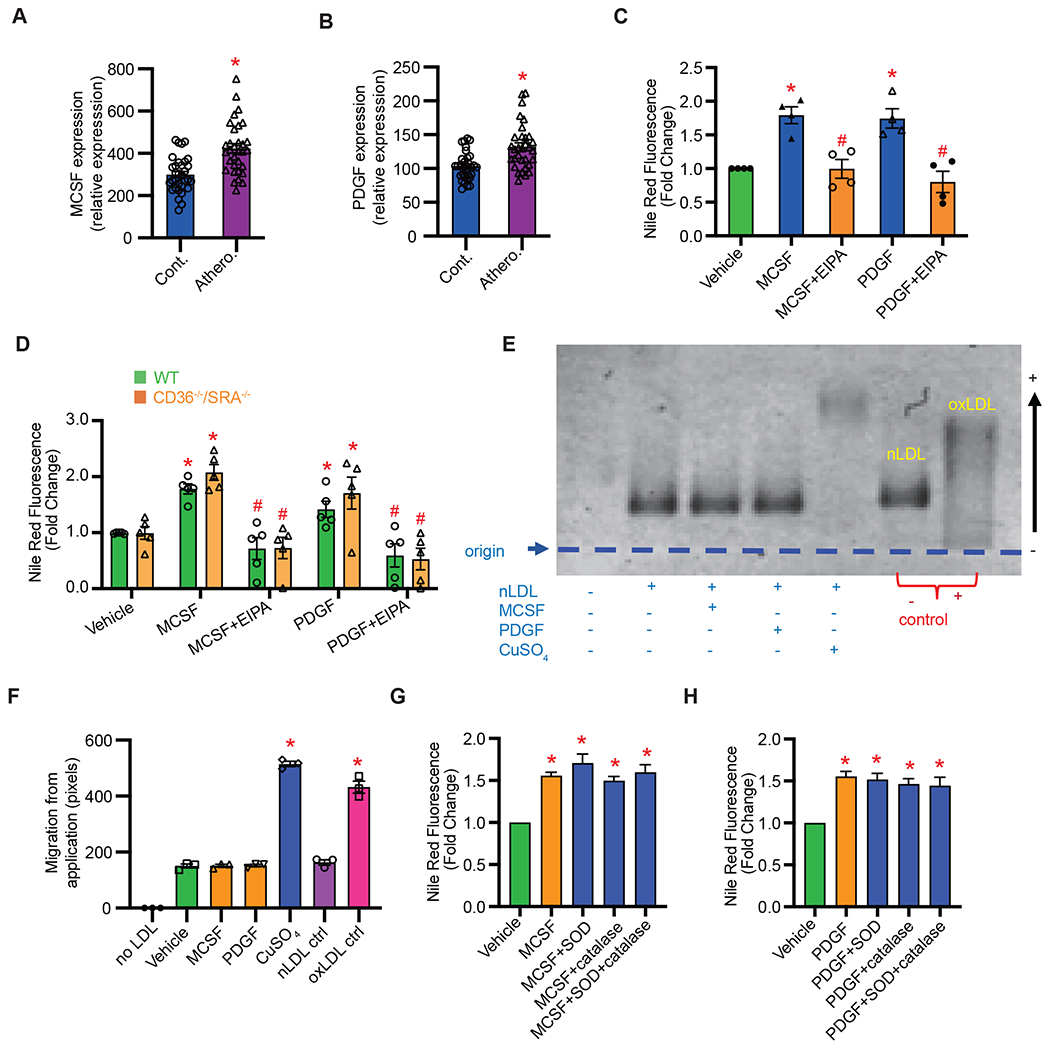
A & B, Analysis of publicly available database demonstrating increased expression of MCSF (A) and PDGF (B) in human atherosclerotic arteries compared with control tissue (n = 32). C, MCSF (100 ng/ml) and PDGF (200 ng/ml) stimulate nLDL internalization in THP-1 macrophages via macropinocytosis (n = 4). D, MCSF and PDGF stimulate cholesterol accumulation in WT and CD36−/−/SR-A−/− BMDM incubated with nLDL (50 μg/ml) (n = 5). E, THP-1 macrophages were treated with vehicle, MCSF (100 ng/ml) or PDGF (200 ng/ml) in the presence of 50 μg/ml nLDL for 24 hrs. LDL oxidation was determined by agarose gel electrophoresis of collected conditioned media. Positive controls: nLDL + CuSO4 (50 μM, 72 hrs) and oxLDL. Negative control: nLDL. F, Quantification of electrophoretic mobility (n = 3). G & H, THP-1 macrophages were pretreated with ± SOD (100 U/ml) and/or catalase (250 U/ml) for 1 hr, incubated with 50 μg/ml nLDL for 24 hrs in the presence or absence of MCSF (G) or PDGF (H). Cells were incubated with 50 ng/ml Nile Red for 7 min and FACS quantification for Nile Red fluorescence was performed. Data are mean ± SEM. *p < 0.05 vs. vehicle, #p < 0.05 vs. MCSF or PDGF.
Visualization of foam cell macropinocytosis in human and murine atherosclerotic arteries
To our knowledge, no previous studies have provided direct visual evidence of macrophage macropinocytosis in atherosclerotic arteries. We used serial section Transmission Electron Microscopy (ssTEM) imaging to analyze the 3D ultrastructure of the subendothelial layer in atherosclerotic arteries. For these experiments, lipid-laden macrophages in the atherosclerotic aorta of ApoE−/− mice (12 weeks Western diet) were followed through ~200 ultra-thin (60 nm) serial sections. Analysis of subendothelial ultrastructure identified foam cells that develop large, sheet-like membrane protrusions (red arrows) that curve back (blue asterisk) to form parallel membrane protrusions (orange arrows) resembling circularized C-shaped ruffles (Fig. 4A). Three-dimensional reconstruction of ssTEM images provides important details on the physical characteristic of membrane ruffles, including total surface area, ruffle volume, and tip-base distance (Fig. 4, B–C and Video S1). A reconstructed curved ruffle in 3D and corresponding 2D ssTEM image on the surface of foam cell are shown in Fig. 4D. Quantification of membrane ruffles, macropinocytic cups and cup closure in atherosclerotic arteries is shown in Fig. S7. Next, we analyzed the inner curvature (IC) of human atherosclerotic aorta for evidence of macropinocytic membrane activities. Importantly, ssTEM images identified cells in the subendothelial layer of atherosclerotic IC that demonstrate the full cycle of macropinocytosis, including single membrane protrusions (red arrows), C-shaped ruffles or macropinocytotic cups (red asterisk), and formation of membrane-derived vesicles, ranging from 170 nm to 700 nm in diameter, located at the base of membrane protrusions in the cytosol (orange arrows) (Fig. 4, E–G). Characterization of tissue donors, including age, sex, cardiovascular risk factors, medication, co-morbidities and cause of death is shown in Table S1.
Figure 4. Visualization of macrophage macropinocytosis in human and murine atherosclerotic arteries.

A, Serial section TEM imaging demonstrating the presence of plasma membrane protrusions on the surface of lipid-laden macrophages in atherosclerotic ApoE−/− aorta. LD: lipid droplets, N: nucleus. Red arrows - membrane protrusion, asterisk – curving ruffle, orange arrows – parallel side of identified ruffle. Scale bar, 1 μm. B, 3D reconstruction of a foamy macrophage in ApoE−/− aorta. Scale cube, 1 μm3. C, Calculated total surface area, ruffle volume and tip-base distance of identified membrane ruffles. D, 3D reconstruction of curved ruffle. Inset: TEM image of curved ruffle in 2D. H = height, W = width and D = depth. E, Human heart and aortic tissue isolated from a cadaveric donor with a history of cardiovascular disease (patient # 1). IC = atheroprone Inner Curvature. F & G, TEM imaging demonstrating formation of single membrane protrusions (red arrows), parallel ruffles (red asterisk) and membrane-derived vesicles (orange arrows) in IC segments of human atherosclerotic aorta. H, Left: Cross section of human atherosclerotic LAD (patient # 2). Right: Immunogold-labeling of LDL (blue arrowheads). Open cups – red arrows, macropinosomes: orange arrows. I, A schematic diagram illustrating the design of in vivo LDL tracking experiments. J, Representative gating strategy identifying isolated F4/80+ macrophages from atherosclerotic ApoE−/− aorta. FACS analysis of DiI fluorescence in isolated F4/80+ cells. K, Quantification of mean fluorescence intensity, n = 3, *p < 0.05. Data are mean ± SEM.
Impaired efferocytosis of apoptotic cells has been demonstrated to contribute to the pathogenesis of atherosclerosis (38). Solid particles, microbes or fragments of apoptotic cells were not visualized near the plasma membrane of macrophages, indicating efferocytosis-independent plasma membrane activities (Fig. 4, F & G). As coronary artery atherosclerosis is a leading cause of death in both men and women (4), we next sectioned human atherosclerotic left anterior descending (LAD) coronary arteries [type Vb lesion (39)] from cadaveric donors (Fig. 4H) and performed IEM imaging to detect LDL in the shoulder region of the lesion. Immunolabeling localized LDL in the cytosol of lesional cells and near the plasma membrane, within open macropinocytic cups (red arrows) and closed macropinosomes (orange arrows) in human atherosclerotic LAD (Fig. 4H). Although aggregated LDL (aggLDL) has been shown to induce phagocytosis in macrophages (40), diameter of phagocytic cups surrounding aggLDL is expected to be in the lower nm range [50-300 nm (41)] and significantly smaller than the observed cups that are 1 μm in diameter or larger.
To date, no previous studies have investigated macrophage macropinocytosis of LDL in atherosclerotic arteries. Next, we treated atherosclerotic ApoE−/− mice (12 weeks Western diet) with vehicle or EIPA (25 mg/kg/day, i.p. for 3 days prior to sacrifice) and retro-orbitally injected DiI-nLDL (100 μg) to quantify lipoprotein macropinocytosis by macrophages in atherosclerotic lesions (Fig. 4I). Flow cytometry analysis demonstrated that isolated F4/80+ cells internalized high amounts of DiI-LDL in atherosclerotic arteries and the uptake was substantially inhibited by pharmacological inhibition of macropinocytosis (Fig. 4, J & K).
Genetic inhibition of macropinocytosis selectively in myeloid cells inhibits atherosclerosis
Next, we generated mice lacking NHE1 selectively in myeloid cells (LysMCre+ NHE1fl/fl, hereafter referred to as NHE1ΔM) to inhibit macrophage macropinocytosis in vitro and in vivo (Fig. S8, A & B). RT-PCR data demonstrate that NHE1 is the most highly expressed NHE isoform in macrophages and deletion of NHE1 in NHE1ΔM mice did not induce compensatory changes in the expression of other NHE isoforms (Fig. S8, C & D). As shown in Fig. S8E, PDGF- and MCSF-induced nLDL macropinocytosis was significantly inhibited in NHE1ΔM bone marrow-derived macrophages (BMDM) compared with NHE1fl/fl controls. Atherosclerosis was induced in NHE1ΔM and NHElfl/fl mice by AAV-mediated PCSK9 overexpression, partial LCA ligation and 4 weeks of Western diet feeding. We observed significantly smaller atherosclerotic lesions in the middle (80.7% decrease) and distal (90.8% decrease) LCA segments of NHE1δM mice compared with NHE1f/f controls (Fig. 5, A–C). No differences in body weight, fat mass, plasma cholesterol, blood glucose and blood pressure were measured between the groups (Fig. 5, D–J). Flow cytometry analysis demonstrated significantly reduced internalization of exogenously administered DiI-nLDL by peritoneal NHE1-deficient macrophages compared with NHE1f/f controls, confirming inhibition of macrophage macropinocytosis in NHE1ΔM mice in vivo (Fig. 5, K & L). To investigate the effect of NHE1 deletion on LDL uptake by lesional macrophages, we injected CFDA-labeled (green-fluorophore cell tracker) BMDM from NHE1ΔM and NHE1f/f mice intravenously to Western diet-fed (16 weeks) LDLR−/− mice. Twenty-four hours after Dil-nLDL injection, CFDA+/CD11b+ macrophages were isolated from the atherosclerotic aorta and Dil fluorescence quantified. As shown in Fig. 5M, CFDA-labeled NHE1 knockout macrophages accumulated significantly less Dil-nLDL compared with NHE1f/f controls.
Figure 5. Genetic deletion of NHE1 in myeloid cells reduces atherosclerosis development in hypercholesterolemic mice.

A-J, NHE1f/f and NHE1ΔM mice were injected with PCSK9-AAV, subjected to partial LCA ligation and fed a Western diet for 3 weeks to induce atherosclerosis. A, Representative images of isolated LCA (yellow arrows), scale bar: 1 mm. B, Oil Red O staining for proximal, middle and distal LCA segments, scale bar: 100 μm. C, Quantification of ORO positive area (n = 15 and 13 for NHE1f/f and NHE1ΔM, respectively). D, Total plasma cholesterol. E, Lipid profile of pooled samples (n = 5). F, blood glucose (n = 9 and 7 for NHE1f/f and NHE1ΔM mice, respectively). G, systolic blood pressure (n = 5 and 4 for NHE1f/f and NHE1ΔM mice, respectively). H, body weight measured prior to sacrifice (n = 9 and 7 for NHE1f/f and NHE1ΔM mice, respectively). I, fat mass and J, fluid content measured using NMR (n = 9 and 7 for NHE1f/f and NHE1ΔM mice, respectively). K & L, internalization of Dil-nLDL by NHE1f/f and NHE1ΔM macrophages in peritoneal cavity. Confocal images of harvested peritoneal macrophages (K), FACS analysis of Dil fluorescence (L) (n = 5). M, LDLR−/− mice were injected with CFDA-labeled BMDM from NHE1f/f and NHE1ΔM mice and Dil-LDL fluorescence in CD11b+ CFDA+ aortic macrophages isolated from LDLR−/− mice determined (n = 4). Data are mean ± SEM. *p < 0.05.
The LCA model represents a combination of vascular injury, disturbed flow and a relatively short-term hypercholesterolemia. To address this limitation and confirm results, we used a more traditional atherosclerosis model of chronic hypercholesterolemia. The atherosclerotic lesion area in the aorta of NHE1ΔM mice was significantly reduced (75.1%) following 16 weeks of Western diet compared to NHE1f/f mice (Fig. 6, A & C). Similar results were observed when atherosclerotic lesion area in the aortic sinus was quantified (Fig. 6, B & D). Further, the neointima area and collagen content in the aortic sinus were significantly decreased in NHE1ΔM mice compared with NHE1f/f controls (Fig. 6, E & F). Plasma cholesterol, systolic blood pressure, body weight, fat mass, fluid content and fasting blood glucose in NHE1ΔM mice were not different from control animals (Fig. 6, G–L). These results suggest that genetic inhibition of macropinocytosis selectively in myeloid cells attenuates development of atherosclerosis in hypercholesterolemic mice.
Figure 6. Genetic deletion of myeloid cell NHE1 inhibits atherosclerosis development in atheroprone PCSK9-overexpressing mice following 16 weeks Western diet feeding.

A, En face Oil Red O (ORO) staining of the thoracic aorta. Scale bar: 2 mm. B, Representative images of ORO, H&E and Masson’s trichrome staining of the aortic sinus. Scale bar 0.2 mm. C, Quantification of aortic ORO positive area in (A), D-F, Quantification of ORO positive area (D), lesion area (E) and collagen deposition (F) in the aortic sinus (n = 9 and 10 for NHE1f/f and NHE1ΔM, respectively). G, Total cholesterol (n = 9 and 10 for NHE1f/f and NHE1ΔM, respectively) H, systolic blood pressure (n = 6 and 7 for NHE1f/f and NHE1ΔM, respectively). I, body weight (n = 9 and 10 for NHE1f/f and NHE1ΔM, respectively). J, fat mass (n = 9 and 10 for NHE1f/f and NHE1ΔM, respectively) and K, fluid content (n = 9 and 10 for NHE1f/f and NHE1ΔM, respectively). L, blood glucose (n = 9 and 10 for NHE1f/f and NHE1ΔM, respectively). Data are mean ± SEM. *p < 0.05.
A “repurposed” FDA-approved drug that inhibits macrophage macropinocytosis attenuates atherosclerosis development in hypercholesterolemic mice
Amiloride and its analogues that selectively block NHE1 are considered to be the best choices for pharmacological inhibition of macropinocytosis in animal models (31). A previous study demonstrated that amiloride monotherapy improves pulse wave velocity (PWV), a surrogate marker for arterial stiffness and atherosclerosis, in pre-hypertensive patients independent of its blood pressure lowering effect (42). Despite this information, no prior studies have investigated whether pharmacological inhibition of macropinocytosis would be a viable therapeutic strategy for patients with atherosclerotic vascular disease. We have recently performed a large unbiased-screen of an FDA-approved drug library and identified a potent (IC50 = 131 nM), non-toxic [selectivity index (CC50/IC50) > 300] low MW compound (imipramine) that inhibits macropinocytosis in macrophages, independent of NHE1 regulation (43). Imipramine is a tricyclic antidepressant (TCA) with high oral bio-availability (95%) and a half-life of nearly 20 hrs that is clinically used in children to treat enuresis and adults with depression (44). As shown in Fig. 7A, preincubation of macrophages with imipramine inhibited MCSF- and PDGF-induced intracellular lipid accumulation following nLDL treatment. We next investigated the efficacy of imipramine to attenuate atherosclerosis development in hypercholesterolemic mice (Fig. 7B). Combined Orcein and Martius Scarlet Blue (OMSB) staining followed by automatic segmentation and pseudo-coloring (Fig. 7C; Algorithm) was used for qualitative and quantitative analyses of atherosclerotic lesions as described previously (45). Results from OMSB staining demonstrated that imipramine significantly attenuates atherosclerotic lesion formation compared with vehicle treatment (Fig. 7C–E). Imipramine inhibited plaque area, relative internal vessel area and collagen content compared with vehicle treatment (Fig. 7F–I). Additionally, we tested the effects of imipramine on internalization of Dil-nLDL by lesional macrophages in vivo. Consistent with our in vitro results (Fig. 7A) and inhibition of atherosclerosis in imipramine-treated animals, imipramine suppressed accumulation of Dil-nLDL in CD11b+ cells isolated from atherosclerotic arteries (Fig. 7J). Finally, we observed no significant differences in blood glucose, total cholesterol, blood pressure, body weight, fat mass and whole-body fluid content between imipramine-treated and control mice (Fig. 7K–P). Taken together, the results of these experiments support the use of repurposed macropinocytosis inhibitors for pharmacological treatment of atherosclerosis.
Figure 7. Treatment with a “repurposed” FDA-approved drug that inhibits macropinocytosis attenuates atherosclerosis development in hypercholesterolemic mice.

A, THP-1 macrophages were pretreated with 5 μM imipramine for 1 hr, then treated with vehicle, MCSF (100 ng/ml) or PDGF (200 ng/ml) in the presence of 50 μg/ml nLDL for 24 hrs. Cells were incubated with Nile Red for 7 min and FACS quantification for Nile Red fluorescence was performed (n = 4). B-O, Wild-type mice were injected with PCSK9-AAV, underwent partial LCA ligation and fed a Western diet for 4 weeks to induce atherosclerosis. Mice were treated with vehicle or imipramine (s.c.). B, Representative images of LCA (yellow arrows), scale bar: 1 mm. C, Representative Orcein and Martius Scarlet Blue (OMSB) staining for proximal, middle and distal segments and segmentation algorithm of LCA. L: lumen, P: plaque area, VWA: Vessel wall area. Scale bar: 100 μm (Red). The inset shows zoomed-in images for distal segments of LCA, scale bar: 50 μm (Black). D, Quantification of plaque area (n = 5), E, area under the curve (AUC). F-I, Quantification of plaque area (F), vessel wall area (G), relative internal vessel area (IVA %, H) and relative collagen area (Collagen %, I) (n = 5). J, Quantification of mean Dil fluorescence intensity in isolated CD11b+ lesional cells, (n = 4). K-P, blood glucose (K, n = 9). L, systolic blood pressure (n = 5). M, body weight (n = 9). N, fat mass (n = 9) and O, fluid content (n = 9). P, Total cholesterol (n = 9). Data are mean ± SEM. *p < 0.05 vs. vehicle, #p < 0.05 vs. MCSF or PDGF.
Discussion
The role of SR-mediated modified lipid uptake in macrophage foam cell formation was first described four decades ago (5). These observations have been confirmed by numerous studies and led to the classical dogma that subendothelial modification of LDL plays a critical role in the pathogenesis of atherosclerosis and unmodified lipids are not atherogenic (5–8). The central goal of this study was to investigate the role of SR-independent macrophage LDL macropinocytosis in the pathogenesis of atherosclerosis. The rationale for investigating SR- and modification (i.e. oxidation)-independent pathways of LDL uptake is predicated on the observations that i) SR-knockout mice are partially protected from atherosclerosis development (5, 10), ii) atherosclerotic coronary artery disease-related mortality is higher in CD36-deficient patients compared to the general population (12), and iii) antioxidant therapies have failed in clinical trials (15). Although, these results imply the existence of an alternative, SR-independent pathway of macrophage lipid internalization in the pathogenesis of atherosclerosis, the precise mechanisms remain unclear.
To our knowledge, only two studies have investigated macrophage macropinocytosis in atherosclerotic arteries (46, 47). These studies demonstrated that aortic macrophages internalize Angiospark fluorescent nanoparticles via macropinocytosis. Despite these earlier observations, no previous studies have determined the effects of macropinocytosis inhibition selectively in myeloid cells on atherosclerosis development, investigated macrophage LDL macropinocytosis in atherosclerotic arteries, or analyzed atherosclerotic vessels (human or murine) for evidence of macropinocytosis-associated plasma membrane activities. Here, we report the role of macrophage LDL macropinocytosis in atherosclerosis development via performing pharmacological studies in male and female mice, LDLR-knockdown and ApoE−/− mice as well as genetic inhibition of macropinocytosis selectively in myeloid cells. We would like to add that each animal models have its limitations, therefore, we utilized various atherosclerosis models including ApoE−/− mice, PCSK9-AAV-induced LDLR downregulation, accelerated surgical and chronic hyperlipidemia-induced atherosclerosis models. It is possible that the extent of macropinocytosis stimulation may be different in these models, between arterial beds and stage of atherosclerosis. For instance, surgical manipulation and associated disturbed flow may regulate expression of certain cytokines that stimulate macropinocytosis in the LCA model differently compared to chronic hypercholesterolemia-induced atherosclerosis in the thoracic aorta. It is, however, important to add that physiologically relevant stimulators of macropinocytosis (thrombospondin-1, MCSF, PDGF) have been shown to be upregulated in the arterial wall in response to injury, disturbed flow and chronic hyperlipidemia-induced inflammation (48–50).
To date, no signaling molecules that selectively or exclusively mediate macropinocytosis have been identified. We acknowledge that NHE1 inhibition may affect pathways other than macropinocytosis. Importantly, we used both pharmacological (EIPA) and genetic (myeloid cell-specific NHE1 deletion) approaches to inhibit macropinocytosis in vivo. Further, we identified a structurally and mechanistically distinct small molecule compound [FDA-approved drug in a screen of > 640 drugs; (43)] that inhibits macrophage macropinocytosis (and not other endocytic mechanisms) in an NHE1 independent manner. Imipramine treatment inhibited macrophage macropinocytosis in vitro and attenuated development of atherosclerosis in vivo.
In the present study, EIPA-induced inhibition of atherosclerosis in wild-type and SR knockout mice suggest an important role for macropinocytosis in macrophage LDL uptake in vivo. These results are consistent with a previous report demonstrating that amiloride reduces lesion development in lipopolysaccharide (LPS)-treated ApoE−/− mice (51). However, it is important to add that amiloride also inhibits T-type calcium channels (52), epithelial sodium channels (42) and NHE (53) in multiple cell types and block urokinase plasminogen activator activity (54). Although CD36 and SR-A mediate 80-90% modified LDL uptake by macrophages (7), compensatory upregulation of alternative SR-mediated pathways is possible, and it is a limitation of the present study.
In vitro experiments using scanning electron microscopy demonstrated that foam cells form membrane ruffles that may circularize and close, leading to macropinosome formation and macropinocytosis (55, 56). However, it was unknown whether macrophages can form membrane protrusions in the extracellular matrix against proteoglycans, collagen and other fibrous proteins and despite increased arterial stiffness observed in atherosclerotic arteries (57). Using high-resolution TEM imaging, we identified cells in the subendothelial layer of atherosclerotic arteries that demonstrate the full cycle of macropinocytosis, including single membrane protrusions, curved ruffles, macropinocytotic cups and formation of intracellular vesicles resembling macropinosomes. Immunoelectron microscopy localized LDL particles in cup-like structures and closed macropinosomes suggesting that lipid macropinocytosis may occur in human atherosclerotic arteries. Importantly, ultrastructural analysis of atherosclerotic intima did not identify solid particles, microbes or fragments of apoptotic cells near plasma membrane protrusions, indicating phagocytosis/efferocytosis-independent stimulation of plasma membrane activities. Quantification of DiI-nLDL uptake by lesional CD11b+ cells are consistent with these results and suggest macropinocytosis-mediated lipid internalization in atherosclerotic arteries. A limitation of the study is that we did not perform lineage tracing to investigate macropinocytosis in macrophages, differentiated smooth muscle cells or dedifferentiated macrophage-like smooth muscle cells.
In a large cohort of patients hospitalized with atherosclerotic coronary artery disease (ST- and non–ST-segment elevation myocardial infarction and unstable angina), approximately half had admission concentration of LDL lower than 100 mg/dl and almost 20% of patients had LDL < 70 mg/dl (58). Although these results indicate that the therapeutic goal of circulating LDL levels needs to be even lower, this may not be feasible due to increased rate of adverse effects associated with more aggressive lipid-lowering therapies and their limitations to further lower LDL concentration. Previous studies from our laboratory and others demonstrated that macrophage foam cell formation can be induced when macropinocytosis is stimulated in the presence of 5 mg/dl nLDL in vitro (18, 56). These results indicate that macrophage macropinocytosis-mediated foam cell formation may occur even in patients with normal or low LDL concentration. Taken together, these results suggest the need for a combinatorial therapy that lowers circulating LDL concentration, improves the barrier function of endothelial cells to attenuate LDL uptake into the arterial wall and inhibits macrophage macropinocytosis of sub-endothelially retained lipids. Testing this new therapeutic approach in a preclinical model, we provided evidence that a repurposed FDA-approved drug that inhibits macropinocytosis in vitro and in vivo reduces atherosclerosis development in hypercholesterolemic mice.
Excessive uptake of LDL by macrophages in the arterial wall is a critical process in the development and progression of atherosclerosis. The precise mechanisms, however, that mediate macrophage internalization of lipids, leading to foam cell formation and development of atherosclerotic lesions remain incompletely understood. In summary, the results of the present work support an important role for macrophage macropinocytosis in LDL uptake and development of atherosclerosis. These results identify macropinocytosis as a potential therapeutic target in atherosclerosis.
Materials and Methods
Study design
This study was designed to investigate the role of receptor-independent LDL macropinocytosis in arterial lipid accumulation and atherosclerosis development. We treated hypercholesterolemic wild-type (WT) and CD36−/−/SRA−/− mice with the gold standard macropinocytosis inhibitor EIPA (NHE1 blocker) and utilized myeloid cell-specific NHE1 deletion (LysMCre+ NHE1fl/fl) to investigate the role of macropinocytosis in atherosclerotic lesion development. Further, we repurposed an FDA-approved drug (imipramine) that inhibits macrophage macropinocytosis in an NHE1-independent manner for the treatment of atherosclerosis. We used physiologically relevant macropinocytosis stimulators (MCSF and PDGF) and overexpression of oncogenic Ras proteins (H-RasG12V) to stimulate macropinocytosis in WT and CD36−/−/SRA−/− BMDM to investigate SR- and macropinocytosis-mediated lipid accumulation in macrophages. We performed serial section high-resolution imaging of murine and human atherosclerotic arteries to identify foam cells and examine plasma membrane ruffling, cup formation and macropinosome internalization. The number of animals utilized for each group is specified in the figure legends. The in vitro experimental data included in this manuscript are representative of multiple independent experiments as indicated. All animal studies were performed according to procedures and protocols approved by the Institutional Animal Care and Use Committee at Augusta University.
Statistical analysis
All data are presented as mean ± SEM. Data were analyzed by GraphPad InStat software (GraphPad Software, Inc.). Data normality was investigated employing the Shapiro-Wilk normality test. Student’s t-test was performed to analyze parametric data and differences between two groups. Comparisons among multiple groups were made using one/two-way ANOVA with an appropriate post hoc test (Tukey’s). A p < 0.05 was considered to be statistically significant.
Supplementary Material
Supplementary Video S1. 3D reconstruction of a foamy macrophage in ApoE−/− aorta. Nucleus (blue), Lipid droplets (yellow) and cell membrane (red). Scale cube: 1 μm3
Data file S1. Individual data points (Microsoft Excel format).
Acknowledgments:
The authors wish to thank Brendan Marshall and Libby Perry (Augusta University) for their help with the TEM and SEM imaging experiments. Drs. Neal Weintraub and David Stepp (Augusta University) assisted us with blood pressure and NMR experiments, respectively. NHE1 floxed mice were kindly provided by Dr. Dandan Sun (University of Pittsburgh) under a Material Transfer Agreement. The authors wish to thank Jessica Holland, MSMI, CMI, Medical Illustrator at Augusta University, for her assistance in preparing the illustrations in this publication.
Some of the material has been included in a pending US patent application (serial number: 17/191,398 filed on 3/3/2021, “Compositions and Methods for Treating Atherosclerotic Vascular Disease”).
Sources of Funding:
This work was supported by NIH grants (R01HL139562 and R00HL114648 awarded to G.C., R01HL138410 awarded to R.L., and K99HL146954 and R00HL146954 given to B.S.) and American Heart Association predoctoral fellowship (PRE34080052) awarded to H.-P.L. The work was also supported by NCN grant no. 2021/42/A/NZ4/00273 awarded to S.C.
Footnotes
Competing interest declaration: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be taken as a potential conflict of interest.
Data availability:
All figures have associated raw data, which are available with permission from the corresponding author.
References
- 1.Chiu JJ, Chien S, Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91, 327–387 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghosh S, Zhao B, Bie J, Song J, Macrophage cholesteryl ester mobilization and atherosclerosis. Vascul Pharmacol 52, 1–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silverman MG et al. , Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA 316, 1289–1297 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Lloyd-Jones D et al. , Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 119, e21–181 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Goldstein JL, Ho YK, Basu SK, Brown MS, Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci U S A 76, 333–337 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuchibhotla S et al. , Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res 78, 185–196 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunjathoor VV et al. , Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 277, 49982–49988 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Febbraio M et al. , Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 105, 1049–1056 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kodama T et al. , Type I macrophage scavenger receptor contains alpha-helical and collagen-like coiled coils. Nature 343, 531–535 (1990). [DOI] [PubMed] [Google Scholar]
- 10.Makinen PI et al. , Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc Res 88, 530–538 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Manning-Tobin JJ et al. , Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol 29, 19–26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuasa-Kawase M et al. , Patients with CD36 deficiency are associated with enhanced atherosclerotic cardiovascular diseases. J Atheroscler Thromb 19, 263–275 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Silverstein RL, Febbraio M, CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal 2, re3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishi K et al. , Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol 22, 1649–1654 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Lonn E, Do antioxidant vitamins protect against atherosclerosis? The proof is still lacking*. J Am Coll Cardiol 38, 1795–1798 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Steinhubl SR, Why have antioxidants failed in clinical trials? Am J Cardiol 101, 14D–19D (2008). [DOI] [PubMed] [Google Scholar]
- 17.Barthwal MK et al. , Fluid-phase pinocytosis of native low density lipoprotein promotes murine M-CSF differentiated macrophage foam cell formation. PLoS One 8, e58054 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruth HS, Huang W, Ishii I, Zhang WY, Macrophage foam cell formation with native low density lipoprotein. J Biol Chem 277, 34573–34580 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Kruth HS et al. , Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem 280, 2352–2360 (2005). [DOI] [PubMed] [Google Scholar]
- 20.van Rheenen J et al. , EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J Cell Biol 179, 1247–1259 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mellstrom K, Heldin CH, Westermark B, Induction of circular membrane ruffling on human fibroblasts by platelet-derived growth factor. Exp Cell Res 177, 347–359 (1988). [DOI] [PubMed] [Google Scholar]
- 22.Bosedasgupta S, Pieters J, Inflammatory stimuli reprogram macrophage phagocytosis to macropinocytosis for the rapid elimination of pathogens. PLoS Pathog 10, e1003879 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Racoosin EL, Swanson JA, M-CSF-induced macropinocytosis increases solute endocytosis but not receptor-mediated endocytosis in mouse macrophages. J Cell Sci 102 (Pt 4), 867–880 (1992). [DOI] [PubMed] [Google Scholar]
- 24.BoseDasgupta S, Moes S, Jenoe P, Pieters J, Cytokine-induced macropinocytosis in macrophages is regulated by 14-3-3zeta through its interaction with serine-phosphorylated coronin 1. FEBS J 282, 1167–1181 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Araki N, Johnson MT, Swanson JA, A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol 135, 1249–1260 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stenina OI, Plow EF, Counterbalancing forces: what is thrombospondin-1 doing in atherosclerotic lesions? Circ Res 103, 1053–1055 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajavashisth TB et al. , Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature 344, 254–257 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Kazberuk W, Mysliwiec M, Galar B, [Hepatocyte growth factor (HGF) and atherosclerosis]. Pol Merkur Lekarski 17, 88–91 (2004). [PubMed] [Google Scholar]
- 29.Hoff HF, Gaubatz JW, Gotto AM Jr., Apo B concentration in the normal human aorta. Biochem Biophys Res Commun 85, 1424–1430 (1978). [DOI] [PubMed] [Google Scholar]
- 30.van der Kooij MA, Morand OH, Kempen HJ, van Berkel TJ, Decrease in scavenger receptor expression in human monocyte-derived macrophages treated with granulocyte macrophage colony-stimulating factor. Arterioscler Thromb Vasc Biol 16, 106–114 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Ivanov AI, Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods Mol Biol 440, 15–33 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Commisso C et al. , Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar S, Kang DW, Rezvan A, Jo H, Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab Invest 97, 935–945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kruth HS, Receptor-independent fluid-phase pinocytosis mechanisms for induction of foam cell formation with native low-density lipoprotein particles. Curr Opin Lipidol 22, 386–393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rohrer L, Freeman M, Kodama T, Penman M, Krieger M, Coiled-coil fibrous domains mediate ligand binding by macrophage scavenger receptor type II. Nature 343, 570–572 (1990). [DOI] [PubMed] [Google Scholar]
- 36.Schmees C et al. , Macropinocytosis of the PDGF beta-receptor promotes fibroblast transformation by H-RasG12V. Mol Biol Cell 23, 2571–2582 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Racoosin EL, Swanson JA, Macrophage colony-stimulating factor (rM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J Exp Med 170, 1635–1648 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kojima Y et al. , CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stary HC et al. , A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol 15, 1512–1531 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Suits AG, Chait A, Aviram M, Heinecke JW, Phagocytosis of aggregated lipoprotein by macrophages: low density lipoprotein receptor-dependent foam-cell formation. Proc Natl Acad Sci U S A 86, 2713–2717 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maor I, Hayek T, Hirsh M, Iancu TC, Aviram M, Macrophage-released proteoglycans enhance LDL aggregation: studies in aorta from apolipoprotein E-deficient mice. Atherosclerosis 150, 91–101 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Bhagatwala J et al. , Epithelial sodium channel inhibition by amiloride on blood pressure and cardiovascular disease risk in young prehypertensives. J Clin Hypertens (Greenwich) 16, 47–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin HP et al. , Identification of novel macropinocytosis inhibitors using a rational screen of Food and Drug Administration-approved drugs. Br J Pharmacol 175, 3640–3655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dean L, in Medical Genetics Summaries, Pratt VM et al. , Eds. (Bethesda (MD), 2012). [PubMed]
- 45.Gajda M, Jasztal A, Banasik T, Jasek-Gajda E, Chlopicki S, Combined orcein and martius scarlet blue (OMSB) staining for qualitative and quantitative analyses of atherosclerotic plaques in brachiocephalic arteries in apoE/LDLR(−/−) mice. Histochem Cell Biol 147, 671–681 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buono C, Anzinger JJ, Amar M, Kruth HS, Fluorescent pegylated nanoparticles demonstrate fluid-phase pinocytosis by macrophages in mouse atherosclerotic lesions. J Clin Invest 119, 1373–1381 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anzinger JJ et al. , Measurement of Aortic Cell Fluid-Phase Pinocytosis in vivo by Flow Cytometry. J Vasc Res 54, 195–199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mishra V, Sinha SK, Rajavashisth TB, Role of macrophage colony-stimulating factor in the development of neointimal thickening following arterial injury. Cardiovasc Pathol 25, 284–292 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Pang S et al. , Correlation between the Serum Platelet-Derived Growth Factor, Angiopoietin-1, and Severity of Coronary Heart Disease. Cardiol Res Pract 2020, 3602608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao C, Isenberg JS, Popel AS, Human expression patterns: qualitative and quantitative analysis of thrombospondin-1 under physiological and pathological conditions. J Cell Mol Med 22, 2086–2097 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cui GM et al. , Amiloride attenuates lipopolysaccharide-accelerated atherosclerosis via inhibition of NHE1-dependent endothelial cell apoptosis. Acta Pharmacol Sin 34, 231–238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopez-Charcas O, Rivera M, Gomora JC, Block of human CaV3 channels by the diuretic amiloride. Mol Pharmacol 82, 658–667 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Chambrey R, Achard JM, St John PL, Abrahamson DR, Warnock DG, Evidence for an amiloride-insensitive Na+/H+ exchanger in rat renal cortical tubules. Am J Physiol 273, C1064–1074 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Vassalli JD, Belin D, Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett 214, 187–191 (1987). [DOI] [PubMed] [Google Scholar]
- 55.Swanson JA, Phorbol esters stimulate macropinocytosis and solute flow through macrophages. J Cell Sci 94 (Pt 1), 135–142 (1989). [DOI] [PubMed] [Google Scholar]
- 56.Ghoshal P et al. , Nox2-Mediated PI3K and Cofilin Activation Confers Alternate Redox Control of Macrophage Pinocytosis. AntioxidRedox Signal 26, 902–916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farrar DJ, Bond MG, Riley WA, Sawyer JK, Anatomic correlates of aortic pulse wave velocity and carotid artery elasticity during atherosclerosis progression and regression in monkeys. Circulation 83, 1754–1763 (1991). [DOI] [PubMed] [Google Scholar]
- 58.Sachdeva A et al. , Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J 157, 111–117 e112 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video S1. 3D reconstruction of a foamy macrophage in ApoE−/− aorta. Nucleus (blue), Lipid droplets (yellow) and cell membrane (red). Scale cube: 1 μm3
Data file S1. Individual data points (Microsoft Excel format).
Data Availability Statement
All figures have associated raw data, which are available with permission from the corresponding author.