

Abstract
Background
Inclusion body myositis (IBM) is a late‐onset inflammatory muscle disease (myopathy) associated with progressive proximal and distal limb muscle atrophy and weakness. Treatment options have attempted to target inflammatory and atrophic features of this condition (for example with immunosuppressive and immunomodulating drugs, anabolic steroids, and antioxidant treatments), although as yet there is no known effective treatment for reversing or minimising the progression of inclusion body myositis. In this review we have considered the benefits, adverse effects, and costs of treatment in targeting cardinal effects of the condition, namely muscle atrophy, weakness, and functional impairment.
Objectives
To assess the effects of treatment for IBM.
Search methods
On 7 October 2014 we searched the Cochrane Neuromuscular Disease Group Specialized Register, the Cochrane Central Register for Controlled Trials (CENTRAL), MEDLINE, and EMBASE. Additionally in November 2014 we searched clinical trials registries for ongoing or completed but unpublished trials.
Selection criteria
We considered randomised or quasi‐randomised trials, including cross‐over trials, of treatment for IBM in adults compared to placebo or any other treatment for inclusion in the review. We specifically excluded people with familial IBM and hereditary inclusion body myopathy, but we included people who had connective tissue and autoimmune diseases associated with IBM, which may or may not be identified in trials. We did not include studies of exercise therapy or dysphagia management, which are topics of other Cochrane systematic reviews.
Data collection and analysis
We used standard Cochrane methodological procedures.
Main results
The review included 10 trials (249 participants) using different treatment regimens. Seven of the 10 trials assessed single agents, and 3 assessed combined agents. Many of the studies did not present adequate data for the reporting of the primary outcome of the review, which was the percentage change in muscle strength score at six months. Pooled data from two trials of interferon beta‐1a (n = 58) identified no important difference in normalised manual muscle strength sum scores from baseline to six months (mean difference (MD) ‐0.06, 95% CI ‐0.15 to 0.03) between IFN beta‐1a and placebo (moderate‐quality evidence). A single trial of methotrexate (MTX) (n = 44) provided moderate‐quality evidence that MTX did not arrest or slow disease progression, based on reported percentage change in manual muscle strength sum scores at 12 months. None of the fully published trials were adequately powered to detect a treatment effect.
We assessed six of the nine fully published trials as providing very low‐quality evidence in relation to the primary outcome measure. Three trials (n = 78) compared intravenous immunoglobulin (combined in one trial with prednisone) to a placebo, but we were unable to perform meta‐analysis because of variations in study analysis and presentation of trial data, with no access to the primary data for re‐analysis. Other comparisons were also reported in single trials. An open trial of anti‐T lymphocyte immunoglobulin (ATG) combined with MTX versus MTX provided very low‐quality evidence in favour of the combined therapy, based on percentage change in quantitative muscle strength sum scores at 12 months (MD 12.50%, 95% CI 2.43 to 22.57). Data from trials of oxandrolone versus placebo, azathioprine (AZA) combined with MTX versus MTX, and arimoclomol versus placebo did not allow us to report either normalised or percentage change in muscle strength sum scores. A complete analysis of the effects of arimoclomol is pending data publication. Studies of simvastatin and bimagrumab (BYM338) are ongoing.
All analysed trials reported adverse events. Only 1 of the 10 trials interpreted these for statistical significance. None of the trials included prespecified criteria for significant adverse events.
Authors' conclusions
Trials of interferon beta‐1a and MTX provided moderate‐quality evidence of having no effect on the progression of IBM. Overall trial design limitations including risk of bias, low numbers of participants, and short duration make it difficult to say whether or not any of the drug treatments included in this review were effective. An open trial of ATG combined with MTX versus MTX provided very low‐quality evidence in favour of the combined therapy based on the percentage change data given. We were unable to draw conclusions from trials of IVIg, oxandrolone, and AZA plus MTX versus MTX. We need more randomised controlled trials that are larger, of longer duration, and that use fully validated, standardised, and responsive outcome measures.
Plain language summary
Therapy for inclusion body myositis
Review question
We reviewed the evidence from clinical trials about the effects of drug treatments for inclusion body myositis (IBM). We did not include trials of exercise or management of swallowing difficulties, as these are subjects of other Cochrane reviews.
Background
IBM is a disease that mainly affects older people. The main symptoms are increasing muscle wasting and weakness of the arms and legs. Some people experience swallowing difficulties. As yet no therapy has been shown to alter the course of the disease. Treatments that have been tested include agents that suppress or alter the immune response, drugs that promote muscle growth, and antioxidants.
Study characteristics
This review included 10 trials (249 participants). One of these trials (24 participants) is completed but has not yet been published. Seven trials compared treatments with placebo (inactive treatment): three of intravenous immunoglobulin (IVIg), two of interferon beta‐1a (IFN beta‐1a), and one each of oxandrolone, methotrexate (MTX), and arimoclomol (not yet published). A further two trials compared MTX with combined immunosuppressive therapy (MTX with anti‐T lymphocyte immunoglobulin (ATG) (an agent that destroys white blood cells) and MTX with azathioprine). In these two trials, participants and investigators knew which treatment participants were receiving, which could have biased the results.
Results and quality of the evidence
For our primary outcome, which was muscle strength, we were only able to combine the results for the two trials of IFN beta‐1a therapy versus placebo. This treatment did not appear to offer a benefit in terms of muscle strength. MTX also did not stop or retard loss of muscle strength when compared to placebo. We considered the evidence from these trials to be of moderate quality because the trials were too small to rule out a possible benefit for these drugs. For the other trials, the evidence was of very low quality. Three trials compared IVIg (combined in one trial with prednisone) to a placebo, but we were unable to perform meta‐analysis because the available data were not suitable. One trial of ATG combined with MTX versus MTX alone provided very low‐quality evidence of an effect on muscle strength in favour of MTX plus ATG at 12 months. The other comparisons, of MTX versus placebo, oxandrolone versus placebo, azathioprine combined with MTX versus MTX, and arimoclomol versus placebo were reported in single trials that did not provide enough data for analysis of the effect on muscle strength.
Due to their small size and short duration, the trials we studied were generally unable to give definitive answers as to whether the treatments tested were effective or ineffective. All of the interventions we studied had some adverse effects and are known to cause potentially serious adverse events. We need larger trials of longer duration, using robust ways of measuring the effects of treatments that are meaningful to people with IBM. Agreeing on common trial measurements will also make it easier to compare trial results and assess potential treatments.
The evidence is current to October 2014.
Summary of findings
Summary of findings for the main comparison. Intravenous immunoglobulin versus placebo for inclusion body myositis.
| Intravenous immunoglobulin versus placebo for inclusion body myositis | ||||||
| Patient or population: people with inclusion body myositis Settings: Intervention: intravenous immunoglobulin Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Intravenous immunoglobulin | |||||
| Change in muscle strength at 6 months (%) | Not estimable | Not estimable | Not estimable | 78 (3 studies) | See comment | 2 of the 3 studies assessed change in muscle strength at 3 months and the other at 6 months. Data were not suitable for meta‐analysis. |
| Change in muscle strength at 12 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in muscle mass at 6 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in handgrip strength at 6 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Change in timed walk at 6 months (%) e.g. 10‐metre or 6‐minute walk test at 6 months ‐ not measured |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Significant adverse events | Not estimable | Not estimable | Not estimable | 78 (3 studies) | See comment | Dalakas 1997: unclear whether dropouts from treatment or placebo group. Dalakas 2001: treatment group withdrawals = 0; placebo group withdrawals = 1. Walter 2000: 2 withdrawals but unspecified whether from treatment or placebo group |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Summary of findings 2. Interferon beta‐1a compared to placebo for inclusion body myositis.
| Interferon beta‐1a (IFN beta‐1a) compared to placebo for inclusion body myositis | ||||||
| Patient or population: people with inclusion body myositis Settings: Intervention: IFN beta‐1a Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | IFN beta‐1a | |||||
| Change in muscle strength at 6 months (normalised MMT score ‐ see Characteristics of Studies table) | The mean changes in normalised muscle strength in the control groups was ‐0.03 and ‐0.08 | The mean change in normalised muscle strength in the intervention groups was 0.06 lower (0.15 lower to 0.03 higher) | ‐ | 58 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Change in muscle strength at 12 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in muscle mass at 6 months (%) | The mean change in muscle mass (%) in the control groups was ‐0.85 and ‐1.77. | The mean change in muscle mass (%) in the intervention groups was 0.22 higher (1.69 lower to 2.13 higher) | ‐ | 58 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Change in handgrip strength at 6 months (%) | Not estimable | Not estimable | Not estimable | 58 (2 studies) | See comment | Baseline measurements were not reported, so % change was not calculable. There was a non‐significant difference in handgrip strength (kg) between IFN beta‐1a and placebo in 1 trial (MD ‐0.16, 95% CI ‐1.55 to 1.22). In the other the MD was 1.84 kg with a 95% CI of 0.46 to 3.21 in favour of IFN beta‐1a |
|
Change in timed walk at 6 months (%) e.g. 10‐metre or 6‐minute walk test at 6 months ‐ not measured |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Significant adverse events | Not estimable | Not estimable | Not estimable | 58 (2 studies) | See comment | Muscle Study Group 2001: treatment group withdrawals = 1; placebo group withdrawals = 0. Muscle Study Group 2004: treatment group withdrawals = 2; placebo group withdrawals = 1 |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; MMT: manual muscle testing | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 The CI includes the possibility of an effect favouring either IFN beta‐1a or placebo when data from the two trials are combined.
Summary of findings 3. Methotrexate compared to placebo for inclusion body myositis.
| Methotrexate compared to placebo for inclusion body myositis | ||||||
| Patient or population: people with inclusion body myositis Settings: Intervention: methotrexate Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Methotrexate | |||||
| Change in muscle strength at 6 months (%) | Not estimable | Not estimable | Not estimable | 44 (1 study) | See comment | Data collected at 12 months only |
| Change in muscle strength at 12 months (%) ‐ MMT | The mean change in muscle strength (%) in the control group was ‐2.0% | The mean change in muscle strength (%) in the intervention group was 1.5% higher (1.0 lower to 3.9 higher) |
‐ | 44 (1 study) | ⊕⊕⊝⊝ moderate1 | MD in mean change based on intention‐to‐treat data. Per protocol: MD 1.6%, 95% CI ‐2.3 to 5.4. This study was assessed as having a low overall risk of bias. |
| Change in muscle mass at 6 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in handgrip strength at 6 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Change in timed walk at 6 months (%) e.g. 10‐metre or 6‐minute walk test at 6 months ‐ not measured |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Significant adverse events | 43 per 1000 | 381 per 1000 (52 to 1000) | RR 8.76 (1.19 to 64.28) | 44 (1 study) | ⊕⊕⊝⊝ moderate2 |
Badrising 2002: treatment group withdrawals = 8/21; placebo group withdrawals = 1/23 |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MMT: manual muscle testing; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded for imprecision. The single randomised controlled trial of methotrexate was insufficiently powered to exclude a possible benefit from methotrexate.
2 Downgraded for imprecision.
Summary of findings 4. Methotrexate and anti‐T lymphocyte immunoglobulin compared to methotrexate for inclusion body myositis.
| Methotrexate (MTX) and anti‐T lymphocyte immunoglobulin (ATG) compared to MTX for inclusion body myositis | ||||||
| Patient or population: people with inclusion body myositis Settings: Intervention: methotrexate and anti‐T lymphocyte immunoglobulin Comparison: methotrexate | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| MTX | MTX and ATG | |||||
| Change in muscle strength at 6 months (%) | Not estimable | Not estimable | Not estimable | 11 (1 study) | See comment | Data collected at 12 months only |
| Change in muscle strength at 12 months (%) ‐ QMT | The mean change in muscle strength (%) in the control group was ‐11.1% | The mean change in muscle strength (%) in the intervention groups was 12.50 higher (2.43 to 22.57 higher) |
Not estimable | 11 (1 study) | ⊕⊝⊝⊝ very low1 | |
| Change in muscle mass at 12 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in handgrip strength at 6 months (%) | Not estimable | Not estimable | Not estimable | 11 (1 study) | See comment | Data collected at 12 months only; rescaling of data not performed due to uncertainty in the assumption of linear change |
|
Change in timed walk at 6 months (%) e.g. 10‐metre or 6‐minute walk test at 6 months ‐ not measured |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Significant adverse events | Not estimable | Not estimable | Not estimable | 11 (1 study) | See comment | Lindberg 2003: combined ATG and MTX group withdrawals = 0; MTX group withdrawals = 1 |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; QMT: quantitative muscle testing; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded for study design: as an open‐label study, there is a high risk of bias. 2 Downgraded twice for imprecision: 11 participants. The minimum clinically important difference is not known, and the MD has wide CI.
Summary of findings 5. Methotrexate and azathioprine compared to methotrexate (plus leucovorin) for inclusion body myositis.
| Methotrexate (MTX) and azathioprine (AZA) compared to MTX (plus leucovorin) for inclusion body myositis | ||||||
| Patient or population: people with inclusion body myositis Settings: Intervention: methotrexate and azathioprine (plus leucovorin) Comparison: methotrexate | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Methotrexate | Methotrexate and azathioprine | |||||
| Change in muscle strength at 6 months (%) | Not estimable | Not estimable | Not estimable | 11 (1 study) | See comment | Categorical change in MMT only reported. This study was graded as having a high risk of bias. |
| Change in muscle strength at 12 months (%) (using MMT or QMT) at 12 months ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in muscle mass at 6 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in handgrip strength at 6 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Change in timed walk at 6 months (%) e.g. 10‐metre or 6‐minute walk test at 6 months ‐ not measured |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Significant adverse events | Not estimable | Not estimable | Not estimable | 11 (1 study) | See comment | Leff 1993: Combined AZA and MTX = 3 withdrawals; MTX (plus leucovorin) = 0 withdrawals |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MMT: manual muscle testing; QMT: quantitative muscle testing | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Summary of findings 6. Oxandrolone compared to placebo for inclusion body myositis.
| Oxandrolone compared to placebo for inclusion body myositis | ||||||
| Patient or population: people with inclusion body myositis Settings: Intervention: oxandrolone Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Oxandrolone | |||||
| Change in muscle strength at 6 months (%) | Not estimable | Not estimable | Not estimable | 16 (1 study) | See comment | Data collected at 12 months only; re‐scaling of data not performed due to uncertainty in the assumption of linear change. There were also insufficient data to calculate percentage change in muscle strength. This study was graded as having an unclear risk of bias |
| Change in muscle strength at 12 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in muscle mass at 6 months (%) | Not estimable | Not estimable | Not estimable | 16 (1 study) | See comment | Data collected at 12 months only; rescaling of data not performed due to uncertainty in the assumption of linear change. |
| Change in handgrip strength at 6 months (%) ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Change in timed walk at 6 months (%) e.g. 10‐metre or 6‐minute walk test at 6 months ‐ not measured |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Significant adverse events | Not estimable | Not estimable | Not estimable | Not estimable | See comment | Rutkove 2002: treatment group withdrawals = 1; placebo group withdrawals = 2 |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Background
Description of the condition
Inclusion body myositis (IBM) is a late‐onset inflammatory muscle disease (myopathy) with a distinctive pattern of proximal and distal limb atrophy and weakness. IBM is considered to be the most common acquired myopathy in those over age 50. In early published series, IBM has accounted for up to 28% of all idiopathic inflammatory myopathies, although the true proportion could be much higher (Lotz 1989). In the Netherlands, prevalence has been estimated at 4.9 per million inhabitants (Badrising 2000). Between 2000 and 2008, prevalence in Western Australia rose from 9.3 to 14.9 per million inhabitants, a change attributed to improved case identification (Needham 2008; Phillips 2000). Prevalence adjusted for age over 50 years is higher, up to 51.3 per million population (Needham 2008).
IBM is usually a sporadic and isolated disorder that can be associated with secondary mitochondrial DNA abnormalities in excess of those seen with normal ageing (Oldfors 1995). In rare instances, typical IBM occurs in families (Amato 1998; Tateyama 2003); this familial IBM should not be confused with hereditary inclusion body myopathy, which is not usually associated with inflammation and in which there may be mutations in the GNE gene (Huizing 2009). IBM is sometimes associated with a variety of connective tissue and autoimmune diseases, including rheumatoid arthritis (Soden 1994), vitamin B12 deficiency (Khraishi 1992), Sjögren's syndrome (Gutmann 1985; Khraishi 1992), chronic immune thrombocytopenia (Riggs 1984), sarcoidosis (Danon 1986), collagen vascular disease (Lane 1985), and common variable immune deficiency (Dalakas 1995; Lindberg 1990).
To date, clinicopathologically defined criteria for sporadic IBM, as proposed by Griggs 1995 and revised by Benveniste 2010 and Hilton‐Jones 2010, have formed the basis for diagnostic criteria. Improved case ascertainment through clinical assessment has supported the application of clinically defined diagnostic criteria. However, for the purpose of reviewing the existing clinical trials, we included those trials using clinicopathologically defined sporadic IBM, as defined by Griggs 1995, Benveniste 2010, or Hilton‐Jones 2010.
Potential outcome measures
The muscle atrophy and weakness of IBM usually follow a slowly progressive course. In the absence of established effective drug treatment, the mainstay of treatment is supportive. Natural history studies suggest that people with IBM can experience a 3.5% to 5.2% annual decline in compound muscle strength graded manually (Cortese 2013; Cox 2011). Because of its characteristic pattern of muscle involvement, IBM may cause predictable functional difficulties. For example, weakness of the long (extrinsic) finger flexors can impair handgrip, while quadriceps weakness can cause knee instability, making rising from chairs, managing stairs, and walking increasingly difficult. For the purpose of this review, it seemed reasonable to choose those outcome measures assessing the cardinal effects of IBM, namely muscle atrophy, weakness, and functional impairment.
In terms of atrophy, muscle mass can be measured in a number of ways, including:
urinary creatinine excretion under controlled conditions;
potassium isotope counting;
magnetic resonance imaging (MRI);
computerised tomography (CT) muscle imaging; and
dual energy X‐ray absorptiometry (DEXA).
Muscle strength can be measured manually (referred to as manual muscle testing, or MMT) and is commonly scored using the six‐point Medical Research Council (MRC) strength scale or modified MRC scale. Alternatively, the maximal voluntary isometric contraction or isokinetic muscle strength can be measured using a hand‐held or fixed myometer (referred to as quantitative muscle testing (QMT)) and the results expressed in units of force (newtons, kilograms, or pounds). However, as with MMT, the selected muscles tested by QMT can vary; QMT sum scores are often expressed as the z‐score, which is the sum of the standard deviations from the mean for each muscle tested.
In terms of measuring functional impairment, single‐item tests include timed walking tests, stair climb, and rise from chair; we have chosen timed walking tests as appropriate measures for the patient population in this review. There are also questionnaire based, multi‐item rating scales, such as the IBM Functional Rating Scale, that provide an overall score across different functional tasks (Jackson 2008). In choosing appropriate outcome measures for this review, we also wanted to consider other patient reported outcome measures, including symptom severity and quality of life, as well as adverse events associated with treatment. However, these outcome measures are not consistently applied across different muscle disease centres and trials at present.
We can specify a minimum time period for the primary outcome measure in relation to its sensitivity to change in a slowly progressive condition. Power calculations based upon available natural history data for muscle strength in IBM support a minimum trial length of six months (Rose 2001).
Description of the intervention
Interventions included pharmacological agents, dietary supplements, and musculoskeletal surgery. We excluded the management of dysphagia and the role of exercise therapy for muscle disease, which other Cochrane systematic reviews of interventions discuss (Hill 2004; Voet 2013).
Why it is important to do this review
As the first systematic review of treatment for IBM, this Cochrane review aimed to determine the evidence base for current treatment and to identify dose‐responses where possible. Our findings may also be relevant to other neuromuscular and age‐related conditions in which muscle weakness, atrophy, and inflammation are prominent symptoms.
Objectives
To systematically assess the effects of treatment for IBM.
Methods
Criteria for considering studies for this review
Types of studies
We considered for inclusion randomised or quasi‐randomised trials, including cross‐over trials, of treatment for IBM (except for exercise therapy and dysphagia management). We included comparisons of treatment versus placebo or any other treatment. We included studies reported as full text, those published as abstract only, and unpublished data, with no language restrictions.
Types of participants
All participants were over 18 years of age and had a clinicopathologically defined diagnosis of IBM (Benveniste 2010; Griggs 1995; Hilton‐Jones 2010). We specifically excluded people with familial IBM and hereditary inclusion body myopathy, but we included people who had connective tissue and autoimmune diseases associated with IBM, which may or may not be identified in trials.
Types of interventions
We included the following interventions.
Immunosuppressive agents, e.g. prednisolone (and other corticosteroids), cyclophosphamide, chlorambucil, azathioprine, methotrexate (MTX), and ciclosporin.
Immunomodulatory interventions, e.g. intravenous immunoglobulin, leukopheresis, plasma exchange, and immune‐targeted monoclonal antibodies.
Antioxidants, e.g. vitamin E.
Mitochondrial substrates, e.g. carnitine and ubiquinone.
Anabolic steroids and muscle supplements.
Other interventions except for exercise therapy and dysphagia management.
Therapies were administered using various protocols including as single agents, combined therapy, or sequential therapy. We analysed different interventions separately.
Types of outcome measures
Primary outcomes
The primary outcome measure for this review was percentage change in muscle strength (using MMT or QMT) from baseline at six months.
Secondary outcomes
Secondary outcome measures for this review were as follows.
Percentage change from baseline in muscle strength (using MMT or QMT) at 12 months.
Percentage change from baseline in handgrip strength at 6 months.
Percentage change from baseline in muscle mass (by whatever method, e.g. MRI) at 6 months.
Percentage change from baseline in timed walk (e.g. 10‐metre or 6‐minute walk) at 6 months.
Significant adverse events from the intervention.*
We evaluated QMT only when the trial did not use MMT for muscle testing.
Where relevant data were available we considered the costs of interventions in the Discussion.
*An `adverse event' is defined as an adverse outcome that occurs during or after the use of a drug or other intervention but is not necessarily caused by it (Cochrane 2015).
Search methods for identification of studies
Electronic searches
On 7 October 2014, the Trials Search Co‐ordinator searched the Cochrane Neuromuscular Disease Group Specialized Register, the Cochrane Central Register for Controlled Trials (CENTRAL, 2014, Issue 9 in the Cochrane Library), MEDLINE (January 1966 to September 2014), and EMBASE (January 1947 to September 2014). Detailed search strategies are in the appendices: Cochrane Neuromuscular Disease Group Specialized Register (Appendix 1), CENTRAL (Appendix 2), MEDLINE (Appendix 3), and EMBASE (Appendix 4).
We searched all databases from their inception to the present, and we imposed no restriction on language of publication.
In November 2014, we searched ClinicalTrials.gov (clinicaltrials.gov) and the World Health Organization International Clinical Trials Registry Platform (ICTRP) (who.int/trialsearch/) for ongoing trials and completed unpublished studies using the search term `inclusion body myositis'.
Searching other resources
We checked references in the identified trials and contacted trial authors to identify any additional published or unpublished data. In 1999, the review authors scanned conference abstracts including those of the American Academy of Neurology, the International Conference on Neuromuscular Diseases, the World Muscle Society, and the European Neurological Society for relevant studies but did not update this search.
Data collection and analysis
At least two review authors (from among JM, KJ, KL, MW, MR and RB) extracted data and resolved any discrepancies by discussion. We contacted the authors of the trials to provide missing data where possible.
Selection of studies
At least two review authors (from among MR, KJ, MW, and JM) examined the papers identified by the search strategy for studies eligible for inclusion. The review authors independently confirmed that studies were randomised or quasi‐randomised trials and that diagnostic criteria for IBM had been met.
Data extraction and management
At least five review authors (from among MR, KJ, KL, MW, JM, and RB) independently performed data extraction using a specially designed data extraction form. The review authors contacted authors of included trials to provide missing data where possible. One or two review authors checked and entered data into the Cochrane authoring and statistical software, Review Manager (RevMan) 5 (MR and KL) (RevMan 2014); another review author checked the data entry (KJ). A non‐conflicted review author performed data extraction independently if any review author had potential conflicts of interest, for example through involvement in an included study.
Assessment of risk of bias in included studies
Two review authors (MR and KJ) independently assessed the risk of bias in included studies using the following criteria: sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessors, incomplete outcome data, selective outcome reporting, and other risk of bias. The review authors identified high, low, or unclear risk of bias for each trial according to criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We used the `unclear' rating when there was insufficient information to reach a judgement or when, despite knowing what occurred in the study, the risk of bias remained unclear. We also examined whether studies included explicit diagnostic criteria, validation of outcome measurements, and power calculations to detect statistical benefit. Where we were uncertain, we contacted trial authors for clarification. The review authors resolved any disagreements over `Risk of bias' assessment by consensus. We conducted the review according to the published protocol (Rose 2014), reporting any deviations from it in Differences between protocol and review.
Measures of treatment effect
We analysed dichotomous data as a risk ratio with a 95% confidence interval (CI). We analysed continuous data as the mean difference, or standardised mean difference with 95% CI when outcomes were conceptually the same but measured in different ways. We calculated a treatment effect through random‐effects meta‐analysis, using RevMan 5 (RevMan 2014).
Unit of analysis issues
Included studies determined compound muscle strength using different muscle groups, which represented a potential unit of analysis error. The carry‐over effect of sequential intervention in cross‐over trials was another potential source of unit of analysis error. There was also a possible learning effect in the primary outcome of interest, muscle strength, that we would have considered in particular for cross‐over trials if they had provided data suitable for analysis.
Dealing with missing data
We contacted investigators in order to verify key study characteristics and to obtain missing numerical outcome data where possible (such as when a study was only available as an abstract). Where this was not possible, and we thought that missing data introduced serious bias, we explored the impact of including such studies.
Assessment of heterogeneity
We used the I² statistic to measure heterogeneity among the trials in each analysis. If we identified any substantial unexplained heterogeneity, we planned to report this and to explore the possible causes by prespecified subgroup analysis.
Assessment of reporting biases
We were not able to pool enough trials (that is more than 10) to create and examine a funnel plot to explore possible small‐study biases.
Data synthesis
We used a random‐effects model on the assumption that the included studies estimated different but related intervention effects. We applied a fixed‐effect model to further consider the presence of any heterogeneity among included studies. As the review included several comparisons that could not be combined in the same analysis, we reported the results for each comparison separately.
`Summary of findings' table
We created a `Summary of findings' table using the following outcomes.
Percentage change in muscle strength (using MMT or QMT) from baseline at 6 months.
Percentage change from baseline in muscle strength (using MMT or QMT) at 12 months.
Percentage change from baseline in muscle mass (by whatever method, e.g. MRI) at 6 months.
Percentage change from baseline in handgrip strength at 6 months.
Percentage change from baseline in timed walk (e.g. 10‐metre or 6‐minute walk) at 6 months.
Significant adverse events from the intervention.
We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of a body of evidence (studies that contribute data for the prespecified outcomes). We employed methods and recommendations described in Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), using GRADEpro software (GRADEpro 2014). We justified all decisions to down‐ or upgrade the quality of studies using footnotes and made comments to aid readers' understanding of the review where necessary. Three review authors (MR, KJ, and RB) completed this evidence grading, which all review authors checked and agreed on.
Subgroup analysis and investigation of heterogeneity
We were unable to perform subgroup analysis to investigate treatment effects in particular groups of participants because the included studies provided insufficient detail about the different participant groups. We were unable to use meta‐regression techniques to investigate heterogeneity.
Sensitivity analysis
We would have completed a sensitivity analysis on the basis of risk of bias and to further explore heterogeneity in the results if sufficient data (trials) had been available.
Results
Description of studies
The search strategies in the Appendices produced the following results: Cochrane Neuromuscular Disease Group Specialized Register 23 records, CENTRAL 26 records, MEDLINE 273 records, and EMBASE 95 records. In total, the review authors identified 24 studies, with 10 fulfilling the inclusion criteria; see Characteristics of included studies. We excluded 14 studies (4 ongoing) because they were not randomised controlled trials (RCTs); see Characteristics of excluded studies.
The interventions used among the 10 trials identified for inclusion were: intravenous immunoglobulin (IVIg) (Dalakas 1997; Dalakas 2001; Walter 2000); interferon beta‐1a (IFN beta‐1a) (Muscle Study Group 2001; Muscle Study Group 2004); methotrexate (MTX) (Badrising 2002); an anabolic steroid (oxandrolone) (Rutkove 2002); and arimoclomol (Machado 2013). All of these trials compared the intervention, used on its own, to placebo. The remaining two trials compared combination immunosuppressive therapy (MTX and anti‐T lymphocyte immunoglobulin (ATG) (Lindberg 2003); and MTX and azathioprine (AZA) (Leff 1993)) to an MTX treatment regimen.
As a protocol deviation, we included three studies that used clinicopathological diagnostic criteria not specified in Types of participants (Badrising 2002; Leff 1993; Walter 2000).
We identified three ongoing studies (one of which had three associated trial registrations) from ClinicalTrials.gov and the World Health Organization International Clinical Trials Registry Platform (EUCTR2007‐004359‐12‐IT; NCT00001265; NCT01423110). See Characteristics of ongoing studies.
Intravenous immunoglobulin versus placebo
Three trials compared IVIg with placebo (Dalakas 1997; Dalakas 2001; Walter 2000).
Dalakas 1997 block‐randomised 22 participants fulfilling the diagnostic criteria according to Griggs 1995 either to placebo (consisting of dextrose in half‐normal saline) or to 2 g/kg body weight IVIg monthly for 3 months. After a washout period of at least one month, the participants had the option of crossing over to the alternative treatment for a further three months. Nineteen of the participants were also being treated with prednisone or another immunosuppressant (MTX, AZA) prior to the study; only three participants continued with a low‐to‐moderate dose of prednisone during the trial. Assessments were made at baseline and at the end of each three‐month period, with both investigators and participants blinded to the treatment intervention. The study authors reported baseline mean total Medical Research Council (MRC) scores to be comparable.
Walter 2000 block‐randomised 22 participants (11 in each group) fulfilling diagnostic criteria similar to Griggs 1995. Participants were randomised to either placebo (1% human albumin in 2.5% glucose) or to IVIg at 2 g/kg body weight given over two to five days per month for six months, before crossing over to the other regimen for another six months. Nineteen of the participants had received various drugs prior to the study, including corticosteroids, AZA, and combined MTX and AZA. Six participants in each group remained on a constant, pretreatment medication, with nine receiving 2.5 mg to 15 mg daily corticosteroids and three receiving 4 mg to 10 mg daily corticosteroids and 100 mg to 150 mg daily AZA. All participants in both groups received physiotherapy once or twice weekly. The trial was double blinded with monthly outcome measurement from baseline to 12 months. Although the groups differed in baseline mean age (67 ± 12 years in the IVIg‐placebo group and 51 ± 11 years in the placebo‐IVIg group, the MRC sum scores and Neuromuscular Symptom and Disability Functional Scores showed no statistically significant between‐group differences at baseline.
Dalakas 2001 block‐randomised 37 participants (according to the flow chart in the study report) who fulfilled the diagnostic criteria for IBM according to Griggs 1995 either to placebo (consisting of dextrose in half‐normal saline) or two daily doses of 1g/kg body weight IVIg (2 g/kg bodyweight IVIg) monthly for three months. In addition, all participants in both groups received prednisone (tapered from 60 mg daily to 60 mg every other day). Assessments were made at baseline and at the end of each month of treatment for three months, with investigators and participants both blinded to intervention. At baseline, age and muscle strength (QMT and MRC sum scores) were comparable between treatment and placebo groups.
Interferon beta‐1a versus placebo
Two trials compared the use of IFN beta‐1a with placebo.
Muscle Study Group 2001 randomised 30 participants who had definite or probable IBM according to the criteria of Griggs 1995 into a double‐blinded, parallel‐group study. Sample size was chosen "to provide 85% power to detect a difference in tolerability of 95% in the placebo group versus 50% in the IFN beta‐1a group". The participants received either IFN beta‐1a or a matching placebo (lyophilised powder reconstituted with sterile saline). The dose of IFN beta‐1a was 15 μg/week initially and 30 μg/week from 4 weeks, administered by intramuscular injection once weekly for 24 weeks. Participants also received a 650 mg dose of paracetamol at the time of injection and repeated six‐hourly with a total of four doses. Trialists made assessments at baseline and at 4, 12, and 24 weeks. Baseline characteristics suggested between‐group similarity for age and average motor function; however, trial authors acknowledge a predominance of men in the treatment group.
The second trial performed by the same group recruited 30 participants (Muscle Study Group 2004). The study design and outcome measures were consistent with Muscle Study Group 2001, but the dosage of IFN beta‐1a was increased to 60 μg/week. Assessments were reported at baseline and at 4, 12, and 24 weeks; no primary outcome measure was specified. All reported baseline characteristics suggested that the two treatment groups were clinically similar. There was a predominance of men in the placebo group.
Methotrexate versus placebo
One double‐blinded trial randomised 44 participants to receive either MTX or an identical‐looking placebo (Badrising 2002). Sample size was chosen "to detect a difference of 100 Newtons (N) in mean [strength] changes or a clinically important stabilisation". Over a treatment period of 48 weeks, participants started on an initial dose of 5 mg a week that increased by 5 mg every 6 weeks up to 20 mg/week. To enhance blinding, the investigators reduced the dose by 2.5 mg without explanation for three months, and restored doses to 20 mg/week thereafter. The average dose was 14.6 mg/week in those who completed the trial and 14.0 mg/week in all treated participants. Forty‐two participants had a diagnosis of definite IBM and two a diagnosis of probable IBM, according to the European Neuromuscular Centre (ENMC) diagnostic criteria for IBM (Verschuuren 1997), elements of which are similar to those of Griggs 1995. Assessments were made at baseline, 22, and 48 weeks or immediately after withdrawal. The baseline characteristics suggested between‐group similarity. The study authors performed both an intention‐to‐treat analysis, carrying forward the last assessment, and a per protocol analysis of those who completed the study.
Combined immunosuppressive therapy: methotrexate and anti‐T lymphocyte immunoglobulin versus methotrexate
Lindberg 2003 was an open, randomised trial of 11 participants who fulfilled the morphological diagnostic criteria for IBM according to Griggs 1995. This trial compared 12 months' treatment with oral MTX 7.5 mg/week (MTX group, n = 5) with 12 months' MTX treatment preceded by 7 days of intravenous anti‐T lymphocyte immunoglobulin treatment (MTX + ATG group, n = 6). ATG doses were fixed for the first two days (5 mg/kg and 4 mg/kg body weight, respectively) and varied thereafter to keep the T lymphocyte counts between 50 x 106/L and 150 x 106/L. The total amount of ATG given over seven days varied between 24.8 mg/kg and 30.2 mg/kg body weight. Five participants in the MTX + ATG group and four participants in the MTX group also took prednisone (10 mg to 30 mg every second day) during the trial. Participants received methylprednisolone (125 mg injection) before the first ATG infusion. Assessments were made 2 months before commencement of treatment and at 0, 2, 4, 6, 9, and 12 months of treatment. Mean between‐group muscle strength was reported to be nearly equal at baseline.
Combined immunosuppressive therapy: methotrexate and azathioprine versus methotrexate (plus leucovorin)
Leff 1993 was an open, randomised trial of 11 participants who were diagnosed as having definite myositis by the Bohan and Peters' criteria (Bohan 1975), together with light microscopic changes consistent with biopsy‐proven IBM.
The trial authors compared six months of intravenous MTX with a combination treatment of oral azathioprine and MTX (AZA + MTX). For the MTX regimen, the investigators infused 0.5 g/m2 intravenously over one hour every two weeks; participants on intravenous MTX also received oral doses of 50 mg/m2 leucovorin rescue. Oral doses of AZA and MTX began at 50 mg/day and 7.5 mg/week, respectively, and gradually increased to a maximum of 150 mg/day and 25 mg/week, respectively, over the course of the first 12 weeks unless adverse events occurred. In addition, both treatment groups received prednisone, which was tapered to a small, alternate‐day dose of 0.25 mg/kg after one month. Participants only crossed over regimens if worsening or stabilisation occurred after six months of drug therapy. If improvement occurred after the first six months, the participants continued prednisolone and discontinued the trial therapy; such participants were kept under observation, and only when worsening occurred did they begin the other therapeutic regimen. The trial authors defined change in clinical status by categorical changes in MMT and activities of daily living (ADL) between the start and end of treatment. The trial authors did not report whether participants' baseline characteristics were similar.
Oxandrolone versus placebo
One trial compared oxandrolone, an anabolic steroid (a synthetic androgen), with placebo (Rutkove 2002). This double‐blinded study randomised participants to 20 mg/day oxandrolone orally (10 mg twice daily) (n = 10) or placebo (n = 9) for 12 weeks. After a washout period of two to four months, the groups crossed over to the alternate intervention for another 12 weeks. All participants fulfilled the Griggs 1995 criteria for definite IBM. Assessments were performed at baseline, at the end of the first intervention period, at the end of the washout period, and at the end of the second 12‐week intervention period (with additional serologic evaluation at 6 weeks of each treatment). Only 13 of the 19 participants completed both periods of the study, although the changes reported on oxandrolone were based on the 15 participants who completed active treatment, and the changes reported on placebo were based on the 14 participants who completed a placebo period. Baseline median muscle strength (whole‐body maximal voluntary isometric contraction testing and MMT scores) were significantly higher in the group receiving placebo for the first study period (P = 0.03 and P = 0.008, respectively).
Arimoclomol versus placebo
Machado 2013 compared the safety and tolerability of arimoclomol with placebo in participants who fulfilled the diagnostic criteria for IBM according to Griggs 1995. This double‐blinded trial randomised one group to 100 mg three times daily and the other to placebo for four months, with an eight‐month blinded follow‐up phase. Available data were insufficient to determine the between‐group similarity in baseline characteristics.
Risk of bias in included studies
We have reported `Risk of bias' assessments for each included study in Characteristics of included studies. Figure 1 summarises review authors' assessments for each `Risk of bias' domain for all trials. The overall risk of bias in the included studies was unclear in 6 of the 10 trials. The review authors evaluated one trial of MTX as at low risk of bias (Badrising 2002). We judged three other trials to be at high risk of bias: two open studies of multi‐agent treatment, Leff 1993 and Lindberg 2003, and Dalakas 1997, due to incomplete outcome data, selective reporting, and breaking of randomisation by elective cross‐over.
1.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study. Red (‐) = high risk of bias, yellow (?) = unclear risk of bias and green (+) = low risk of bias.
Insufficient information on blinding procedures was a common source of unclear risk of bias; this finding was particularly important because the primary outcome was a measure of muscle strength performance, likely to be influenced by a lack of blinding. Most studies also did not clearly demonstrate minimisation of the potentially confounding effects of previous or concurrent treatments.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
Intravenous immunoglobulin versus placebo
Three studies contributed data for this comparison (Dalakas 1997; Dalakas 2001; Walter 2000).
Percentage change from baseline in muscle strength at 6 months
In Dalakas 1997 (19 analysed participants), the investigators assessed muscle strength change after three months of the intervention (cross‐over study). The paper provided no statistical comparison between muscle strength in the IVIg and placebo groups at baseline. Since participants had the option to cross over, only the first period of the trial was randomised. We considered rescaling trial data (0 to 10 scale; maximum sum score = 200) to estimate the percentage change in muscle strength at six months despite an assumption of linear change. However, we were not able to perform secondary analysis because no standard deviation (SD) values were given. Primary data were not available for re‐analysis, and we could not accurately regenerate primary data from the graphs and tables. At three months, trial authors found a non‐significant gain in mean MRC points with IVIg and a non‐significant loss with placebo. The mean change in the IVIg group was 4.2 MRC points (range ‐16 to 39.8), whereas the mean change in the placebo group was ‐2.7 MRC points (range ‐10 to 8). Trial authors reported "the effect of IVIg did not differ significantly compared with placebo in overall muscle strength" using a per protocol analysis.
In Dalakas 2001 (36 analysed participants; 19 in treatment group), trialists assessed muscle strength change following three months of the intervention (parallel‐group study). We considered whether to rescale trial data (0 to 10 scale) to estimate the percentage change in muscle strength at six months. However, the maximum sum score was unclear, as the study authors described 12 muscle group actions but reported 13 muscle groups as assessed. Also, they reported muscle strength change from baseline separately for upper and lower extremities, despite the primary outcome measure being total‐body MRC sum scores. At three months, trial authors found no significant difference in mean muscle strength scores with IVIg compared with placebo. Trial authors concluded "the lack of improvement in strength, the primary endpoint of our study, was disappointing" based on a per protocol analysis. We intended to pool the data for upper and lower extremities, but primary data were unavailable for secondary analysis.
In Walter 2000 (20 analysed participants), the investigators assessed muscle strength change following six months of the intervention (cross‐over study). They reported comparable baseline muscle strength between IVIg and placebo groups based on MRC sum scores (P = 0.49). In the first period of the trial, there was a mean increase of 2.5 MRC points (0 to 6 scale; maximum sum score = 180) with IVIg from a baseline of 137.4 (± 28.3) to 139.9 (± 30.6); in the placebo group there was a mean increase of 4 MRC points from a baseline of 141.5 (± 33.8) to 145.5 (± 26.6) points. In the second period of intervention following cross‐over, there was a mean increase of 4.5 MRC points, from 145.5 (± 26.6) to 150 (± 25.3) with IVIg and 6.8 MRC points with placebo, from 139.9 (±30.6) to 146.7 (± 28.1). The trial publication did not clearly state whether the figures in parentheses are SD. Trial authors also provided the overall comparative data for IVIg‐placebo versus placebo‐IVIg groups, although this does not measure IVIg efficacy. Trial authors reported "there were no significant changes in MRC scales during IVIg treatment" based on a per protocol analysis of efficacy data. We intended to pool the data for percentage change in muscle strength with IVIg versus placebo, but primary data were unavailable for secondary analysis.
Percentage change from baseline in muscle strength at 12 months
No available data.
Percentage change from baseline in handgrip at 6 months
No available data.
Percentage change from baseline in muscle mass at 6 months
No available data.
Percentage change from baseline in timed walk at 6 months
No available data.
Significant adverse events from the intervention
In Dalakas 1997, two participants dropped out of the study before completing the first period, and a third completed the first period but refused to continue for reasons not stated. It is not clear if these dropouts were from the treatment or placebo group. In Dalakas 2001, there was one dropout from the placebo group due to death following a heart attack after the first infusion. In Walter 2000, the trial authors recorded two dropouts, but did not provide reasons for their discontinuation.
Dalakas 1997 and Dalakas 2001 did not report adverse events experienced by participants in sufficient detail for the review authors to analyse the data. In Dalakas 1997, trial authors reported that there were no serious side effects; some participants experienced a mild headache, but without specification of the intervention group. In Dalakas 2001, trial authors again noted no serious side effects in the IVIg‐randomised participants. Walter 2000 reported no serious adverse events during the trial, although two participants had headaches with raised body temperature (38°C), and two participants developed allergic exanthema of the skin.
Interferon beta‐1a versus placebo
Two studies contributed data for this comparison (Muscle Study Group 2001; Muscle Study Group 2004).
Percentage change from baseline in muscle strength at 6 months
In Muscle Study Group 2001 (29 analysed participants but 30 reported with an intention‐to‐treat (ITT) principle; 14 in treatment group), investigators assessed the muscle strength change following 24 weeks of the intervention (parallel‐group study). They tested a total of 34 muscle groups, and the text suggested a maximum MRC sum score of 170 (based on an MRC scale of 0 to 5). Trial authors generated a `composite' score, which is defined in relation to QMT strength scores as "the average number of SD units from predicted normal strength, given age, gender and height of the subject". The mean MMT changes of 0.03 (SD 0.16) increment with placebo and 0.06 (SD 0.15) decline with IFN beta‐1a appear to be consistent with a non‐significant change in mean number of SD units plus SD, rather than mean muscle strength scores plus SD. The results were in favour of placebo (MD ‐0.09, 95% CI ‐0.20 to 0.02) based on an ITT principle with no computation of missing data. However, the formulation of the composite MMT score was not clear. We intended to ascertain the percentage change in muscle strength for IFN beta‐1a versus placebo, but primary data were unavailable for secondary analysis.
In Muscle Study Group 2004 (28 analysed participants including carry‐forward of 1 participant's data and no computation of missing data from 2 participants; 15 in treatment group), the trialists assessed muscle strength change similarly following 24 weeks of the intervention (parallel‐group study). The dosage of IFN beta‐1a was double that used in the earlier trial, but study methodology was otherwise consistent. The mean MMT decreases of 0.08 (SD 0.21) with placebo and 0.08 (SD 0.22) with IFN beta‐1a again appeared to be consistent with a non‐significant change in mean number of SD units plus SD, although this was not clearly stated. The results were neither in favour of placebo nor IFN beta‐1a (MD 0.00, 95% CI ‐0.16 to 0.16).
As we were unable to ascertain percentage change in muscle strength, we decided to complete a pooled analysis of data from the two trials for change in normalised muscle strength sum scores. Overall, there was no significant benefit of IFN beta‐1a over placebo; the combined data (58 participants) produced an MD of ‐0.06 (95% CI ‐0.15 to 0.03) in favour of placebo, but non‐significant (Analysis 1.1; Figure 2). The result was not sensitive to the use of a fixed‐effect versus a random‐effects analysis.
1.1. Analysis.
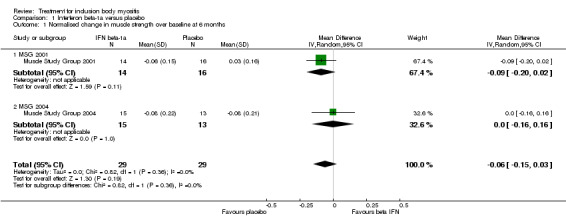
Comparison 1 Interferon beta‐1a versus placebo, Outcome 1 Normalised change in muscle strength over baseline at 6 months.
2.

Forest plot of comparison: 1 Interferon beta‐1a versus placebo, outcome: 1.1 Normalised change in muscle strength over baseline at 6 months.
Percentage change from baseline in muscle strength at 12 months
No available data.
Percentage change from baseline in handgrip at 6 months
Data were unavailable for secondary analysis of handgrip strength; no baseline data were provided to estimate percentage change in handgrip strength based on the mean change in each participant. However, the mean change in grip strength scores was reported: ‐0.72 kg (SD 1.99) with IFN beta‐1a and ‐0.72 kg (SD 1.46) with placebo (MD ‐0.16 kg, 95% CI ‐1.55 to 1.22) for Muscle Study Group 2001. Muscle Study Group 2004 reported a change in grip strength of +0.23 kg (SD 1.66) with IFN beta‐1a and ‐1.45 kg (SD 1.44) with placebo (MD 1.84 kg, 95% CI 0.46 to 3.21) in favour of IFN beta‐1a. In the absence of normalised or percentage change data, we did not perform pooled analysis of the data from these two trials.
Percentage change from baseline in muscle mass at 6 months
Muscle Study Group 2001 and Muscle Study Group 2004 measured lean mass as a surrogate for muscle mass.
In Muscle Study Group 2001, there was an increase in lean mass of 0.1% with IFN beta‐1a and a decrease of 0.85% with placebo. The mean reported change was +0.04 kg (SD 1.10) from a baseline of 40.1 kg (SD 7.5) with IFN beta‐1a, and ‐0.34 kg (SD 1.96) from a baseline of 39.8 kg (SD 9.4) with placebo (MD 0.64 kg, 95% CI ‐0.71 to 1.99, with MD adjusted for investigator effects in an analysis‐of‐variance model). When calculated by percentage change, the MD was 0.95%, 95% CI ‐1.86 to 3.76.
In Muscle Study Group 2004, there was a decrease in lean mass with IFN beta‐1a (‐2.18%) and placebo (‐1.77%). There was a decrease of 0.94 kg (SD 1.32) from a baseline of 43.2 kg (SD 10.2) in the IFN beta‐1a group, and a decrease of 0.82 kg (SD 1.79) from a baseline of 46.3 kg (SD 9.9) in the placebo group (MD ‐0.10, 95% CI ‐1.40 to ‐1.19, with MD adjusted as previously). When calculated by percentage change, the MD was ‐0.41%, 95% CI ‐3.02 to 2.20.
Overall, the positive gain in lean mass was negligible in the first study relative to the large SD values and an undefined dual energy X‐ray absorptiometry precision error; there was no positive gain of lean mass in the second study. Subsequently, IFN beta‐1a did not appear to influence lean mass after six months of treatment, irrespective of the therapeutic dose. The combined percentage change data from these 2 trials (58 participants) produced an MD of 0.22% (95% CI ‐1.69 to 2.13) in favour of IFN beta‐1a; the CI includes the possibility of an effect favouring either IFN beta‐1a or placebo (Analysis 1.2; Figure 3). The result was not sensitive to the use of a fixed‐effect versus a random‐effects analysis.
1.2. Analysis.

Comparison 1 Interferon beta‐1a versus placebo, Outcome 2 Percentage change over baseline in muscle mass at 6 months.
3.

Forest plot of comparison: 1 Interferon beta‐1a versus placebo, outcome: 1.2 Percentage change over baseline in muscle mass at 6 months.
Percentage change from baseline in timed walk at 6 months
No available data.
Significant adverse events from the intervention
In Muscle Study Group 2001, 1 participant from the IFN beta‐1a group withdrew from the study owing to death post‐surgery for colon cancer; although 29 people completed the trial, the study authors based subsequent analysis on all 30 participants (16 placebo, 14 IFN beta‐1a), carrying forward the results of the deceased. In Muscle Study Group 2004, three participants dropped out of the trial, two from the IFN beta‐1a group and one from the placebo group. The analyses carried forward data from one of the dropouts in the IFN beta‐1a group to the endpoint of the study. The reason given for one participant dropping out of the IFN beta‐1a group was post‐injection flu‐like reaction; the reason for the other IFN beta‐1a group dropout is not clear from the report.
Adverse events reported with IFN beta‐1a included one death post‐intervention with cause unlikely to be related to medication, flu‐like symptoms, arthralgia/myalgias, skin rash, injection site reaction, diarrhoea, headache, depression, chills/fever, and abdominal pain or classified as other/unspecified. Trial authors reported numbers of participants experiencing each type of adverse event and mean numbers of adverse events per participant. Muscle Study Group 2001 recorded a mean of 2.65 (no SD provided) participant‐reported adverse events with IFN beta‐1a compared with 2.25 (no SD) in the placebo group. In Muscle Study Group 2004, the trial authors recorded a mean of 4.00 (SD 2.83) participant‐reported adverse events with IFN beta‐1a, compared with 2.36 (SD 1.98) with placebo. Only Muscle Study Group 2004 reported the proportion of participants experiencing any type of adverse event following intervention: 79% of participants reported adverse events with placebo, and 81% with IFN beta‐1a.
Methotrexate versus placebo
Percentage change from baseline in muscle strength at 6 months
One study contributed data for this comparison (Badrising 2002). No six‐month data were available, although ITT and per protocol analyses shown in graphs indicated declines in compound QMT at 22 weeks in both MTX and placebo groups.
Percentage change from baseline in muscle strength at 12 months
Badrising 2002 (44 analysed participants; 21 in the treatment group using ITT analysis) reported mean changes in muscle strength sum scores using percentages, thus we presented the results as reported by trial authors. For the primary outcome measure of the trial, mean QMT sum scores declined in both groups by 48 weeks (‐0.2% in MTX and ‐3.4% in placebo) (MD 3.2%, 95% CI ‐2.5% to 9.1%). By limiting analysis to only those participants who fully completed the study (per protocol analysis), the trial authors found 0.9% improvement with MTX compared to a decline of 2.7% with placebo (MD 3.6%, 95% CI ‐3.3% to 10.7% in favour of placebo).
Mean MMT sum scores also declined in MTX and placebo groups: ‐0.5% for MTX and ‐2.0% for placebo (MD 1.5%, 95% CI ‐1.0% to 3.9%). Using the per protocol analysis, MMT sum score changes were ‐2.2% for MTX and ‐3.8% for placebo (MD 1.6%, 95% CI ‐2.3% to 5.4%).
Percentage change from baseline in handgrip at 6 months
No available data.
Percentage change from baseline in muscle mass at 6 months
No available data.
Percentage change from baseline in timed walk at 6 months
No available data.
Significant adverse events from the intervention
Eight of the 21 participants in the MTX group dropped out, compared with 1 of 23 in the placebo group (risk ratio (RR) 8.76, 95% CI 1.19 to 64.28, Analysis 2.1). The significantly higher dropout rate reported with MTX was due to nausea (n = 3), hair loss (n = 2), arthralgia (n = 2), and progressive muscle weakness (n = 1). One participant on placebo treatment discontinued trial medication because of progressive muscle weakness. Both cases of dropout for progressive muscle weakness could have been the result of disease progression, but trial authors classified them as adverse events. Trial authors also reported that four participants in the MTX group and one participant in the placebo group required dose reductions, although it is not clear whether these were the same people who discontinued with the trial.
2.1. Analysis.
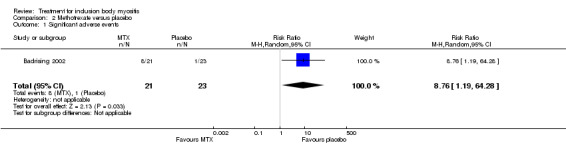
Comparison 2 Methotrexate versus placebo, Outcome 1 Significant adverse events.
Combined immunosuppressive therapy: methotrexate and anti‐T lymphocyte immunoglobulin versus methotrexate
One study contributed data for this comparison (Lindberg 2003).
Percentage change from baseline in muscle strength at 6 months
No available data.
Percentage change from baseline in muscle strength at 12 months
At 12 months, the MTX + ATG group showed a change of 1.4% (SD 9.8%) in compound QMT compared with ‐11.1% (SD 7.2%) in the MTX group (P = 0.021). In the MTX + ATG group, mean strength increased from 90.3 N (SD 15.3) to 91.6 N (SD 18.2), as compared to the MTX group, in which there was a decrease from 78.4 N (SD 33.3) to 71.4 N (SD 34.5). The MD was 12.50% (95% CI 2.43 to 22.57) in favour of MTX (Analysis 3.1).
3.1. Analysis.

Comparison 3 Anti‐T lymphocyte + methotrexate versus methotrexate, Outcome 1 Percentage change from baseline (QMT) at 12 months.
Percentage change from baseline in handgrip at 6 months
No available data. The review authors opted not to rescale trial data from 12 to 6 months due to the uncertainty of assuming a linear change between these time points.
Percentage change from baseline in muscle mass at 6 months
No available data.
Percentage change from baseline in timed walk at 6 months
No available data.
Significant adverse events from the intervention
Ten participants completed the trial, but the trial authors based the analysis on 11 participants by carrying forward the data taken from the last assessment of the 1 person who dropped out. The use of ATG in this trial was not complicated by any serum sickness or anaphylaxis. However, one participant in the MTX group developed severe pneumonia after 118 days of treatment and was withdrawn from the study.
Combined immunosuppressive therapy: methotrexate and azathioprine versus methotrexate (plus leucovorin)
One study contributed data for this comparison (Leff 1993). The trial authors measured change in muscle strength based on MMT but presented results categorically (improved, stabilised, or worse), which prevented any data extraction.
Percentage change from baseline in muscle strength at 6 months
No available data.
Percentage change from baseline in muscle strength at 12 months
No available data.
Percentage change from baseline in handgrip at 6 months
No available data.
Percentage change from baseline in muscle mass at 6 months
No available data.
Percentage change from baseline in timed walk at 6 months
No available data.
Significant adverse events from the intervention
Two of 11 participants discontinued combined AZA and MTX treatment due to side effects that were described as transient, reversible, and mainly gastrointestinal; these participants both underwent cholecystectomies for acute cholecystitis and were unable to resume drug treatment. A third participant did not cross over to MTX (plus leucovorin) due to pneumonitis. Another complication reported during the trial was a flareup of pre‐existing gout in one participant during MTX (plus leucovorin) treatment. Trial authors reported that most participants received the full six months of each regimen. Insufficient data were available to give actual numbers of participants from each group who experienced significant adverse events.
Oxandrolone versus placebo
One study contributed data for this comparison (Rutkove 2002). This was a cross‐over trial that reported the effects of 12 weeks' treatment with oxandrolone versus placebo. Overall changes were reported at the end of the trial for 15 participants who completed the oxandrolone phase and 14 participants who completed the placebo phase. The trial authors reported median values and interquartile ranges "because of concerns about the normality of the data". The trial authors expressed results as GEE (generalised estimating equation) estimates and standard errors, with the result that the available data were insufficient to calculate percentage change in outcome measures.
Percentage change from baseline in muscle strength at 6 months
Insufficient data.
Percentage change from baseline in muscle strength at 12 months
Insufficient data.
Percentage change from baseline in handgrip at 6 months
No available data.
Percentage change from baseline in muscle mass at 6 months
Insufficient data.
The analyses only excluded those participants who dropped out prior to completion of the first treatment period.
Arimoclomol versus placebo
One trial investigated the use of arimoclomol for treating IBM (Machado 2013). However, only the abstract was available at the time of this review. In the pilot study, 16 participants used arimoclomol (100 mg 3 times daily) and 8 participants received a placebo for 4 months. The abstract did not report numerical data suitable for inclusion in this review.
Discussion
Intravenous immunoglobulin versus placebo
We planned to perform a meta‐analysis of the IVIg trials using data reported for our primary outcome, muscle strength, albeit at different time points. However, none of the IVIg studies reported data in a form that could be combined at 3, 6, or 12 months. Dalakas 1997 presented change in MRC sum scores for participants at three months in graphical form; we did not feel confident to accurately convert the graphical data to numerical values for meta‐analysis. Additionally, we expected the need to extrapolate three‐month data to six months to amplify any existing inaccuracies. In order to perform our analysis, we would have needed to rescale the original three‐month trial data to six months, assuming a linear rate of change in strength over time. Dalakas 2001 provided summary data for upper and lower limbs, but calculation of a percentage change from these summary scores and extrapolation to six months would again be subject to large assumptions in terms of the effect size. The Walter 2000 cross‐over trial similarly did not provide data in a form suitable for meta‐analysis.
Walter 2000 included 2 groups of 11 participants of different mean ages (67 years and 51 years); in theory, the cross‐over design of the trial should negate any baseline differences seen between the two groups. However, we were unable to perform subgroup analysis with respect to age because of insufficient data. Subsequently, we cannot rule out the influence of faster disease progression in the older subgroup in relation to sarcopenia and enhanced mitochondrial dysfunction, as proposed previously (Dalakas 2001; Santorelli 1996). Exploring the relationship between inflammation, mitochondrial dysfunction, and muscle atrophy and determining the rate of IBM progression could be essential for understanding any clinically relevant change with treatment (Rygiel 2014); additionally, we may need to consider the sexual dimorphic effect of ageing in relation to muscle protein synthesis in IBM, as postulated in the study of older adults (Smith 2012). While an ageing effect on our reviewed outcomes cannot be confirmed, the small, non‐significant improvements in muscle strength with both IVIg and placebo may indicate a general learning effect in strength measurement as a proxy measure for disease progression; Neuromuscular Symptom and Disability Functional Score changes showed statistically significant improvement with IVIg only. We planned to perform subgroup meta‐analyses for factors such as age and carry‐over effect, but insufficient data were available.
In terms of the intervention, Dalakas 2001, unlike the other two trials, provided the IVIg and non‐IVIg groups with equal doses of prednisone. The supplementation with prednisone was provided on the basis that the combination of steroid with IVIg might have a synergistic effect in improving muscle strength, based on studies in dermatomyositis and Guillain‐Barré syndrome (Dalakas 1993; Dutch GBS 1994). However, unlike for IBM, for dermatomyositis and Guillain‐Barré syndrome, IVIg alone is known to be effective. For the purposes of meta‐analysis, we would have to assume that prednisone did not contribute any effect for either the IVIg or non‐IVIg treatment group.
In terms of study design, the three trials used two different MRC scales, and they did not assess the same muscle groups or muscle group actions. Dalakas 1997 involved elective cross‐over in the second phase of the trial, such that we could consider only the first phase of intervention for meta‐analysis (Elbourne 2002). The inconsistency in methodology between trials is particularly important because IBM affects different muscles to a variable extent. As a result of the variable muscle involvement in IBM, IVIg might be beneficial for some muscles more than others at any given time point in the disease.
When summarising our findings, we assessed the quality of evidence for the effects of IVIg on muscle strength at six months as very low due to selective reporting and other issues of trial design (high risk of bias in one of the trials and an unclear overall risk of bias in the other two trials).
In conclusion, while marginal increments in muscle strength were identified in two of three trials of IVIg (Dalakas 1997; Walter 2000), we could not determine an overall effect of IVIg versus placebo due to inconsistencies in trial methodology and reporting (see Table 1).
None of the IVIg trials included a statistical analysis of the incidence of significant adverse events with intervention to facilitate full evaluation of treatment effect. According to Meyler's Side Effects of Drugs (Chalker 2000; Dukes 2000), current Ig preparations cause about 3% to 4% of people to experience adverse reactions. Other adverse events associated with IVIg include mild influenza‐like illness, sweating, hypotension, chills, fever, nausea, and vasomotor reactions. More serious adverse effects include anaphylactic reactions; however, these are very rare and may occur in as few as 1 in 6000 people (Aronson 2006). Stroke and myocardial infarction have been reported after high‐dose IVIg, as a result of increased plasma viscosity. At high doses, neutropenia and disseminated intravascular coagulation have also been reported, and very rarely, acute renal failure (Aronson 2006). Walter 2000 reported some participants developing headache or raised body temperature, and two participants developed an allergic reaction.
Interferon beta‐1a versus placebo
Neither of the IFN beta‐1a studies showed a significantly greater benefit with IFN beta‐1a over placebo. We were able to perform a meta‐analysis of parallel‐group trials comparing different doses of IFN beta‐1a with placebo. The standardised trial procedures and reporting across both trials made pooling of data from Muscle Study Group 2001 and Muscle Study Group 2004 possible. The pooled analysis for normalised muscle strength change from baseline produced a MD in compound MMT in favour of placebo (moderate‐quality evidence), but with CIs including the possibility of an effect in either direction (Analysis 1.1; Figure 2; Table 2). The forest plot for lean mass, as surrogate for muscle mass (Figure 3), also did not show an effect in favour of IFN beta‐1a (moderate‐quality evidence).
Despite some reported improvement in grip strength with IFN beta‐1a treatment, we could not assess the quality of this evidence because the available data were incomplete.
Neither of the IFN beta‐1a trials included a statistical analysis of the incidence of significant adverse events to facilitate full evaluation of the treatment effect. Among the participants who took 30 µg doses of IFN beta‐1a, 84% reported adverse events (Muscle Study Group 2001); this figure was approximately 81% in participants who took 60 µg doses of IFN beta‐1a (Muscle Study Group 2004). Flu‐like reactions following injection were the most commonly specified complaint with high‐dose treatment, while diarrhoea was the most commonly specified adverse event with low‐dose treatment (Muscle Study Group 2001; Muscle Study Group 2004). Rarer adverse events associated with IFN beta‐1a can include mood and personality changes, suicide attempts, hepatitis, and thyroid dysfunction (Aronson 2006). Such an adverse event profile and the large percentage of users experiencing more minor adverse events may give rise to problems with compliance over a longer period. However, despite the high frequency of adverse events seen in these two trials, none was assessed as significant enough to require dose reduction (Muscle Study Group 2001; Muscle Study Group 2004).
Methotrexate versus placebo
A single RCT provided moderate‐quality evidence of no significant effect of MTX on muscle strength at 12 months; the trial was insufficiently powered to exclude a possible benefit from MTX (see Table 3). Approximately 38% of participants who were on MTX reported adverse events (Analysis 2.1), and there was a statistically significant dropout rate in the MTX treatment group (moderate‐quality evidence). A larger or longer trial of MTX could be problematic in terms of compliance (Badrising 2002). The most common adverse events caused by MTX are nausea, vomiting, alopecia, oral mucositis, or effects of myelosuppression (Aronson 2006). While there are pharmacological agents to reduce or alleviate common adverse events, polypharmacy may also affect compliance.
Combination therapy in the treatment of IBM
The benefit of combination therapy with MTX and ATG, Lindberg 2003, or MTX and AZA, Leff 1993, remains unclear in the treatment of IBM. Combined MTX and ATG appeared to show some benefit, but the evidence, based on a small, open‐label trial, is very low quality and should be interpreted with caution (see Table 4) (Lindberg 2003). Aside from risk of bias issues, any positive effect of ATG combined with MTX versus MTX alone could either be due to the effects of ATG alone or to its effects when combined with MTX. Adverse effects known to be associated with ATG include "leukopenia and thrombocytopenia, fever, arthralgia, rash, urticaria, hepatotoxicity, hyperglycaemia, hypertension, and diarrhoea" (Aronson 2006). Serum sickness can also occur later. However, none of these potential adverse effects was reported in the trial; only a single case of pneumonia was reported.
In the only trial that looked at AZA and MTX in combination (Leff 1993), we could obtain no quantitative data (see Table 5). Similar to Lindberg 2003, this trial was open label and had a small number of participants (very low‐quality evidence). As a combination therapy, there was a risk of exposure to the effects of both drugs in the combination. Adverse events on the regimen that included AZA were gastrointestinal symptoms, acute cholecystitis, and a case of pneumonitis. In the MTX alone group, the authors reported only a flareup of pre‐existing gout; other adverse events known to occur with AZA alone include fever, nausea, leukopenia, thrombocytopenia, and anaemia (Aronson 2006). Neither Lindberg 2003 nor Leff 1993 provided a statistical analysis of the incidence of significant adverse events with the intervention to facilitate a full evaluation of the treatment effect.
Oxandrolone versus placebo
In comparing oxandrolone with placebo, the trial authors highlighted that small numbers of participants and a relatively short trial duration restricted the interpretation of results (Rutkove 2002). The trial authors also considered that between‐group differences in baseline characteristics may have had some confounding effects. The trial authors reported a close‐to‐significant treatment effect for improving whole body strength with oxandrolone at 12 weeks. However, we noted that the methods of analysis (generalised estimating equation) were unusual for this trial design, and the power of the test for carry‐over effects was low. Taking into account these and the other study limitations, results should be interpreted with caution (very low‐quality evidence).
In terms of adverse events, anabolic steroids have some androgenic activity that can give rise to acne and other signs of virilisation and may affect lipoprotein profiles. As the associated androgenic activity is weak, these adverse events are not common. However, gynaecomastia has occurred with the long‐term use of anabolic steroids as a growth promoter in boys (Aronson 2006). Withdrawal of high doses of anabolic steroids can give rise to menopause‐like symptoms (Chalker 2000; Dukes 2000). Rutkove 2002 reported no significant adverse events, which was perhaps related to the low doses used over a relative short period of time but, as with all other analysed trials, the investigators included no definition of what constituted an adverse event in order to evaluate treatment effect. If anabolic steroids are associated with few or no significant adverse events in practice, possible therapeutic potential might justify further trials to explore their use in treating IBM. Conversely, more adverse events and issues with compliance may be anticipated in a longer trial of anabolic steroids.
Arimoclomol versus placebo
At the time of review, we did not attempt to evaluate the effects of arimoclomol for treating IBM because the relevant data were not available for systematic review. The trial was only available in abstract (Machado 2013), and it was powered to assess safety and tolerability, not treatment effect.
The lessons from these trials
This systematic review of treatment for IBM identified nine analysable RCTs and one RCT published in an abstract only. In terms of determining a treatment effect, only 2 of the included trials reported power calculations, and we evaluated all 10 trials as underpowered to detect a statistically significant effect. One trial that included power calculations was primarily a safety and tolerability study, rather than an efficacy study (Muscle Study Group 2001). The other trial indicated multiple reasons for the lower‐than‐expected power for their study, including rate of decline in the placebo group; variability in QMT strength measures; and a higher‐than‐expected participant dropout rate (Badrising 2002). The largest included RCT had only 22 participants in the treatment group (Walter 2000). The largest analysed treatment group included only 21 participants, using an intention‐to‐treat analysis (Badrising 2002), and 20 participants with a per protocol analysis of the primary outcome measure (Walter 2000). Rose 2001 calculated that each group of a placebo‐controlled trial needed 94 participantsto have 90% power to detect a 4% difference in mean change in muscle strength between two groups over a 6‐month period. This 4% larger difference over placebo was determined to be equivalent to arresting disease progression with the drug on trial. Based on trial experience, Muscle Study Group 2004 calculated that a 2‐arm, 6‐month intervention would require 208 participants per group to detect the arrest of disease progression using QMT with 90% power, a 2‐tailed t‐test, and significance level of 5%.
Multicentre recruitment, pooling of trial results, or both, might allow for the accumulation of sufficient data for a more robust answer as to the efficacy of treatment for IBM. However, we still expect differences in both methodologies and the type of intervention to affect treatment effect estimates and the validity of results. Across the studies included in this review, meta‐analysis was compromised by the fact that outcome measurement was not standardised to measure the cardinal effects of interest, namely muscle weakness, atrophy, and functional impairment. Additionally, some studies used normalised MDs for assessing change in muscle strength.
For muscle strength testing, it would be helpful to standardise appropriate test methodology (for example, manual or quantitative measurements (or both), specification and number of muscles or muscle actions tested, and the detection of a minimal clinically important difference) for use in research trials. The potential for harm resulting from treatment also needs to be carefully considered; none of the included studies defined what constituted an adverse effect, although drug treatments were associated with adverse effects of variable severity. Most of the studies also did not specify the methods used to monitor adverse effects. Analysis of treatment effect can risk bias towards a focus on favourable outcome measures in the absence of sufficient information on conduct and reporting of adverse events (Loke 2007).
None of the completed trials made reference to responsiveness or a minimal clinically important difference for any outcome measure in relation to disease progression. The validation of trial outcome measures, where reported, was also not performed specifically in an IBM population.
In terms of comparing future clinical trials in IBM, it will be important to minimise the differences in inclusion and exclusion criteria applied; this review found that trials used a range of criteria regarding comorbidity and concomitant treatment that could have a fundamental impact on study outcomes. Some have argued that insistence on pathological criteria for the inclusion of IBM in clinical trials may result in such trials attempting to treat participants who have more advanced, and therefore inherently less treatable, disease. Currently proposed diagnostic criteria reduce the emphasis on pathological criteria with the aim of allowing recruitment of participants earlier in the course of their disease. However, this strategy is subject to verification, and currently there are no trials that apply these newer criteria for IBM.
Costs
UK costs for treatment with the interventions used in the included studies would be:
Interferon beta‐1a (Avonex): injection, 60 μg (12 million units)/mL, net price 0.5 mL (30 μg, 6 million‐unit) prefilled syringe = GBP 163.50 (BNF 2014).
Methotrexate: tablets, 2.5 mg, net price 24‐tablet pack = GBP 2.22; 10 mg, net price 100‐tablet pack = GBP 37.06 (BNF 2014).
Azathioprine: tablets, 50 mg, 56‐tablet pack = GBP 3.42 (BNF 2014).
Intravenous immunoglobulin: based on 2 g/kg in 70 kg man = GBP 3906 to 4900 (DH 2011).
Oxandrolone: a price from 2000 of USD 4 per 2.5 mg tablet (Beaston‐Blaakman 2007). In the UK, injection, nandrolone decanoate 50 mg/L, net price 1 mL ampoule = GBP 3.17 by deep intramuscular injection, 50 mg every 3 weeks (BNF 2014).
Intervention treatment costs per person over a six‐month period were estimated to be greatest for IVIg (approximately GBP 15,000) followed by IFN beta‐1a therapy, and with markedly lower costs for oxandrolone by intramuscular injection, AZA, and MTX treatment.
Potential biases in the review process
We chose to express the primary outcome as the percentage change in compound MMT over time because it is a widely used approach to muscle strength assessment in clinical research. However, percentage change calculation was rarely feasible using the available data, and two trials reported normalised data instead. Our percentage figures related to group‐level changes, but these are likely to be different from those calculated using absolute data from individuals. We expected the data to be subject to uncertainty in relation to clinical and statistical heterogeneity and significance. Using absolute changes would be preferable, but these were not retrievable across all trials in the review. Also, to calculate mean change in absolute MMT scores might have required that only those muscles tested by all studies and scaled to the same MRC grading were included in analysis.
Another potential bias in the review process was in the type of selected outcome measures. Our selected functional impairment outcome measures were restricted to specific single‐item tasks, while inflammatory and pathological biomarkers were not analysed at all. Nevertheless, our data extraction did suggest that included trials inconsistently assessed or reported those additional functional, inflammatory, and pathological outcomes.
Some of the review authors were investigators in included trials. A non‐conflicted review author performed independent data extraction if any review author had potential conflicts of interest, for example through involvement in an included study.
A further limitation of the review was that the review methods were unlikely to adequately detect serious, rare adverse events. We therefore discussed adverse events described in other sources in the Discussion.
Agreements and disagreements with other studies or reviews
The results of this systematic review confirmed previous observational study and review findings that there are no established, evidence‐based treatments for IBM as yet (Benveniste 2011; Breithaupt 2013; Machado 2013b). The small sample sizes and short duration of clinical trials are recognised as major limitations in the evaluation of treatment efficacy in this muscle condition (Breithaupt 2013; Fergusson 2005). In terms of assessment, MMT scores of muscle strength are widely used in research and clinical practice, but further development of sensitive outcome measures is advocated (Breithaupt 2013; Machado 2013). Studies included in this review used different methodological and analytical approaches to assess muscle strength and applied a range of secondary outcome measures. Such variations in methods and outcome measures hindered the pooling of trial data to contribute to the evidence base. As part of a systematic review in neurological conditions, Fergusson 2005 analysed the same three IVIg trials for IBM as in our review; like us, they were unable to come to a conclusion about the efficacy of IVIg from the available data. Fergusson 2005 similarly elected not to perform pooled analysis due to between‐trial differences in methodology. Interestingly, the benefit of IVIg in chronic inflammatory demyelinating polyneuropathy (CIDP) was identified using a disability scale, as was the benefit Walter 2000 showed for IVIg in IBM, using the Neuromuscular Symptom and Disability Functional Score. Such evidence perhaps argues for the use of disability scales in future IBM trials. However, Fergusson 2005 also emphasised that evidence of benefit still does not necessarily support IVIg as a first‐line treatment owing to other factors, including adverse events and cost.
Evidence of mitochondrial abnormalities in IBM that are in excess of those seen with normal ageing may suggest another range of therapeutic options, but there are currently no known effective treatments for primary mitochondrial disease (Pfeffer 2012). Similarly, currently there are no drugs with the therapeutic potential to arrest or slow down the degenerative pathology seen in IBM (Breithaupt 2013). A common issue highlighted across Cochrane intervention reviews in muscle disease is the quality of study design and paucity of RCTs, emphasising ongoing problems with sample size and risk of bias (Hill 2004; Pfeffer 2012; Voet 2013). The validity of the evidence base also appears to be limited by a lack of standardisation in the collection, evaluation, and reporting of data. This systematic review identified specific quality issues in clinical trials of people with IBM, which should encourage investigators to validate outcome measures and ensure standardisation of trial procedures.
Only one of the studies included in this review measured swallowing function, which we did not include as a predefined outcome measure. In the future a Cochrane review of treatment for swallowing difficulties in chronic muscle disease will include IBM (Hill 2004). In terms of ongoing drug trials for IBM, we identified an RCT of bimagrumab (BYM338), which has been developed to target molecular pathways involved in muscle growth, and which we may include in a future review. An RCT of simvastatin is also pending completion.
Authors' conclusions
Implications for practice.
Trials of interferon beta‐1a and methotrexate provided moderate‐quality evidence of having no effect on the progression of sporadic inclusion body myositis although we cannot exclude clinically relevant effects. An open trial of anti T‐lymphocyte immunoglobulin combined with methotrexate versus methotrexate provided very low‐quality evidence in favour of the combined therapy, based on the percentage change data given. We were unable to draw conclusions from trials of intravenous immunoglobulin, oxandrolone, and azathioprine plus methotrexate versus methotrexate. Overall trial design limitations and selective reporting made it difficult to say whether or not any of the drug treatments were effective in arresting or slowing disease progression. Any decision to prescribe these treatment regimens in the absence of evidence for or against benefit will need to consider the potential adverse effects and the cost of medication.
Implications for research.
More randomised controlled trials are needed on which to base treatment decisions. Such trials will need to recruit larger numbers and be of longer duration than has hitherto been the case, in order to derive definite conclusions as to the benefit or otherwise of any intervention. Standardisation of the trial protocols using agreed diagnostic criteria and validated, responsive outcome measures for such trials is also necessary. Finally, we recommend a cost‐utility analysis to assist decision‐making in the treatment of IBM.
What's new
| Date | Event | Description |
|---|---|---|
| 7 July 2015 | Amended | Minor typographical corrections |
History
Protocol first published: Issue 2, 1999 Review first published: Issue 6, 2015
| Date | Event | Description |
|---|---|---|
| 30 June 2015 | Amended | Conflict of interest corrected |
| 8 February 1999 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
Angela Gunn, the Trials Search Co‐ordinator of the Cochrane Neuromuscular Disease Group conducted database searches.
This project was supported by the National Institute for Health Research via Cochrane Infrastructure funding to the Cochrane Neuromuscular Disease Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health. The Cochrane Neuromuscular Disease Group is also supported by the MRC Centre for Neuromuscular Disease.
Appendices
Appendix 1. Cochrane Neuromuscular Disease Group Specialized Register (CRS) search strategy
#1 myositis NEAR "inclusion body" [REFERENCE] [STANDARD] #2 (myositis NEAR "inclusion body") AND (INREGISTER) [REFERENCE] [STANDARD]
Appendix 2. CENTRAL search strategy
#1 "inclusion body" NEAR myositis #2 MeSH descriptor Myositis, Inclusion Body, this term only #3 (#1 OR #2)
Appendix 3. MEDLINE (OvidSP) search strategy
Database: Ovid MEDLINE(R) <1946 to September Week 4 2014> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 randomized controlled trial.pt. (389226) 2 controlled clinical trial.pt. (89867) 3 randomized.ab. (285185) 4 placebo.ab. (151031) 5 drug therapy.fs. (1748071) 6 randomly.ab. (201598) 7 trial.ab. (296312) 8 groups.ab. (1285103) 9 or/1‐8 (3290339) 10 exp animals/ not humans.sh. (4016034) 11 9 not 10 (2802960) 12 exp Myositis, Inclusion Body/ or inclusion body myositis.tw. (1362) 13 11 and 12 (285) 14 remove duplicates from 13 (273)
Appendix 4. EMBASE (OvidSP) search strategy
Database: Embase <1980 to 2014 Week 40> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 crossover‐procedure.sh. (40306) 2 double‐blind procedure.sh. (115609) 3 single‐blind procedure.sh. (18869) 4 randomized controlled trial.sh. (350916) 5 (random$ or crossover$ or cross over$ or placebo$ or (doubl$ adj blind$) or allocat$).tw,ot. (1065137) 6 trial.ti. (163276) 7 or/1‐6 (1198699) 8 (animal/ or nonhuman/ or animal experiment/) and human/ (1290984) 9 animal/ or nonanimal/ or animal experiment/ (3256545) 10 9 not 8 (2727334) 11 7 not 10 (1100982) 12 limit 11 to embase (913119) 13 exp Inclusion Body Myositis/ or inclusion body myositis.tw. (1908) 14 12 and 13 (97) 15 remove duplicates from 14 (95)
Appendix 5. Trials registry search strategy
inclusion body myositis
Data and analyses
Comparison 1. Interferon beta‐1a versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Normalised change in muscle strength over baseline at 6 months | 2 | 58 | Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.15, 0.03] |
| 1.1 MSG 2001 | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐0.20, 0.02] |
| 1.2 MSG 2004 | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.16, 0.16] |
| 2 Percentage change over baseline in muscle mass at 6 months | 2 | 58 | Mean Difference (IV, Random, 95% CI) | 0.22 [‐1.69, 2.13] |
| 2.1 MSG 2001 | 1 | 30 | Mean Difference (IV, Random, 95% CI) | 0.95 [‐1.86, 3.76] |
| 2.2 MSG 2004 | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐0.41 [‐3.02, 2.20] |
Comparison 2. Methotrexate versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Significant adverse events | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 8.76 [1.19, 64.28] |
Comparison 3. Anti‐T lymphocyte + methotrexate versus methotrexate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Percentage change from baseline (QMT) at 12 months | 1 | 11 | Mean Difference (IV, Fixed, 95% CI) | 12.5 [2.43, 22.57] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Badrising 2002.
| Methods | Randomised, placebo‐controlled, parallel‐group, double‐blind trial | |
| Participants | 44 randomised participants (11 female) Treated group mean age: 68 ± 8 years; mean duration of symptoms: 9 ± 5 years Placebo group mean age: 69 (± 7) years; mean duration of symptoms: 11 (± 7) years Inclusion criteria: diagnostic criteria for definite or probable IBM; sufficient residual muscle strength to evaluate changes; absence of risk factors for MTX‐induced toxicity; no use of immunosuppressive therapy for at least 6 weeks before the study; no previous use of MTX; no use of medication interfering with MTX pharmacokinetics or pharmacodynamics; absence of severe dysphagia interfering with oral medication use. Exclusion criteria: unspecified. |
|
| Interventions | MTX versus placebo for 48 weeks | |
| Outcomes |
Primary outcome measure(s)
14 muscle groups were tested, but the actual muscles tested was not reported. Secondary outcome measure(s)
32 muscle groups were tested by MMT using a 5‐point MRC scale, but the actual muscles tested was not reported.
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The investigators describe a random component in the sequence generation process: "Patients were randomly assigned, using a computer‐generated schedule...The randomization schedule used random numbers in permuted blocks of 4". |
| Allocation concealment (selection bias) | Low risk | Central allocation: "The code was concealed by the pharmacy and broken after assessment of all patients". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding procedures undertaken following allocation concealment: "To enhance blinding, all patients were requested to decrease their 20mg dosage by 2.5mg without explanation after routine laboratory evaluations for 3 months. After blood assessments, the dosage was restored to 20mg per week". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment described following allocation concealment: "A blinded assessor (JV) monitored patients with regard to treatment schedules...Another blinded assessor (UB) evaluated the QMT and MMT measurements and patients' opinions concerning the state of muscle weakness". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of participants randomised and reasons for missing data stated; all subjects including dropouts analysed with average values and data variance clearly specified. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but outcomes prespecified in the methods are all reported in the results. |
| Other bias | Low risk | Minimisation of other treatment effects clearly specified: "no use of immunosuppressive therapy for at least 6 weeks before the study, no previous use of MTX, no use of medication interfering with MTX pharmacokinetics or pharmacodynamics, and absence of severe dysphagia interfering with oral medication use". |
Dalakas 1997.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over study | |
| Participants | 19 randomised participants (gender unspecified) IVIg group mean age: 61.2 (42 to 74) years; mean disease duration: 5.6 (3 to 10) years Placebo group mean age: 66.1 (35 to 76) years; mean disease duration: 7.4 (4 to 16) years Inclusion criteria: diagnostic criteria of IBM; active disease characterised by progressive muscle weakness; impaired ability to perform fully the ADL; absence of another systemic illness. Exclusion criteria: coronary artery disease; immunoglobulin A deficiency; kidney dysfunction; bedridden patients. |
|
| Interventions | IVIg versus placebo for 3 months | |
| Outcomes |
Primary outcome measure(s)
MMT was completed for 26 muscle groups and muscle group actions bilaterally: deltoid; biceps brachii; triceps brachii; brachioradialis; wrist extensors; wrist flexors; iliopsoas; gluteus maximus; quadriceps femoris; hamstrings; neck, finger and foot extension and flexion. However, analysis was completed on only 10/26 muscle groups as data were not always available. The 10 selected muscle groups were graded using a modified (0 to 10) MRC scale (Brooke 1983), with a total maximal MRC score of 200. Secondary outcome measure(s)
Swallowing function was also investigated by ultrasound assessment of the duration of both wet and dry swallowing. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The investigators describe a random component in the sequence generation process: "The patients were assigned to receive IVIg or placebo by a block‐randomization procedure". |
| Allocation concealment (selection bias) | Low risk | Central allocation: "Randomization was performed at the pharmacy". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement: "The principal investigator, the physicians, nurses, physical therapists, and statistician were unaware of which treatment was administered" but "Sixteen of the 19 patients correctly identified the period during which they received placebo or IVIg". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement: blinding not described fully following allocation concealment, as above. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No SD values included to assess for a clinically relevant bias in observed effect size. No statistical analysis for the incidence of adverse events was completed. |
| Selective reporting (reporting bias) | High risk | Data largely presented graphically and with discrepancies between data points and figures reported elsewhere. No study protocol available. |
| Other bias | High risk | Randomisation was broken by giving participants the option to cross over intervention. There was also a potential carry‐over effect of previous treatment(s): "Nineteen of the patients had been treated previously with high‐dose prednisone or therapeutic doses of another immunosuppressant (methotrexate, azathioprine) for at least 4 to 6 months." There was also a minimum washout period of 1 month, which may not be long enough to exclude a carry‐over effect. |
Dalakas 2001.
| Methods | Randomised, double‐blind, placebo‐controlled study | |
| Participants | 37 randomised participants (gender unspecified) Treated group mean age: 68.21 (no SD or range) years Placebo group mean age: 68.35 (no SD or range) years. Duration of symptoms not given. Inclusion criteria: diagnostic criteria for sporadic IBM; active disease characterised by progressive muscle weakness that impaired ability to perform independently various activities of daily living such as walking without falls, dressing, buttoning, or climbing up stairs; ambulatory independently or with assistance Exclusion criteria: wheelchair‐bound patients; coronary artery disease; immunoglobulin A deficiency; kidney dysfunction; any systemic illness. |
|
| Interventions | IVIg and prednisone versus placebo and prednisone for 3 months | |
| Outcomes |
Primary outcome measure(s)
MMT was completed for 13 muscle groups and muscle group actions bilaterally (although 12 specified): deltoid; biceps brachii; triceps brachii; brachioradialis; wrist extensors; wrist flexors; iliopsoas; gluteus maximus; quadriceps femoris; hamstrings; foot extension or flexion. Muscle strength was graded on a modified (0 to 10) MRC scale (Brooke 1983).
QMT was examined for the following muscle group actions: shoulder abduction; forearm flexion and extension; hip flexion; leg extension and flexion; foot extension. Secondary outcome measure(s)
Histological features were also assessed in repeated biopsies. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The investigators describe a random component in the sequence generation process: "The patients were assigned to receive IVIg or placebo by a block‐randomization procedure". |
| Allocation concealment (selection bias) | Low risk | Central allocation: "Randomization was performed at the pharmacy". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement: "The principal investigator, the physicians, nurses, physical therapists, and statisticians were unaware of which type of the IV infusion was administered" and based on participants' own assessment "no apparent signs to unblind any of the patients were observed." However, only the assessor of QMT and not MMT is confirmed to have remained blinded, suggesting that blinding of key personnel could have been broken. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement: as above, only the assessor of QMT is confirmed to have "remained blinded". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of participants randomised and reasons for missing data stated; 1 dropout (died from myocardial infarction on placebo infusion) not analysed. No statistical analysis for the incidence of adverse events was completed. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but outcomes prespecified in the methods are all reported in the results. |
| Other bias | Unclear risk | Insufficient information to permit judgement, but potential carry‐over effect of previous treatment(s): "Several patients had been treated previously with high‐dose prednisone or therapeutic doses of another immunosuppressant (methotrexate, azathioprine), but they had not been taking any such medicine up to 3 months before enrollment". |
Leff 1993.
| Methods | Open, randomised, cross‐over trial | |
| Participants | 11 randomised participants (2 female) Mean age: 54 ± 8 years (calculated from table). Duration of symptoms not given. Inclusion criteria: Bohan and Peter's criteria for definite myositis and biopsy‐proven IBM with typical light microscopic changes; signs of active inflammatory muscle disease; progressive weakness refractory to therapy; weak enough to have at least 2 muscle groups graded 3 or less on a scale of 0 to 5 by manual muscle testing; reduction of at least 1 functional level below normal in at least 1 activity group on an assessment of the ADL for functional capabilities. Exclusion criteria: severe intercurrent illness; cancer; infection; past history of alcohol abuse; pregnancy; abnormal renal or hepatic function. "Concurrent use of alcohol or nonsteroidal anti‐inflammatory drugs was forbidden". |
|
| Interventions | Azathioprine and MTX versus MTX for 6 months | |
| Outcomes |
Investigators graded MMT using a 6‐point MRC scale. They examined 7 muscle groups or muscle group actions bilaterally: gluteus maximus and medius; iliopsoas; quadriceps; deltoid; trapezius; biceps brachialis; as well as neck flexors and extensors unilaterally. The maximum MRC sum score was reported to be 80. Trial authors defined "improvement" and "worsening" as a change of at least 1 MMT grade in 2 muscle groups and a net change in score of at least 2 points on MMT. They defined "stabilisation" as any result other than "improvement" or "worsening".
ADL were assessed using a modified Convery Assessment Scale (Convery 1977); 4 items relating to feeding, grooming, wheelchair use, and perineal care were deleted from the original questionnaire, and a question about reaching above eye level was added into the scale. "Improvement" was defined as a net increase of at least 1 functional level in at least 1 category in the ADL score. A "worsening" clinical status was defined as similar decreases in ADL scores, and "stabilisation" was any result other than "improvement" or "worsening". No primary outcome measure was specified. Other pathophysiological measures included laboratory investigations (CK, aldolase, lactate dehydrogenase, aspartate aminotransferase and alanine aminotransferase levels), gradings for MRI and inflammatory changes in muscle biopsies. |
|
| Notes | Single‐centre study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information given. |
| Allocation concealment (selection bias) | Unclear risk | No information given. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reason for missing outcome data likely to be related to true outcome: "All patients received 6 months of each regime unless: a) worsening was noted after 3 months; b) after 3 months of the second (or cross‐over) therapy no improvement was noted; or c) unacceptable side effects occurred". While "most patients received the full 6 months of each regime" this is not quantified; there is also evidence of adverse effects leading to exclusion that might have been underestimated in seriousness and without intention‐to‐treat analysis: "...2 patients underwent cholecystectomies during the study for acute cholecystitis and subsequently were unable to continue because they experienced gastrointestinal intolerance and/or abnormal liver function tests when they resumed treatment with the drugs". No statistical analysis for the incidence of adverse events was completed. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement: numerical strength scale interpreted only by subjective improvement, stabilisation, or worsening. |
| Other bias | High risk | After the first treatment period, participants were observed for worsening before beginning the second period, breaking randomisation. There was also a potential carry‐over effect of previous treatment(s): "For 2 weeks before protocol entry, the prednisone dose was held stable, and apheresis or immunosuppressive therapy was not given" |
Lindberg 2003.
| Methods | Open randomised controlled trial | |
| Participants | 11 randomised participants (3 female) ATG group mean age: 72 ± 7.0 years; mean duration of disease: 7.2 ± 3.4 years MTX group mean age: 64.8 ± 4.0 years; mean duration of disease: 9.8 ± 5.0 years Inclusion criteria: the morphological criteria of IBM diagnosis included inflammation, rimmed vacuoles and intracellular amyloid deposits or 15 to 20 nm filaments Exclusion criteria: unspecified. |
|
| Interventions | MTX and ATG versus MTX (plus leucovorin) for 12 months | |
| Outcomes |
Primary outcome measure(s)
The primary outcome measure was the relative change of mean overall muscle strength, expressed as a percentage. Maximal voluntary muscle strength was measured with a handheld myometer and was used to assess the following muscle group actions: elbow flexion and extension; wrist dorsal extension; hip flexion; knee extension. Handgrip mean volumetric contraction was also quantitated using hand‐held myometry. Other outcome measures were blood levels of CK and T lymphocyte subsets. Muscle biopsies were done at the start and end of the trial, and inflammation in the tissue was graded visually. MVIC was measured using a hand‐held myometer. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information given. |
| Allocation concealment (selection bias) | Unclear risk | No information given. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of participants randomised and reasons for missing data stated; last data carried forward with average values and variance clearly specified. No statistical analysis for the incidence of adverse events was completed. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement: percentage change in muscle strength data given but no actual scores. |
| Other bias | Unclear risk | Insufficient information to permit judgement, but potential carry‐over effect of previous treatment(s). |
Machado 2013.
| Methods | Randomised, double‐blind, placebo‐controlled trial | |
| Participants | 24 participants (7 female) fulfilling Griggs criteria for definite or probable IBM | |
| Interventions | Arimoclomol versus placebo for 16 weeks | |
| Outcomes |
Primary outcome(s)
Secondary outcomes(s)
|
|
| Notes | Abstract only. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised. No further information. |
| Allocation concealment (selection bias) | Unclear risk | Abstract only. Not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double‐blind, placebo‐controlled". No further information. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Double‐blind, placebo‐controlled". No further information. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only. Unable to assess. |
| Selective reporting (reporting bias) | Unclear risk | No protocol. Unable to assess. |
| Other bias | Unclear risk | None identified. |
Muscle Study Group 2001.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group trial | |
| Participants | 30 randomised participants (11 female) IFN beta‐1a (Avonex)‐treated group: mean age: 65.7 ± 9.3 years; age at onset: 57.0 ± 10.0 years Placebo‐treated group mean age: 65.9 ± 10.3 years; age at onset: 57.3 ± 9.3 years Inclusion criteria: diagnostic criteria for definite or probable IBM; able to walk independently 15 feet (cane, walkers, orthoses allowed); age > 30 to ≤ 80 years; women of childbearing potential must have a reliable method of birth control; must not have received immunosuppressive agents for at least 3 months prior to enrolment; able to give informed consent. Exclusion criteria: presence of any one of the following medical conditions: uncontrolled diabetes mellitus; congestive heart disease; symptomatic cardiomyopathy; symptomatic coronary artery disease; cancer other than skin cancer < 5 years previously; multiple sclerosis or other chronic serious medical illness; presence of any of the following on routine blood screening: white blood cell count < 3000, platelets < 100,000, hematocrit < 30%, blood urea nitrogen > 30 mg%, symptomatic liver disease with serum albumin < 3 g/dL, prothrombin time or partial thromboplastin time > upper range of normal; presence of major depression on day of screening or history of attempted suicide; forced vital capacity < 50% of predicted; pregnancy or lactating; history of noncompliance with other therapies; other disease that required immunosuppressive therapy within the last 12 months; coexistence of other neuromuscular disease. |
|
| Interventions | IFN beta‐1a versus placebo for 24 weeks | |
| Outcomes |
QMT was performed using the QMA system; 5 muscle groups or muscle group actions were tested on each side: biceps; triceps; quadriceps; hamstrings, and ankle dorsiflexion. These results were expressed as the average number of SD units from predicted normal strength, given the age, gender, and height of the participant. Handgrip MVIC was also quantitated using myometry but with a hand‐held device rather than the fixed myometer used for other muscle tests. MMT tested a total of 34 (unspecified) muscle groups using a 6‐point MRC scale.
Measurements of biologic effects of treatment were also performed: serum neutralising antibodies and serum neopterin levels. No primary outcome measure was specified. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The investigators describe a random component in the sequence generation process: "a computer‐generated randomization plan developed by the Biostatistics Centre at the University of Rochester (NY). The randomization was stratified by center and included blocking" |
| Allocation concealment (selection bias) | Low risk | Central allocation: "Only the biostatistics programmer and the pharmacist at each site...had access to the treatment assignment" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement: active injections may have been more likely to cause local reactions; all participants treated with non‐steroidal anti‐inflammatory drug to mask systemic reactions from active compound. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficent information to permit judgement: blinding of assessors not described fully following allocation concealment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of participants randomised and reasons for missing data stated; intention‐to‐treat principle stated with no computation of missing data from 1 dropout. No statistical analysis for the incidence of adverse events was completed. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but outcomes prespecified in the methods are all reported in the results. |
| Other bias | Unclear risk | Insufficient information to permit judgement, but potential carry‐over effect of previous treatment(s), although inclusion criteria state that participants "must not have received immunosuppressive agents for at least 3 months prior to enrollment" |
Muscle Study Group 2004.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group trial | |
| Participants | 30 randomised participants (11 female) IFN beta‐1a (Avonex)‐treated group mean age: 64.9 ± 6.9 years; age at onset: 57.5 ± 8.4 years Placebo‐treated group mean age: 64.9 ± 7.3 years; age at onset: 55.5 ± 7.1 years Inclusion and Exclusion criteria as per Muscle Study Group 2001 |
|
| Interventions | IFN beta‐1a versus placebo for 24 weeks | |
| Outcomes | As per Muscle Study Group 2001 | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The investigators reference a random component in the sequence generation process: "Randomization procedures were identical to those used in our previous trial" |
| Allocation concealment (selection bias) | Low risk | Central allocation: "...only the biostatistics programmer and site pharmacists had access to the treatment assignments" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement: active treatment injections may have been more likely to cause local reactions; all participants treated with non‐steroidal anti‐inflammatory drug to mask systemic reactions from active compound. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement: blinding of assessors not described fully following allocation concealment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of participants randomised and reasons for missing data stated; intention‐to‐treat principle stated, although data from only 1 of 3 dropouts were carried forward for analysis. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but outcomes prespecified in the methods are all reported in the results. |
| Other bias | Unclear risk | Insufficient information to permit judgement, but potential carry‐over effect of previous treatment(s), although inclusion criteria as per their previous study specified that participants must not have received immunosuppressive agents for at least 3 months prior to enrolment. |
Rutkove 2002.
| Methods | Block‐randomised, double‐blind, placebo‐controlled, cross‐over trial | |
| Participants | 19 randomised participants, but baseline characteristics provided for 16 participants who completed the trial (2 female). Overall mean age: 68.5 years (no SD or range). Mean duration of disease unspecified. Inclusion criteria: pathological criteria for a definite diagnosis of IBM; consistent clinical and laboratory features including prominent weakness of quadriceps, weakness of wrist flexors greater than extensors, age older than 40 years, and elevated serum CK, without evidence of other significant neurologic problems. Exclusion criteria: history of prostate or breast cancer; haemodialysis; congestive heart failure; atypical or restricted forms of IBM; a coexisting neuromuscular condition; uncontrolled hypertension; history of substance abuse; currently taking warfarin; IVIg use within 2 months or currently on an immunosuppressive drug regimen including corticosteroids. |
|
| Interventions | Oxandrolone versus placebo for 12 weeks | |
| Outcomes |
Primary outcome measure(s)
Using a fixed myometer, whole body MVICT was measured bilaterally for muscle group actions including: shoulder flexion and extension; elbow flexion and extension; knee flexion and extension; foot dorsiflexion. Handgrip strength was also evaluated quantitatively by MVICT but using a hand‐held myometer. Secondary outcome measure(s)
Whole body MMT was graded using an expanded 1‐5 MRC scale. 13 bilateral muscle group actions were assessed: shoulder external rotation and abduction; elbow flexion and extension; wrist flexion and extension; hip flexion, abduction and presumed adduction (written as "abduction" in publication); knee flexion and extension; foot plantar flexion and dorsiflexion. 2 unilateral muscle group actions were also assessed: head flexion and extension. The maximum reported MRC score was 140.
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The investigators describe a random component in the sequence generation process: "...a four‐person block‐randomization procedure" |
| Allocation concealment (selection bias) | Low risk | Central allocation: pharmacy‐generated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement: blinding not described fully following allocation concealment: "the principal investigator, coinvestigators, nurses and physical therapists were blinded to the treatment assignments...and the randomization code was maintained in the hospital research pharmacy", but 8/13 participants "...correctly reported that they were on drug..." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Insufficient information to permit judgement: possible unblinding of outcome assessment following allocation concealment indicated, as above |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement: baseline characteristics not available for all randomised participants, although reasons for missing data given. Also, non‐parametric data are reported due to concerns about the normality of the data, but no normality test results are given. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but outcomes prespecified in the methods are all reported in the results. |
| Other bias | Unclear risk | Insufficient information to permit judgement, but potential carry‐over of previous treatment(s), although "Patients were...excluded if they had received IVIg within 2 months or were currently on an immunosuppressive drug regimen including corticosteroids" |
Walter 2000.
| Methods | Block‐randomised, double‐blind, placebo‐controlled, cross‐over trial | |
| Participants | 22 randomised participants (8 female). Mean age: 59 ± 14 years; disease duration: 1 to 14 years Inclusion criteria: clinical and histological criteria for definite sporadic IBM. Exclusion criteria: stabilisation or improvement on current immunosuppressive therapy; severe coronary heart disease; renal insufficiency; intolerance to homologous immunoglobulins or human serum proteins; other relevant neuromuscular disorders; those confined to bed or wheelchair for longer than 1 year. |
|
| Interventions | IVIg versus placebo for 6 months | |
| Outcomes |
Primary outcome measure(s)
MMT was completed for the following muscle groups and muscle group actions bilaterally: deltoid; biceps brachii; triceps brachii; brachioradialis; hand flexion and extension; finger flexion and extension; knee flexion and extension; foot flexion and extension. 2 muscle group actions were tested unilaterally: neck flexion and extension. Muscle strength was graded on a 0‐6 MRC scale, with a total maximal score of 180. The change in upper and lower extremity strength was also analysed as a subgroup.
Secondary outcome measure(s)
Electromyographic tests to assess graded change in spontaneous activity were also performed. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The investigators describe a random component in the sequence generation process: "block‐randomization procedure" |
| Allocation concealment (selection bias) | Low risk | Method of allocation concealment not described, but "the randomization code was not broken until all patients completed the study". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement: blinding not described fully following allocation concealment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement: blinding not described fully following allocation concealment, although authors retrospectively state that assessors were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement: reasons not given for missing data from 2 dropouts; all participant data carried forward for safety analysis, but only completed data used for efficacy analysis; also non‐parametric statistical analyses performed, although both mean and median data are reported in results and without specification of SD or interquartile range. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement: the primary outcomes specified in the methods are not fully reported in the results; authors do not compare IVIg versus placebo periods. |
| Other bias | Unclear risk | Insufficient information to permit judgement, but potential carry‐over effect from previous treatment(s): "Of the 22 patients 19 patients had previously been treated with various drugs". Also, "Physiotherapy was administered to all patients in both groups once or twice per week throughout the entire trial" but was not assessed as part of the therapeutic intervention. |
ADL: activities of daily living
ALS‐FRS: Amyotrophic Lateral Sclerosis Functional Rating Scale
ATG: anti‐T lymphocyte immunoglobulin
CK: creatine kinase
DEXA: dual energy X‐ray absorptiometry
HSP70: 70 kilodalton heat shock protein
IFN: interferon
IVIg: intravenous immunoglobulin
MTX: methotrexate
MMT: manual muscle testing
MRC: Medical Research Council
MRI: magnetic resonance imaging
MVICT: maximal voluntary isometric contraction testing
QMA system: Quantitative Muscle Assessment system
QMT: quantitative muscle testing
SD: standard deviation
SF‐36: Short Form‐36 Health Survey
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ACTRN12614000082606 | Not a randomised trial |
| Amato 1994 | Not a randomised trial |
| Arnardottir 2003 | Not a randomised trial. Exercise‐based intervention |
| Danon 1982 | Not a randomised trial |
| Heikkillä 2001 | Not a randomised trial. Exercised‐based intervention |
| Joffe 1993 | Not a randomised trial |
| Kosmidis 2013 | Not a randomised trial |
| Lindberg 1994 | Not a randomised trial |
| Mastaglia 1998 | Non randomised trial |
| Mowzoon 2001 | Not a randomised trial (1 participant with IBM) |
| NCT00079768 | Not a randomised trial |
| NCT00917956 | Not a randomised trial |
| NCT01519349 | Not a randomised trial |
| Soueidan 1993 | Not a randomised trial |
IBM: inclusion body myositis
Characteristics of ongoing studies [ordered by study ID]
EUCTR2007‐004359‐12‐IT.
| Trial name or title | Simvastatin treatment in inclusion body myositis (IBM) |
| Methods | Randomised, controlled, open, parallel‐group trial |
| Participants | People affected by IBM (diagnosis based on clinical, radiological, and pathological data); males and females; aged 18 to 80 years; patients able to give informed consent |
| Interventions | Oral simvastatin |
| Outcomes | Main objective: safety and tolerability of simvastatin Primary endpoint(s): improvement in functional indices |
| Starting date | 2007 |
| Contact information | Italian Medical Agency |
| Notes | ‐ |
NCT00001265.
| Trial name or title | Study and treatment of inflammatory muscle diseases |
| Methods | Observational study |
| Participants | People aged 16 and over with known or suspected idiopathic inflammatory myopathies or people with other connective tissue diseases with weakness or myalgia for an inflammatory myopathy or other muscle process |
| Interventions | None specified |
| Outcomes | None specified |
| Starting date | August 1991 |
| Contact information | National Institutes of Health Clinical Center |
| Notes |
NCT01423110.
| Trial name or title | Efficacy, safety and tolerability of BYM338 in patients with sporadic inclusion body myositis |
| Methods | Double‐blind, parallel assignment, placebo‐controlled study |
| Participants | People aged 40 to 80 years old with sporadic IBM |
| Interventions | BYM338 or placebo |
| Outcomes | Primary outcome measures: effect of BYM338 on thigh muscle volume by MRI (after 8 weeks' treatment): change in thigh muscle volume Secondary outcome measures: effect of BYM338 on muscle function by timed get‐up‐and‐go test (after 8 weeks' treatment): change in muscle function measured on scale by test results |
| Starting date | August 2011 |
| Contact information | Novartis Pharmaceuticals |
| Notes | Study identifier: CBYM338X2205 |
IBM: inclusion body myositis; MRI: magnetic resonance imaging
Differences between protocol and review
We used The Cochrane `Risk of bias' tool (Higgins 2011). We commented on the use of explicit diagnostic criteria, the quality of outcome measures, and the power of the study to detect benefit where appropriate in the text.
We searched clinical trials registries for ongoing trials but did not carry out an up‐to‐date handsearch of conference abstracts.
We referred to Griggs 1995, Benveniste 2010, and Hilton‐Jones 2010 for clinicopathologically defined IBM, but two trials referred to clinicopathologically defined criteria using alternative references.
We renamed our outcome `significant side‐effects' as `significant adverse events'.
We defined significant adverse events as the incidence of withdrawals.
We clarified significant adverse events as the incidence of participant withdrawal from trials, with reasons explained where possible. In line with the definition of an adverse event, we accepted that withdrawal might occur for reasons unspecified or unrelated to the intervention.
Contributions of authors
For the protocol: MR and KL drafted earlier versions of the protocol with input from co‐authors (Rose 1999; Rose 2007). KJ updated the protocol in 2014. All review authors reviewed drafts and agreed on the final text.
For the review stage:
MR, KJ, MW, and JM assessed studies for inclusion.
MR, KJ, KL, MW, and JM extracted data.
MR and KL entered data; KJ checked data entry.
MR and KJ assessed the risk of bias in included studies.
RB assisted with analyses and drafted `Summary of findings' tables.
MR, KJ, and RB drafted text.
All review authors reviewed drafts and agreed on the final text.
Sources of support
Internal sources
Wissenschaftliches Institut, Private Europäische Medizinische Akademie der Klinik Bavaria in Kreischa GmbH, An derWolfsschlucht 1‐2, 01731 Kreischa, Germany.
Lehrstuhl Therapiewissenschaften, SRH Fachhochschule für Gesundheit Gera gGmbH, Hermann‐Drechsler‐Str. 2, 07548 Gera, Germany.
Gesundheitswissenschaften/Public Health, Medizinische Fakultät Carl Gustav Carus der TU Dresden, Fetscherstr. 74, 01307 Dresden, Germany.
University of Southampton, UK.
-
University College London Hospital (UCLH) NHS Foundation Trust, UK.
Ruth Brassington is employed as Managing Editor of the Cochrane Neuromuscular Disease Group, which is hosted by UCLH. The National Institute for Health Research (NIHR) is the largest single funder of this Cochrane Review Group.
External sources
-
Association Française contre les Myopathies, France.
Grant: Katherine Jones
Declarations of interest
MR is a member of the Muscle Study Group that published the two trials of IFN beta‐1a for IBM. These trials were grant funded with a proportion of those funds paid to my institution for the conduct of the trial only and with no personal financial benefit ensuing. For these trials the drug (Avonex) and matching placebo were supplied free of charge by the manufacturer Biogen. The trial protocols, data entry, data analysis, and publications were in the hands of the investigators with no input from Biogen.
KJ's research contribution has been paid for by a grant from the Association Française contre les Myopathies.
KL has no known financial conflicts of interest.
MW has published one randomised trial of IVIg in IBM. She contributed to the Novartis EU Local Advisory Board Meeting (IBM BYM338 trial) on 15 April 2013.
JM has been a member of advisory boards for CSL Behring, Octapharma, and Grifols, companies that produce IVIg. He received meeting expenses to attend Peripheral Nerve Society meetings in 2011, 2012, and 2013 from Baxter and CSL Behring, which produce IVIg products. He is a local principal investigator for the RESILIENT study, an ongoing study of bimagrumab in sporadic IBM sponsored by Novartis.
MD has published three randomised trials of IVIg in IBM. He has accepted institutional grants unrelated to the present review from: CSL, Genesis, Merck, Novartis, and Genzyme. He has also received personal compensation for lectures or consultancies from Novartis, Dysimmune Diseases Foundation, Therapath, Genzyme, Octapharma, and Baxter.
RB has no known financial conflicts of interest. She is Managing Editor of the Cochrane Neuromuscular Disease Group.
RG is a member of the Muscle Study Group that published two trials of IFN beta‐1a for IBM. He has no other known conflicts of interest.
Edited (no change to conclusions)
References
References to studies included in this review
Badrising 2002 {published data only}
- Badrising UA, Maat‐Schieman ML, Ferrari MD, Zwinderman AH, Wessels JA, Breedveld FC, et al. Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Annals of Neurology 2002;51(3):369‐72. [PUBMED: 11891832] [DOI] [PubMed] [Google Scholar]
Dalakas 1997 {published data only}
- Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K. Treatment of inclusion‐body myositis with IVIg: a double‐blind, placebo‐controlled study. Neurology 1997;48(3):712‐6. [PUBMED: 9065553] [DOI] [PubMed] [Google Scholar]
- Fujii M, Koffman B, Sivakumar K, Dalakas MC. Histologic characteristics in repeated muscle biopsies of patients with inclusion body myositis treated in a control trial with high‐dose intravenous immunoglobulin and prednisone. Neurology 1997;48 Suppl 2:A332‐3. [Google Scholar]
Dalakas 2001 {published data only}
- Dalakas MC, Koffman B, Fujii M, Spector S, Sivakumar K, Cupler E. A controlled study of intravenous immunoglobulin combined with prednisolone in the treatment of IBM. Neurology 2001;56(3):323‐7. [PUBMED: 11171896] [DOI] [PubMed] [Google Scholar]
Leff 1993 {published data only}
- Leff RL, Miller FW, Hicks J, Fraser DD, Plotz PH. The treatment of inclusion body myositis: a retrospective review and a randomised prospective trial of immunosuppressive therapy. Medicine 1993;72(4):225‐35. [PUBMED: 8393509] [DOI] [PubMed] [Google Scholar]
Lindberg 2003 {published data only}
- Lindberg C, Trysberg E, Tarkowski A, Oldfors A. Anti‐T‐lymphocyte globulin treatment in inclusion body myositis: a randomized pilot study. Neurology 2003;61(2):260‐2. [PUBMED: 12874415] [DOI] [PubMed] [Google Scholar]
Machado 2013 {published and unpublished data}
- Machado P, Miller A, Herbelin L, He J, Noel J, Wang Y, et al. Safety and tolerability of arimoclomol in patients with sporadic inclusion body myositis: a randomised, double‐blind, placebo‐controlled, phase IIA proof‐of‐concept trial. Annals of the Rheumatic Diseases 2013;72(Suppl 3):A164. [Google Scholar]
- Machado P, Miller A, Herbelin L, He J, Wang Y, McVey AL, et al. Arimoclomol study in IBM: [personal communication]. Email to P Machado 4 November 2014.
Muscle Study Group 2001 {published data only}
- The Muscle Study Group. Randomized pilot trial of betaINF1a (Avonex) in patients with inclusion body myositis. Neurology 2001;57(9):1566‐70. [PUBMED: 11706093] [DOI] [PubMed] [Google Scholar]
Muscle Study Group 2004 {published data only}
- The Muscle Study Group. Randomized pilot trial of high‐dose betaINF‐1a in patients with inclusion body myositis. Neurology 2004;63(4):718‐20. [PUBMED: 15326251] [DOI] [PubMed] [Google Scholar]
Rutkove 2002 {published data only}
- Rutkove SB, Parker RA, Nardin RA, Connolly CE, Felice KJ, Raynor EM. A pilot randomized trial of oxandrolone in inclusion body myositis. Neurology 2002;58(7):1081‐7. [PUBMED: 11940697] [DOI] [PubMed] [Google Scholar]
Walter 2000 {published data only}
- Pongratz D, Lochmuller H, Toepfer M, Schlotter B, Schroder M, Muller‐Felber W, et al. High dose intravenous immunoglobulin (IVIG) in the treatment of inclusion body myositis ‐ a double‐blind placebo‐controlled study. Muscle & Nerve 1998;21(Suppl 7):S131. [DOI] [PubMed] [Google Scholar]
- Walter MC, Lochmuller H, Toepfer M, Schlotter B, Reilich P, Schroder M, et al. High‐dose immunoglobulin therapy in sporadic inclusion body myositis: a double blind, placebo‐controlled study. Journal of Neurology 2000;247(1):22‐8. [PUBMED: 10701893] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
ACTRN12614000082606 {published data only}
- ACTRN12614000082606. Pilot study of the treatment of patients with sporadic inclusion body myositis with the anaplerotic medication Triheptanoin. https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=365625 (accessed 6 November 2014).
Amato 1994 {published data only}
- Amato AA, Barohn RJ, Jackson CE, Pappert EJ, Sahenk Z, Kissel JT. Inclusion body myositis: treatment with intravenous immunoglobulin. Neurology 1994;44(8):1516‐8. [PUBMED: 8058161] [DOI] [PubMed] [Google Scholar]
Arnardottir 2003 {published data only}
- Arnardottir S, Alexanderson H, Lundberg IE, Borg K. Sporadic inclusion body myositis: pilot study on the effects of a home exercise program on muscle function, histopathology and inflammatory reaction. Journal of Rehabilitation Medicine 2003;35(1):31‐5. [PUBMED: 12610846 ] [DOI] [PubMed] [Google Scholar]
Danon 1982 {published data only}
- Danon MJ, Reyes MG, Perurena OH, Masdeu JC, Manaligod JR. Inclusion body myositis. A corticosteroid‐resistant idiopathic inflammatory myopathy. Archives of Neurology 1982;39(12):760‐4. [PUBMED: 6291495] [DOI] [PubMed] [Google Scholar]
Heikkillä 2001 {published data only}
- Heikkillä S, Viitanen JV, Kautiainen H, Rajamäki T, Mäntyvuo P, Harju T. Rehabilitation in myositis: preliminary study. Physiotherapy 2001;87(6):301‐9. [Google Scholar]
Joffe 1993 {published data only}
- Joffe MM, Love LA, Leff RL, Fraser DD, Targoff IN, Hicks JE, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. American Journal of Medicine 1993;94(4):379‐87. [PUBMED: 8386437] [DOI] [PubMed] [Google Scholar]
Kosmidis 2013 {published data only}
- Kosmidis ML, Alexopoulos H, Tzioufas AG, Dalakas MC. The effect of anakinra, an IL1 receptor antagonist, in patients with sporadic inclusion body myositis (sIBM): a small pilot study. Journal of the Neurological Sciences 2013;334(1‐2):1235. [DOI] [PubMed] [Google Scholar]
- NCT01165008. Anakinra in myositis. http://www.clinicaltrials.gov/ct2/show/NCT01165008 (accessed 6 November 2014).
Lindberg 1994 {published data only}
- Lindberg C, Persson LI, Bjorkander J, Oldfors A. Inclusion body myositis: clinical, morphological, physiological and laboratory findings in 18 cases. Acta Neurologica Scandinavica 1994;89(2):123‐31. [PUBMED: 8191875] [DOI] [PubMed] [Google Scholar]
Mastaglia 1998 {published data only}
- Mastaglia FL, Phillips BA, Zilko PJ. Immunoglobulin therapy in inflammatory myopathies. Journal of Neurology, Neurosurgery and Psychiatry 1998;65(1):107‐10. [PUBMED: 9667570] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mowzoon 2001 {published data only}
- Mowzoon N, Sussman A, Bradley WG. Mycophenolate (CellCept) treatment of myasthenia gravis, chronic inflammatory polyneuropathy and inclusion body myositis. Journal of the Neurological Sciences 2001;185(2):119‐22. [PUBMED: 11311292] [DOI] [PubMed] [Google Scholar]
NCT00079768 {published data only}
- NCT00079768. Alemtuzumab to treat sporadic inclusion body myositis. http://www.clinicaltrials.gov/ct2/show/NCT00079768 (accessed 6 November 2014).
NCT00917956 {published data only}
- NCT00917956. Lithium in inclusion body myositis (IBM) (Li‐IBM). http://www.clinicaltrials.gov/ct2/show/NCT00917956 (accessed 6 November 2014).
NCT01519349 {published data only}
- NCT01519349. Follistatin gene transfer to patients with Becker muscular dystrophy and sporadic inclusion body myositis. http://www.clinicaltrials.gov/ct2/show/NCT01519349 (accessed 6 November 2014).
Soueidan 1993 {published data only}
- Soueidan SA, Dalakas M. Treatment of inclusion‐body myositis with high‐dose intravenous immunoglobulin. Neurology 1993;43(5):876‐9. [PUBMED: 8492940] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
EUCTR2007‐004359‐12‐IT {published data only}
- EUCTR2007‐004359‐12‐IT. Simvastatin treatment in inclusion body myositis (IBM) ‐ ND. http://apps.who.int/trialsearch/Trial.aspx?TrialID=EUCTR2007‐004359‐12‐IT (accessed 6 November 2014). [EUCTR2007‐004359‐12‐IT]
NCT00001265 {published data only}
- NCT00001265. Study and treatment of inflammatory muscle diseases. http://www.clinicaltrials.gov/show/NCT00001265 (accessed 6 November 2014).
NCT01423110 {published data only}
- NCT01423110. Efficacy and safety of bimagrumab/BYM338 at 52 weeks on physical function, muscle strength, mobility in sIBM patients. http://www.clinicaltrials.gov/ct2/show/NCT01423110 (accessed 6 November 2014).
- NCT01423110. Efficacy, safety and tolerability of BYM338 in patients with sporadic inclusion body myositis. http://www.clinicaltrials.gov/ct2/show/NCT01423110 (accessed 6 November 2014).
- NCT02250443. Study of long‐term safety, efficacy tolerability of BYM338 in patients with sporadic inclusion body myositis. http://www.clinicaltrials.gov/ct2/show/NCT02250443 (accessed 6 November 2014).
Additional references
Amato 1998
- Amato AA, Shebert RT. Inclusion body myositis in twins. Neurology 1998;51(2):598‐600. [DOI] [PubMed] [Google Scholar]
Aronson 2006
- Aronson JK (editor). Meyler's Side Effects of Drugs: The International Encyclopedia of Adverse Drug Reactions and Interactions. Meyler's Side Effects of Drugs: The International Encyclopedia of Adverse Drug Reactions and Interactions. 15th Edition. Amsterdam: Elsevier BV, 2006. [Google Scholar]
Badrising 2000
- Badrising UA, Maat‐Schieman M, Duinen SG, Breedveld F, Doorn P, Engelen B, et al. Epidemiology of inclusion body myositis in the Netherlands: a nationwide study. Neurology 2000;55(9):1385‐8. [DOI] [PubMed] [Google Scholar]
Beaston‐Blaakman 2007
- Beaston‐Blaakman A, Shepard DS, Stone N, Shevitz AH. Cost‐effectiveness of clinical interventions for AIDS wasting. AIDS Care 2007;19(8):996‐1001. [DOI] [PubMed] [Google Scholar]
Benveniste 2010
- Benveniste O, Hilton‐Jones D. International Workshop on Inclusion Body Myositis held at the Institute of Myology, Paris, on 29 May 2009. Neuromuscular Disorders 2010;20(6):414‐21. [DOI] [PubMed] [Google Scholar]
Benveniste 2011
- Benveniste O, Guiget M, Freebody J, Dubourg O, Squier W, Maisonobe T, et al. Long‐term observational study of sporadic inclusion body myositis. Brain 2011;134(Pt 1):3176‐84. [DOI] [PubMed] [Google Scholar]
BNF 2014
- Joint Formulary Committee. British National Formulary. Vol. 68, London: British Medical Association and Royal Pharmaceutical Society of Great Britain, 2014. [Google Scholar]
Bohan 1975
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). The New England Journal of Medicine 1975;292(7):344‐7. [PUBMED: 1090839] [DOI] [PubMed] [Google Scholar]
Breithaupt 2013
- Breithaupt M, Schmidt J. Update on treatment of inclusion body myositis. Current Rheumatology Reports 2013;15(5):329. [DOI] [PubMed] [Google Scholar]
Brooke 1983
- Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, Miller JP, et al. Clinical investigation in Duchenne dystrophy 2: determination of the "power" of therapeutic trials based on the natural history. Muscle & Nerve 1983;6(2):91‐103. [PUBMED: 6343858] [DOI] [PubMed] [Google Scholar]
Chalker 2000
- Chalker J, Leuwer M, Lunde PKM, McInnes GT, Thelle D, Velo GP, et al. Meyler's Side Effects of Drugs. 14th Edition. Amsterdam: Elsevier Science B.V., 2000. [Google Scholar]
Cochrane 2015
- The Cochrane Collaboration. Cochrane glossary. http://www.cochrane.org/glossary (accessed 27 January 2015).
Convery 1977
- Convery FR, Minteer MA, Amiel D, Connett KL. Polyarticular disability: a functional assessment. Archives of Physical Medicine and Rehabilitation 1977;58(11):494‐9. [PubMed] [Google Scholar]
Cortese 2013
- Cortese A, Machado P, Morrow J, Dewar L, Hiscock A, Miller A, et al. Longitudinal observational study of sporadic inclusion body myositis: implications for clinical trials. Neuromuscular Disorders 2013;23(5):404‐12. [DOI] [PubMed] [Google Scholar]
Cox 2011
- Cox FM, Titulaer MJ, Sont JK, Wintzen AR, Verschuuren JJ, Badrising UA. A 12‐year follow‐up in sporadic inclusion body myositis: an end stage with major disabilities. Brain 2011;134(Pt 11):3167‐75. [DOI] [PubMed] [Google Scholar]
Dalakas 1993
- Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high‐dose intravenous immunoglobulin infusions as treatment for dermatomyositis. The New England Journal of Medicine 1993;329(27):1993‐2000. [PUBMED: 8247075] [DOI] [PubMed] [Google Scholar]
Dalakas 1995
- Dalakas MC, Illa I. Common variable immunodeficiency and inclusion body myositis ‐ a distinct myopathy mediated by natural killer cells. Annals of Neurology 1995;37(6):806‐10. [DOI] [PubMed] [Google Scholar]
Danon 1986
- Danon MJ, Perurena OH, Ronan S, Manaligod JR. Inclusion body myositis associated with systemic sarcoidosis. Canadian Journal of Neurological Sciences 1986;13(4):334‐6. [DOI] [PubMed] [Google Scholar]
DH 2011
- Department of Health. Clinical guidelines for immunoglobulin use: update to second edition. http://www.ivig.nhs.uk/documents/dh_129666.pdf (accessed 27 January 2015) 2011.
Dukes 2000
- Dukes MNG, Aaronson JK. Meyler's Side Effects of Drugs. 14th Edition. Oxford: Elsevier, 2000. [Google Scholar]
Dutch GBS 1994
- Dutch Guillain‐Barre Syndrome Study Group. Treatment of Guillain‐Barre syndrome with high‐dose immune globulins combined with methylprednisolone: a pilot study. Annals of Neurology 1994;35(6):749‐52. [PUBMED: 8210234] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Fergusson 2005
- Fergusson D, Hutton B, Sharma M, Tinmouth A, Wilson K, Cameron DW, et al. Use of intravenous immunoglobulin for treatment of neurologic conditions: a systematic review. Transfusion 2005;45(10):1640‐57. [DOI] [PubMed] [Google Scholar]
GRADEpro 2014 [Computer program]
- McMaster University. GRADEpro. Hamilton, Ontario: McMaster University, 2014. www.gradepro.org.
Griggs 1995
- Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, et al. Inclusion body myositis and myopathies. Annals of Neurology 1995;38(5):705‐13. [PUBMED: 7486861] [DOI] [PubMed] [Google Scholar]
Gutmann 1985
- Gutmann L, Govindan S, Riggs JE, Schochet SSJ. Inclusion body myositis and Sjogren's syndrome. Archives of Neurology 1985;42(10):1021‐2. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hill 2004
- Hill M, Hughes T, Milford C. Treatment for swallowing difficulties (dysphagia) in chronic muscle disease. Cochrane Database of Systematic Reviews 2004, Issue 2. [DOI: 10.1002/14651858.CD004303.pub2] [DOI] [PubMed] [Google Scholar]
Hilton‐Jones 2010
- Hilton‐Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG. Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London, 13 June 2008. Neuromuscular Disorders 2010;20(2):142‐7. [DOI] [PubMed] [Google Scholar]
Huizing 2009
- Huizing M, Krasnewich DM. Hereditary inclusion body myopathy: a decade of progress. Biochimica et Biophysica Acta 2009;1792(9):881‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jackson 2008
- Jackson CE, Barohn RJ, Gronseth G, Pandya S, Herbelin L. Inclusion body myositis functional rating scale: a reliable and valid measure of disease severity. Muscle & Nerve 2008;37(4):473–6. [DOI] [PubMed] [Google Scholar]
Khraishi 1992
- Khraishi MM, Jay V, Keystone EC. Inclusion body myositis in association with vitamin B12 deficiency and Sjögren's syndrome. Journal of Rheumatology 1992;19(2):306‐9. [PubMed] [Google Scholar]
Lane 1985
- Lane RJ, Fulthorpe JJ, Hudgson P. Inclusion body myositis: a case with associated collagen vascular disease responding to treatment. Journal of Neurology, Neurosurgery & Psychiatry 1985;48(3):270‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lindberg 1990
- Lindberg C, Persson L, Oldfors A, Soderstrom T, Hedstrom A, Bjorkander J. Inclusion body myositis: association with immunodeficiency. Journal of the Neurological Sciences 1990;98 (Suppl):178. [Google Scholar]
Loke 2007
- Loke YK, Price D, Herxheimer A, Cochrane Adverse Events Methods Group. Systematic reviews of adverse effects: framework for a structured approach. BMC Medical Research Methodology 2007;7:32. [DOI: 10.1186/1471-2288-7-32] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lotz 1989
- Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis. Observations in 40 patients. Brain 1989;112(Pt 3):727‐47. [PUBMED: 2543478] [DOI] [PubMed] [Google Scholar]
Machado 2013b
- Machado P, Brady S, Hanna MG. Update in inclusion body myositis. Current Opinion in Rheumatology 2013;25(6):763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Needham 2008
- Needham M, Corbett A, Day T, Christiansen F, Fabian V, Mastaglia F. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. Journal of Clinical Neuroscience 2008;15(12):1350‐3. [DOI] [PubMed] [Google Scholar]
Oldfors 1995
- Oldfors A, Moslemi AR, Fyhr IM, Holme E, Larsson NG, Lindberg C. Mitochondrial DNA deletions in muscle fibers in inclusion body myositis. Journal of Neuropathology & Experimental Neurology 1995;54(4):581‐7. [DOI] [PubMed] [Google Scholar]
Pfeffer 2012
- Pfeffer G, Majamaa K, Turnbull, DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD004426.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Phillips 2000
- Phillips BA, Zilko PJ, Mastaglia FL. Prevalence of sporadic inclusion body myositis in Western Australia. Muscle & Nerve 2000;23(6):970‐2. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Riggs 1984
- Riggs JE, Schochet SSJ, Gutmann L, McComas CF, Rogers JS. Inclusion body myositis and chronic immune thrombocytopenia. Archives of Neurology 1984;41(1):93‐5. [DOI] [PubMed] [Google Scholar]
Rose 2001
- Rose MR, McDermott MP, Thornton CA, Palenski C, Martens WB, Griggs RC. A prospective natural history study of inclusion body myositis: implications for clinical trials. Neurology 2001;57(3):548‐50. [PUBMED: 11502935] [DOI] [PubMed] [Google Scholar]
Rygiel 2014
- Rygiel KA, Miller J, Grady, JP, Rocha MC, Taylor RW, Turnbull DM. Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathology and Applied Neurobiology 2015;41(3):288‐303. [DOI: 10.1111/nan.12149] [DOI] [PMC free article] [PubMed] [Google Scholar]
Santorelli 1996
- Santorelli FM, Sciacco M, Tanji K, Shanske S, Vu TH, Golzi V, et al. Multiple mitochondrial DNA deletions in sporadic inclusion body myositis: a study of 56 patients. Annals of Neurology 1996;39(6):789‐95. [DOI] [PubMed] [Google Scholar]
Smith 2012
- Smith GI, Reeds DN, Hall AM, Chambers KT, Finck BN, Mittendorfer B. Sexually dimorphic effects of aging on skeletal muscle protein synthesis. Biology of Sex Differences 2012;3(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Soden 1994
- Soden M, Boundy K, Burrow D, Blumbergs P, Ahern M. Inclusion body myositis in association with rheumatoid arthritis. Journal of Rheumatology 1994;21:344‐6. [PubMed] [Google Scholar]
Tateyama 2003
- Tateyama M, Saito N, Fujihara K, Shiga Y, Takeda A, Narikawa K, et al. Familial inclusion body myositis: a report on two Japanese sisters. Internal Medicine (Tokyo, Japan) 2003;42(10):1035‐8. [DOI] [PubMed] [Google Scholar]
Verschuuren 1997
- Verschuuren JJ, Badrising UA, Wintzen AR, Engelen BGM, Hoeven H, et al. Inclusion body myositis. In: Emery AEH editor(s). Diagnostic Criteria for Neuromuscular Disorders. London: Royal Society of Medicine Press, 1997:81–4. [Google Scholar]
Voet 2013
- Voet NBM, Kooi EL, Riphagen II, Lindeman E, Engelen BGM, Geurts ACH. Strength training and aerobic exercise training for muscle disease. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD003907.pub4] [DOI] [Google Scholar]
References to other published versions of this review
Rose 1999
- Rose MR, Griggs R, Dalakas M. Immunotherapy for inclusion body myositis. Cochrane Database of Systematic Reviews 1999, Issue 2. [DOI: 10.1002/14651858.CD001555] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rose 2007
- Rose MR, Dalakas M, Griggs R, Leong K, Miller J, Walter MC. Treatment for inclusion body myositis. Cochrane Database of Systematic Reviews 2007, Issue 1. [DOI: 10.1002/14651858.CD001555.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rose 2014
- Rose MR, Dalakas M, Griggs R, Leong K, Miller J, Walter MC, Jones K. Treatment for inclusion body myositis. Cochrane Database of Systematic Reviews 2014, Issue 11. [DOI: 10.1002/14651858.CD001555.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]