

Abstract
Concepts regarding etiology and pathophysiology of sarcoidosis have changed remarkably within the past five years. Sarcoidosis is now viewed as complex multi-causation disease related to a diverse collection of external environmental or infectious signals. It is generally accepted that the cause of sarcoidosis is unknown. Moreover, concepts of the inflammatory pathway have been modified by the realization that intrinsic genetic factors and innate immunity may modify adaptive immune responses to external triggers. With those potential regulatory pathways in mind, we will attempt to discuss the current understanding of the inflammatory response in sarcoidosis with emphasis on development of pulmonary granulomatous pathology. In that context, we will emphasize that both macrophages and T lymphocytes play key roles, with sometimes overlapping cytokine production (i.e. TNFα and IFN-γ) but also with unique mediators that influence the pathologic picture. Numerous studies have shown that in a sizable number of sarcoidosis patients, granulomas spontaneously resolve, usually within three years. Other sarcoidosis patients, however, may develop a chronic granulomatous disease which may subsequently lead to fibrosis. This chapter will outline our current understanding of inflammatory pathways in sarcoidosis which initiate and mediate granulomatous changes or onset of pulmonary fibrosis.
Keywords: alveolar macrophages, lymphocytes, innate immunity, granuloma mediators, Th17, PPARγ
INTRODUCTION.
Concepts regarding etiology and pathophysiology of sarcoidosis have changed remarkably within the past five years (1). Sarcoidosis is now viewed as complex multi-causation disease related to a diverse collection of external environmental or infectious signals (2, 3). It is generally accepted that the cause of sarcoidosis is unknown. Moreover, concepts of the inflammatory pathway have been modified by the realization that intrinsic genetic factors and innate immunity may modify adaptive immune responses to external triggers. With those potential regulatory pathways in mind, we will attempt to discuss the current understanding of the inflammatory response in sarcoidosis with emphasis on development of pulmonary granulomatous pathology. In that context, we will emphasize that both macrophages and T lymphocytes play key roles, with sometimes overlapping cytokine production (i.e. TNFα and IFN-γ) but also with unique mediators that influence the pathologic picture. Numerous studies have shown that in a sizable number of sarcoidosis patients, granulomas spontaneously resolve, usually within three years (4). Other sarcoidosis patients, however, may develop a chronic granulomatous disease which may subsequently lead to fibrosis. This chapter will outline our current understanding of inflammatory pathways in sarcoidosis which initiate and mediate granulomatous changes or onset of pulmonary fibrosis.
I. INDUCTION OF INFLAMMATION.
1.1. Environmental Factors.
Inorganic Materials.
Pulmonary Inflammation associated with sarcoidosis has been linked to multiple environmental factors including inorganic and organic substances. Some of the putative organic substances are antigens of infectious agents although the disease itself is not infectious. Inorganic factors may include particulate matter such as dusts and silicates, as were observed in the “sarcoid like” granulomatous pulmonary disease found in responders to the World Trade Center disaster (5). Sarcoidosis cases have also been associated with occupational exposures to various inorganic agents such as encountered in fire-fighting, construction, and machining [reviewed in (3)}.
Infectious Agents.
The possibility of an infectious origin for sarcoidosis has been suggested by its similarity to tuberculosis. Infectious organisms associated with sarcoidosis include Mycobacteria and Cutibacterium acnes (formerly named Propionibacterium acnes). DNA residues from both agents have been reported in sarcoidosis granulomas but no live bacteria have been found [reviewed in (2)]. Moreover, in both cases, related proteins (mycobacterial catalase-peroxidase and C. acnes catalase) were found to elicit elevated T lymphocyte responses in sarcoidosis patients, suggesting that sarcoidosis might represent an immune-mediated disease to bacterial components (2). The concept of sarcoidosis-associated immune reactivity stems from early studies in which intradermal injection of a pasteurized suspension of sarcoid lymphoid tissue was utilized by Kveim, a Norwegian pathologist, as a diagnostic test for sarcoidosis (reviewed in (6). This “Kveim test” found that after 4–6 weeks, granulomatous responses were elicited in skin from sarcoidosis patients but not from controls. Later proteonomic analyses of Kveim “antigen” reported the presence of the mycobacterial catalase-peroxidase protein (7) has been shown to trigger elevated Interferon gamma (IFN-γ) responses in sarcoidosis patients (8). Currently, association of prior mycobacterial infection is present in many sarcoidosis cases but there is no direct evidence for an infection-initiated etiology [reviewed in (9)].
1.2. Intrinsic Factors
Genetic Profiles and Autoimmunity.
Autoimmune mechanisms have been considered in sarcoidosis because of its complex diversity of symptoms. Proteonomic studies have identified numerous sarcoidosis-related proteins in Kveim preparations, and have shown that one of them (vimentin) can elicit elevated IFN-γ responses in peripheral blood mononuclear cells (PBMC) of sarcoidosis patients but not healthy controls (6). Vimentin is an intermediate filament protein secreted by alveolar macrophages and is important to cellular interactions (reviewed in (10)). Antibodies specific to vimentin have been detected in bronchial lavage fluid (BALF) from sarcoidosis patients (11). Immune reactivity to vimentin is not specific to sarcoidosis, however, and antibodies to vimentin have been found in autoimmune conditions such as systemic lupus erythematosis (SLE) and rheumatoid arthritis (RA) (10). In addition, low levels have been detected in BALF from healthy individuals (11). In sarcoidosis, additional studies are needed to define the role of vimentin as it is unclear whether it is a cause or effect of inflammation.
Analysis of gene markers in sarcoidosis have focused on CD4+ T lymphocytes which accumulate in the lungs prior to granuloma formation [reviewed in (12)]. Characterization of these cells indicates the presence of adaptive immune responses in sarcoidosis, with antigen-driven activation involving antigen-presenting cells such as macrophages. Several human leukocyte antigen (HLA) gene patterns have emerged from sarcoidosis analyses, with HLA-DRB1*01 and HLA-DRB1*04 appearing to be protective in Caucasian populations while risk factors for sarcoidosis are represented by HLA-DRB1*03, *11, *12, *14, and *15 (12). Interestingly, HLA-DRB1*03 is also present in patients with Lofgren syndrome, a self-limiting form of sarcoidosis characterized by acute onset, fever, and clinical symptomology such as bilateral hilar lymphadenopathy (11) [reviewed in (2)]. Clinical disease course in Lofgren syndrome correlates with DRB1*03 presence: disease resolution occurs in 95% of DRB1*03-positives but in only 49% of DRB1*03 negatives (12). With respect to vimentin antibodies in BALF, higher titers have been found in HLA-DRB1*03-positive sarcoidosis patients compared to HLA-DRB1*03-negatives (11). The significance of these findings requires further study.
Genetic studies in sarcoidosis have also analyzed candidate genes associated with sarcoidosis such as the immune-related genes, tumor necrosis factor (TNF), and the Interleukin 23 receptor (IL23R) (12). TNF meta-analysis studies revealed a significant association of the −308 polymorphism with sarcoidosis compared to controls, while other studies also found a −307 haplotype associated with good prognosis and a −857T containing one associated with persistent disease (reviewed in (12)). Studies of IL-23R variants have noted an association with Crohn disease (CD) as well as sarcoidosis. The IL23R gene codes for a subunit of the IL23 receptor which is important to T-helper 17 (Th17)-mediated processes and several alleles have been found with significant associations to sarcoidosis [reviewed in (12)]. More recent approaches to the genetics of sarcoidosis have utilized genome-wide association studies (GWAS) [reviewed in (13)]. The major findings from these studies have confirmed the role of TH1 adaptive immune responses in sarcoidosis with emphasis on IFN-γ functions and signaling pathways. The authors concluded that future GWAS studies in sarcoidosis need to utilize more cutting-edge approaches to allow analyses of single cell subpopulations as well as sequential monitoring of patients for changes in disease status (13).
Innate Immunity: Overview.
This section will consider several avenues of innate immune reactivity that have been reported in sarcoidosis. Innate immune responses are considered to be direct contributors to granulomatous inflammation in sarcoidosis (reviewed in (14)). Relevant innate immune factors to be discussed include the following: (a) Pattern Recognition Receptors (PRRs); (b) the NOD-like receptor NLRP3 inflammasome network (15); (c) the mTOR signaling pathway; (d) serum amyloid A (SAA), an acute phase protein, and (e) chitotriosidase, an enzyme involved in pathogen defense (16) (Table 1).
TABLE 1.
MEDIATORS OF INNATE IMMUNITY IN SARCOIDOSIS LUNG
| INNATE IMMUNE MEDIATORS | ROLE IN PATHOPHYSIOLOGY | PULMONARY LOCATION |
|---|---|---|
| Pattern Recognition Receptors (PRRs) (17) | Host defense against pathogens; mediate inflammatory responses. | Expressed on alveolar macrophages, epithelial cells, endothelial cells, others |
| Pathogen-associated molecular patterns (PAMPs) (17) | Conserved microbial molecules recognized by PRRs; activate release of inflammatory cytokines. | Responders to PAMPs include alveolar macrophages, epithelial cells, endothelial cells, others. |
| Danger-associated molecular patterns (DAMPs) (17) | Endogenous intracellular molecules from injured cells; mediate inflammatory responses. | Responders to DAMPs include alveolar macrophages, epithelial cells, endothelial cells, others. |
| Toll-like Receptors (TLRs) (14, 17) | Bind both microbial and endogenous ligands; mediate inflammatory responses. | Expressed on Lymphocytes, alveolar macrophages, epithelial cells, dendritic cells. |
| NLRP3 inflammasome network (15) (18) (19) (20) | Host defense against pathogens; initiates pyroptosis and release of proinflammatory cytokines. | Granuloma tissues. |
| MTOR signalling pathway (21) (22) | Regulates rapid responses of innate immune cells; involved in initiation and progression of granulomas. | Granuloma tissues. |
| Serum Amyloid A (SAA) (23) (24, 25) | Acute phase reactant; constituent of granulomas. | Macrophages and giant cells within granuloma tissues. |
| Chitotriosidase (16) (26) | Host defense against chitin-containing fungi and protozoa. | Produced by activated macrophages; detectable in sarcoidosis sera. |
Innate Immunity: Pattern Recognition Receptors (PRRs).
Within the lung, PRRs represent an innate defense against infections within the alveolus and are expressed on alveolar macrophages, epithelial cells, endothelial cells, and other cell types [reviewed in (17)]. PRRs sense microbial invaders by recognition of conserved microbial molecules classically defined as “pathogen-associated molecular patterns” (PAMPs). PRR encounter activates cellular production of inflammatory cytokines and chemokines which in turn recruit and activate macrophages and neutrophils. In the case of non-infectious injury elicited by large, inert particles such as silica crystals, some PRRs may also become active. In addition, release of endogenous intracellular molecules from injured cells, defined as “danger-associated molecular patterns” (DAMPs) also mediate further cellular inflammatory responses to non-infectious injuries (17).
Among the PRRs are Toll-like receptors (TLRs) and the cytosolic NOD-like receptors (NLRs) which include NLRP3. These molecules are expressed in alveolar macrophages, lung epithelial cells and dendritic cells as well as on lymphocytes (reviewed in (17)). There are ten members of the human TLR family located either at the cell surface (TLRs, 2, 4–6, 10) or in lysosomal/endosomal membranes (TLR3, 7–9). Ligands include both microbial and endogenous factors which can induce production of antimicrobial peptides and proinflammatory mediators such as TNFα, IL-8, and proIL-1β. Further processing of proIL-1β is required by caspase activation via NLRP3 (discussed below). Mycobacterial ligands may recognize TLR2 and TLR9 and polymorphisms in both have been associated with susceptibility to mycobacterial infection and granulomatous pathobiology [reviewed in (14)]. In sarcoidosis, enhanced TNFα responses to TLR2 stimulation have been found in cells from both blood and lungs (14). A positive feedback loop has been suggested by the capacities of TNFα and IFN-γ to enhance TLR2 expression on pulmonary epithelial cells (14).
Innate Immunity: The NLR Inflammasome Network.
NLRs comprise another innate immune receptor family of 22 members (in humans) but few of these have been functionally characterized. Five of these NLRs have been shown to assemble “inflammasomes” which are high-molecular-weight protein complexes present in the cytosol of stimulated immune cells (reviewed in (18)). These complexes have a critical role in host defense against pathogens and can be triggered by diverse stimuli. Basic functions of the NLRP3 inflammasome (which has been more intensely studied than other NLRs), are to initiate an inflammatory form of cell death (pyroptosis) and to release proinflammatory cytokines IL-1β and IL-18 [reviewed in (19)]. In macrophages, NLRP3 activity begins with a priming signal which can be supplied by TLR4 agonists that induce expression of NLRP3 and proIL-1β. Next, an activation signal may be triggered by the above described PAMPs and DAMPs, which promote inflammasome assembly, caspase-1-mediated IL-1β and IL-18 secretion, and pyroptosis. Interestingly, the priming step alone has been found to be sufficient for human monocytes to mediate caspase-1 activation and IL-1β release (19). In sarcoidosis, the NLRP3 inflammasome network has been shown to be constitutively activated and involved in granuloma formation (20). Upregulation of NLRP3 components including cleaved caspase-1 and IL-1β has been found in sarcoid pulmonary granulomas, and NLRP3 mRNA was elevated in cell-sorted sarcoid alveolar macrophages compared to controls (20). Of interest were findings in a mouse granuloma model which showed that NLRP3 KO mice exhibited decreased granuloma formation compared to wildtype. Additional murine studies also showed that microRNA miR-223 acted as a down-regulator of NLRP3 and in miR-223 KO mice, granulomas were increased in size (20). In sarcoidosis alveolar macrophages, miR-223 levels were decreased in contrast to the elevation of NLRP3. These findings illustrate the importance of NLRP3 inflammasomes as an active component of innate immunity in sarcoidosis pathogenesis.
Innate Immunity: the mTOR pathway.
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase regulatory-associated protein which acts as a central regulator of cellular metabolism [reviewed in (21)]. MTOR forms a part of two complexes: mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2) which primarily shape rapid responses of innate immune cells represented by monocytes, macrophages, dendritic cells, neutrophils, mast cells, and innate-like NK cells. Rapamycin inhibits these mTOR complexes and has been used as an immunosuppressive drug to prevent kidney allograft rejection (21). MTOR pathways become activated by various extracellular signals such as growth factors, TLR ligands, and cytokines. Activation controls a wide range of cellular functions including translation, protein synthesis, cell growth, metabolism, and anabolic processes. Studies in mice have shown that mTORC1 can also become activated by deletion of the gene encoding tuberous sclerosis 2 (Tsc2) resulting in induced hypertrophy and proliferation culminating in excessive pulmonary granuloma formation in vivo (22). Inhibition of mTORC1 induced apoptosis and completely resolved granulomas in Tsc2-deficient mice (22). In studies of sarcoidosis patients, clinical disease progression was found to correlate with mTORC1 activation, macrophage proliferation, and glycolysis (22). Findings support a role for the innate immune mTOR pathway in initiation and progression of granuloma pathophysiology.
Innate Immunity: Serum Amyloid A.
Serum amyloid A (SAA) is a highly conserved acute phase reactant primarily synthesized by the liver. Circulating SAA levels can increase by as much as 1000-fold during inflammation [reviewed in (23)]. In sarcoidosis, staining for SAA revealed high levels of expression in granulomatous tissues compared to tissues from patients with other granulomatous disorders (24, 25). SAA is not specific to sarcoidosis, however, and statistical analyses indicate SAA staining is not sensitive enough for use in diagnostic testing (25). SAA expression in sarcoid granulomas localizes to macrophages and giant cells and was found to correlate with numbers of CD3+ T cells within granulomas, suggesting an SAA linkage to local Th1 immune responses (24). Findings from a murine granuloma model indicated that SAA regulated granuloma size, in part via TLR2 signaling, with production of IFN-γ, TNF, and IL-10 accompanying increased granuloma size (24). Anti-TLR2 antibodies attenuated these effects (24). Authors suggest that data points to SAA as a constituent and innate regulator of chronic granulomatous inflammation in sarcoidosis (24).
Innate Immunity: Chitotriosidase.
Chitotriosidase is the major active chitinase enzyme in humans and is produced mainly by activated macrophages [reviewed in (26)]. The enzyme is an innate immune defense against chitin-containing pathogens such as fungi and protozoa. In several human diseases, including sarcoidosis, chitotriosidase is elevated in serum and is a marker of disease severity (26). A recent study of 694 sarcoidosis patients and 101 healthy controls confirmed the presence of significantly elevated values for serum chitotriosidase in sarcoidosis (16). Values were also found to be increased in patients with extrapulmonary involvement, and in patients requiring increased steroid dosage (16). The mechanisms by which chitotriosidase is implicated in sarcoidosis granulomatous pathophysiology, however, remain to be determined.
2. Inflammatory Granuloma Formation.
Initiation of Granuloma Structure.
Granuloma structure is the product of coordinated responses from both innate and adaptive immunity to poorly degradable antigens [reviewed in (27)]. Foreign antigenic materials deposited in the lungs are acquired by macrophages which release many pro-inflammatory cytokines (TNFα, IL-1β, IL-6) that persistently stimulate immune response pathways. Dendritic cells pick up the degraded antigens and migrate to regional lymph nodes where antigens are presented to naïve CD4+ T cells. Clonal T cell differentiation and proliferation occur in the nodes and activated CD4+ T cells, via chemokines, migrate back to inflammatory sites within the lung. Both induction and maintenance of granuloma formation in sarcoidosis require CD4+ cell activation [reviewed in (28)]. Once in the lungs CD4+ T cells release cytokines (IFN-γ, TNFα, IL-12 and IL-18) which stimulate macrophages to organize into giant cells and granulomas. Evidence from sarcoidosis studies suggests that mediastinal lymph nodes constitute the initial site of granuloma formation prior to pulmonary granuloma development [Reviewed by (29)]. Unlike granulomas of infectious origin, sarcoid granulomas rarely have significant necrotic areas. A mature sarcoid granuloma is composed of both epithelioid and multinucleated giant cells in a tight configuration surrounded mainly by CD4+ T helper cells. Historically, sarcoidosis granulomas have been considered to be driven by Th1 lymphocytes but recent data suggest Th-17 cells have major roles in granuloma formation and persistence [reviewed in (2, 30)].
Granuloma Mediators.
Macrophages are one of the major producers of the enzyme, Matrix Metalloproteinase12 (MMP12) (31). MMP12 is one of a family of proteolytic enzymes that degrades extracellular matrix elastin and enables infiltration of immune cells responsible for inflammation and granuloma formation. In sarcoidosis, MMP12 has been found to be one of the most highly expressed (> 25-fold) enzymes in lung tissues and MMP12 protein is elevated in BAL fluids (32). Strikingly, MMP12 expression was highest near areas of active granulomatous inflammation, and MMP12 levels in BALF correlated with disease severity. Inhibition studies with a global MMP inhibitor (Marimastat) have reported reduced granuloma formation in lung tissue models of mycobacterial infection (33). More recently, a comparative study of mmp12 KO and wild-type mice was carried out in a carbon nanotube-induced granuloma model (34). Results indicated that granulomas formed in wildtype mice were detected as early as 10 days post instillation. These early granulomas were poorly formed, but by 60 days post-instillation, granulomas appeared to be well defined. Surprisingly, no histological differences in granuloma formation were noted acutely in mmp12 KO mice compared to wildtype at day 10, but by 60 days the granulomas were smaller and less well-formed. These results suggest that MMP12 is required to maintain chronic granuloma pathology (34). Currently, pursuit of novel MMP inhibitors continues with efforts to define MMP roles in specific disease-related immunological responses and inflammation (35).
Serum Markers of Inflammation in Sarcoidosis: Chemokines.
Inflammatory chemokines CXCL9, CXCL10, and CXCL11, are produced in the sarcoid granulomatous lung by several cell types, including macrophages, endothelial cells, and fibroblasts [reviewed in (36)]. The primary induction signal for these chemokines is IFN-γ produced by activated CD4+ T helper lymphocytes [reviewed in (37)]. The major effect of these chemokines is to promote an influx of T cells into inflammatory tissues by binding and signaling through the CXCR3 receptor present on activated T cells (37). Analyses of IFN-γ inducible chemokines in sarcoidosis patients have shown that serum levels are higher than those of healthy controls and correlate with each other (36, 38). Additional analyses in sarcoidosis have shown that CXCL9 levels correlate best with systemic organ involvement, and both CXCL10 and CXCL11 levels correlate with pulmonary function decline.
Serum Markers of Inflammation in Sarcoidosis: sIL-2R.
Quantitation of serum levels of the soluble Interleukin 2 receptor (sIL-2R) represents a standardized clinical assay for assessment of T lymphocyte activation in various immune disorders (39). T cells express the receptor for IL-2 and during activation, begin to secrete the receptor in soluble form [reviewed in (40)]. As in other immune-related disorders, elevated blood sIL-2R levels correlate with disease activity in sarcoidosis patients and have been shown to be predictive of spontaneous remission (41). In a recent study of undiagnosed patients whose sIL-2R results were available before a definitive diagnosis had been made, sensitivity and specificity of serum sIL-2R for detection of sarcoidosis were 88% and 85%, respectively. Additional analyses revealed that the sIL-2R assay was superior to serum angiotensin converting enzyme (ACE) measurement used previously in diagnosis of sarcoidosis cases (62% sensitivity and 76% specificity) (40).
Serum Markers of Inflammation in Sarcoidosis: Chitotriosidase.
As reviewed previously above, chitotriosidase, a component of innate immunity, has been shown to be elevated in sera from sarcoidosis patients with active disease (26). In a study population of some 232 sarcoidosis patients, sensitivity and specificity of serum chitotriosidase for detection of sarcoidosis compared to healthy controls was calculated to be 88.6% and 92.8%, respectively (42).
Granuloma Components: Macrophages.
Macrophages represent the basic building blocks of granulomas. Early events in granuloma construction involve aggregation of macrophages for transformation into epitheloid cells (reviewed in (27)). The influence of CD4-driven inflammatory cytokines (IFN-γ, TNFα, IL-12, IL-18) further promotes cell-cell fusion between macrophages and dendritic cells or monocytes, creating multinucleated giant cells which form a large portion of mature granuloma core structure (43). Outer portions of granulomas contain large numbers of T lymphocytes and a few B lymphocytes.
Alveolar Macrophages: Lipid Homeostasis and Inflammation.
Alveolar macrophages exhibit unique lipid metabolic properties compared to other tissue macrophages because of the complex lung microenvironment (43, 44). The lung is constantly bombarded with foreign material and further, is coated with a lipid-rich surfactant that serves to prevent pulmonary collapse (45). The lung is the most active lipid-secreting organ because of surfactant production (46). Alveolar macrophages represent an essential component of surfactant clearance and lipid homeostasis within the lung (47). Surfactant contains phospholipids and neutral lipids, the bulk of which is cholesterol (48). Alveolar macrophage ATP-binding cassette (ABC) lipid transporters, ABCA1 and ABCG1 participate in clearance of cholesterol (49). Deficiencies of lipid transporters result in increased intracellular cholesterol together with over-production of pro-inflammatory cytokines and chemokines [reviewed in (50)]. Overloading macrophages with cholesterol has been shown to activate inflammasome pathways, and cytokine production [reviewed (51)].
The transcription factor, PPARγ directly or indirectly regulates many genes involved in cholesterol metabolism and transport, including the ATP lipid transporters [reviewed in (43)]. PPARγ also antagonizes transcription of many pro-inflammatory genes via mechanisms that may include direct association with co-activators, or transrepression of transcription factors (43). Healthy alveolar macrophages, unlike other tissue macrophages, display high levels of PPARγ [reviewed in (44)]. Alveolar macrophages from macrophage-specific PPARγ KO mice exhibit impaired surfactant lipid metabolism characterized by accumulation of intracellular neutral lipids, dysregulated lipid transporters, and elevated inflammatory cytokines (52, 53). In addition, PPARγ deficiency also resulted in dysregulation of alveolar macrophage Liver X Receptor pathways which are critical to the promotion of cellular cholesterol export (54). Dysregulation of PPARγ, ABCA1/ABCG1 lipid transporters, and pro-inflammatory cytokines are significant findings in alveolar macrophages from sarcoid patients (55).
In addition to cholesterol regulation in the lung, lipid transporters play key roles in host innate immunity; deficiencies lead to impaired immune cell homeostasis, further aggravating pulmonary inflammation (51).
Macrophage Profiles in the Lung
Studies of macrophage activation have led to a concept of classic and alternative activation termed M1 and M2, analogous to that of Th1 and Th2 cells (reviewed in (56)). More recently, however, it has become clear that whereas M1 and M2 phenotypes were derived from in vitro studies, tissue macrophages in vivo may display mixed responses (57).
Macrophage plasticity allows participation in both promotion and resolution of inflammation [reviewed in (58)]. Generally, sarcoidosis data illustrate a model of persistent inflammation with macrophages exhibiting an M1 profile induced by pro-inflammatory cytokines such as IFNγ (28). Alveolar macrophages from pulmonary sarcoidosis patients have also been shown to produce IFN-γ which may provide further stimulation for granuloma formation (59). The M1 or “classical activation” phenotype renders macrophages efficient killers of bacteria as well as transmitters of pro-inflammatory cytokines (IL-1β, IL-12, TNFα) [reviewed in (60)]. An M2 or “alternative activation” phenotype, in contrast, allows macrophages to promote tissue repair which is a necessary function for the later resolution phase of inflammation [reviewed in (60)]. Inducers of the M2 macrophage phenotype include cytokines IL-4, IL-13, IL-10, and TGF-β (60). The participation of M2 macrophages in fibrosis is still poorly understood but M2 macrophages and giant cells have been noted within fibrotic areas of muscle specimens from sarcoidosis patients (61). Additional studies are needed to more specifically define macrophage functions in sarcoidosis, particularly with respect to changes from a chronic granulomatous status into fibrosis.
Granuloma Components: Lymphocytes.
Lymphocytes infiltrating sarcoid granulomas were once considered to be mostly Th1-polarized CD4+ cells but current findings have established the presence of elevated Th17 phenotype lymphocytes in sarcoid granulomas and lymph nodes (62, 63) (Table 2). Th17 cells are a subgroup of CD4+ T lymphocytes that secrete IL-17A, a cytokine which can induce IFN-γ and TNFα production in macrophages [reviewed in (2, 30)]. These Th17 products stimulate macrophages and promote both giant cell and granuloma formation. Th17 cells are not found in all granulomatous diseases, for example they are not present in chronic beryllium disease (64). IL-17A itself, however, can be produced in the lung by other cells such as NK cells, and has an important role in mucosal immunity, including the respiratory tract (65). Th17 cells are generated from CD4+ T cells exposed to TGFβ and IL-6 via a signaling cascade which culminates in tyrosine phosphorylation of STAT3 and STAT1 [reviewed in (66)]. STAT3 induces Th17-related genes such as IL-17A and IL-23R as well as transcription factor retinoic acid receptor-related orphan nuclear receptor γt (RORγt) which is a negative regulator of alternative lineage phenotypes.
TABLE 2.
T LYMPHOCYTES PRESENT IN SARCOIDOSIS LUNG
| INITIAL PHENOTYPE | INDUCING MOLECULES | INDUCED PHENOTYPE | SECRETED CYTOKINES | ROLE IN PATHOPHYSIOLOGY |
|---|---|---|---|---|
| CD4+ | TGFβ, IL-6 | Th17 (CCR6+, CCR4+, CXCR3−, RORγt+) | IL-17A, IL-22, IL-23. | Maintains mucosal surfaces (74); stimulates granuloma-forming cells (2). |
| Th17 | IL-1β, IL-12, IL-23 | Th1/Th17 (RORγt+, T-bet+) | IFN-γ, IL-17A | Promotes granuloma formation (30). |
| Th1/Th17 | IFN-γ, IL-12 | Th17.1 (CCR6+, CCR4−, CXCR3+) | IFN-γ | Possible drivers of sarcoid immune response (64). |
| CD4+ | Antigen affinity; ICOS* binding to macrophage ICOS* ligand (30) | Regulatory T cells (Tregs) (CD4+/CD25+, FoxP3+, CCR6+, CTLA-4+) | IL-10, TGFβ | Immunosuppressive activity (2). |
| CD4+ | IFN-γ, IL-1, IL-12, CCL5, others (30, 61) | Th1 (T-bet+, CCR6−, CCR4−, CXCR3+) | IFN-γ, IL-2 | Th1 % < Th17.1 in sarcoid lung (64). |
Inducible co-stimulator.
Th17 cells are remarkable for their plasticity in adopting proinflammatory or regulatory functions depending upon the microenvironment. Exposure to IL-1β, IL-12, and IL-23, transforms Th17 cells into a dual phenotype of Th1/Th17 which further drives inflammation with secretion of IFN-γ in addition to IL-17A. Further exposure of Th1/Th17 cells to Th1 cytokines IFN-γ and IL-12, can induce a Th17.1 phenotype which secretes mainly IFN-γ. Expression of the transcription factor, T-bet has also been detected on some Th17 cells and thought to represent a transition state leading to the Th17.1 phenotype (38). Chemokine receptors CCR6, CXCR3, and CCR4 have been helpful in differentiating among these CD4+ T helper cells with (1) Th1 expressing CXCR3+; (2) Th17 expressing CCR6+ and CCR4 +; and (3) Th17.1 expressing CCR6+ and CXCR3+ (62) (Table 2). Studies indicate that CCR6-expressing Th17 cells are recruited to lungs in pulmonary sarcoidosis by the macrophage chemokine CCL20. Newly diagnosed sarcoidosis patients with stage 2 pulmonary disease display elevated serum levels of IL-6, CCL20, IL-17A, and TGFβ (30, 67). Ultimately, transformed Th17.1 cells appear in the lungs and do not proliferate, but have been considered to be end-stage cells which continue involvement in chronic pulmonary sarcoidosis (62)
Conventional regulatory T cells (Tregs) (CD4+/CD25+) are also present in sarcoid granulomas and some studies suggest that sarcoidosis disease progression or regression is determined by the balance of Th17 and Treg cells [reviewed in (2, 30)]. These Tregs also express FoxP3 and secrete both IL-10 and TGFβ. Data cited from newly diagnosed sarcoidosis patients in the study mentioned above indicated an imbalance of Th17/Tregs that coincided with the diagnosis of active disease (67). Interestingly, murine studies have shown that Th17 cells may further differentiate via TGFβ and SMAD3 signaling pathways into IL-10 secreting, Foxp3+ Treg cells which no longer produce IL-17 (30). These CD4+ Th17 Tregs appear to suppress inflammation by direct contact with pro-inflammatory cells. The immunosuppressive ability of conventional Tregs, however, appears to be reduced in sarcoidosis patients (2).
INFLAMMATION AND FIBROSIS.
Less than 20% of sarcoidosis patients develop fibrosis which strongly associates with non-resolving pulmonary granulomatous inflammation [reviewed in (4, 28)]. More research is needed to improve our understanding of how fibrosis can begin in a sarcoidosis pulmonary milieu characterized by elevated IFN-γ which is known to inhibit collagen expression (28). Recent studies in human patients (including sarcoidosis) and murine models have cited evidence that expression of the programmed cell death-1 (PD-1) marker on CD4+ T cells promotes pulmonary fibrosis via STAT3-mediated secretion of IL-17A and TGF-β1 ((68). PD-1, a marker of cell exhaustion, is frequently displayed by chronically activated T effector cells and has been noted in patients with chronic sarcoidosis [reviewed in (69)]. Blockade of PD-1 has been shown to restore CD4+ T cell proliferative function in sarcoidosis patients (70) and to significantly reduce fibrosis in murine models (68). Currently, there are no clinically validated biomarkers for identifying sarcoidosis patients at risk of pulmonary fibrosis.
SUMMARY:
Sarcoidosis remains a heterogeneous disease of unknown cause(s). The pathophysiology of sarcoidosis granuloma formation begins when alveolar macrophages contact inhaled particulate matter that is antigenic and capable of initiating immune activation of macrophages, dendritic cells and lymphocytes. Initial studies in sarcoidosis focused on Th1-polarized lymphocytes but current studies have shown the vigorous presence of Th17.1 phenotypic lymphocytes in granuloma stimulation and formation (62). Moreover, several forms of innate immune mechanisms have also been found to participate in pulmonary inflammatory changes (14). Both lymphocytes and alveolar macrophages generate numerous inflammatory signals (chemokines, cytokines, enzymes) that organize macrophages into giant cells and granuloma structures. Many of these products can be detected in sera and provide clinically relevant data with respect to sarcoidosis disease status (36, 40). Figure 1 summarizes our current understanding of mechanisms of granuloma formation in sarcoidosis. In most patients, granulomas spontaneously resolve but questions remain regarding markers and pathways leading to fibrosis in sarcoidosis.
FIGURE 1.
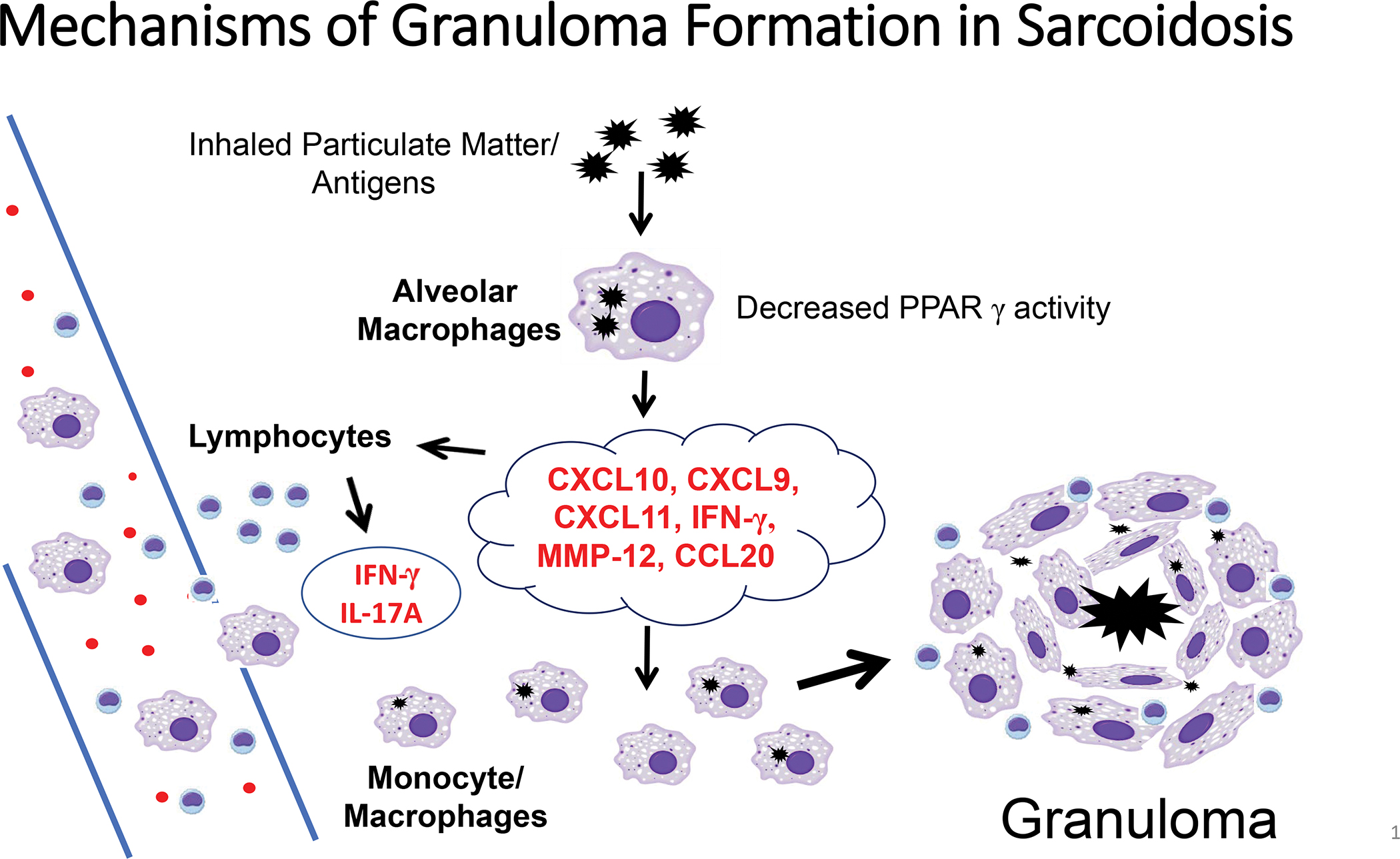
The pathophysiology of sarcoidosis granuloma formation begins when alveolar macrophages contact inhaled particulate matter that is antigenic and capable of initiating immune activation of macrophages, dendritic cells and lymphocytes. Lymphocytes and macrophages generate products (chemokines CXCL9, CSCL10, CSCL11, CCL20; cytokines IFN-γ, IL-17A; enzymes MMP-12) that organize macrophages into giant cells and granuloma structures.
NEW DIRECTIONS.
The interplay between macrophages and lymphocytes in granuloma formation and resolution has not been fully defined. In the last decade, advances in flow cytometry, lineage tracing systems, and single-cell transcriptomics, are making it possible to define the spectrum of macrophages phenotypes in different microdomains within healthy and diseased tissue (44, 71). Questions regarding the role of resident macrophages and monocyte derived-macrophages have not been addressed in sarcoidosis and application of these newer techniques may provide insight into factors which drive progressive versus resolving disease. Furthermore, the varied complexity and plasticity of T effector cells currently reported in sarcoidosis has opened additional questions regarding pathways of sarcoid disease progression or remission. The roles of Th1 and Th17 regulatory cells in sarcoidosis are unclear because of some confusing findings such as elevated Th17.1 in the self-limiting Lofgren’s syndrome compared to non-Lofgren’s patients [reviewed in (30)]. It has been suggested that initial findings at diagnosis may provide clues to later sarcoidosis outcomes and therefore careful monitoring of patients is required in larger long-term studies. Unfortunately, current knowledge of cellular immune pathways in sarcoidosis is not yet sufficient to allow prediction of final sarcoid disease status from findings obtained at diagnosis (reviewed in (69). Application of newer analytical technologies are needed to improve our understanding of mechanisms in disease progression or remission and possibly, to provide better clinical treatment for sarcoidosis patients (72, 73).
ABBREVIATIONS
- ABCA1
ATP-binding cassette lipid transporter-A1
- ABCG1
ATP-binding cassette lipid transporter-G1
- CCL
C-C Motif Chemokine Ligand
- CCR
C-C chemokine receptor
- CXCL
C-X-C motif ligand
- DAMPs
danger-associated molecular patterns
- GWAS
genome-wide association studies
- HLA
human leukocyte antigen
- IFN-γ
interferon-gamma
- IL
interleukin
- IL23R
Interleukin 23 receptor
- mTOR
mammalian target of rapamycin
- MMP12
Matrix Metalloproteinase12
- NLRs
NOD-like receptors
- PAMPs
pathogen-associated molecular patterns
- PRRs
Pattern Recognition Receptors
- PBMC
peripheral blood mononuclear cells
- PPARγ
peroxisome proliferator-activated receptor gamma
- PD-1
programmed cell death-1
- RORγt
retinoic acid receptor-related orphan nuclear receptor γt
- SAA
serum amyloid A
- sIL-2R
soluble Interleukin 2 receptor
- STAT
signal transducer and activator of transcription
- Th17
T-helper 17
- TLRs
Toll-like receptors
- Tregs
regulatory T cells
- TGFβ
transforming growth factor beta
- TNFα
tumor necrosis factor alpha
- Tsc2
tuberous sclerosis 2
REFERENCES
- 1.Crouser ED, Maier LA, Wilson KC, Bonham CA, Morgenthau AS, Patterson KC, et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;201(8):e26–e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett D, Bargagli E, Refini RM, Rottoli P. New concepts in the pathogenesis of sarcoidosis. Expert Rev Respir Med. 2019;13(10):981–91. [DOI] [PubMed] [Google Scholar]
- 3.Judson MA. Environmental Risk Factors for Sarcoidosis. Frontiers in Immunology. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Culver DA, Judson MA. New advances in the management of pulmonary sarcoidosis. BMJ. 2019;367:l5553. [DOI] [PubMed] [Google Scholar]
- 5.Crowley LE, Herbert R, Moline JM, Wallenstein S, Shukla G, Schechter C, et al. “Sarcoid like” granulomatous pulmonary disease in World Trade Center disaster responders. Am J Ind Med. 2011;54(3):175–84. [DOI] [PubMed] [Google Scholar]
- 6.Eberhardt C, Thillai M, Parker R, Siddiqui N, Potiphar L, Goldin R, et al. Proteomic Analysis of Kveim Reagent Identifies Targets of Cellular Immunity in Sarcoidosis. PLoS One. 2017;12(1):e0170285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song Z, Marzilli L, Greenlee BM, Chen ES, Silver RF, Askin FB, et al. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J Exp Med. 2005;201(5):755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen ES, Wahlstrom J, Song Z, Willett MH, Wiken M, Yung RC, et al. T cell responses to mycobacterial catalase-peroxidase profile a pathogenic antigen in systemic sarcoidosis. J Immunol. 2008;181(12):8784–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen ES, Moller DR. Etiologic role of infectious agents. Semin Respir Crit Care Med. 2014;35(3):285–95. [DOI] [PubMed] [Google Scholar]
- 10.Kaiser Y, Eklund A, Grunewald J. Moving target: shifting the focus to pulmonary sarcoidosis as an autoimmune spectrum disorder. Eur Respir J. 2019;54(1). [DOI] [PubMed] [Google Scholar]
- 11.Kinloch AJ, Kaiser Y, Wolfgeher D, Ai J, Eklund A, Clark MR, et al. In Situ Humoral Immunity to Vimentin in HLA-DRB1*03(+) Patients With Pulmonary Sarcoidosis. Front Immunol. 2018;9:1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer A, Grunewald J, Spagnolo P, Nebel A, Schreiber S, Muller-Quernheim J. Genetics of sarcoidosis. Semin Respir Crit Care Med. 2014;35(3):296–306. [DOI] [PubMed] [Google Scholar]
- 13.Schupp JC, Vukmirovic M, Kaminski N, Prasse A. Transcriptome profiles in sarcoidosis and their potential role in disease prediction. Curr Opin Pulm Med. 2017;23(5):487–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen ES. Innate immunity in sarcoidosis pathobiology. Curr Opin Pulm Med. 2016;22(5):469–75. [DOI] [PubMed] [Google Scholar]
- 15.Riteau N, Bernaudin JF. In addition to mTOR and JAK/STAT, NLRP3 inflammasome is another key pathway activated in sarcoidosis. Eur Respir J. 2020;55(3). [DOI] [PubMed] [Google Scholar]
- 16.Bennett D, Cameli P, Lanzarone N, Carobene L, Bianchi N, Fui A, et al. Chitotriosidase: a biomarker of activity and severity in patients with sarcoidosis. Respir Res. 2020;21(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Opitz B, van Laak V, Eitel J, Suttorp N. Innate Immune Recognition in Infectious and Noninfectious Diseases of the Lung. American Journal of Respiratory and Critical Care Medicine. 2010;181(12):1294–309. [DOI] [PubMed] [Google Scholar]
- 18.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–20. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10(2):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huppertz C, Jager B, Wieczorek G, Engelhard P, Oliver SJ, Bauernfeind FG, et al. The NLRP3 inflammasome pathway is activated in sarcoidosis and involved in granuloma formation. Eur Respir J. 2020;55(3). [DOI] [PubMed] [Google Scholar]
- 21.Weichhart T, Hengstschlager M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. 2015;15(10):599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linke M, Pham HT, Katholnig K, Schnoller T, Miller A, Demel F, et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol. 2017;18(3):293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265(2):501–23. [DOI] [PubMed] [Google Scholar]
- 24.Chen ES, Song Z, Willett MH, Heine S, Yung RC, Liu MC, et al. Serum amyloid A regulates granulomatous inflammation in sarcoidosis through Toll-like receptor-2. Am J Respir Crit Care Med. 2010;181(4):360–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huho A, Foulke L, Jennings T, Koutroumpakis E, Dalvi S, Chaudhry H, et al. The role of serum amyloid A staining of granulomatous tissues for the diagnosis of sarcoidosis. Respir Med. 2017;126:1–8. [DOI] [PubMed] [Google Scholar]
- 26.Elmonem MA, van den Heuvel LP, Levtchenko EN. Immunomodulatory Effects of Chitotriosidase Enzyme. Enzyme Res. 2016;2016:2682680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakthivel P, Bruder D. Mechanism of granuloma formation in sarcoidosis. Curr Opin Hematol. 2017;24(1):59–65. [DOI] [PubMed] [Google Scholar]
- 28.Patterson KC, Chen ES. The Pathogenesis of Pulmonary Sarcoidosis and Implications for Treatment. Chest. 2018;153(6):1432–42. [DOI] [PubMed] [Google Scholar]
- 29.Broos CE, van NM, Hoogsteden HC, Hendriks RW, Kool M, van den Blink B. Granuloma formation in pulmonary sarcoidosis. Front Immunol. 2013;4:437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crouser ED. Role of imbalance between Th17 and regulatory T-cells in sarcoidosis. Curr Opin Pulm Med. 2018;24(5):521–6. [DOI] [PubMed] [Google Scholar]
- 31.Fingleton B Matrix metalloproteinases as regulators of inflammatory processes. Biochim Biophys Acta Mol Cell Res. 2017;1864(11 Pt A):2036–42. [DOI] [PubMed] [Google Scholar]
- 32.Crouser ED, Culver DA, Knox KS, Julian MW, Shao G, Abraham S, et al. Gene Expression Profiling Identifies MMP-12 and ADAMDEC1 as Potential Pathogenic Mediators of Pulmonary Sarcoidosis. American Journal of Respiratory and Critical Care Medicine. 2009;179(10):929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parasa VR, Muvva JR, Rose JF, Braian C, Brighenti S, Lerm M. Inhibition of Tissue Matrix Metalloproteinases Interferes with Mycobacterium tuberculosis-Induced Granuloma Formation and Reduces Bacterial Load in a Human Lung Tissue Model. Front Microbiol. 2017;8:2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohan AN N; Malur A; Soliman E; McPeek M; Leffler N; Ogburn D; Tokarz; Knudson W; Gharib SA; Schnapp LM; Barna BP; Thomassen MJ Matrix Metalloproteinase-12 is Required for Granuloma Progression. Frontiers in Immunology. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fields GB. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells. 2019;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arger NK, Ho M, Woodruff PG, Koth LL. Serum CXCL11 correlates with pulmonary outcomes and disease burden in sarcoidosis. Respir Med. 2019;152:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su R, Nguyen ML, Agarwal MR, Kirby C, Nguyen CP, Ramstein J, et al. Interferon-inducible chemokines reflect severity and progression in sarcoidosis. Respir Res. 2013;14:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arger NK, Machiraju S, Allen IE, Woodruff PG, Koth LL. T-bet Expression in Peripheral Th17.0 Cells Is Associated With Pulmonary Function Changes in Sarcoidosis. Front Immunol. 2020;11:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rubin LA, Nelson DL. The soluble interleukin-2 receptor: biology, function, and clinical application. Ann Intern Med. 1990;113(8):619–27. [DOI] [PubMed] [Google Scholar]
- 40.Eurelings LEM, Miedema JR, Dalm V, van Daele PLA, van Hagen PM, van Laar JAM, et al. Sensitivity and specificity of serum soluble interleukin-2 receptor for diagnosing sarcoidosis in a population of patients suspected of sarcoidosis. PLoS One. 2019;14(10):e0223897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou Y, Zhang Y, Zhao M, Li Q, Li H. sIL-2R levels predict the spontaneous remission in sarcoidosis. Respir Med. 2020;171:106115. [DOI] [PubMed] [Google Scholar]
- 42.Bargagli E, Bennett D, Maggiorelli C, Di Sipio P, Margollicci M, Bianchi N, et al. Human chitotriosidase: a sensitive biomarker of sarcoidosis. J Clin Immunol. 2013;33(1):264–70. [DOI] [PubMed] [Google Scholar]
- 43.Wilson JL, Mayr HK, Weichhart T. Metabolic Programming of Macrophages: Implications in the Pathogenesis of Granulomatous Disease. Front Immunol. 2019;10:2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. 2019;129(7):2619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawgood S, Poulain FR. The pulmonary collectins and surfactant metabolism. Ann Rev Physiol. 2001;63:495–519. [DOI] [PubMed] [Google Scholar]
- 46.Tarling EJ, Edwards PA. Dancing with the sterols: critical roles for ABCG1, ABCA1, miRNAs, and nuclear and cell surface receptors in controlling cellular sterol homeostasis. Biochim Biophys Acta. 2012;1821(3):386–95. [DOI] [PubMed] [Google Scholar]
- 47.Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med. 2002;347(26):2141–8. [DOI] [PubMed] [Google Scholar]
- 48.Veldhuizen R, Nag K, Orgeig S, Possmayer F. The role of lipids in pulmonary surfactant. Biochim Biophys Acta. 1998;1408(2–3):90–108. [DOI] [PubMed] [Google Scholar]
- 49.Tarling EJ, de Aguiar Vallim TQ, Edwards PA. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol Metab. 2013;24(7):342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fessler MB. A New Frontier in Immunometabolism. Cholesterol in Lung Health and Disease. Ann Am Thorac Soc. 2017;14(Supplement_5):S399–S405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gowdy KM, Fessler MB. Emerging roles for cholesterol and lipoproteins in lung disease. Pulm Pharmacol Ther. 2013;26(4):430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malur A, Mccoy AJ, Arce S, Barna BP, Kavuru MS, Malur AG, et al. Deletion of PPARγ in alveolar macrophages is associated with a Th-1 pulmonary inflammatory response. J Immunol. 2009;182:5816–22. [DOI] [PubMed] [Google Scholar]
- 53.Baker AD, Malur A, Barna BP, Ghosh S, Kavuru MS, Malur AG, et al. Targeted PPAR{gamma} deficiency in alveolar macrophages disrupts surfactant catabolism. Journal of Lipid Research. 2010;51(6):1325–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baker AD, Malur A, Barna BP, Kavuru MS, Malur AG, Thomassen MJ. PPARgamma regulates the expression of cholesterol metabolism genes in alveolar macrophages. Biochem Biophys Res Commun. 2010;393(4):682–7. [DOI] [PubMed] [Google Scholar]
- 55.Barna BP, McPeek M, Malur A, Fessler MB, Wingard CJ, Dobbs L, et al. Elevated MicroRNA-33 in Sarcoidosis and a Carbon Nanotube Model of Chronic Granulomatous Disease. Am J Respir Cell Mol Biol. 2016;54(6):865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smigiel KS, Parks WC. Macrophages, Wound Healing, and Fibrosis: Recent Insights. Curr Rheumatol Rep. 2018;20(4):17. [DOI] [PubMed] [Google Scholar]
- 59.Robinson BW, McLemore TL, Crystal RG. Gamma interferon is spontaneously released by alveolar macrophages and lung T lymphocytes in patients with pulmonary sarcoidosis. J Clin Invest. 1985;75(5):1488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun. 2014;6(6):716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prokop S, Heppner FL, Goebel HH, Stenzel W. M2 polarized macrophages and giant cells contribute to myofibrosis in neuromuscular sarcoidosis. Am J Pathol. 2011;178(3):1279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramstein J, Broos CE, Simpson LJ, Ansel KM, Sun SA, Ho ME, et al. IFN-gamma-Producing T-Helper 17.1 Cells Are Increased in Sarcoidosis and Are More Prevalent than T-Helper Type 1 Cells. Am J Respir Crit Care Med. 2016;193(11):1281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Broos CE, Koth LL, van NM, In ‘t Veen JCCM, Paulissen SMJ, van Hamburg JP, et al. Increased T-helper 17.1 cells in sarcoidosis mediastinal lymph nodes. Eur Respir J. 2018;51(3). [DOI] [PubMed] [Google Scholar]
- 64.Greaves SA, Atif SM, Fontenot AP. Adaptive Immunity in Pulmonary Sarcoidosis and Chronic Beryllium Disease. Front Immunol. 2020;11:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iwanaga N, Kolls JK. Updates on T helper type 17 immunity in respiratory disease. Immunology. 2019;156(1):3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harbour SN, DiToro DF, Witte SJ, Zindl CL, Gao M, Schoeb TR, et al. TH17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity. Sci Immunol. 2020;5(49). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ding J, Dai J, Cai H, Gao Q, Wen Y. Extensively disturbance of regulatory T cells - Th17 cells balance in stage II pulmonary sarcoidosis. Int J Med Sci. 2017;14(11):1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Celada LJ, Kropski JA, Herazo-Maya JD, Luo W, Creecy A, Abad AT, et al. PD-1 up-regulation on CD4(+) T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-beta1 production. Sci Transl Med. 2018;10(460). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonham CA, Strek ME, Patterson KC. From granuloma to fibrosis: sarcoidosis associated pulmonary fibrosis. Curr Opin Pulm Med. 2016;22(5):484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Braun NA, Celada LJ, Herazo-Maya JD, Abraham S, Shaginurova G, Sevin CM, et al. Blockade of the programmed death-1 pathway restores sarcoidosis CD4(+) T-cell proliferative capacity. Am J Respir Crit Care Med. 2014;190(5):560–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alexander MJ, Budinger GRS, Reyfman PA. Breathing fresh air into respiratory research with single-cell RNA sequencing. Eur Respir Rev. 2020;29(156). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garman L, Montgomery CG, Rivera NV. Recent advances in sarcoidosis genomics: epigenetics, gene expression, and gene by environment (G x E) interaction studies. Curr Opin Pulm Med. 2020;26(5):544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bhargava M, Viken KJ, Barkes B, Griffin TJ, Gillespie M, Jagtap PD, et al. Novel protein pathways in development and progression of pulmonary sarcoidosis. Sci Rep. 2020;10(1):13282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma Q Polarization of Immune Cells in the Pathologic Response to Inhaled Particulates. Front Immunol. 2020;11:1060. [DOI] [PMC free article] [PubMed] [Google Scholar]