

Abstract
A seven-month-old girl presented with bilateral roving nystagmus, hyperopia, and retinal dystrophy, and was brought to our ophthalmology clinic. Visual-evoked potentials (VEPs) were non-recordable in both the eyes. No other systemic symptoms or abnormalities were observed. Whole exome sequencing (WES) identified a compound heterozygous mutation in the IFT140 gene: c.1990G > A (p. Glu664Lys) and c.2214_2217del (p.Asp738GlufsTer47). The genetic results support a diagnosis of Mainzer-Saldino syndrome (MSS). Importantly, c.2214_2217del is a novel mutation in the IFT140 gene. Although the patient presents with isolated retinal dystrophy, it is crucial to monitor renal function overtime. Taken together, our results reinforce the role of IFT140 in syndromic ciliopathies. This report also highlights the role of combined WES approaches in identifying underlying mutations in infants presenting with isolated retinal dystrophy, considering MSS may present differently over time.
Keywords: Mainzer-Saldino syndrome, Retinal dystrophy, Whole exome sequencing
Highlights
-
•
A novel mutation in IFT140 was found to cause Mainzer-Saldino syndrome (MSS).
-
•
MSS may present as isolated retinal dystrophy in infant, and genetic testing plays a crucial role in diagnosis.
-
•
Patients with MSS should receive systemic follow-up, particularly renal function.
1. Introduction
Ciliopathies are a heterogeneous group of diseases that affect intraflagellar transport (IFT) and alter cilia function [1]. Given the widespread presence of IFT in the cellular cilia and flagella of eukaryotic organisms, its mutations can give rise to a wide range of symptoms in multiple systems. IFT particles are multiprotein complexes that can be biochemically grouped into two subcomplexes, IFTA and IFTB [2], having distinct functions. IFTA is crucial in the retrograde transport whereas and IFTB is crucial in the anterograde trafficking, along ciliary axonemes [3]. Mutations in IFTA form ciliary bulges due to protein accumulation at the distal tip, whereas mutations in IFTB often lead to failure in cilia formation due to defects in importing cilia-building components [4,5].
Mainzer-Saldino syndrome (MSS) is a rare autosomal recessive ciliopathy, clinically characterized by cone-shaped phalangeal epiphyses, chronic renal disease, and retinal dystrophy. Less common characteristics, such as short stature, cerebellar ataxia, and hepatic fibrosis [6]. MSS is most commonly attributed to the dysfunction of the IFT140 protein, which encodes a subunit of IFTA [7]. Pathogenic variation in IFT140 has not only been reported in patients with MSS, but isolated retinitis pigmentosa (RP), cranioectodermal dysplasia (CED), Jeune syndrome and opitz trigonocephaly syndrome (OTCS) [8,9].
Here, we report a pediatric patient carrying a novel pathogenic mutation in the IFT140 gene, presenting with isolated retinal dystrophy. Although there was no evident systemic manifestation on initial presentation, the genetic results supported a diagnosis of MSS.
2. Materials and methods
We collected the patient's medical records, family history, and clinical presentation, along with blood samples for hemogram and biochemistry profiles. Bone age study, renal ultrasound, liver ultrasound, and detailed ophthalmic examinations were performed. Whole exome sequencing (WES) was performed to detect possible DNA alterations. First, 100 ng genomic DNA from the sample to be tested was fragmented to an average size of 180–280 bp. Library preparation and enrichment capture were performed using Illumina platform-compatible Roche KAPA HyperExome kit. Enriched captured DNA fragments were amplified and sequenced using 2 × 150 bp Paired-End formats on a NovaSeq sequencer. Low-quality bases (Q < 30) were trimmed. Sequence data were aligned based on GRCh38 using DRAGEN (SW:05.021.595.3.7.5) and variant site searching was performed using DRAGEN (SW:05.021.595.3.7.5). Confirmed variant sites were annotated using the Variant Effect Predictor (v101) and Jannovar (0.35). Human phenotype ontology and Online Mendelian Inheritance in Man (OMIM) databases were used to identify candidate genes based on patient phenotypes. Variants previously reported as benign or likely to be benign in the ClinVar database and variants with a prevalence of >5% in the Common Variation Databases (dbSNP version 150 and Taiwan Biobank) were excluded. Genetic variants in the CDS region were analyzed for changes in protein structure using prediction softwares, including SIFT, PolyPhen-2, and MutationTaster. The variants were then clinically interpreted according to the ACMG/AMP 2015 guidelines. We also performed Sanger sequencing with further trio analysis, for the patient and her parents. All collected data were de-identified. The corresponding author has full access to all the data and bears the final responsibility for the decision to submit the data for publication.
3. Case presentation
A seven-month-old female infant was born at full term as the first child of a non-consanguineous couple. Her parents reported that she was unable to follow objects and she was referred to our ophthalmology department. On initial examination, she was blinking with light but not following objects. Roving nystagmus presented in all positions of gaze was evident, and Hirschberg test showed orthotropia. Hyperopic refractive errors of approximately +6.0 Diopter were noted in both the eyes. Anterior segment evaluation and intraocular pressure measurements were insignificant. Fundus examination revealed attenuated vessels with vascular sheathing and marbleized diffuse retinal pigment dispersion (Fig. 1C). Visual evoked potentials (VEPs) were non-recordable bilaterally (Fig. 2). Brain magnetic resonance imaging (MRI) showed no structural abnormalities. Ultrasonography at the initial presentation revealed no significant abnormalities in the kidneys, liver, or pancreas. No clinical or radiographic evidence of skeletal dysplasia was observed. We noted that this patient had some facial features, like frontal bossing, high forehead and broad nasal bridge (Fig. 1A). However, she did not have narrow thorax, shorter ribs or any other skeletal abnormalities. No hearing impairment was observed.
Fig. 1.
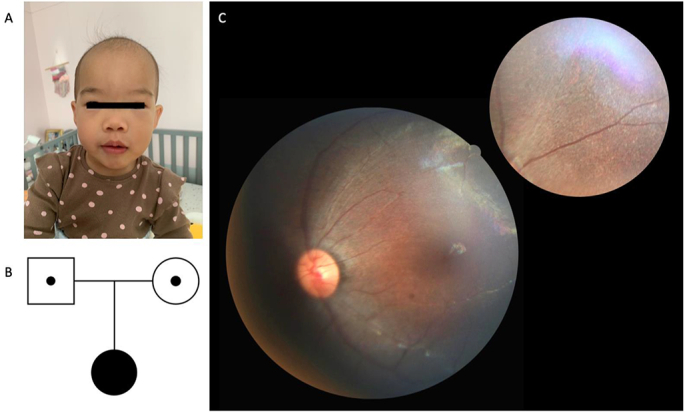
The photograph of the patient, the fundus image, and the pedigree.
(A) Front view photograph of seven-month-old patient. (B) Pedigree of patient's family. (C) Color photographs of fundus showing attenuated and sheathing vessels with pigment mottling of the retina pigment epithelium throughout.
Fig. 2.

Flash visual evoked potentials (VEPs) showing nonrecordable (NR) in both eyes. (A) Right eye stimulation. (B) Left eye stimulation.
Notably, WES illustrated two important compound heterozygous mutations affecting the IFT140 (intraflagellar transport 140) gene. A paternal missense mutation NM_014714.4:c.1990G > A, NP_055529.2.1:p. Glu664Lys (located at chr16:1564074) and a maternal frameshift mutation NM_014714.4:c.2214_2217del, NP_055529.2: p.Asp738GlufsTer47 (located at ch16:1558117–1558120) are shown in Fig. 1B and Fig. 3. The former mutation has been reported to be pathogenic in the ClinVar database (variant ID:31679) with a global allele frequency of 0.004% (gnomAD – Exomes) and an East Asian frequency of 0.004% (gnomAD – Genomes). This mutation has been correlated with Mainzer-Saldino syndrome (MSS) in previous studies (PMID:26359340, PMID:26216056, PMID:22503633). The c.1990G > A (p.Glu664Lys) changes showed approximately 80% disorganization of IFT140 and loss of basal body localization associated with an increase in the cytoplasmic portion (PMID:22503633). The second mutation observed, c. 2214_2217del (Asp738GlufsTer47), was a novel mutation. Currently, there is no incidence record in commonly used databases, including ExAC, 1000 genomes, gnomAD, and the Taiwan Biobank. This variant has neither been documented in ClinVar nor been reported in PubMed. Since this mutation causes premature termination of protein translation, the prediction software MutationTaster predicts that the effect of this mutation on the protein results is “Disease causing,” and the point mutation is classified as “Pathogenic” according to the ACMG guidelines. Based on these findings, the patient was diagnosed with MSS.
Fig. 3.

(A) Scheme of IFT140 showing the positions of two mutations. (B–C) BAM file snapshots of the two mutation spots.
4. Results and discussion
Ciliopathies are genetic disorders associated with the dysfunction of primary cilia which affect multiple organ systems [10]. Primary cilia operate as cellular signaling centers that modulate various extracellular stimuli, and they are essential in regulating cellular responses during early vertebrate and specific tissue development [11]. Ciliopathies can be organ-specific, as in the case of polycystic kidney disease, nephronophthisis, or LCA [12,13], or can be syndromic, as in the case of Jeune syndrome, Bardet-Biedl syndrome, and MSS. As research indicates that the cumulative prevalence of ciliopathies is approximately 1 in every 2000 individuals, accurate and timely diagnosis is essential to avoid significant morbidity and mortality [14].
Currently, IFT140 mutations have been identified in several syndromic ciliopathies, including MSS. Khan et al. studied 11 subjects with confirmed homozygous IFT140-related retinopathy and found that although some patients had frank skeletal or renal disease, not all patients showed obvious extraocular findings [15]. This is consistent with the presentation in our patient, where ophthalmic phenotypes including infantile nystagmus, hyperopia, and retinal dystrophy were apparent with no systemic manifestations observed to date. LCA and retinitis pigmentosa are commonly described in a series of MSS cases. In these reports, early onset severe visual impairment may appear earlier than systemic symptoms [7,16,17]. This finding can partly be explained by the distinct role of IFT140 in maintaining cilia function in photoreceptors [18]. In light of these findings, it is important to note that variable expressivity for ciliopathies is common, and that careful systemic work-up and monitor need to be emphasized.
In this study, the frameshift mutation c.2214_2217del (p.Asp738GlufsTer47) detected in our patient is a novel variant located in the IFT140. This novel mutation has not been previously reported as a cause of MSS, and seems to be related to early ophthalmic involvement rather than renal or hepatic impairment in our patient. The classic MSS phenotype often includes renal failure [19], which was not observed in our patient. However, given the nature of IFT140, it is important to arrange a systemic follow-up for this patient to monitor extraocular diseases, particularly renal function [20].
In summary, we identified compound heterozygous IFT140 variants in a patient with MSS. Given the pathogenic variation in IFT140, MSS must be considered as a differential diagnosis in patients with isolated retinal dystrophy. The present study strengthens the rationale that early monitoring of systemic diseases including renal function is crucial in patients with MSS.
Funding
This study was supported by the Ministry of Science and Technology, Taiwan, grant number: 110-2628-B-075-010; Taipei Veterans General Hospital, grant number: V111C-102; Yen Tjing Ling Medical Foundation, grant number: CI-111-29.
Declaration of Competing Interest
There are no conflicts of interest to disclose.
Acknowledgements
We wish to thank the patient and her family in this study for their generous participation.
Data availability
The data that has been used is confidential.
References
- 1.Waters A.M., Beales P.L. Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 2011;26:1039–1056. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kannabiran C. Review: Intraflagellar transport proteins in the retina. Mol. Vis. 2020;26:652–660. [PMC free article] [PubMed] [Google Scholar]
- 3.Engel B.D., Ishikawa H., Wemmer K.A., Geimer S., Wakabayashi K., Hirono M., Craige B., Pazour G.J., Witman G.B., Kamiya R., Marshall W.F. The role of retrograde intraflagellar transport in flagellar assembly, maintenance, and function. J. Cell Biol. 2012;199:151–167. doi: 10.1083/jcb.201206068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blacque O.E., Cevik S., Kaplan O.I. Intraflagellar transport: from molecular characterisation to mechanism. Front. Biosci. 2008;13:2633–2652. doi: 10.2741/2871. [DOI] [PubMed] [Google Scholar]
- 5.Liem K.F., Jr., Ashe A., He M., Satir P., Moran J., Beier D., Wicking C., Anderson K.V. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J. Cell Biol. 2012;197:789–800. doi: 10.1083/jcb.201110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beals R.K., Weleber R.G. Conorenal dysplasia: a syndrome of cone-shaped epiphysis, renal disease in childhood, retinitis pigmentosa and abnormality of the proximal femur. Am. J. Med. Genet. A. 2007;143A:2444–2447. doi: 10.1002/ajmg.a.31948. [DOI] [PubMed] [Google Scholar]
- 7.Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A., Gerber S., Delphin N., Bigot K., Orssaud C., Silva E., Baudouin V., Oud M.M., Shannon N., Le Merrer M., Roche O., Pietrement C., Goumid J., Baumann C., Bole-Feysot C., Nitschke P., Zahrate M., Beales P., Arts H.H., Munnich A., Kaplan J., Antignac C., Cormier-Daire V., Rozet J.M. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 2012;90:864–870. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walczak-Sztulpa J., Posmyk R., Bukowska-Olech E.M., Wawrocka A., Jamsheer A., Oud M.M., Schmidts M., Arts H.H., Latos-Bielenska A., Wasilewska A. Compound heterozygous IFT140 variants in two polish families with Sensenbrenner syndrome and early onset end-stage renal disease. Orphanet. J. Rare Dis. 2020;15:36. doi: 10.1186/s13023-020-1303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pena-Padilla C., Marshall C.R., Walker S., Scherer S.W., Tavares-Macias G., Razo-Jimenez G., Bobadilla-Morales L., Acosta-Fernandez E., Corona-Rivera A., Mendoza-Londono R., Corona-Rivera J.R. Compound heterozygous mutations in the IFT140 gene cause Opitz trigonocephaly C syndrome in a patient with typical features of a ciliopathy. Clin. Genet. 2017;91:640–646. doi: 10.1111/cge.12924. [DOI] [PubMed] [Google Scholar]
- 10.Baker K., Beales P.L. Making sense of cilia in disease: the human ciliopathies. Am. J. Med. Genet. C: Semin. Med. Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 11.Goetz S.C., Anderson K.V. The primary cilium: a signalling Centre during vertebrate development. Nat. Rev. Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zerres K., Volpel M.C., Weiss H. Cystic kidneys. Genetics, pathologic anatomy, clinical picture, and prenatal diagnosis. Hum. Genet. 1984;68:104–135. doi: 10.1007/BF00279301. [DOI] [PubMed] [Google Scholar]
- 13.Grochowsky A., Gunay-Aygun M. Clinical characteristics of individual organ system disease in non-motile ciliopathies. Transl. Sci. Rare. Dis. 2019;4:1–23. doi: 10.3233/TRD-190033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kagan K.O., Dufke A., Gembruch U. Renal cystic disease and associated ciliopathies. Curr. Opin. Obstet. Gynecol. 2017;29:85–94. doi: 10.1097/GCO.0000000000000348. [DOI] [PubMed] [Google Scholar]
- 15.Bifari I.N., Elkhamary S.M., Bolz H.J., Khan A.O. The ophthalmic phenotype of IFT140-related ciliopathy ranges from isolated to syndromic congenital retinal dystrophy. Br. J. Ophthalmol. 2016;100:829–833. doi: 10.1136/bjophthalmol-2015-307555. [DOI] [PubMed] [Google Scholar]
- 16.Oud M.M., Latour B.L., Bakey Z., Letteboer S.J., Lugtenberg D., Wu K.M., Cornelissen E.A.M., Yntema H.G., Schmidts M., Roepman R., Bongers E. Cellular ciliary phenotyping indicates pathogenicity of novel variants in IFT140 and confirms a Mainzer-Saldino syndrome diagnosis. Cilia. 2018;7:1. doi: 10.1186/s13630-018-0055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montolio-Marzo S., Catala-Mora J., Madrid-Aris A., Armstrong J., Diaz-Carcajosa J., Carreras E. IFT144 and mild retinitis pigmentosa in Mainzer-Saldino syndrome: a new association. Eur. J. Med. Genet. 2020;63 doi: 10.1016/j.ejmg.2020.104073. [DOI] [PubMed] [Google Scholar]
- 18.Khanna H. Vol. 4. 2015. Photoreceptor Sensory Cilium: Traversing the Ciliary Gate Cells; pp. 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidts M., Vodopiutz J., Christou-Savina S., Cortes C.R., McInerney-Leo A.M., Emes R.D., Arts H.H., Tuysuz B., D'Silva J., Leo P.J., Giles T.C., Oud M.M., Harris J.A., Koopmans M., Marshall M., Elcioglu N., Kuechler A., Bockenhauer D., Moore A.T., Wilson L.C., Janecke A.R., Hurles M.E., Emmet W., Gardiner B., Streubel B., Dopita B., Zankl A., Kayserili H., Scambler P.J., Brown M.A., Beales P.L., Wicking C., Uk10K, Duncan E.L., Mitchison H.M. Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 2013;93:932–944. doi: 10.1016/j.ajhg.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidts M., Frank V., Eisenberger T., Al Turki S., Bizet A.A., Antony D., Rix S., Decker C., Bachmann N., Bald M., Vinke T., Toenshoff B., Di Donato N., Neuhann T., Hartley J.L., Maher E.R., Bogdanovic R., Peco-Antic A., Mache C., Hurles M.E., Joksic I., Guc-Scekic M., Dobricic J., Brankovic-Magic M., Bolz H.J., Pazour G.J., Beales P.L., Scambler P.J., Saunier S., Mitchison H.M., Bergmann C. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney. Dis. Hum. Mutat. 2013;34:714–724. doi: 10.1002/humu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that has been used is confidential.