

Abstract
Human neutrophil elastase (HNE) is a proteolytic enzyme belonging to the serine protease family and is involved in a variety of pathologies. Thus, compounds able to inhibit HNE represent promising therapeutics for the treatment of inflammatory diseases. Here, we report the further elaboration of our previously reported 3-methylisoxazolone derivatives, synthesizing a new series of 3-nor-derivatives bearing different substituents at the 4-phenyl ring. The most potent compounds 3a, 3g, and 3h, had IC50 values of 16, 11, and 18 nM, respectively. Molecular modeling studies and molecular dynamic (MD) simulations demonstrated no substantial differences between the 3-methylisoxazole derivatives previously tested and the corresponding 3-unsubstituted derivatives in the snapshot conformations sampled during the MD simulations, which is consistent with their similar levels of HNE inhibitory activity. Thus, we conclude that the isoxazolone scaffold is a good scaffold for developing HNE inhibitors, as it tolerates several modifications when adhering to basic scaffold requirements, and the resulting derivatives are quite potent HNE inhibitors.
Keywords: human neutrophil elastase inhibitors, molecular and crystal structures, molecular modeling
1 |. INTRODUCTION
Neutrophils are one of primary immune cell populations in humans and produce inflammatory mediators that target invading microorganisms and foreign materials (Korkmaz, Horwitz, Jenne, & Gauthier, 2010; Stapels, Gijsbrecht, & Rooijakkers, 2015). Neutrophils adhere to the endothelium, migrate to sites of infection/inflammation, and release granule components, such as human neutrophil elastase (HNE). HNE is a small, basic, and soluble glycoprotein of about 30 kDa (Sinha et al., 1987) and is stored in neutrophil azurophilic granules with other serine proteases. HNE and other serine proteases are key enzymes involved in inflammation, coagulation, and host defense (Massberg et al., 2010; Pham, 2006). On the other hand, HNE also plays a central role in several inflammatory diseases, such as chronic obstructive pulmonary disease (Lucas, Costa, Guedes, & Moreira, 2013; Pandey, De, & Mishra, 2017), cystic fibrosis (Twigg et al., 2015), rheumatoid arthritis (Hilbert, Schiller, Arnhold, & Arnold, 2002), cancer (Akizuki et al., 2007; Lerman et al., 2017; Lerman & Hammes, 2017; Vaguliene, Zemaitis, Lavinskiene, Miliauskas, & Sakalauskas, 2013), and other disorders with an inflammatory component (Bronze-da-Rocha & Santos-Silva, 2018; Marto et al., 2018). Because of their potential for proteolytic tissue damage, neutrophil protease activity is regulated by specific endogenous (α-1 antitrypsin, secretory leucocyte protease inhibitor and elafin) and exogenous inhibitors, thereby preventing several inflammatory diseases with severe impact on organ tissue integrity (von Nussbaum et al., 2016; von Nussbaum & Li, 2015). Therefore, HNE is an attractive therapeutic target, and the design of new small molecule inhibitors of this enzyme is an area of extensive investigation. Nevertheless, only two HNE inhibitors are commercially available: Sivelestat (Elaspol® 100), which is in use for the treatment of ALI and ARDS only marketed in Korea and Japan (Iwata et al., 2010), and Prolastin (purified α1-AT), which is use for the treatment of α1-antitrypsin deficiency (Bayer Corporation, 2002). In order to develop new compounds that modulate the HNE proteolytic activity, our research group has investigated several molecular scaffolds, such as indazoles (Crocetti et al., 2011, 2013), indoles (Crocetti et al., 2016), azaindoles (Crocetti et al., 2018; Giovannoni et al., 2019), cinnolinones (Giovannoni et al., 2016), and thiazolones (Crocetti et al., 2017). Most recently, we focused on the synthesis isoxazol-5(2H)-one derivatives (Giovannoni et al., 2018; Vergelli et al., 2017). These new isoxazolone-based derivatives exhibited HNE inhibitory activity in the nanomolar range (IC50 = 20–96 nM), and molecular modeling studies allowed us to determine that the endocyclic carbonyl at Position 5 of the isoxazolone was the group attacked by Ser195 of the catalytic triad. The most potent isoxazolones were further evaluated for chemical stability, and the results showed that these derivatives were more stable than our previously described HNE inhibitors with a bicyclic scaffold. Finally, kinetic experiments demonstrated that these compounds acted as competitive HNE inhibitors, and structure–activity relationship (SAR) analyses established that the amide function at Position 2 was important for the activity, and the best substituents at this position were m-tolylcarbonyl and cyclopropylcarbonyl groups (Giovannoni et al., 2018). In order to complete SAR analysis for this class of HNE inhibitors, we designed and synthesized a new series of 4-phenyl-3-unsubstituted derivatives (Figure 1) bearing different substituents on the 4-phenyl ring while maintaining the best fragments at Position 2 of the isoxazolone scaffold (i.e., m-tolylcarbonyl or cyclopropylcarbonyl groups).
FIGURE 1.
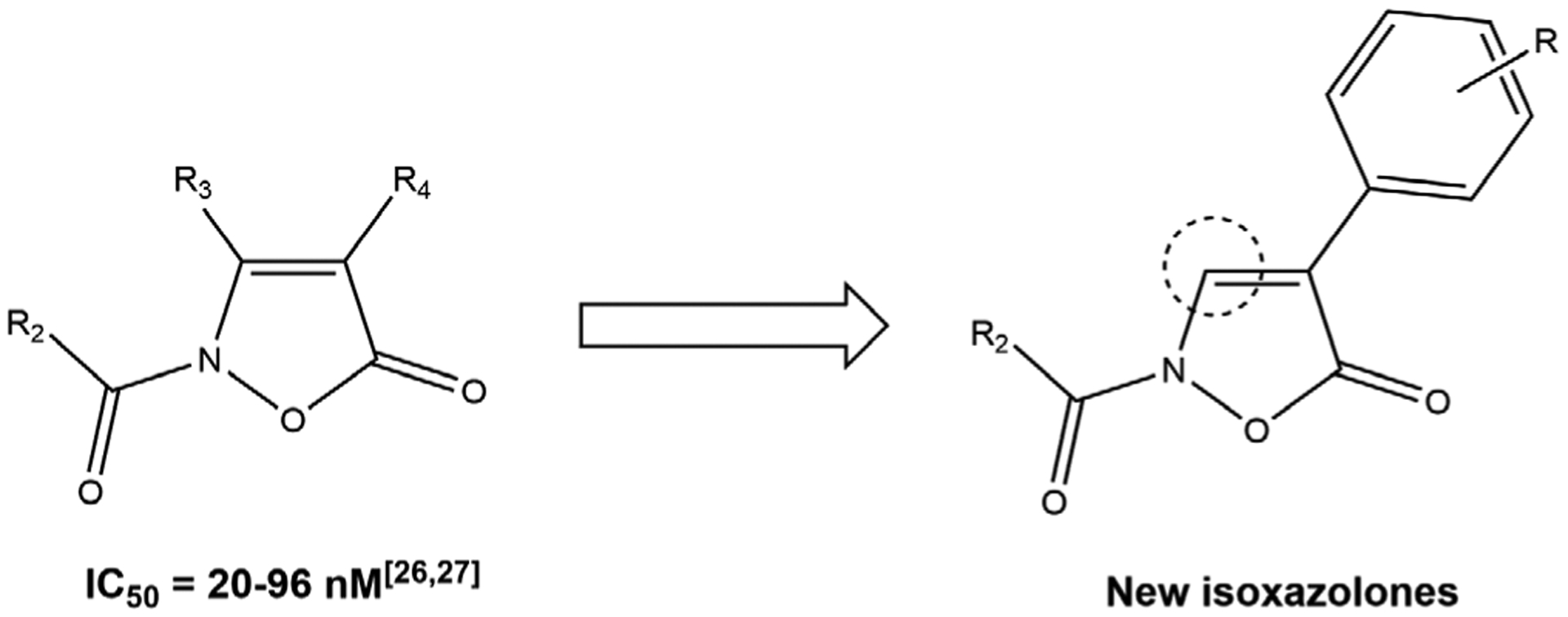
Previously synthesized isoxazolones (Giovannoni et al., 2018; Vergelli et al., 2017) and new derivatives
2 |. EXPERIMENTAL SECTION
2.1 |. Material and methods
All melting points were determined on a Büchi apparatus (New Castle, DE) and are uncorrected. Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. Merck F-254 commercial plates (Merck, Durham, NC) were used for analytical TLC to follow the course of the reactions. Silica gel 60 (Merck 70–230 mesh, Merck, Durham, NC) was used for column chromatography. 1H-NMR, 13C-NMR, 1H-13C HSQC, and 1H-13C HMBC bidimensional spectra were recorded on an Avance 400 instrument (Bruker Biospin Version 002 with SGU, Bruker Inc., Billerica, MA). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm using the solvent as an internal standard. Coupling constants (J values) are given in Hz and were calculated using the TopSpin 1.3 software (Nicolet Instrument Corp., Madison, WI) and are rounded to the nearest 0.1 vHz. Mass spectra (m/z) were recorded on an ESI-TOF mass spectrometer (Brucker Micro TOF, Bruker Inc., Billerica, MA), and reported mass values are within the error limits of ±5 ppm mass units. Microanalyses indicated by the symbols of the elements or functions were performed with a Perkin–Elmer 260 elemental analyzer (PerkinElmer, Inc., Waltham, MA) for C, H, and N, and the results were within ±0.4% of the theoretical values, unless otherwise stated. Reagents and starting material were commercially available.
2.2 |. Chemistry
2.2.1 |. Synthesis of 4-(5-Oxo-2,5-dihydroisoxazol-4-yl)benzonitrile (2h)
A suspension of intermediate 1h (Alves, Dazinger, Veiros, & Kirchner, 2010; 1.38 mmol) in about 6 ml of MeOH/H2O (10:1) was heated at 100°C, and 2.76 mmol of hydroxylamine hydrochloride was added. The mixture was stirred at reflux for 4 hr. After cooling, the solvent was evaporated under vacuum to obtain the crude compound, which was purified by column chromatography using dichloromethane/methanol 8:2 as the eluent. Yield = 82%; mp = 141–143°C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 8.72 (s, 1H, CH), 7.79 (d, 2H, Ar, J = 8.0 Hz), 7.58 (d, 2H, Ar, J = 8.0 Hz). 13C-NMR (100 MHz, CDCl3) δ 169.2 (C), 147.1 (CH), 136.8 (C), 132.1 (2 × CH), 127.1 (2 × CH), 111.8 (C), 111.6 (C), 96.4 (C). ESI-MS calcd. For C10H6N2O2, 186.17; found: m/z 187.05 [M + H]+. Anal. C10H6N2O2 (C, H, N).
2.2.2 |. General procedure for compounds 3a-i and 4a-i
To a suspension of the appropriate substrate 2a-i (Beccalli, La Rosa, & Marchesini, 1984; Snyder et al., 2004; 0.86 mmol) in 10 ml of anhydrous THF, 1.72 mmol of sodium hydride (60% dispersion in mineral oil) and 1.03 mmol of cyclopropanecarbonyl chloride or m-toluoyl chloride were added. The mixture was stirred at room temperature overnight. The solvent was concentrated in vacuo and the resulting mixture was quenched by adding cold water (10 ml). The suspension was extracted with ethyl acetate (3 × 15 mL), dried on anhydrous sodium sulfate, and then filtered to obtain the final compounds 3a-i and 4a-i, which were purified by crystallization from ethanol (3a, 3c, 3e-3i, 4d-f, and 4h) or by column chromatography using cyclohexane/ethyl acetate 6:1 (3b, 3d, 4b, 4c, 4g, and 4i) or hexane/acetone 4:1 (4a) as eluents.
2-(Cyclopropanecarbonyl)-4-phenylisoxazol-5(2H)-one (3a).
Yield = 28%; mp = 171−172°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.72 (s, 1H, CH), 7.78 (d, 2H, Ar, J = 7.2 Hz), 7.42 (t, 1H, Ar, J = 7.2 Hz), 7.36 (d, 2H, Ar, J = 6.0 Hz), 2.45−2.35 (m, 1H, CH cC3H5), 1.31−1.26 (m, 2H, CH2 cC3H5), 1.20−1.15 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.7 (C), 165.9 (C), 136.0 (CH), 128.9 (CH), 128.6 (CH), 127.4 (C), 125.8 (CH), 108.1 (C), 11.8 (CH), 11.2 (CH2). IR ѵ (cm−1): 1,591 (C=C isox), 1,693 (C=O amide), 1,759 (C=O lactone), 3,103 (CH isox). ESI-MS calcd. for C13H11NO3, 229.24; found: m/z 230.08 [M + H]+. Anal. C13H11NO3 (C, H, N).
2-(Cyclopropanecarbonyl)-4-(p-tolyl)isoxazol-5(2H)-one (3b).
Yield = 41%; mp = 130–133°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.67 (s, 1H, CH), 7.67 (d, 2H, Ar, J = 8.2 Hz), 7.22 (d, 2H, Ar, J = 8.2 Hz), 2.45−2.35 (m, 4H, 1H CH cC3H5 + 3H CH3), 1.31−1.26 (m, 2H, CH2 cC3H5), 1.19−1.14 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.7 (C), 166.0 (C), 138.7 (C), 135.5 (CH), 129.6 (CH), 125.7 (CH), 124.4 (C), 108.3 (C), 21.3 (CH3), 11.7 (CH), 11.0 (CH2). IR ѵ (cm−1): 1,600 (C=C isox), 1,678 (C=O amide), 1,770 (C=O lactone), 3,095 (CH isox). ESI-MS calcd. for C14H13NO3, 243.26; found: m/z 244.09 [M + H]+. Anal. C14H13NO3 (C, H, N).
2-(Cyclopropanecarbonyl)-4-(m-tolyl)isoxazol-5(2H)-one (3c).
Yield = 22%; mp = 155–157°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.69 (s, 1H, CH), 7.61 (s, 1H, Ar), 7.57 (d, 1H, Ar, J = 8.0 Hz), 7.30 (t, 1H, Ar, J = 7.6 Hz), 7.16 (d, 1H, Ar, J = 7.2 Hz), 2.44−2.38 (m, 4H, 1H CH cC3H5 + 3H CH3), 1.28−1.23 (m, 2H, CH2 cC3H5), 1.21−1.16 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.7 (C), 166.0 (C), 138.7 (C), 135.9 (CH), 129.4 (CH), 128.8 (CH), 127.3 (C), 126.4 (CH), 122.9 (CH), 108.2 (C), 21.4 (CH3), 11.8 (CH), 11.1 (CH2). IR ѵ (cm−1): 1,589 (C=C isox), 1,647 (C=O amide), 1,768 (C=O lactone), 3,103 (CH isox). ESI-MS calcd. for C14H13NO3, 243.26; found: m/z 244.09 [M + H]+. Anal. C14H13NO3 (C, H, N).
2-(Cyclopropanecarbonyl)-4-(o-tolyl)isoxazol-5(2H)-one (3d).
Yield = 60%; mp = 90–92°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.49 (s, 1H, CH), 7.48 (d, 1H, Ar, J = 7.2 Hz), 7.28−7.22 (m, 3H, Ar), 2.45−2.35 (m, 4H, 1H CH cC3H5 + 3H CH3), 1.30−1.25 (m, 2H, CH2 cC3H5), 1.20−1.15 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.8 (C), 165.2 (C), 138.5 (CH), 136.7 (C), 130.8 (CH), 129.6 (CH), 129.0 (CH), 126.4 (C), 126.1 (CH), 108.5 (C), 20.6 (CH3), 11.8 (CH), 11.0 (CH2). IR ѵ (cm−1): 1,602 (C=C isox), 1,697 (C=O amide), 1,761 (C=O lactone), 3,097 (CH isox). ESI-MS calcd. for C14H13NO3, 243.26; found: m/z 244.09 [M + H]+. Anal. C14H13NO3 (C, H, N).
4-(4-Bromophenyl)-2-(cyclopropanecarbonyl)isoxazol-5(2H)-one (3e).
Yield = 30%; mp = 143–145°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.72 (s, 1H, CH), 7.66 (d, 2H, Ar, J = 8.0 Hz), 7.55 (d, 2H, Ar, J = 8.0 Hz), 2.40−2.35 (m, 1H, CH cC3H5), 1.34−1.28 (m, 2H, CH2 cC3H5), 1.22−1.17 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.1 (C), 165.8 (C), 136.0 (CH), 132.1 (CH), 127.2 (CH), 126.4 (C), 122.6 (C), 109.5 (C), 11.8 (CH), 11.3 (CH2). IR ѵ (cm−1): 1,606 (C=C isox), 1,685 (C=O amide), 1,759 (C=O lactone), 3,086 (CH isox). ESI-MS calcd. for C13H10BrNO3, 308.13; found: m/z 308.98 [M + H]+. Anal. C13H10BrNO3 (C, H, N).
4-(4-Chlorophenyl)-2-(cyclopropanecarbonyl)isoxazol-5(2H)-one (3f).
Yield = 64%; mp = 144–146°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.71 (s, 1H, CH), 7.73 (d, 2H, Ar, J = 8.4 Hz), 7.38 (d, 2H, Ar, J = 8.4 Hz), 2.40−2.35 (m, 1H, CH cC3H5), 1.34−1.29 (m, 2H, CH2 cC3H5), 1.21−1.16 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.7 (C), 165.8 (C), 136.0 (CH), 134.5 (C), 129.2 (CH), 127.0 (CH), 125.9 (C), 107.0 (C), 11.8 (CH), 11.3 (CH2). IR ѵ (cm−1): 1,606 (C=C isox), 1,693 (C=O amide), 1,759 (C=O lactone), 3,099 (CH isox). ESI-MS calcd. for C13H10ClNO3, 263.68; found: m/z 264.04 [M + H]+. Anal. C13H10ClNO3 (C, H, N).
2-(Cyclopropanecarbonyl)-4-(4-methoxyphenyl)isoxazol-5(2H)-one (3g).
Yield = 25%; mp = 198–200°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.62 (s, 1H, CH), 7.72 (d, 2H, Ar, J = 8.2 Hz), 6.94 (d, 2H, Ar, J = 8.2 Hz), 3.83 (s, 3H, OCH3), 2.40−2.35 (m, 1H, CH cC3H5), 1.30−1.25 (m, 2H, CH2 cC3H5), 1.20−1.14 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.7 (C), 166.2 (C), 159.9 (C), 134.8 (CH), 127.2 (CH), 119.7 (C), 114.4 (CH), 108.2 (C), 55.3 (CH3), 11.7 (CH), 11.0 (CH2). IR ѵ (cm−1): 1,597 (C=C isox), 1,676 (C=O amide), 1,762 (C=O lactone), 3,095 (CH isox). ESI-MS calcd. for C14H13NO4, 259.26; found: m/z 260.09 [M + H]+. Anal. C14H13NO4 (C, H, N).
4-(2-(Cyclopropanecarbonyl)-5-oxo-2,5-dihydroisoxazol-4-yl)benzonitrile (3h).
Yield = 31%; mp = 208–210°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.82 (s, 1H, CH), 7.92 (d, 2H, Ar, J = 8.2 Hz), 7.70 (d, 2H, Ar, J = 8.2 Hz), 2.43−2.38 (m, 1H, CH cC3H5), 1.35−1.30 (m, 2H, CH2 cC3H5), 1.23−1.18 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.7 (C), 165.8 (C), 136.8 (C), 132.1 (CH), 127.1 (CH), 126.0 (CH), 118.6 (C), 111.8 (C), 109.6 (C), 11.9 (CH), 11.6 (CH2). IR ѵ (cm−1): 1,587 (C=C isox), 1,712 (C=O amide), 1,753 (C=O lactone), 2,223 (CN), 3,082 (CH isox). ESI-MS calcd. for C14H10N2O3, 254.24; found: m/z 255.07 [M + H]+. Anal. C14H10N2O3 (C, H, N).
2-(Cyclopropanecarbonyl)-4-(4-(trifluoromethyl)phenyl)isoxazol-5(2H)-one (3i).
Yield = 31%; mp = 210–212°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.80 (s, 1H, CH), 7.92 (d, 2H, Ar, J = 8.0 Hz), 7.67 (d, 2H, Ar, J = 8.0 Hz), 2.44−2.39 (m, 1H CH cC3H5), 1.35−1.30 (m, 2H, CH2 cC3H5), 1.24−1.19 (m, 2H, CH2 cC3H5). 13C-NMR (100 MHz, CDCl3) δ 166.8 (C), 165.7 (C), 136.8 (CH), 134.9 (C), 131.1 (C), 125.9 (CH), 124.1 (C), 106.5 (C), 11.8 (CH), 11.5 (CH2). IR ѵ (cm-1): 1,598 (C=C isox), 1,693 (C=O amide), 1,759 (C=O lactone), 3,089 (CH isox). ESI-MS calcd. for C14H10F3NO3, 297.23; found: m/z 298.06 [M + H]+. Anal. C14H10F3NO3 (C, H, N).
2-(3-Methylbenzoyl)-4-phenylisoxazol-5(2H)-one (4a).
Yield = 27%; mp = 120–121°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.89 (s, 1H, CH), 7.85−7.80 (m, 4H, Ar), 7.45−7.37 (m, 5H, Ar), 2.45 (s, 3H, m-CH3-Ph). 13C-NMR (100 MHz, CDCl3) δ 166.1 (C), 160.7 (C), 138.7 (C), 138.0 (CH), 134.6 (CH), 130.5 (CH), 129.6 (C), 129.2 (CH), 129.0 (CH), 128.8 (CH), 128.6 (CH), 127.3 (CH), 126.9 (CH), 125.9 (CH), 124.2 (C), 108.7 (C), 21.3 (CH3). IR ѵ (cm−1): 1,589 (C=C isox), 1,672 (C=O amide), 1,768 (C=O lactone), 3,099 (CH isox). ESI-MS calcd. for C17H13NO3, 279.29; found: m/z 280.09 [M + H]+. Anal. C17H13NO3 (C, H, N).
2-(3-Methylbenzoyl)-4-(p-tolyl)isoxazol-5(2H)-one (4b).
Yield = 47%; mp = 90–92°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.83 (s, 1H, CH), 7.80 (d, 2H, Ar, J = 8.2 Hz), 7.71 (d, 2H, Ar, J = 8.2 Hz), 7.46−7.39 (m, 2H, Ar), 7.23 (d, 2H, Ar, J = 7.6 Hz), 2.44 (s, 3H, m-CH3-Ph), 2.37 (s, 3H, p-CH3-Ph). 13C-NMR (100 MHz, CDCl3) δ 166.2 (C), 160.6 (C), 138.9 (C), 138.6 (C), 137.5 (CH), 134.5 (CH), 130.5 (CH), 129.6 (CH), 129.3 (C), 128.5 (CH), 127.2 (CH), 125.8 (CH), 124.3 (C), 108.8 (C), 21.4 (CH3), 21.3 (CH3). IR ѵ (cm−1): 1,597 (C=C isox), 1,660 (C=O amide), 1,759 (C=O lactone), 3,099 (CH isox). ESI-MS calcd. for C18H15NO3, 293.32; found: m/z 294.11 [M + H]+. Anal. C18H15NO3 (C, H, N).
2-(3-Methylbenzoyl)-4-(m-tolyl)isoxazol-5(2H)-one (4c).
Yield = 42%; mp = 135–137°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.86 (s, 1H, CH), 7.81 (d, 2H, Ar, J = 8.0 Hz), 7.64 (s, 1H, Ar), 7.61 (d, 1H, Ar, J = 8.0 Hz), 7.46−7.39 (m, 2H, Ar), 7.31 (t, 1H, Ar, J = 7.6 Hz), 7.17 (d, 1H, Ar, J = 7.2 Hz), 2.44 (s, 3H, m-CH3-Ph-CO), 2.39 (s, 3H, m-CH3-Ph). 13C-NMR (100 MHz, CDCl3) δ 165.8 (C), 157.6 (C), 138.6 (C), 138.0 (CH), 134.5 (CH), 132.4 (C), 130.5 (CH), 129.6 (CH), 129.3 (C), 128.8 (CH), 128.5 (CH), 127.2 (CH), 126.5 (CH), 125.0 (C), 123.0 (CH), 109.6 (C), 21.4 (CH3), 21.3 (CH3). IR ѵ (cm−1): 1,610 (C=C isox), 1,672 (C=O amide), 1,759 (C=O lactone), 3,105 (CH isox). ESI-MS calcd. for C18H15NO3, 293.32; found: m/z 294.11 [M + H]+. Anal. C18H15NO3 (C, H, N).
2-(3-Methylbenzoyl)-4-(o-tolyl)isoxazol-5(2H)-one (4d).
Yield = 60%; mp = 95–96°C (EtOH). 1H-NMR (400 mHz, CDCl3) δ 8.68 (s, 1H, CH), 7.83 (d, 2H, Ar, J = 8.4 Hz), 7.51 (d, 1H, Ar, J = 7.2 Hz), 7.45−7.40 (m, 2H, Ar), 7.34−7.29 (m, 3H, Ar), 2.45 (s, 3H, m-CH3-Ph), 2.42 (s, 3H, o-CH3-Ph). 13C-NMR (100 MHz, CDCl3) δ 166.8 (C), 160.8 (C), 140.6 (CH), 138.6 (C), 136.3 (C), 134.6 (CH), 130.9 (CH), 130.5 (CH), 129.6 (C), 129.1 (CH), 128.5 (CH), 127.3 (CH), 126.3 (C), 126.2 (CH), 102.2 (C), 21.3 (CH3), 20.7 (CH3). IR ѵ (cm−1): 1,605 (C=C isox), 1,681 (C=O amide), 1,766 (C=O lactone), 3,097 (CH isox). ESI-MS calcd. for C18H15NO3, 293.32; found: m/z 294.11 [M + H]+. Anal. C18H15NO3 (C, H, N).
4-(4-Bromophenyl)-2-(3-methylbenzoyl)isoxazol-5(2H)-one (4e).
Yield = 62%; mp = 159–162°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.90 (s, 1H, CH), 7.81 (d, 2H, Ar, J = 8.0 Hz), 7.71 (d, 2H, Ar, J = 8.4 Hz), 7.56 (d, 2H, Ar, J = 8.4 Hz), 7.46−7.40 (m, 2H, Ar), 2.45 (s, 3H, CH3). 13C-NMR (100 MHz, CDCl3) δ 165.8 (C), 160.6 (C), 138.7 (C), 138.1 (CH), 134.7 (CH), 132.2 (CH), 130.5 (CH), 129.0 (C), 128.6 (CH), 127.4 (CH), 127.3 (CH), 126.3 (C), 122.8 (C), 109.6 (C), 21.3 (CH3). IR ѵ (cm−1): 1,604 (C=C isox), 1,670 (C=O amide), 1,757 (C=O lactone), 3,105 (CH isox). ESI-MS calcd. for C17H12BrNO3, 358.19; found: m/z 359.00 [M + H]+. Anal. C17H12BrNO3 (C, H, N).
4-(4-Chlorophenyl)-2-(3-methylbenzoyl)isoxazol-5(2H)-one (4f).
Yield = 32%; mp = 135–137°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.90 (s, 1H, CH), 7.81−7.76 (m, 4H, Ar), 7.46−7.39 (m, 4H, Ar), 2.45 (s, 3H, CH3). 13C-NMR (100 MHz, CDCl3) δ 165.9 (C), 160.6 (C), 138.7 (C), 138.1 (CH), 134.7 (CH), 132.1 (CH), 130.5 (CH), 129.2 (CH), 129.1 (C), 128.6 (CH), 127.3 (CH), 127.1 (CH), 125.8 (C), 107.5 (C), 21.3 (CH3). IR ѵ (cm−1): 1,605 (C=C isox), 1,660 (C=O amide), 1,770 (C=O lactone), 3,096 (CH isox). ESI-MS calcd. for C17H12ClNO3, 313.74; found: m/z 314.05 [M + H]+. Anal. C17H12ClNO3 (C, H, N).
4-(4-Methoxyphenyl)-2-(3-methylbenzoyl)isoxazol-5(2H)-one (4g).
Yield = 28%; mp = 168–170°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.82 (s, 1H, CH), 7.84−7.77 (m, 4H, Ar), 7.49−7.44 (m, 2H, Ar), 6.98 (d, 2H, Ar, J = 8.8 Hz), 3.87 (s, 3H, OCH3), 2.48 (s, 3H, m-CH3-Ph). 13C-NMR (100 MHz, CDCl3) δ 165.8 (C), 157.0 (C), 159.8 (C), 138.5 (C), 134.4 (CH), 130.4 (CH), 129.3 (CH), 129.0 (CH), 128.6 (C), 128.5 (CH), 127.4 (CH), 127.2 (CH), 114.4 (C), 114.1 (CH), 109.8 (C), 55.3 (CH3), 21.4 (CH3). IR ѵ (cm−1): 1,604 (C=C isox), 1,668 (C=O amide), 1,763 (C=O lactone), 3,093 (CH isox). ESI-MS calcd. for C18H15NO4, 309.32; found: m/z 310.10 [M + H]+. Anal. C18H15NO4 (C, H, N).
4-(2-(3-Methylbenzoyl)-5-oxo-2,5-dihydroisoxazol-4-yl)benzonitrile (4h).
Yield = 28%; mp = 165–167°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 9.02 (s, 1H, CH), 7.96 (d, 2H, Ar, J = 8.0 Hz), 7.83 (d, 2H, Ar, J = 8.4 Hz), 7.72 (d, 2H, Ar, J = 8.4 Hz), 7.49 (d, 1H, Ar, J = 7.2 Hz), 7.44 (t, 1H, Ar, J = 7.0 Hz), 2.46 (s, 3H, CH3). 13C-NMR (100 MHz, CDCl3) δ 165.4 (C), 160.6 (C), 139.6 (CH), 138.8 (C), 135.0 (CH), 132.7 (CH), 132.1 (C), 130.6 (CH), 128.7 (CH), 127.4 (CH), 126.2 (CH), 118.4 (C), 112.0 (C), 106.4 (C), 21.3 (CH3). IR ѵ (cm−1): 1,678 (C=C isox), 1,722 (C=O amide), 1,737 (C=O lactone), 2,222 (CN), 3,080 (CH isox). ESI-MS calcd. for C18H12N2O3, 304.30; found: m/z 305.09 [M + H]+. Anal. C18H12N2O3 (C, H, N).
2-(3-Methylbenzoyl)-4-(4-[trifluoromethyl]phenyl)isoxazol-5(2H)-one (4i).
Yield = 72%; mp = 164–166°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 9.00 (s, 1H, CH), 7.96 (d, 2H, Ar, J = 8.0 Hz), 7.83 (d, 2H, Ar, J = 8.8 Hz), 7.69 (d, 2H, Ar, J = 8.0 Hz), 7.49−7.41 (m, 2H, Ar), 2.45 (s, 3H, CH3). 13C-NMR (CDCl3) δ 165.83 (C), 157.0 (C), 139.0 (CH), 138.5 (C), 135.8 (C), 134.4 (CH), 134.1 (C), 131.0 (CH), 130.6 (CH), 130.2 (C), 129.8 (CH), 128.6 (CH), 127.4 (CH), 126.0 (CH), 125.9 (CH), 124.1 (C), 109.6 (C), 21.3 (CH3). IR ѵ (cm−1): 1,598 (C=C isox), 1,693 (C=O amide), 1,759 (C=O lactone), 3,089 (CH isox). ESI-MS calcd. for C18H12F3NO3, 347.29; found: m/z 348.08 [M + H]+. Anal. C18H12F3NO3 (C, H, N).
2.3 |. Pharmacology
Compounds were dissolved in 100% DMSO at 5 mM stock concentrations. The final concentration of DMSO in the reactions was 1%, and this level of DMSO had no effect on enzyme activity. The HNE inhibition assay was performed in black flat-bottom 96-well microtiter plates. Briefly, a solution containing 200 mM Tris–HCl, pH 7.5, 0.01% bovine serum albumin, 0.05% Tween-20, and 20 mU/ml of HNE (Calbiochem) was added to wells containing different concentrations of each compound. The reaction was initiated by addition of 25 μM elastase substrate (N-methylsuccinyl-Ala-Ala-Pro-Val-7-amino-4-methylcoumarin, Calbiochem) in a final reaction volume of 100 μl/well. Kinetic measurements were obtained every 30 s for 10 min at 25°C using a Fluoroskan Ascent FL fluorescence microplate reader (Thermo Electron, MA) with excitation and emission wavelengths at 355 and 460 nm, respectively. For all compounds tested, the concentration of inhibitor that caused 50% inhibition of the enzymatic reaction (IC50) was calculated by plotting % inhibition versus logarithm of inhibitor concentration (at least six points). The data are presented as the mean values of at least three independent experiments with relative standard deviations of <15%.
2.4 |. Single crystal X-ray diffraction
Data for Compound A were collected at 100 K on an Xcalibur PX Ultra CCD four-circle diffractometer using a graphite monochromator and Cu Ka radiation. A reference frame was monitored every 50 frames to control for stability of the crystal, and the system revealed no intensity decay. The data set was corrected for Lorentz, polarization effects, and absorption corrections were performed by the ABSPACK multiscan procedure of the CrysAlis (CrysAlis RED, n.d., version 171.37.35) data reduction package. The structure was solved using the direct method with the SHELXS-2011 (Sheldrick, 1990) software, and refinement was performed using the SHELXL-2013 (Sheldrick, 2008) software package. All nonhydrogen atoms were located from the initial solution or from subsequent electron density difference maps during the initial course of the refinement. After locating the nonhydrogen atoms, the models were refined against F2, first using isotropic and finally anisotropic thermal displacement parameters. Hydrogen atoms have been introduced from subsequent electron density difference maps at the end of the refinement. Programs used in the crystallographic calculations included WinGX (Farrugia, 2012) and ORTEP (Farrugia, 1997) for graphics. Crystal and refinement data are listed in Table S1. Crystal structural data are available from the Cambridge Crystallographic CCDC 1937512.
2.5 |. Molecular modeling procedures
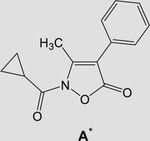
Compounds 3a, 4a, A, and B were considered in the modeling studies. The molecular structure of A, as obtained from X-ray diffraction (details in the related sections), was used as the starting geometry in the computational study for Compound A and as a building block for 3a. The starting geometry for 4a and B was derived by the structure of the closely related isoxazolone derivative 3-ethyl-2-(3-methylbenzoyl)isoxazol-5(2H)-one (Vergelli et al., 2017). The 3D arrangement of all molecules was improved by geometry optimization procedures, followed by molecular dynamic (MD) simulations at 300 and 600 K (time step = 1 fs, equilibration time = 100 ps, production time = 1,000 ps). MD simulations were performed both in vacuum and in implicit solvent, the latter was simulated using distance-dependent dielectric constants set to 4r. Ten snapshot conformations with evenly spaced intervals were extracted from MD trajectories (performed at 600 K with ε = 4r) and subsequently minimized (by using the steepest descent and conjugate gradient algorithms) in order to obtain starting geometries for the subsequent quantum chemical calculations (QC) and MD simulations with HNE. The structure of HNE complexed with a peptide chloromethyl ketone inhibitor was used for the MD simulations (1HNE [Navia et al., 1989] entry of the Protein Data Bank). The bound inhibitor was removed, as well as all of the water molecules, and hydrogen atoms were added prior to starting simulations. MD simulations on each ligand-HNE assembly were performed at 300 K under vacuum (time step = 1 fs, equilibration time = 100 ps, production time 5,000 ps). The programs used for the energy minimization and MD were the simulation protocols Minimization, Standard Dynamics Cascade, and Analyze Trajectory, implemented in Accelrys Discovery Studio 2.1 (Accelrys Discovery, 2019). The Force Field used for all simulations was CHARMm (Brooks et al., 1983). Molecular plots were produced by the program Discovery Studio Visualizer (v 4.5; Dassault Systemes BIOVIA, 2015). GAUSSIAN09 (Rev. C01; Frisch et al., 2016) was used for QC calculations by using the B97D (Grimme, 2006) functional. The basis set was 6–31 + G(d,p) (Hehre, Ditchfield, & Pople, 1972). The Berny algorithm was used (Peng, Ayala, Schlegel, & Frisch, 1996). The reliability of the stationary points was assessed by evaluation of the vibrational frequencies. For each inhibitor, different conformational isomers chosen among the low-energy lying conformers (as found in MD simulations and subsequent geometry optimization) were considered in QC calculations. Finally, CrystalExplorer17 (Turner et al., 2017) was used to compute the Hirshfeld surface (HS).
3 |. RESULTS AND DISCUSSION
3.1 |. Chemistry
All new compounds were synthesized as reported in Figure 2, and the structures were confirmed on the basis of analytical and spectral data. Starting from 2-aryl-2-formylethylacetate derivatives 1a-i (Alves et al., 2010; Beccalli et al., 1984; Liu, Chen, Wu, & Tang, 2016; Rajput et al., 2014), we performed cyclization with hydroxylamine hydrochloride and piperidine in ethanol at reflux temperature (for compounds 2a-g; Beccalli et al., 1984; Snyder et al., 2004) or with hydroxylamine hydrochloride in a mixture MeOH/H2O 10:1 at reflux (for compounds 2h and 2i [Snyder et al., 2004]) to obtain the key intermediates with an isoxazolone core of Type 2. Despite the possibility that this nucleus can exist in three different tautomers (Cencioni, Franchini, & Orienti, 1968; Franchini, 1968; Frolund et al., 2005), we confirmed in our previous work (Giovannoni et al., 2018) and here that acylation of intermediates 2a-i (Beccalli et al., 1984; Snyder et al., 2004) resulted exclusively in the N–CO derivatives 3a-i and 4a-i when used the appropriate acyl/aroyl chloride and sodium hydride in anhydrous tet-rahydrofuran at room temperature. This result indicates that the NH form is the only product formed under these reaction conditions. In order to confirm our findings, assignment of the structure of final compounds of Types 3 and 4 was performed using 1H-NMR, 13C-NMR, IR spectroscopy, and 2D NMR (1H-13C HSQC, 1H-13C HMBC).
FIGURE 2.
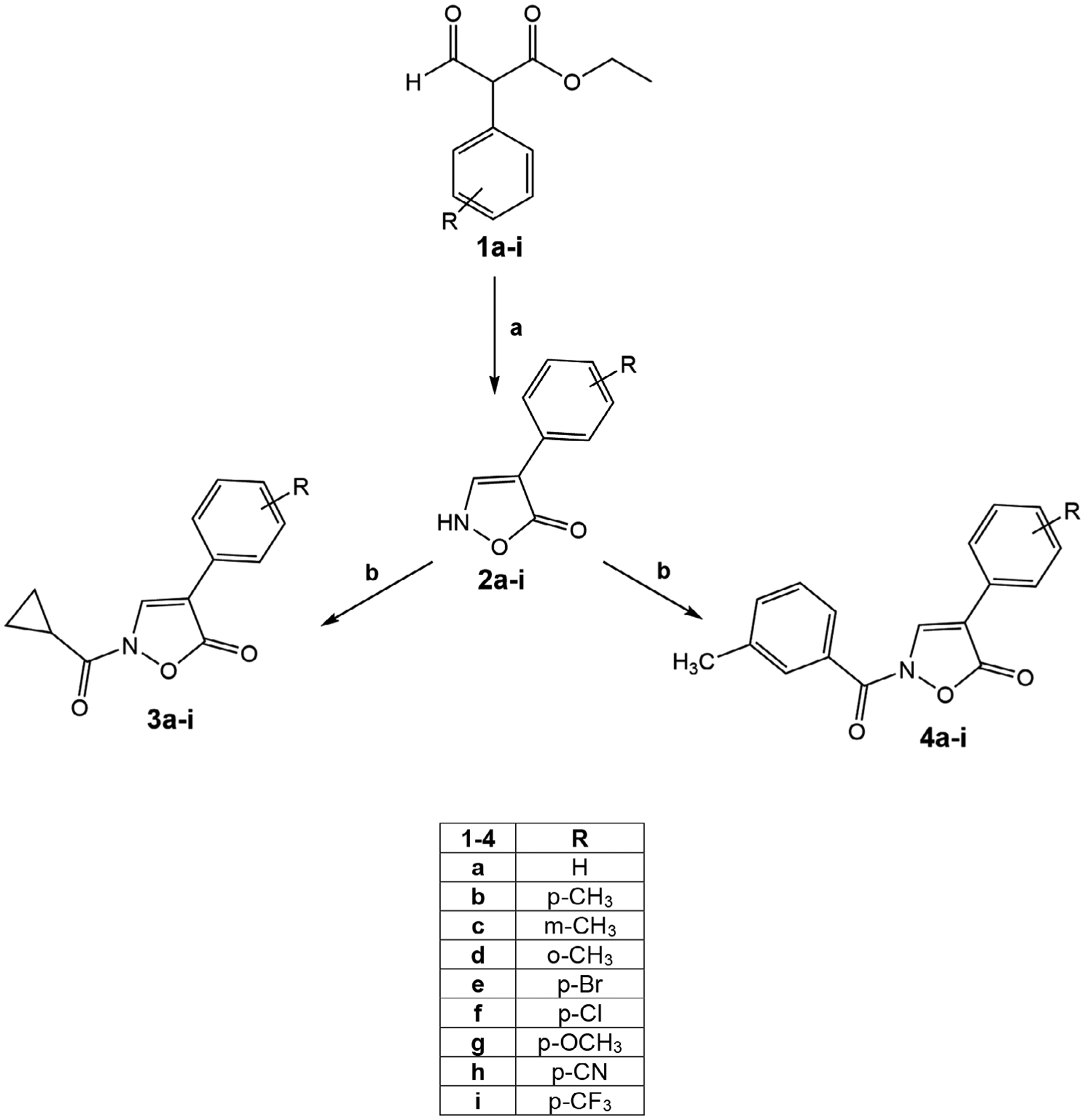
Reagents and conditions. (a) for 2a-g: EtOH, piperidine, NH2OH·HCl, 80°C, 2 hr; for 2h,i: MeOH/H2O (10:1), NH2OH·HCl, 100°C, 4 h; (b) m-CH3-Ph-COCl or cC3H5-COCl, dry THF, NaH (60% dispersion in mineral oil), r.t., 24 hr
3.2 |. Biological evaluation and SAR analysis
All compounds were evaluated for their ability to inhibit HNE in comparison with Sivelestat. All the biological results are reported in Table 1, as compounds of Type 3 (N-cyclopropylcarbonyl derivatives) and of Type 4 (N-m-tolylcarbonyl derivatives) containing the same substituents R in the phenyl ring at Position 4 (R is shown in the central column). For comparison, we also include our previous reported compounds A and B, which belong to the series of the 3-methylisoxazolones (Giovannoni et al., 2018). Our biological results verify that all compounds of Type 3 and 4 are potent HNE inhibitors, as their HNE inhibitory activity was in the low nanomolar range (IC50 = 11–81 nM), with most compounds being more active than Sivelestat (IC50 = 44 nM). Starting from compounds 3a and 4a, which are derivatives of the reference compounds A and B, respectively, elimination of the methyl group at Position 3 increased activity of both compounds ~fourfold for 3a and twofold for 4a (3a vs. A: IC50 = 16 nM vs. 59 nM; 4a vs. B: IC50 = 46 nM vs. 77 nM). On the other hand, the introduction of different substituents on the phenyl ring at Position 4 led to compounds which, regardless of the group at Position 2 of the amide function (i.e., m-tolyl or cyclopropyl) had the same potency, such as when the substituent was a methyl group (3b-d and 4b-d), but also when Cl, Br, or methoxy groups were present (3e-g and 4e-g). Slightly greater differences were observed for 3h and 4h, where the N-cyclopropylcarbonyl derivative 3h was about ~fourfold more potent than the N-m-tolylcarbonyl derivative 4h (IC50 = 18 vs. 78 nM, respectively). The same trend was observed for the p-CF3-phenyl derivatives 3i and 4i (IC50 = 33 and 81 nM, respectively).
TABLE 1.
HNE inhibitory activity of compounds 3a-i and 4a-i
| ||||
|---|---|---|---|---|
| Comp. | IC50 (nM)a | R | IC50 (nM)a | Comp. |
| 3a | 16 ± 5 | H | 46 ± 12 | 4a |
| 3b | 26 ± 4 | p-CH3 | 22 ± 7 | 4b |
| 3c | 22 ± 5 | m-CH3 | 31 ± 3 | 4c |
| 3d | 39 ± 11 | o-CH3 | 28 ± 3 | 4d |
| 3e | 31 ± 3 | p-Br | 47 ± 7 | 4e |
| 3f | 42 ± 8 | p-Cl | 25 ± 2 | 4f |
| 3g | 11 ± 3 | p-OCH3 | 23 ± 7 | 4g |
| 3h | 18 ± 5 | p-CN | 78 ± 2 | 4h |
| 3i | 33 ± 1 | p-CF3 | 81 ± 28 | 4i |
|
59 ± 18 | - | 77 ± 27 |
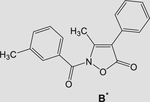
|
Note: Sivelestat IC50 = 44 ± 20 nM.
Abbreviation: HNE, human neutrophil elastase.
The experiments were performed in triplicate using N-methylsuccinyl-Ala-Ala-Pro-Val-7-amino-4-cumarin as substrate for HNE.
Overall, we found that the nature of the substituent (CH3, Br, Cl, OCH3, CN, CF3) and/or their position (para, meta, or ortho) in the phenyl ring do not seem to significantly influence HNE inhibitory potency, indicating that the pocket of the enzyme hosting the aromatic ring is wide and able to hold bulky groups with different chemical and electronic properties.
3.3 |. Molecular structure from single crystal X-ray diffraction and molecular modeling
The solid-state molecular structure of compound A (Giovannoni et al., 2018) was obtained from single crystal X-ray diffraction. A crystallizes in the P212121 with one independent molecule in the asymmetric unit. Figure 3 shows an ORTEP-3 representation of the compound together with atom labeling. Bond distances and angles are within the expected ranges. The conformation of the compound, as defined by the dihedral angles τ1 (−179.1[2]), τ2 (−38.5[5]), and τ3 (−175[2]) (see Figure 4a), can be described as anti-gauche-anti (Klyne & Prelog, 1960). A search in the Cambridge Structural Database 5.40 (CSD; Groom, Bruno, Lightfoot, & Ward, 2016) investigating the usual range of values for τ1, τ2, and τ3 found that 53 hits with the fragment sketched in Figure 4b are present in the CSD; of these, only two (AMIXPO and GUJJIF refcodes) feature a C=O bound to 2-N. The related torsion angle τ1 adopts an eclipsed and anti-conformation, respectively; finally, an anti-conformation for τ1 was also observed in the isoxazolone derivative 3-ethyl-2-(3-methylbenzoyl)isoxazol-5(2H)-one (Vergelli et al., 2017). No comparisons were made for τ2 due to the lack of suitable reference structures. The τ3 dihedral adopted the usual conformation.
FIGURE 3.
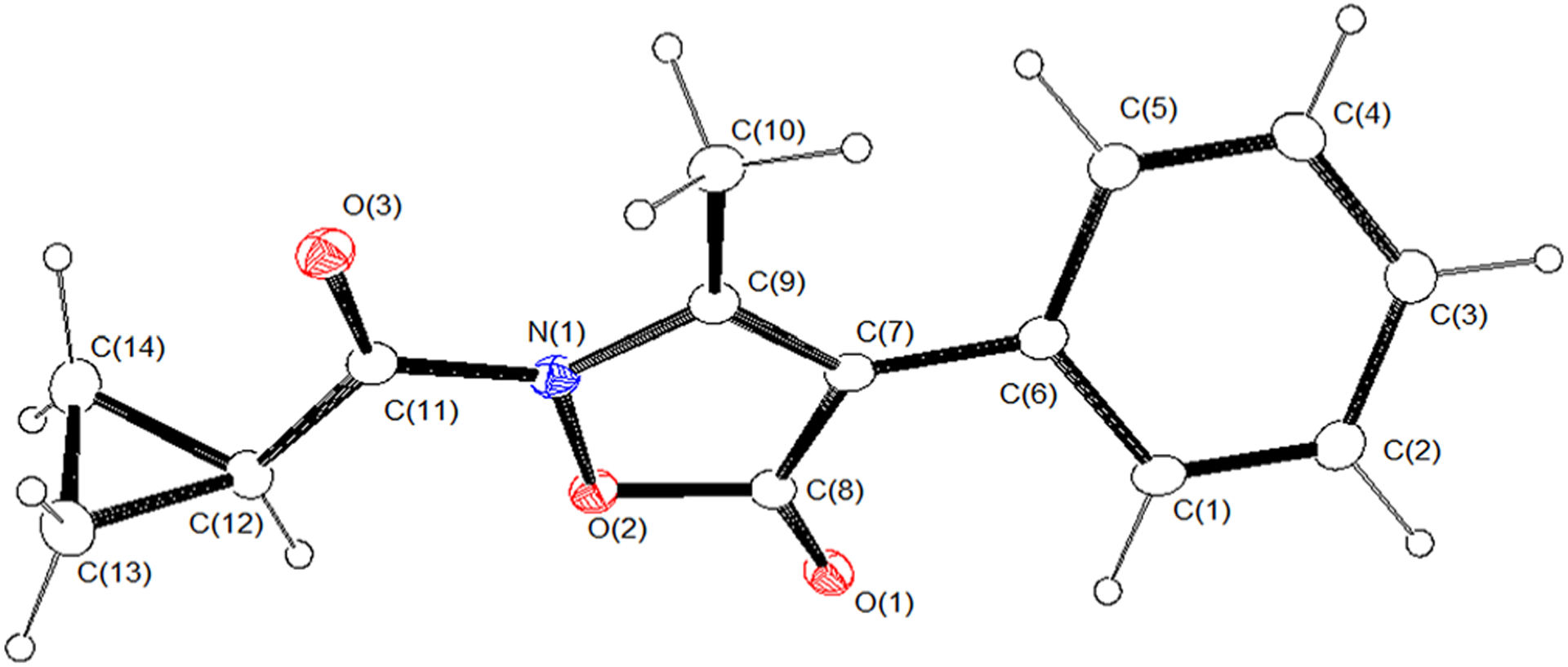
ORTEP-3 view of the asymmetric unit of A (ellipsoid probability 20%)
FIGURE 4.
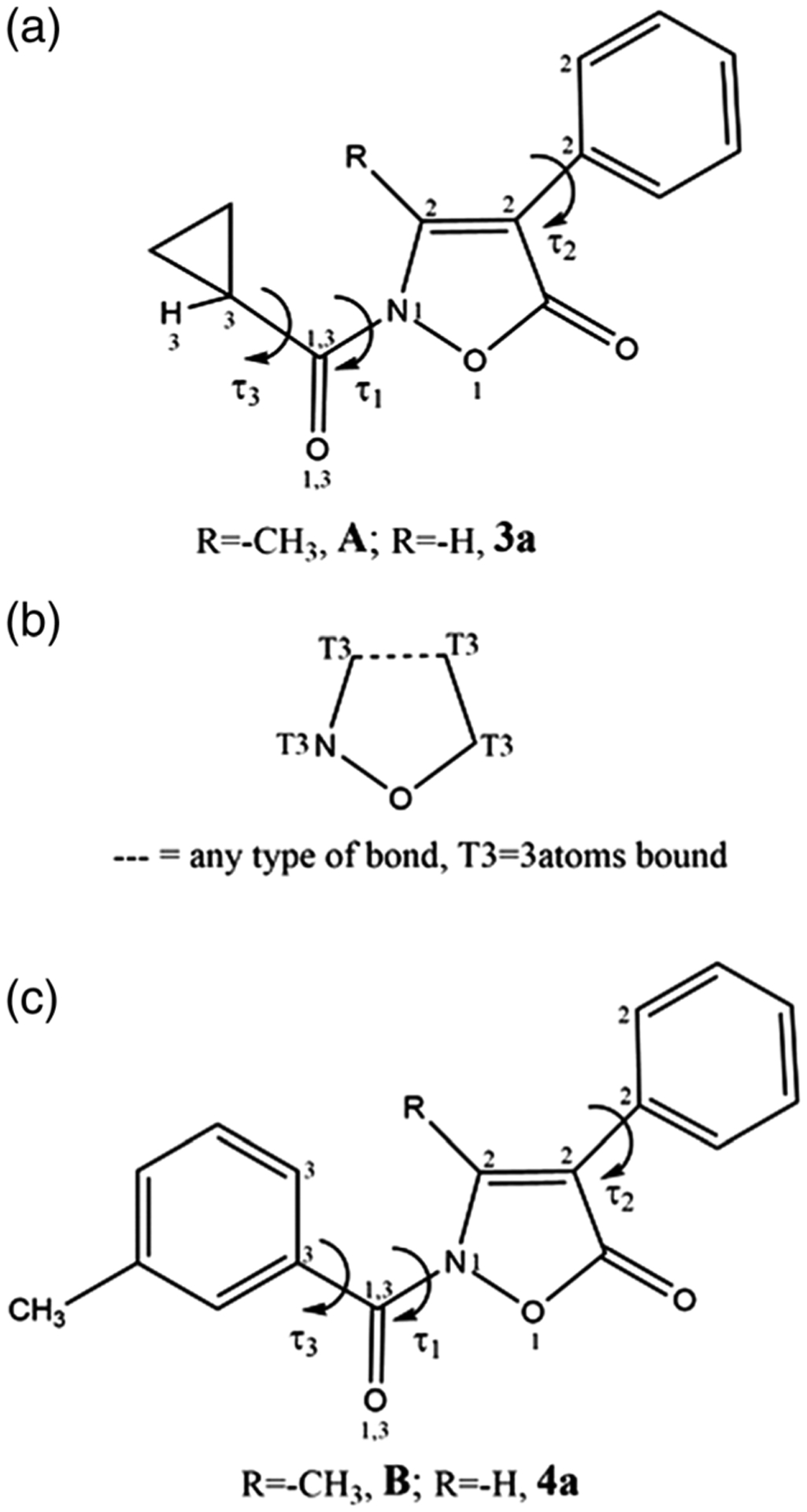
(a) Schematic of the cyclopropyl derivatives A and 3a and the related dihedral angles defining the molecule’s conformation; (b) fragment searched in the Cambridge Structural Database; (c) schematic of the m-tolylcarbonyl derivatives B and 4a and the related dihedral angles defining the molecule’s conformation
Molecular dynamic simulations at 300 and 600 K were performed in order to explore the conformational space accessible to compound A and, eventually, to find other low-energy conformers. For comparison, the same approach (MD at 300 and 600 K) was used to sample the conformational space and identify low-energy conformers of closely related compounds 3a, 4a, and B (Giovannoni et al., 2018; sketched in Table 1).
The overall shape of each molecule was not affected by either the different simulation medium (vacuum and ε = 4r) or the temperature (300 and 600 K), as shown by the values adopted by the dihedral angles τ1, τ2, and τ3 (Figure 4a,c) throughout the MD simulations. In particular, snapshot conformations collected from MD trajectories at 300 and 600 K all had τ1 dihedrals in an anti-conformation, which was maintained throughout the simulations. As expected, a larger variety of conformations was observed for τ2 and τ3 (-Figures S1–S4). In addition, all the molecules had an almost identical overall shape that can be roughly described as an elongated cylinder, as shown in Figure 5 where superimposition of the minimized conformers obtained from a sampling of the corresponding MD trajectories (T = 600 K, ε = 4r) is shown for each molecule. As expected, results for compounds 4a and B correlated well with those of the closely related isoxazolone derivatives, as we reported previously (Giovannoni et al., 2018).
FIGURE 5.
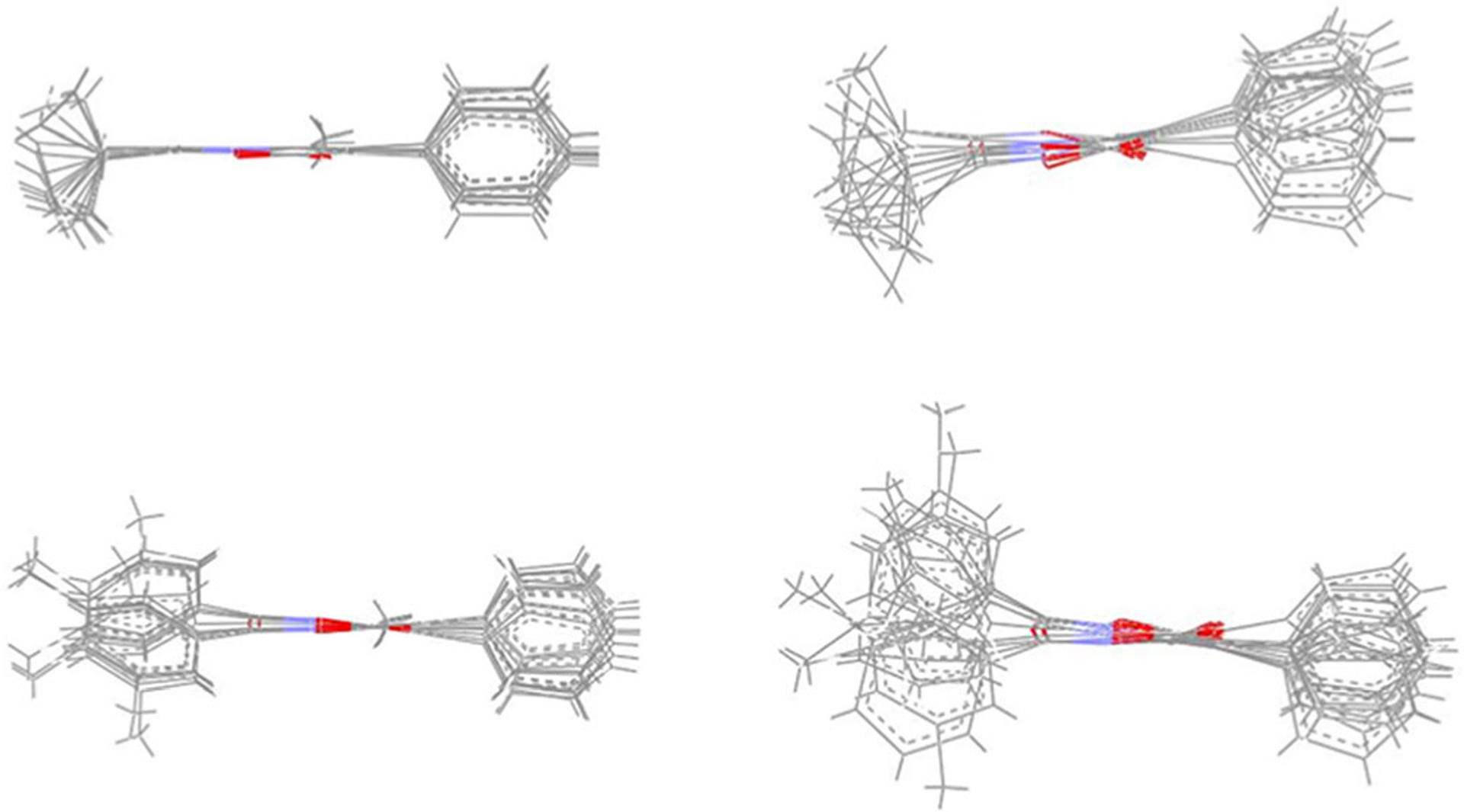
Superimposition of the minimized conformers of A (top left), 3a (top right), B (bottom left), and 4a (bottom right)
The different conformational isomers optimized by the QC method converge to the same conformer for compounds 3a and A (anti-eclipsed-anti and anti-gauche-anti, respectively), which, in the case of compound A, was identical to that found in the solid state (Figure S5a). For the m-toluoyl derivatives, the optimization procedure led to different conformers (Figure S5b) that in both cases (i.e., 4a and B) essentially differed in the conformations adopted by τ3 (τ1 was in all the cases close to 180° and τ2 was gauche). Regardless of the orientation of the m-toluoyl group, differences in the energy content were negligible. Finally, as estimated by the Mullikan and APT charge-derivation schemes, there were not significant differences in atomic charge of the 5-C=O carbon atom, which is involved in the attack of Ser195, on moving from the 3-methylisoxazoles (A and B) to the corresponding nor-derivatives (3a and 4a).
In summary, conformation of the τ1 dihedral in the cyclopropyl (3a and A) and m-tolyl (4a and B) derivatives appears frozen, regardless of the environment (MD simulation in vacuum and ε = 4r, crystal) or the temperature (MD simulations at 300 and 600 K). In contrast, τ2 and τ3 accessed almost all conformations during MD simulations (300 and 600 K). For 4a and B, small differences in orientation of the m-tolouyl group did not affect the relative energy content of the corresponding conformational isomers. All of the compounds had a cylindrical overall shape.
3.4 |. Intermolecular binding propensity from single crystal X-ray diffraction and molecular modeling
Given that N–C=O plays the role of anchoring the molecule to the subpocket of the binding site through H-bond interactions with the receptor, we analyzed the intermolecular contacts involving this group in the crystal lattice of compound A to assess its propensity to act an H-bond acceptor. In the crystalline solid (Figure 6), the oxygen atom belonging to the N–C=O group acts as an acceptor for the hydrogen atom provided by a symmetry-related molecule: O(3)⋯H(4)C(4)iv (iv = 1.5 − x,1 − y, −0.5 + z). This interaction is quite weak (Desiraju & Steiner, 1999) as determined by the O⋯H distance (2.41(3)Å) and the related angle (143(3)°).
FIGURE 6.
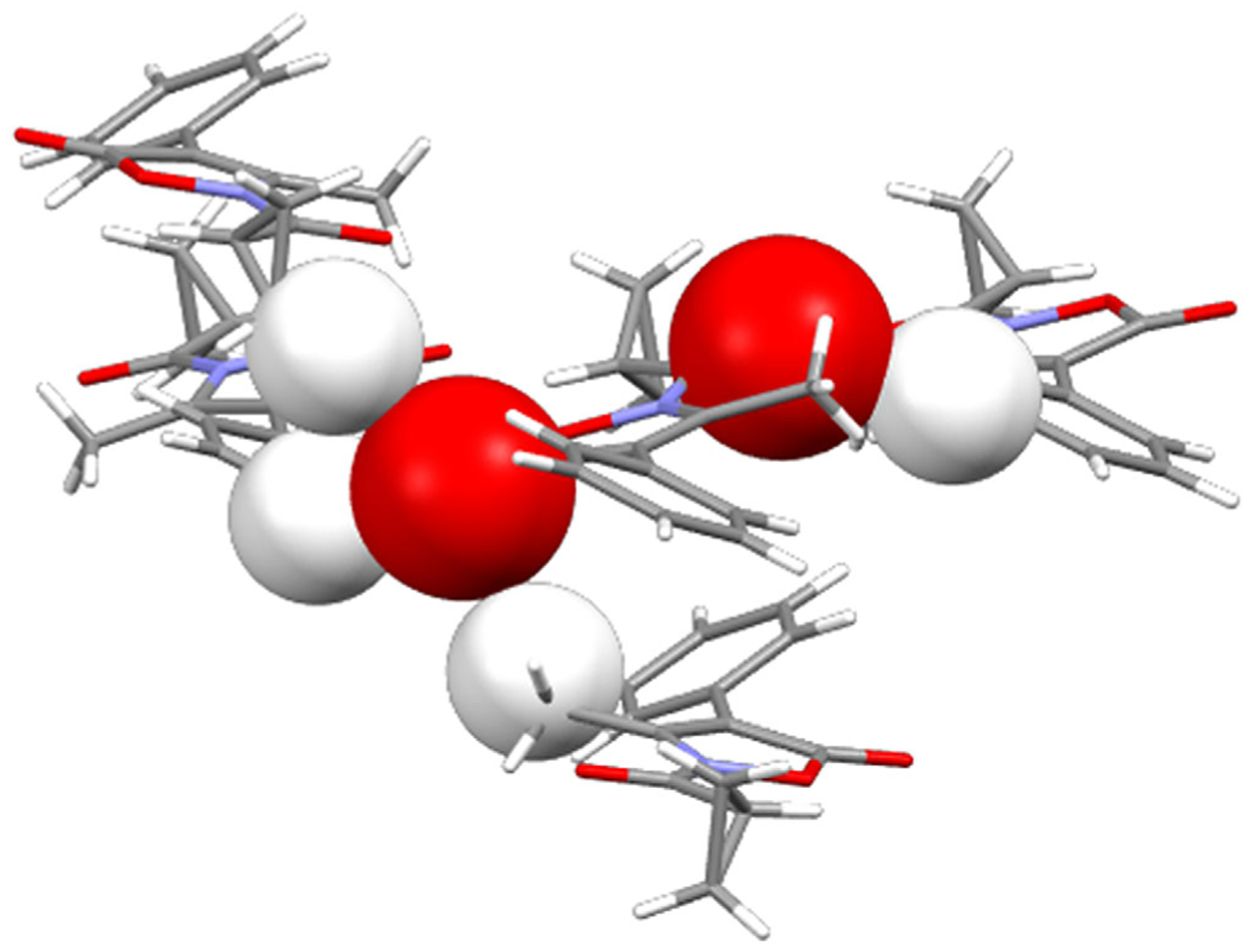
Crystal packing of compound A highlighting the carbonyl oxygen atoms (space filling representation) involved in intermolecular contacts with the hydrogen atoms of neighboring molecules (space filling representation)
The oxygen atom of the 5-C=O group (whose carbon atom appears to be involved in the attack of Ser195) also contributes to the net intermolecular contacts acting as a tri-furcated acceptor for the hydrogen atoms provided by three distinct symmetry-related molecules: O(1)⋯H(13A)C(13)i (i = 2 − x, 0.5 + y, 0.5 − z), O(1)⋯H(10C)C (10)ii (ii = −0.5 + x, 1.5 − y, 1 − z) and O(1)⋯H(3)C(3)iii (iii = 1.5 − x, 2 − y, −0.5 + z). These interactions also are quite weak [39], as determined by the O⋯H distances (2.55(4), 2.65(4), and 2.67(3)Å, respectively, and the related angles (160(3), 124(3), and 136(3)°). The relative strength of these interactions is clearly evident in the HS of A shown in Figure 7 (the brightest red spots are associated with the closest intermolecular contacts), which also shows the closest neighboring molecules involved in the H-bonds.
FIGURE 7.
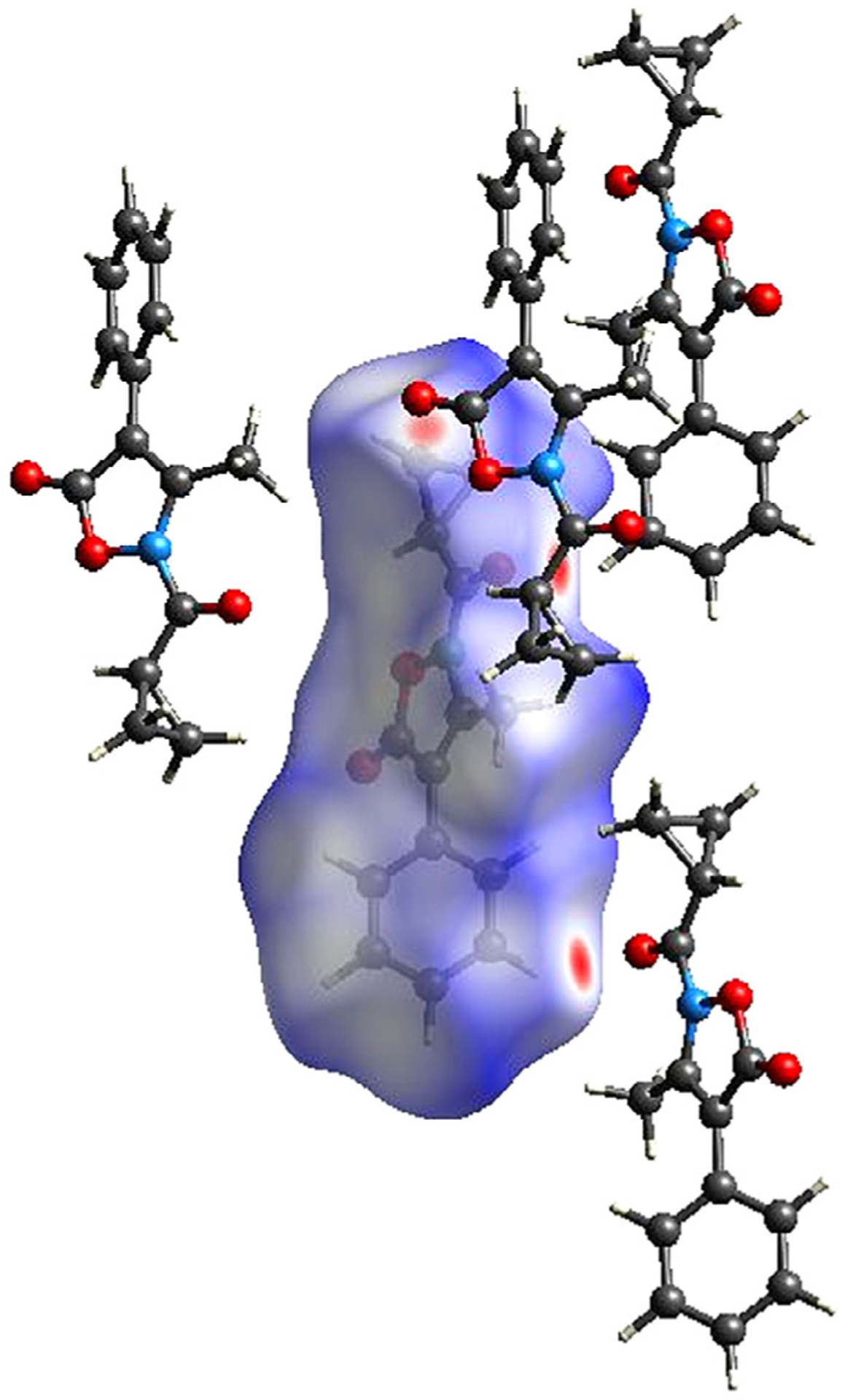
Hirshfeld surface of A and neighboring molecules associated with close contacts
Following the same approach previously adopted (Giovannoni et al., 2018), the exposure to the environment of both carbonyl groups in molecule A and 3a was further assessed by the dimension of two spheres centered on the 5-C=O carbon atom and on the N–C=O oxygen atom. In both cases, a sphere having radius 2.5 Å appears suitable to map the accessible region about the two carbonyl groups (Figure 8A). Thus, as with the N–C=O oxygen atom, the different substituents at the 3-position (–CH3 vs H) did not seem to affect its accessibility to the enzyme. Identical results were found for the m-tolyl derivatives 4a and B (Figure 8B). Finally, we can also conclude that substitution with cyclopropyl or m-tolyl did not appear to have consequences on exposure of the anchoring carbonyl group. In summary, no significant differences were detected both within each class of inhibitors (3a vs A; 4a vs B) and also between the two families (cyclopropyl vs m-toluoyl derivatives) in terms of the accessibility of the two functional groups more specifically involved in the interaction with HNE.
FIGURE 8.
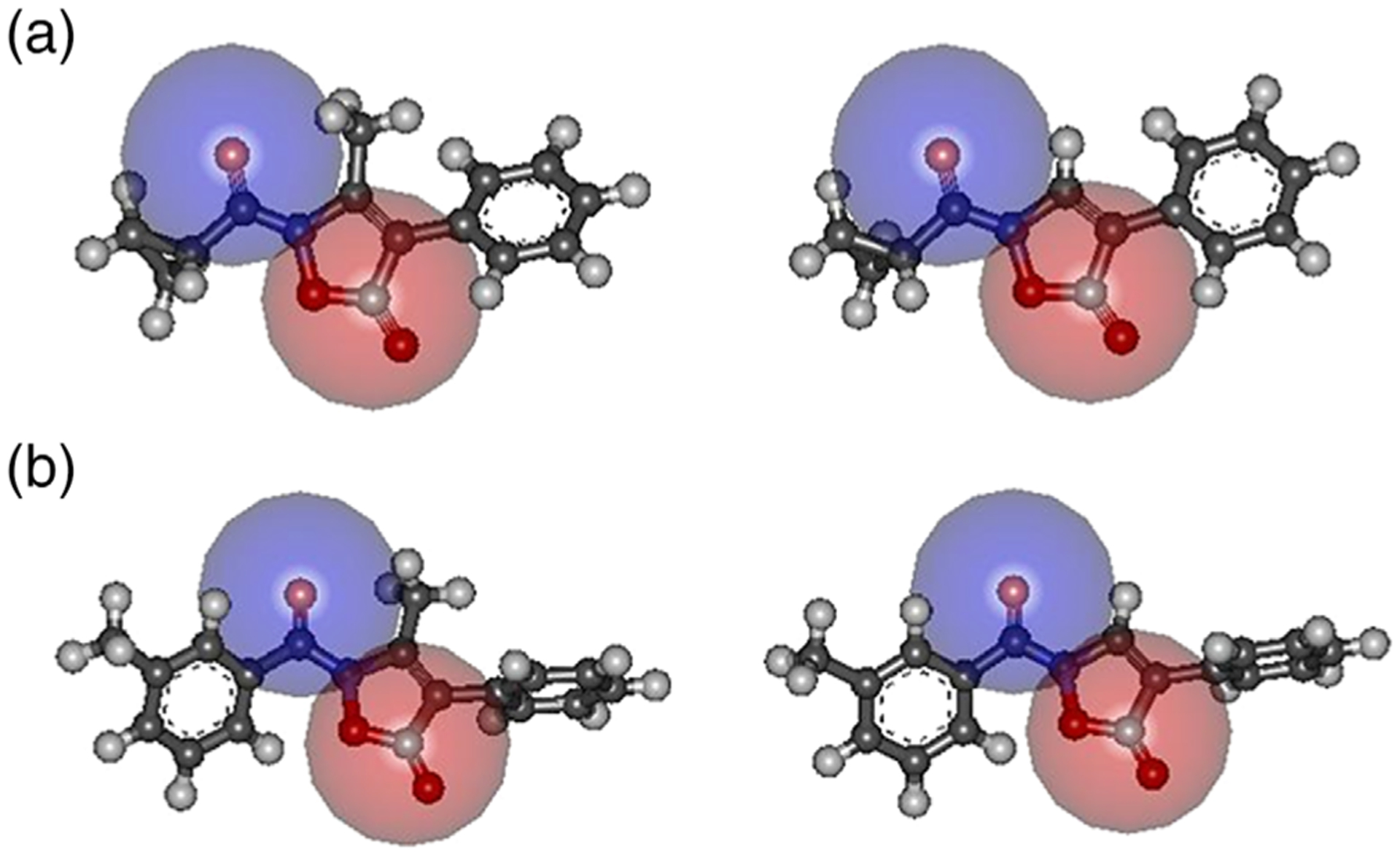
(a) Accessibility of the 5-C O and N–C=O groups estimated by the dimension of a sphere (r = 2.5 Å) centered on the carbon atom (red) and on the oxygen atom (blue). Left: (a) right: 3a; (b) accessibility of the 5-C O and N–C=O groups estimated by the dimension of a sphere (r = 2.5 Å) centered on the carbon atom (red) and on the oxygen atom (blue). Left: (b) right: 4a
According to our previous modeling (Giovannoni et al., 2018), the ligand–receptor binding modes were assessed by MD simulations (in vacuum, simulation time 5 ns, T = 300 K), with the atoms outside of the binding sphere constrained to fixed points in space in order to save computational time.
Regarding the ligand-enzyme interactions, no substantial differences were observed between the 3-methylisoxazole derivatives (A and B) and the corresponding nor-derivatives (3a and 4a) in the snapshot conformations sampled during the MD simulations; the same applied for the cyclopropyl/m-toluoyl families (A vs B and 3a vs 4a). In all cases, the ligand-enzyme interaction involved anchoring of the N–C=O group to Gly193 via H-bonding. Consequently, the 5-CO group is quite close to the HNE active site (distance <5 Å in a large fraction of the snapshot conformations), which is consistent with our previously studies on the closely related isoxazolone derivatives (Giovannoni et al., 2018).
4 |. CONCLUSION
With the aim to understand the importance of substituents at Position 3 of the potent isoxazolones previously published by us (Giovannoni et al., 2018; Vergelli et al., 2017), we synthesized a new series of 3-unsubstituted isoxazolones containing a phenyl group at Position 4 with various substituents. Elimination of the substituent at Position 3, as well as the introduction of various groups (CH3, Br, Cl, OCH3, CN, CF3) at different positions (para, meta, and ortho) of the 4-phenyl ring did not influence HNE inhibitory activity. In fact, all new isoxazolones were highly active, with IC50 values comparable to those of the three-substituted analogues. The most potent compounds were 3a, 3g, and 3h, which had IC50 values of 16, 11, and 18 nM, respectively. The crystal structure of compound A shows that the carbonyl oxygen atoms, which are both involved in the inhibitory activity, act as H-bond acceptors toward symmetry-related molecules. Molecular modeling on the closely related 3a, A, 4a, and B suggests that the exposure of the N–C=O oxygen atom does not appear affected both by the substituent at three-position (–CH3 vs H) and the cyclopropyl versus m-tolyl groups. Moreover the conformational behavior of the four compounds, as tested by MD simulations, is almost identical. Consistently, no significant differences were observed in the ligand–enzyme interactions: MD simulations have confirmed the pivotal role of the N–CO group as anchoring point to Gly193, as reported in our previously works (Giovannoni et al., 2018; Vergelli et al., 2017).
Thus, we conclude that the isoxazolone scaffold is a good scaffold for developing HNE inhibitors, as it tolerates several modifications when the following fundamental points are considered: (a) the N–CO group at Position 2 is important for interaction with HNE; (b) Position 3 can be unsubstituted or substituted with small alkyl groups (such as methyl, ethyl, isopropyl, etc.) that appears not to interact with the enzyme; (c) Position 4 tolerates bulky groups, indicating the presence of a broad lipophilic pocket; and (d) a CO at Position 5 is fundamental for HNE inhibitory activity, as it is the point of attack by Ser195 of the catalytic triad.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by National Institutes of Health IDeA Program COBRE Grant GM110732; USDA National Institute of Food and Agriculture Hatch project 1009546; and the Montana State University Agricultural Experiment Station.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- Accelrys Discovery. (2019). Accelrys Discovery Studio version 2.1. San Diego, CA: Accelrys Software Inc. [Google Scholar]
- Akizuki M, Fukutomi T, Takasugi M, Takahashi S, Sato T, Harao M, … Yamashita J (2007). Prognostic significance of immunoreactive neutrophil elastase in human breast cancer: Long-term follow-up results in 313 patients. Neoplasia, 9(3), 260–264. 10.1593/neo.06808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves LG, Dazinger G, Veiros LF, & Kirchner K (2010). Unusual anion effects in the iron-catalyzed formation of 3-hydroxyacrylates from aromatic aldehydes ethyl diazoacetate. European Journal of Inorganic Chemistry, 20, 3160–3166. 10.1002/ejic.201000240 [DOI] [Google Scholar]
- Bayer Corporation. (2002). Bayer Corp Prolastin, Company World Wide Web Site. [Google Scholar]
- Beccalli EM, La Rosa C, & Marchesini A (1984). Oxidation of 4-aryl-substituted isoxazolin-5-ones. A new synthesis of 2,5-diaryl-1,3-oxazin-6-ones. Journal of Organic Chemistry, 49, 4287–4290. 10.1021/jo00196a034 [DOI] [Google Scholar]
- Bronze-da-Rocha E, & Santos-Silva A (2018). Neutrophil elastase inhibitors and chronic kidney disease. International Journal of Biological Sciences, 14(10), 1343–1360. 10.7150/ijbs.26111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, & Karplus M (1983). CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of Computational Chemistry, 4, 187–217. 10.1002/jcc.540040211 [DOI] [Google Scholar]
- Cencioni R, Franchini PF, & Orienti M (1968). Dipole moments of 5-substitued isoxazoles. I. Dipole moments and tautomerism of isoxazolin-5-one. Tetrahedron, 24, 151–166. 10.1016/0040-4020(68)89017-9 [DOI] [Google Scholar]
- Crocetti L, Bartolucci G, Cilibrizzi A, Giovannoni MP, Guerrini G, Iacovone A, … Vergelli C (2017). Synthesis and analytical characterization of new thiazol-2-(3H)-ones as human neutrophil elastase (HNE) inhibitors. Chemistry Central Journal, 11, 127. 10.1186/s13065-017-0358-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocetti L, Giovannoni MP, Schepetkin IA, Quinn MT, Khlebnikov AI, Cilibrizzi A, … Vergelli C (2011). Design, synthesis and evaluation of N-benzoylindazole derivatives and analogues as inhibitors of human neutrophil elastase. Bioorganic & Medicinal Chemistry, 19, 4460–4472. 10.1016/j.bmc.2011.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocetti L, Giovannoni MP, Schepetkin IA, Quinn MT, Khlebnikov AI, Cantini N, … Vergelli C (2018). 1H-pyrrolo[2,3-b] pyridine: A new scaffold for human neutrophil elastase (HNE) inhibitor. Bioorganic & Medicinal Chemistry, 26, 5583–5595. 10.1016/j.bmc.2018.09.034 [DOI] [PubMed] [Google Scholar]
- Crocetti L, Schepetkin IA, Ciciani G, Giovannoni MP, Guerrini G, Iacovone A, … Vergelli C (2016). Synthesis and pharmacological evaluation of indole derivatives as deaza analogues of potent human neutrophil elastase inhibitors. Drug Development Research, 77(6), 285–299. 10.1002/ddr.21323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocetti L, Schepetkin IA, Cilibrizzi A, Graziano A, Vergelli C, Giomi D, … Giovannoni MP (2013). Optimization of N-benzoylindazole derivatives as inhibitors of human neutrophil elastase. Journal of Medicinal Chemistry, 56, 6259–6272. 10.1021/jm400742j [DOI] [PMC free article] [PubMed] [Google Scholar]
- CrysAlis RED. (n.d.). Oxford diffraction (version 171.37.35). Abingdon: (2015) Oxford Diffraction Ltd. [Google Scholar]
- Dassault Systèmes BIOVIA. (2015). Discovery Studio Visualizer, Release 4.5. San Diego, CA: Dassault Systèmes. [Google Scholar]
- Desiraju GR, & Steiner T (1999). The weak hydrogen bond, IUcr Monographs on Crystallography. Oxford Science Publications, Oxford University press: Oxford and New York. [Google Scholar]
- Farrugia LJ (1997). ORTEP-3 for windows-a version of ORTEP-III with a graphical user interface (GUI). Journal of Applied Crystallography, 30, 565. 10.1107/S0021889897003117 [DOI] [Google Scholar]
- Farrugia LJ (2012). WinGX and ORTEP for windows: An update. Journal of Applied Crystallography, 45, 849–854. 10.1107/S0021889812029111 [DOI] [Google Scholar]
- Franchini OF (1968). Dipole moments and tautomerism of isoxazol-5-ones. Corsie Sem Chimie, 14, 23–25. [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, …, Fox DJ (2016). Wallingford, CT: Gaussian, Inc.
- Frolund B, Jensen LS, Guandalini L, Canillo C, Vestergaard HT, Kristiansen U, … Liljefors T (2005). Potent 4-aryl- or 4-arylalkyl-substituted 3-isoxazolol GABAA antagonists: Synthesis, pharmacology, and molecular modeling. Journal of Medicinal Chemistry, 48, 427–439. 10.1021/jm049256w [DOI] [PubMed] [Google Scholar]
- Giovannoni MP, Cantini N, Crocetti L, Guerrini G, Iacovone A, Schepetkin IA, … Quinn MT (2019). Further modifications of 1H-pyrrolo[2,3-b]pyridine derivatives as inhibitors of human neutrophil elastase. Drug Development Research, 80(5), 617–628. 10.1002/ddr.21539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni MP, Schepetkin IA, Crocetti L, Ciciani G, Cilibrizzi A, Guerrini G, … Vergelli C (2016). Cinnoline derivatives as human neutrophil elastase inhibitors. Journal of Enzyme Inhibition and Medicinal Chemistry, 31(4), 628–639. 10.3109/14756366.2015.1057718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni MP, Schepetkin IA, Quinn MT, Cantini N, Crocetti L, Guerrini G, … Vergelli C (2018). Synthesis, biological evaluation, and molecular modelling studies of potent human neutrophil elastase (HNE) inhibitors. Journal of Enzyme Inhibition and Medicinal Chemistry, 33(1), 1108–1124. 10.1080/14756366.2018.1480615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimme S (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. Journal of Computational Chemistry, 27(15), 1787–1799. 10.1002/jcc.20495 [DOI] [PubMed] [Google Scholar]
- Groom CR, Bruno IJ, Lightfoot MP, & Ward SC (2016). The Cambridge structural database. Acta Crystallographica Section B, B72, 171–179. 10.1107/S2052520616003954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehre WJ, Ditchfield R, & Pople JA (1972). Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecule. The Journal of Chemical Physics, 56, 2257–2261. 10.1063/1.1677527 [DOI] [Google Scholar]
- Hilbert N, Schiller J, Arnhold J, & Arnold K (2002). Cartilage degradation by stimulated human neutrophils: Elastase is mainly responsible for cartilage damage. Bioorganic Chemistry, 30, 119–132. 10.1006/bioo.2002.1242 [DOI] [PubMed] [Google Scholar]
- Iwata K, Doi A, Ohji G, Oka H, Oba Y, Takimoto K, … Shimada T (2010). Effect of neutrophil elastase inhibitors (Sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress (ARDS): A systematic review and meta-analysis. International Medicine, 49, 2423–2432. 10.2169/internalmedicine.49.4010 [DOI] [PubMed] [Google Scholar]
- Klyne W, & Prelog V (1960). Description of steric relationships across single bonds. Experentia, 16, 521–523. 10.1007/BF02158433 [DOI] [Google Scholar]
- Korkmaz B, Horwitz MS, Jenne DE, & Gauthier F (2010). Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacological Reviews, 62, 726–759. 10.1124/pr.110.002733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman I, Garcia-Hernandez ML, Rangel-Moreno J, Chiriboga L, Pan C, Nastiuk KL, … Hammes SR (2017). Infiltrating myeloid cells exert protumorigenic actions via neutrophil elastase. Molecular Cancer Research, 15(9), 1138–1152. 10.1158/1541-7786.MCR-17-0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman I, & Hammes SR (2017). Neutrophil elastase in the tumor microenvironment. Steroids, 133, 96–101. 10.1016/j.steroids.2017.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Chen C, Wu F, & Tang J (2016). Study on the synthesis and biological activities of α-substituted arylacetates derivatives. Bioorganic & Medicinal Chemistry Letters, 26, 1715–1719. 10.1016/j.bmcl.2016.02.055 [DOI] [PubMed] [Google Scholar]
- Lucas SD, Costa E, Guedes RC, & Moreira R (2013). Targeting COPD: Advances on low-molecular-weight inhibitors of human neutrophil elastase. Medicinal Research Reviews, 33, E73–E101. 10.1002/med.20247 [DOI] [PubMed] [Google Scholar]
- Marto J, Ruivo E, Lucas SD, Goncalves LM, Simoes S, Gouveia LF, … Almeida AJ (2018). Starch nanocapsules containing a novel neutrophil elastase inhibitor with improved pharmaceutical performance. European Journal of Pharmaceutics and Biopharmaceutics, 127, 1–11. 10.1016/j.ejpb.2018.01.011 [DOI] [PubMed] [Google Scholar]
- Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, … Engelmann B (2010). Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nature Medicine, 16, 887–896. 10.1038/nm.2184 [DOI] [PubMed] [Google Scholar]
- Navia MA, McKeever BM, Springer JP, Tsau-Yen L, Williams HR, Fluder EM, … Hoogsteen K (1989). Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84- angstrom resolution. Proceedings of the National Academy of Sciences of the United States of America, 86, 7–11. 10.1073/pnas.86.1.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey KC, De S, & Mishra PK (2017). Role of proteases in chronic obstructive pulmonary disease. Frontiers in Pharmacology, 8, 512. 10.3389/fphar.2017.00512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Ayala PY, Schlegel HB, & Frisch MJ (1996). Using redundant internal coordinates to optimize equilibrium geometries and transition states. Journal of Computational Chemistry, 17, 49–56. [Google Scholar]
- Pham CT (2006). Neutrophil serine proteases: Specific regulators of inflammation. Nature Reviews Immunology, 6, 541–550. 10.1038/nri1841 [DOI] [PubMed] [Google Scholar]
- Rajput S, Gardner RC, Failes TW, Arndt GM, Black D, & Kumar N (2014). Synthesis and anticancer evaluation of 3-substituted quinolin-4-ones and 2,3-dihydroquinolin-4-ones. Bioorganic & Medicinal Chemistry, 22, 105–115. 10.1016/j.bmc.2013.11.047 [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (1990). Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr. Sect. A, A46, 467–473. 10.1107/S0108767390000277 [DOI] [Google Scholar]
- Sheldrick GM (2008). A short history of SHELX. Acta Crystallogr. Sect. A, A64, 112–122. 10.1107/S0108767307043930 [DOI] [PubMed] [Google Scholar]
- Sinha S, Watorek W, Karr S, Giles J, Bode W, & Travis J (1987). Primary structure of human neutrophil elastase. Proceedings of the National Academy of Sciences of the United States of America, 84, 2228–2232. 10.1073/pnas.84.8.2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder LB, Meng Z, Mate R, D’Andrea SV, Marinier A, Quesnelle CA, … Bronsona JJ (2004). Discovery of isoxazolinone antibacterial agents. Nitrogen as a replacement for the stereogenic center found in oxazolidinone antibacterials. Bioorganic & Medicinal Chemistry Letters, 14, 4735–4739. 10.1016/j.bmcl.2004.06.076 [DOI] [PubMed] [Google Scholar]
- Stapels DA, Gijsbrecht BV, & Rooijakkers SH (2015). Neutrophil serine proteases in antibacterial defense. Current Opinion in Microbiology, 23, 42–48. 10.1016/j.mib.2014.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MJ, McKinnon JJ, Wolff SK, Grimwood DJ, Spackman PR, Jayatilaka D, & Spackman MA (2017). CrystalExplorer17. University of Western Australia: Crawley, Australia, 2017. http://hirshfeldsurface.net. [Google Scholar]
- Twigg MS, Brockbank S, Lowry P, FitzGerald SP, Taggart C, & Weldon S (2015). The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediators of Inflammation, 2015, 293053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaguliene N, Zemaitis M, Lavinskiene S, Miliauskas S, & Sakalauskas R (2013). Local and systemic neutrophil inflammation in patients with lung cancer and chronic obstructive pulmonary disease. BMC Immunology, 14, 36. 10.1186/1471-2172-14-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergelli C, Schepetkin IA, Crocetti L, Iacovone A, Giovannoni MP, Guerrini G, … Quinn MT (2017). Isoxazol-5(2H)-one: A new scaffold for potent human neutrophil elastase (HNE) inhibitors. Journal of Enzyme Inhibition and Medicinal Chemistry, 32, 821–831. 10.1080/14756366.2017.1326915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Nussbaum F, & Li VMJ (2015). Neutrophil elastase inhibitors for the treatment of (cardio) pulmonary disease: Into clinical testing with pre-adaptive pharmacophores. Bioorganic & Medicinal Chemistry Letters, 25, 4370–4381. 10.1016/j.bmcl.2015.08.049 [DOI] [PubMed] [Google Scholar]
- von Nussbaum F, Li VMJ, Meibom D, Anlauf S, Bechem M, Delbeck M, … Schamberger J (2016). Potent and selective human neutrophil elastase inhibitors with novel equatorial ring topology: in vivo efficacy of the polar pyrimidopyridazine BAY-8040 in a pulmonary arterial hypertension rat model. ChemMedChem, 11(2), 199–206. 10.1002/cmdc.201500269 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.