

Abstract
Lenvatinib, a tyrosine kinase inhibitor (TKI), is a stronger inhibitor of vascular endothelial growth factor receptor, fibroblast growth factor receptors 1 to 4, and platelet-derived growth factor receptor (PDGFR) than other TKIs. We herein report a 77-year-old Japanese woman who received the minimum dose of lenvatinib for treatment of hepatocellular carcinoma. Within one month of starting treatment, she developed severe proteinuria, hypertension, and renal dysfunction. A kidney biopsy showed drug-induced thrombotic microangiopathy, podocytopathy, and polar vasculosis. We also observed damage to the renal tubules, where PDGFR is located. To our knowledge, this is the first report of lenvatinib-induced damage to the renal tubules.
Keywords: thrombotic microangiopathy, podocytopathy, damage to the renal tubules, lenvatinib
Introduction
Lenvatinib inhibits tyrosine kinase, which is involved in tumor angiogenesis and malignant tumor transformation (1). An important target molecule of lenvatinib and other tyrosine kinase inhibitors (TKIs) is vascular endothelial growth factor receptor (VEGFR), and lenvatinib has a novel binding mode to VEGFR2 (one of the multiple VEGFRs). Lenvatinib also binds to fibroblast growth factor receptors 1 to 4, platelet-derived factor receptor (PDGFR), and other receptors (1); hence, it is expected to have therapeutic effects in patients with resistance to other TKIs (2). In general, TKIs may cause hypertension and proteinuria (2).
VEGFRs can normally be found on the surface of endothelial cells, endothelial cell precursors, fibroblasts, and macrophages, as well as on kidney epithelial cells, such as podocytes (3). VEGF is produced in large amounts by glomerular podocytes and guides endothelial cells in the developing kidney; furthermore, it maintains the integrity of the glomerular filtration barrier (4,5). Thus, vascular endothelial damage and podocytopathy in the glomeruli of patients receiving anti-VEGF agents are considered to result in proteinuria (6,7). In some patients, lenvatinib has also been associated with acute kidney injury involving minimal change nephrotic syndrome/focal segmental glomerulosclerosis-like lesions (cFSG), which are caused by impaired podocytes (2,7,8). Damage to the renal tubules by lenvatinib has not been previously reported.
We herein report a case of thrombotic microangiopathy (TMA), podocytopathy and renal tubule damage that developed after 1-month treatment of hepatocellular carcinoma with the minimum dose of lenvatinib (4-8 mg/day). We also discuss possible pathomechanisms of this case.
Case Report
A 77-year-old Japanese woman was referred to our nephrology center for the further examination of kidney disease. Hepatitis B virus had been diagnosed at 54 years old. Radiofrequency ablation, transcatheter arterial chemoembolization, and resection of the superior part of the left lateral segment of the liver had been performed for hepatocellular carcinoma at 76 years old, and she had never undergone any chemical treatment. However, her carcinoma had metastasized to both sides of the lungs, and excision of the metastases was not possible because of her pulmonary function. Treatment with lenvatinib was therefore selected as the next therapeutic option.
Her blood pressure was normal (about 120/70 mmHg), and the blood and biochemical findings before treatment with lenvatinib were as follows: serum creatinine, 1.3 mg/dL; serum urea nitrogen, 41 mg/dL; and estimated glomerular filtration rate (eGFR), 31.8 mL/min/1.73 m2. The urine test was negative for red blood cells (<1/high-power field), the urinary protein excretion was 0.36 g/day, and beta 2-microglobulin was 0.7 mg/gCre. Nephrosclerosis was assumed on the basis of the mild renal dysfunction and findings of urinary abnormalities, although a clear diagnosis was not possible because a renal biopsy had not been performed.
Lenvatinib was started at 8 mg/day (day 1; see Fig. 1). On day 3, the patient developed hypertension that required treatment with olmesartan 20 mg/day, and the dose of lenvatinib was reduced to alternate-day administration of 4 mg and 8 mg. Amlodipine 2.5 mg/day was started on day 10 as an additional treatment for the hypertension, and the next day, the olmesartan dose was reduced to 10 mg/day; the hypertension normalized with this antihypertensive drug. On day 18, the patient developed pneumonia, which required a 12-day interruption of lenvatinib treatment. Amlodipine was discontinued on day 20 and olmesartan on day 22 because of a low blood pressure. Lenvatinib was restarted on day 30 with alternate-day administration of 4 and 8 mg, but the hypertension subsequently reappeared (systolic blood pressure: >180 mmHg). The hypertension worsened despite renewed treatment with olmesartan at 10 mg/day and amlodipine 2.5 mg/day, so the doses were increased to 20 and 5 mg/day, respectively. Urine testing on day 53 showed an increase in the urinary protein excretion to 17.35 g/day, beta 2-microglobulin to 13.1 mg/gCre, and worsening of the serum creatinine level (1.7 mg/dL) and eGFR (23 mL/min/1.73 m2), so we discontinued lenvatinib on day 60. The hypertension immediately improved, and we stopped olmesartan and amlodipine. To confirm the differential diagnosis of kidney disease, a biopsy was performed during the resolving phase (eGFR up to 37 mL/min/1.73 m2) 18 days after lenvatinib was discontinued. At the biopsy, the patient's blood pressure was 140/80 mmHg, and she had bilateral edema of her lower limbs; her urine and blood laboratory data are summarized in Table.
Figure 1.

Clinical course after starting lenvatinib. Alb: albumin, eGFR: estimated glomerular filtration rate, LEN: lenvatinib, sBP: systolic blood pressure, UPCR: urinary protein creatinine ratio
Table.
Clinical Data at Each Time Period.
| At the start of LEN | At the interruption of LEN | At the renal biopsy | Normal range | ||
|---|---|---|---|---|---|
| White blood cells | 6,100 | 7,000 | 5,600 | 3,200-7,900 | /μL |
| Hemoglobin | 8.7 | 9.6 | 7.3 | 11.3-15.0 | g/dL |
| Hematocrit | 27.9 | 30.7 | 22.7 | 34.0-46.3 | % |
| Platelete | 212 | 10.2 | 16.8 | 155-350 | *103/μL |
| Total protein | 8.2 | 6.9 | 6.3 | 6.9-8.4 | g/dL |
| Albumin | 3.5 | 2.9 | 3.4 | 3.9-5.2 | g/dL |
| AST | 19 | 32 | 15 | 13-33 | IU/L |
| ALT | 10 | 19 | 7 | 7-30 | IU/L |
| LDH | 220 | 511 | 209 | 119-229 | IU/L |
| ALP | 402 | 489 | 339 | 117-350 | IU/L |
| γGTP | 15 | 15 | 13 | 9-109 | IU/L |
| Urea nitrogen | 41 | 25 | 16 | 8-21 | mg/dL |
| Creatinine | 1.3 | 1.7 | 1.11 | 0.46-0.78 | mg/dL |
| eGFR | 31.8 | 22.9 | 36.8 | >90 | mL/min/1.73 |
| Uric acid | 8.3 | 7.1 | 7.4 | 2.5-7.0 | mg/dL |
| Na | 138 | 142 | 145 | 139-146 | mmol/L |
| K | 4.4 | 4.5 | 3.8 | 3.7-4.8 | mmol/L |
| Cl | 108 | 111 | 110 | 101-109 | mmol/L |
| Corrected Ca | 9.3 | 9.4 | 9.2 | 8.7-10.1 | mg/dL |
| P | 4.3 | 3.4 | 3.2 | 2.8-4.6 | mg/dL |
| Total bilirubin | 0.7 | 0.9 | 0.6 | 0.3-1.1 | mg/dL |
| CRP | 1.38 | 1.27 | 2.89 | 0.0-0.3 | mg/dL |
| PT | 87.3 | 106 | 90.8 | >75 | % |
| AFP | 2,454.1 | 929.5 | <10.0 | ng/mL | |
| PIVKA-II | 22 | 775 | <40 | mAU/mL | |
| Urine | |||||
| pH | 5.5 | 6.5 | 7.0 | ||
| Specific gravity | 1.024 | 1.007 | |||
| Red blood cell | <1 | <1 | 1-4 | /HPF | |
| Proteinuria | 0.36 | 15.84 | 6.04 | <0.1 | g/day |
| β2-MG | 0.7 | 13.1 | 13.1 | 0.1-1.9 | mg/gCre |
| NAG | 27.3 | 0.8-5.0 | IU/gCre | ||
| α1-MG | 47.73 | mg/gCre |
Kidney biopsy findings
Light microscopy of a kidney biopsy showed global sclerosis in 5 of 42 glomeruli. The preserved glomeruli showed features consistent with TMA, with endothelial swelling, a subendothelial insudative lesion, duplication of the glomerular basement membrane, mesangiolysis, and vacuolar degeneration of the podocytes, although overt thrombosis was not noted in the small arteries or glomeruli (Fig. 2A). Crescent formation and segmental sclerosis were not noted. Massive hyaline degeneration of the podocytes was also evident, suggesting podocytopathy (Fig. 2B). The subendothelial insudative lesion was consistent with fibrin material that was positive for phosphotungstic acid hematoxylin (PTAH) staining (Fig. 2C). In the mesangial areas, immunofluorescence staining was positive for IgM (Fig. 2D) but negative for IgG, IgA, C1q, C3, and C4. Electron microscopy showed marked widening of the subendothelial spaces by electron-lucent material (Fig. 2E) and massive electron-dense material consistent with fibrin in the subendothelial space (Fig. 2F). Podocyte foot process effacement was focal and mild (Fig. 2F). Arteriolar hyalinosis was mild, but polar vasculosis was observed around the glomerular vascular pole (Fig. 2G). Tubular atrophy and tubulointerstitial fibrosis were focal and occupied less than 25% of the total renal cortex; however, brush border loss of proximal tubular epithelial cells (Fig. 2H) and multinuclear formation of distal tubular epithelial cells (Fig. 2I) were noted, suggesting damage to the proximal and distal tubules.
Figure 2.
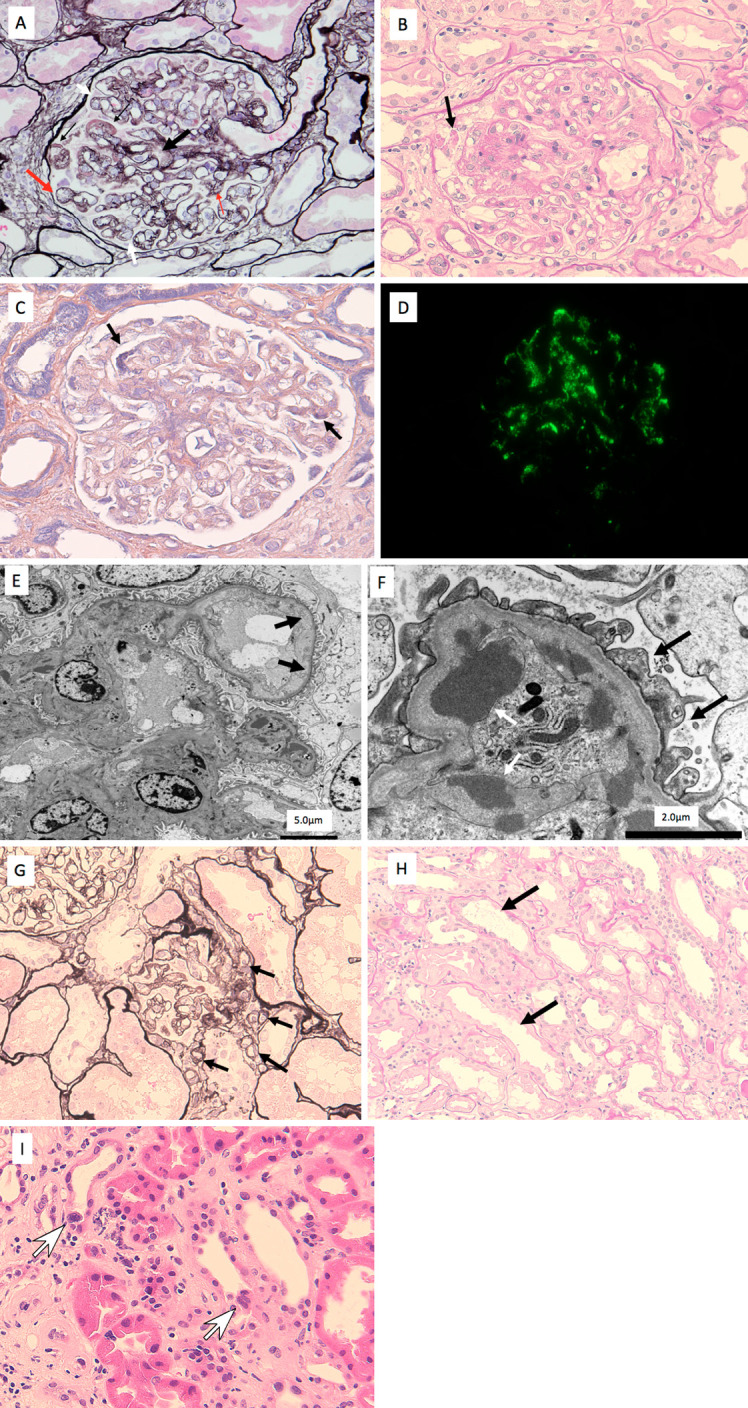
Kidney biopsy findings. A (periodic acid methenamine silver stain): The kidney biopsy showed features consistent with thrombotic microangiopathy, with endothelial swelling, a subendothelial insudative lesion (thin black arrows), duplication of the glomerular basement membrane (white arrow), mesangiolysis (thick black arrow), endothelial swelling (thick red arrow), and vacuolar degeneration of the podocytes (thin red arrows), although overt thrombosis was not noted in the small arteries or glomeruli. B (Periodic acid Schiff stain): Massive hyaline degeneration of the podocytes (black arrow) was also evident, suggesting podocytopathy. C: The subendothelial insudative lesions (black arrows) were considered fibrin material that was positive for phosphotungstic acid hematoxylin (PTAH) staining. D: In the mesangial areas, immunofluorescence staining was positive for IgM. E: Electron microscopy showed marked widening of the subendothelial spaces (black arrows). F: Electron microscopy showed massive electron-dense material in the subendothelial space (white arrow). Podocyte foot process effacement (black arrows) was focal and mild. G (Periodic acid methenamine silver stain): Polar vasculosis was observed around the glomerular vascular pole (black arrows). H (Periodic acid Schiff stain): Brush border loss of proximal tubular epithelial cells (black arrows). I (Hematoxylin and Eosin staining): Multinuclear formation of distal tubular epithelial cells (white arrows) was noted.
Clinical course
At discharge on day 91, the patient's kidney function was even better than before treatment with lenvatinib, as the eGFR had increased from 23 mL/min/1.73 m2 to 38 mL/min/1.73 m2 (pre-treatment eGFR: 31.8 mL/min/1.73 m2). Urinary protein excretion had decreased from 17.35 g/day to 6.84 g/day but remained higher than before treatment (pre-treatment level: 0.36 g/day), and the beta 2-microglobulin had not markedly changed (12.6 mg/gCre).
Discussion
Hypertension, proteinuria, and hematuria are well-known side effects of lenvatinib, with incidence rates of 67.8%, 32.6%, and 5%, respectively (4,9). In Japan, the most common any-grade adverse events are hypertension (42%), diarrhea (39%), decreased appetite (34%), and decreased weight (31%) (7), and they often lead to treatment discontinuation. In the case presented here, severe proteinuria developed after a short treatment period at the minimum dose and resulted in kidney impairment (with an eGFR of 25.0 mL/min/1.73 m2), so we had to discontinue lenvatinib.
A kidney biopsy revealed that severe proteinuria was due to drug-induced thrombotic microangiopathy-characterized by widening of the subendothelial space of the glomerular capillaries, duplication of the glomerular basement membrane, and mesangiolysis (although overt thrombosis was not noted)-and podocytopathy-characterized by massive hyaline degeneration and vacuolar degeneration of podocytes (although foot process effacement was mild). The biopsy also suggested that one cause of the acute kidney dysfunction might have been damage to the renal tubules. We detected damage in both the proximal and distal tubules, but interestingly, regenerated epithelial cells were only detected in the distal tubules. Because PDGFR is needed to regenerate proximal tubules (10), these findings suggest that the PDGFR inhibitor lenvatinib may prevent regeneration of the proximal tubules even after it has been discontinued; however, it is possible also that PDGFR prevented regeneration of the proximal tubules so that regeneration was seen only in the distal tubules.
The PDGF-B/PDGF receptor axis is known to be involved in tubular regeneration after ischemia/reperfusion injury of the kidney (11). Specifically, PDGF-B exerts a wide range of biological effects on renal cells by stimulating cellular proliferation and migration (12). A recent study provided evidence to suggest the importance of active Src kinase in the early phase of PDGF-B-dependent nephrogenic repair after acute ischemia/reperfusion injury (11). In the present case, we found no clinical evidence of renal ischemia, such as hypotension, but the nephrotoxicity of lenvatinib caused tubular damage. We hypothesize that, for some reason, tubular damage occurred before the ischemia/reperfusion injury, and lenvatinib inhibited the regeneration process. The PDGF-B/PDGF receptor axis is present in the proximal tubules but not in the distal tubules and is inhibited by lenvatinib, which explains why no regeneration of proximal tubular epithelial cells was observed.
Delsante et al. described three cases of biopsy-proven renal TMA clinically presenting with proteinuria and stable serum creatinine in patients receiving lenvatinib (13), and Cavalieri et al. (14), Furuto et al. (6), Fleming et al. (15), and Takikita-Suzuki et al. (11) described four other cases. In the cases where a renal biopsy was performed, it invariably showed features of microangiopathic changes. The mechanism underlying glomerular injury in angiogenesis inhibition is likely related to decreased glomerular blood flow, altered endothelial permeability, and reduced endothelial proliferation, which lead to alteration of the filtration barrier and secondary podocyte injury with resultant proteinuria (16). However, none of the cases showed tubular involvement. We believe that proteinuria is affected by glomerular damage caused by VEGFR and PDGFR and that proximal tubule regeneration also may be inhibited by PDGFR.
Izzedine et al. proposed two types of TMA caused by cancer drugs (17): Type 1 TMA is caused by mitomycin C and gemcitabine and presents with microangiopathic hemolytic anemia, thrombocytopenia, and progressive chronic kidney disease requiring dialysis even after drug discontinuation, and microvascular thrombosis in the glomeruli or small arteries is the key event; while Type 2 TMA is caused by VEGF agents, among others, and presents with proteinuria that subsides after drug discontinuation; glomerular capillary thrombosis is usually absent. These two types of TMA have characteristics in common with endotheliosis, including edematous expansion of the subendothelium, duplication of the glomerular basement membrane, and mesangiolysis. Type 2 TMA was established to characterize patients with endotheliosis without thrombosis (18).
In our case, we observed polar vasculosis around the glomerular vascular pole (Fig. 2G). This finding has been reported as a type of vascular lesion in patients with diabetic nephropathy and has been considered to be associated with VEGF-related proliferation and elongation of efferent arterioles and hyperglycemia-related angiogenesis (19). Anti-VEGF agents similar to lenvatinib might contribute to polar vasculosis via pathogenetic mechanisms similar to diabetic nephropathy.
In our patient, the proteinuria values did not return to the levels before treatment, so podocyte injury induced by lenvatinib may have contributed to the persistence of proteinuria.
In conclusion, we encountered a patient who developed proteinuria, hypertension, and renal dysfunction after four weeks of lenvatinib treatment. The proteinuria and renal dysfunction were attributed to type II thrombotic microangiopathy, podocytopathy, and damage to the renal tubules.
Limitations
This patient's kidney biopsy was performed during the resolving phase (eGFR up to 37 mL/min/1.73 m2) 18 days after lenvatinib was discontinued. Because we did not examine the renal histology during the worst phase, podocyte foot process effacement was focal and mild; however, massive hyaline degeneration and vacuolar degeneration of the podocytes were evident, suggesting podocytopathy. Despite this limitation, we believe that our case provides considerable information related to lenvatinib nephropathy.
The authors state that they have no Conflict of Interest (COI).
Acknowledgements
We gratefully acknowledge Yutaka Yamaguchi (Yamaguchi's Pathology Laboratory, Chiba, Japan) for valuable advice regarding the renal pathology.
References
- 1.Yamamoto Y, Matsui J, Matsushima T, et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell 6: 18, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 372: 621-630, 2015. [DOI] [PubMed] [Google Scholar]
- 3.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Med 9: 669-676, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 111: 707-716, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karkkainen MJ, Mäkinen T, Alitalo K. Lymphatic endothelium: a new frontier of metastasis research. Nature Cell Biol 4: E2-E5, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Furuto Y, Hashimoto H, Namikawa A, et al. Focal segmental glomerulosclerosis lesion associated with inhibition of tyrosine kinases by lenvatinib: a case report. BMC Nephrol 19: 273, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391: 1163-1173, 2018. [DOI] [PubMed] [Google Scholar]
- 8.Izzedine H, Mangier M, Ory V, et al. Expression patterns of RelA and c-mip are associated with different glomerular diseases following anti-VEGF therapy. Kidney Int 85: 457-470, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Haddad RI, Schlumberger M, Wirth LJ, et al. Incidence and timing of common adverse events in lenvatinib-treated patients from the SELECT trial and their association with survival outcomes. Endocrine 56: 121-128, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schiessl IM, Grill A, Fremter K, Steppan D, Hellmuth MK, Castrop H. Renal interstitial platelet-derived growth factor receptor-β cells support proximal tubular regeneration. J Am Soc Nephrol 29: 1383-1396, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takikita-Suzuki M, Haneda M, Sasahara M, et al. Activation of Src kinase in platelet-derived growth factor-B-dependent tubular regeneration after acute ischemic renal injury. Am J Pathol 163: 277-286, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abboud HE. Role of platelet-derived growth factor in renal injury. Annu Rev Physiol 57: 297-309, 1995. [DOI] [PubMed] [Google Scholar]
- 13.Delsante M, Monroy-Trujillo JM, Carter-Monroe N, Ball DW, Rosenberg AZ. Lenvatinib-related renal microangiopathy: a case series. Virchows Arch. Forthcoming. [DOI] [PubMed] [Google Scholar]
- 14.Cavalieri S, Cosmai L, Genderini A, et al. Lenvatinib-induced renal failure: two first-time case reports and review of literature. Expert Opin Drug Metab Toxicol 14: 379-385, 2018. [DOI] [PubMed] [Google Scholar]
- 15.Fleming K, McGuinness J, Kipgen D, Glen H, Spiliopoulou P. A case of lenvatinib-induced focal segmental glomerulosclerosis (FSGS) in metastatic medullary thyroid cancer. Case Rep Oncol Med 2018: 6927639, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boor P, van Roeyen CR, Kunter U, et al. PDGF-C mediates glomerular capillary repair. Am J Pathol 177: 58-69, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Izzedine H, Perazella MA. Thrombotic microangiopathy, cancer, and cancer drugs. Am J Kidney Dis 66: 857-868, 2015. [DOI] [PubMed] [Google Scholar]
- 18.Toriu N, Sekine A, Mizuno H, et al. Renal-limited thrombotic microangiopathy due to bevacizumab therapy for metastatic colorectal cancer: a case report. Case Rep Oncol 12: 391-400, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shima N, Sawa N, Yamanouchi M, et al. Characteristic renal histology of a 81-year-old patient with a 30-year history of diabetes mellitus: a case report. CEN Case Rep 9: 338-343, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]