

Summary
CD28 provides the prototypical costimulatory signal required for productive T-cell activation. Known molecular consequences of CD28 costimulation are mostly based on studies of protein signaling molecules. The microRNA cluster miR-17∼92 is induced by T cell receptor stimulation and further enhanced by combined CD28 costimulation. We demonstrate that transgenic miR-17∼92 cell-intrinsically largely overcomes defects caused by CD28 deficiency. Combining genetics, transcriptomics, bioinformatics, and biochemical miRNA:mRNA interaction maps we empirically validate miR-17∼92 target genes that include several negative regulators of T cell activation. CD28-deficient T cells exhibit derepressed miR-17∼92 target genes during activation. CRISPR/Cas9-mediated ablation of the miR-17∼92 targets Pten and Nrbp1 in naive CD28−/− CD4+ T cells differentially increases proliferation and expression of the activation markers CD25 and CD44, respectively. Thus, we propose that miR-17∼92 constitutes a central mediator for T cell activation, integrating signals by the TCR and CD28 costimulation by dampening multiple brakes that prevent T cell activation.
Subject areas: Biological sciences, molecular mechanism of gene regulation, immunology
Graphical abstract
Highlights
-
•
Empirically validated direct miR-17∼92 target gene list in mouse CD4+ T cells
-
•
CD28-deficient CD4+ T cells exhibit derepressed miR-17∼92 targets during activation
-
•
Transgenic miR-17∼92 expression rescues CD28-deficiency in vitro and in vivo
-
•
miR-17∼92 target KO in naive CD28-deficient CD4+ T cells restores distinct phenotypes
Biological sciences; Molecular mechanism of gene regulation; Immunology
Introduction
T cells are critical to protect mammals from infections and tumors. T cell activation, a key event for adaptive immunity, relies on two signals: T cell receptor (TCR) stimulation as well as costimulation by specialized receptors. While the TCR signal provides specificity, costimulation by antigen-presenting cells (APCs) provides the quantitiative and qualitative support for T cell activation (Acuto and Michel, 2003; Esensten et al., 2016). One of the best-studied and prototypical costimulatory molecules is CD28. It promotes multiple processes required for T cell biology such as T cell activation, proliferation, survival, metabolic adaptation, and Interleukin-2 (IL-2) production (Esensten et al., 2016; Riha and Rudd, 2010). These form the basis for clonal expansion and T cell differentiation into a variety of effector T cells that are necessary to mount effective immune responses. Recent data further demonstrates that CD28 expression is not only required for T cell priming but also days later for effector CD4+ T cell responses during infection (Linterman et al., 2014).
Due to its centrality for T cell and immune responses more generally, CD28 is an important target in therapeutic immunology (Sansom and Walker, 2013; Esensten et al., 2016; Edner et al., 2020). CD28 blockade by CTLA4-Ig is clinically used to prevent renal allograft rejection and to treat rheumatic disease (Esensten et al., 2016). In contrast, vaccine adjuvants induce the activation of innate immune cells to express ligands that trigger costimulatory molecules on T cells. Furthermore, immune-activating CTLA-4 blocking antibodies represent the foundation of cancer immunotherapy (Leach et al., 1996) and CD28 is required for cancer immunotherapy with PD-1 blocking antibodies (Kamphorst et al., 2017; Hui et al., 2017; Homet Moreno et al., 2016). Finally, CD28 intracellular signaling domains can provide the required costimulatory signal in second and third generation chimeric antigen receptor (CAR) T cell constructs for adoptive cellular therapies (Gross and Eshhar, 2016). Thus, CD28 is a key clinically relevant immunoregulatory receptor and a precise understanding of molecular events induced by CD28 costimulation has direct therapeutic relevance. However, despite intense research, the understanding of the molecular consequences of CD28 costimulation remains incomplete (Esensten et al., 2016; Tian et al., 2015; Liu et al., 2018). CD28 costimulation acts through pleiotropic effects; it promotes the phosphatidylinositol 3-kinase (PI3K) pathway, amplifies the TCR signal, stabilizes the transcriptome induced by TCR stimulation, and enhances calcineurin/NFAT signaling (Riha and Rudd, 2010; Esensten et al., 2016; Martinez-Llordella et al., 2013). Collectively, CD28 stimulation ultimately leads to transcriptional changes mediated by the transcription factors (TF) NF-κB, AP-1, and NFAT (Esensten et al., 2016). In addition, CD28 also acts through many non-transcriptional mechanisms such as mRNA stabilization and altered mRNA splicing (Esensten et al., 2016). Thus, due to the complexity of CD28 costimulation, substantial controversy remains about the importance of various molecular mechanisms and the relative importance of quantitative versus qualitative CD28-mediated signals (Esensten et al., 2016; Acuto and Michel, 2003; Riha and Rudd, 2010; Sansom and Walker, 2013).
Since a protein-centric view prevailed in most studies, here we investigated the role of the microRNA cluster miR-17∼92, a non-coding RNA, in CD28 costimulation and T cell activation. MicroRNAs (miRNA) are arguably the best-studied class of non-coding genes. These short RNA molecules of ∼22nt length are highly conserved gene repressors that mainly act through base pairing with the 3′ untranslated region (UTR) of target RNAs resulting in their decreased abundance and/or translational inhibition (Baumjohann and Ansel, 2013). Together with transcription factors (TFs), miRNAs are the most important trans-regulators of gene expression, regulating most mRNAs (Bartel, 2018). Target RNAs can be bound by multiple miRNAs and individual miRNAs can bind to and repress multiple genes, often genes found in the same pathway (Baumjohann and Ansel, 2013). Canonical base-pairing is largely determined by miRNA nucleotide positions 2–7, called the “seed” region (Bartel, 2018) but non-canonical miRNA targeting is widespread and can be equally effective (Loeb et al., 2012; Hsin et al., 2018). The interaction of a miRNA and its target mostly results in mild gene repression, often only reducing protein concentration by less than 2-fold. Nevertheless, miRNA-mediated gene regulation is highly consequential for T cell differentiation and function (Xiao and Rajewsky, 2009; O'Connell et al., 2010; Baumjohann and Ansel, 2013; Jeker and Bluestone, 2013).
We noticed that key functions attributed to CD28 such as T cell proliferation, survival, and TFH differentiation are also regulated by miR-17∼92. miR-17∼92, a polycistronic transcript induced by CD28 costimulation (de Kouchkovsky et al., 2013), gets processed into 6 mature miRNAs representing 4 different seed families (Xiao and Rajewsky, 2009). Much like CD28, miR-17∼92 promotes T cell proliferation, survival, and differentiation (Xiao et al., 2008; Jeker and Bluestone, 2013). Moreover, T cell-specific miR-17∼92-deficiency results in severely impaired TFH differentiation and impaired GC formation (Baumjohann et al., 2013; Kang et al., 2013), reminiscent of impaired GC formation observed in CD28-deficient mice (Ferguson et al., 1996). On the contrary, miR-17∼92 overexpression results in a systemic lupus-like syndrome with increased GC formation and autoantibody production (Xiao et al., 2008) most likely due to enhanced TFH generation (Kang et al., 2013; Baumjohann et al., 2013).
Here, we show that miR-17∼92 was necessary to repress genes in CD28−/− T cells and forced miR-17∼92 expression was sufficient to restore an important fraction of the impaired transcriptional regulation and function of CD28-deficiency in murine CD4+ T cells. We used transcriptome analysis, computational predictions and biochemical miRNA/target RNA interaction maps to define a high-confidence set of 68 empirically validated direct miR-17∼92 target genes in T cells. CRISPR/Cas9-mediated deletion of individual miR-17∼92 target genes restored distinct functions in naive CD28−/− T cells. We propose that miR-17∼92 acts as an important mediator of T cell activation/CD28-costimulation by repressing multiple inhibitory proteins that restrain T cells from becoming activated.
Results
T cell receptor and CD28 costimulation synergistically induce miR-17∼92 expression and forced miR-17∼92 expression enables T cell activation of CD28-deficient T cells in vitro
miR-17∼92 expression has previously been linked to T cell activation/CD28 costimulation (Sandberg et al., 2008; de Kouchkovsky et al., 2013) and the CD28 ligands CD80 and CD86 have a dose-dependent effect on miR-17 expression (Wang et al., 2015). However, the relative contribution of TCR versus CD28 is not well understood. To further investigate this relationship for miR-17∼92 induction, we stimulated naive murine CD4 T cells with various combinations of anti-CD3/anti-CD28 monoclonal antibodies (Figure 1A). TCR stimulation alone increased miR-17 expression while isolated CD28 stimulation did not. Compared to low TCR stimulation, high TCR stimulation had a little additional effect on miR-17 but further increased CD69/CD25 expression (Figures 1A and 1B). Adding even a low concentration (0.2 μg/mL) of anti-CD28 antibody to TCR stimulation increased miR-17 expression at both timepoints (24 and 48 h). The induction of miR-17 progressed from 24 h to 48 h. At any concentration of anti-CD3 mAb stimulation and timepoint, CD28 costimulation resulted in a positive interaction for miR-17 expression. At both timepoints, the combination of low anti-CD3 (0.5 μg/mL) and low anti-CD28 (0.2 μg/mL) resulted in higher induction of miR-17 than the highest anti-CD3 stimulation alone (5 μg/mL). Finally, at 48 h, low TCR stimulation combined with high CD28 costimulation increased miR-17 but not CD25/69 expression in comparison to low TCR/low CD28 costimulation. In summary, TCR signaling and CD28 costimulation are intricately linked to miR-17 expression (as a surrogate miRNA for the miR-17∼92 cluster). Although TCR stimulation alone induced miR-17 expression, combined TCR and CD28 stimulation synergistically increased miR-17 resulting in stronger expression than through either signal alone. These results are consistent with and extend previous experiments using natural ligands that demonstrated a graded control of miR-17∼92 by CD28 (Wang et al., 2015).
Figure 1.

TCR and CD28 costimulation synergistically induce miR-17∼92 expression and forced miR-17∼92 expression enables T cell activation of CD28-deficient T cells in vitro
(A and B) Naive murine CD4+ T cells were activated for 24 h or 48 h using the indicated combinations of plate-bound anti-CD3 and anti-CD28 stimulatory mAbs. (A) miR-17 expression assessed by qPCR. (B) Relative frequencies of cells expressing CD25 and CD69, assessed by flow cytometry.
(C–G) wt (black), CD28−/− (purple), rescue (dark blue) CD4+ T cells were stimulated for 48 h with plate-bound anti-CD3 with (+) or without (−) anti-CD28. (C) Blasting of CD4+ T cells shown as MFI of FSC-A of the lymphocyte gate. (D) Proliferation measured by CTV dilution, gated on viable CD4+ T cells. Representative histograms of each genotype activated without (blank) or with (colored) anti-CD28 and quantification of proliferation index. (E and F) Expression of early activation markers CD25/CD69 as well as CD44/CD62L expression. (G) Quantification of flow cytometric intracellular IL-2 staining in CD4+ cells stimulated for 3 h with PMA/Iono/BFA. Data from an experiment with 3 biological replicates per condition (A and B). Data from 3 (C–G) independent experiments with 3–4 biological replicates per group. Error bars represent mean ± SD, Tukey’s or Hom-Sidak’s multiple comparison test; p values: ns = not significant, ∗<0.05 ∗∗ < 0.002 ∗∗∗<0.0002 ∗∗∗∗<0.0001.
In order to further investigate the functional connection between CD28 and miR-17∼92, we analyzed the consequences of T cell-specific loss and gain of miR-17∼92 on key processes regulated by CD28. We compared samples from mice that lack miR-17∼92 in T cells (CD4cre.miR-17∼92lox/lox, designated T1792Δ/Δ hereafter), wildtype (wt) mice, and mice overexpressing miR-17∼92 in T cells (CD4cre.Rosa26loxSTOPloxCAG-miR-17∼92Tg, designated T1792tg/tg hereafter). In comparison to wt T cells, proliferation as well as the production and secretion of the CD28-dependent cytokine interleukin-2 (IL-2) was impaired in T1792Δ/Δ T cells and increased in T1792tg/tg T cells (Figures S1A–S1C) confirming previous findings (Baumjohann et al., 2013; Kang et al., 2013; Steiner et al., 2011). Thus, miR-17∼92-deficiency was reminiscent of phenotypes observed in CD28-deficient mice. In contrast, transgenic miR-17∼92 had the opposite effect. Thus, the results demonstrated that the non-coding RNA miR-17∼92 exerted a dose-dependent regulation of CD4+ T cell proliferation and IL-2 production which are known to be CD28-dependent. We hypothesized that miR-17∼92 could be a downstream mediator or integrator of T cell activation/CD28 costimulation. To test this hypothesis we crossed B6.CD28−/− (CD28−/−) mice (Shahinian et al., 1993) with T1792tg/tg mice, resulting in B6.CD28−/−.CD4cre.Rosa26loxSTOPloxCAG-miR-17∼92Tg designated “rescue” hereafter. T cells in these mice lack CD28 but constitutively express transgenic miR-17∼92. If miR-17∼92 physiologically supported CD28 costimulation then we expected that transgenic miR-17∼92 expressions could restore some of the CD28 defects. First, we investigated how “rescue” cells behaved in vitro. We compared wt, CD28−/−, and “rescue” naive CD4+ T cells stimulated with plate-bound anti-CD3 mAb alone or a combination of anti-CD3 and anti-CD28 mAb. As expected, wt cells blasted (Figure 1C) and increased proliferation (Figure 1D) in response to costimulation compared to anti-CD3 stimulation alone. Compared to wt cells, CD28−/− T cells showed reduced size and proliferation and were unable to respond to anti-CD28 stimulation (Figures 1C and 1D). In contrast, “rescue” T cells stimulated with anti-CD3 alone or combined anti-CD3/anti-CD28 blasted and proliferated like wt T cells fully stimulated with anti-CD3 and anti-CD28 mAb (Figures 1C and 1D).
Next, we turned our attention to surface markers whose expression is either TCR- or CD28-dependent. The early activation marker CD69 is rapidly upregulated during T cell activation and reflects TCR signaling strength (Shang et al., 2018). As a second marker, we used the high affinity IL-2 receptor alpha subunit CD25, a well-known CD28-dependent gene. The relative number of CD69+CD25+ cells and CD69 expression per cell (Figure 1E) was comparable for all genotypes, as predicted for a TCR-dependent marker. In contrast, the signal intensity of CD25 was clearly CD28-dependent. Wildtype T cells increased CD25 MFI after CD28 costimulation while CD28−/− T cells displayed lower CD25 MFI than wt cells and were unable to respond to CD28 stimulation (Figure 1E). In contrast, CD25 expression was fully restored in “rescue” T cells, even after anti-CD3 stimulation alone (Figure 1E). Finally, CD44 expression was also highly CD28-dependent. In concert with the other CD28-dependent parameters (blasting, proliferation, IL-2, and CD25), wt T cells responded to CD28 ligation with CD44 expression but CD28−/− cells lacked CD44 upregulation. In contrast, the frequency of CD44hiCD62Llo “rescue” T cells was at least comparable to fully costimulated (anti-CD3/anti-CD28) wt T cells (Figure 1F). Thus, the miR-17∼92 transgene appeared to efficiently replace CD28 function in vitro, restoring the expression of the CD28-dependent markers CD25 and CD44.
Given the remarkable capacity of the miR-17∼92 transgene to restore discrete functions in CD28-deficient T cells, we measured miR-17 expression upon activation in various genotypes (Figure S1D). T1792Δ/Δ T cells were unable to express miR-17 except for a faint signal at 48 h, likely reflecting incomplete deletion. In contrast, wt T cells increased miR-17 at 24 and 48 h while CD28−/− T cells displayed impaired miR-17 expression. Rescue T cells expressed increased miR-17 already in naive T cells and expression further increased with activation. Expressing one copy of the miR-17∼92 transgene in CD28-sufficient T cells increased miR-17 expression compared to wt cells and adding a second transgene copy further increased miR-17 expression. Thus, at 24 h, rescue T cells displayed slightly increased miR-17 expression compared to wt cells and similar expression to wt cells at 48 h. At 48 h, rescue T cells, T1792 wt/tg and T1792tg/tg T cells overexpressed miR-17∼92. Overall, these results support the suitability of the chosen experimental system. Finally, since IL-2 production constitutes another functionally important consequence of CD28 costimulation and miR-17∼92 correlated strongly with IL-2 production (Figures S1B and S1C), we analyzed this cytokine next. As with blasting or proliferation, CD28-deficient T cells produced less IL-2 but “rescue” T cells produced even supraphysiologic amounts of IL-2 upon activation (Figure 1G). Thus, transgenic miR-17∼92 was sufficient to replace CD28 for several costimulation-dependent processes in vitro.
Transgenic miR-17∼92 enables T cell activation of CD28-deficient T cells in vivo
Next, we investigated whether transgenic miR-17∼92 could also substitute CD28 function in vivo. CD28−/− mice display a severe defect in TFH development and GC formation (Shahinian et al., 1993; Ferguson et al., 1996; Linterman et al., 2009; Walker et al., 1999). Therefore, we infected wt, CD28−/− and “rescue” mice with lymphocytic choriomeningitis virus (LCMV) Armstrong to induce an acute viral infection leading to TH1 and TFH differentiation as well as GC B cell formation. Confirming previous literature, we found severely impaired CD44 upregulation, TFH differentiation, and GC B cell formation in CD28−/− mice compared to wt littermates (Figures 2A–2D). In contrast, all these parameters were restored in “rescue” mice (Figures 2A–2D). This is remarkable given the complexity of TFH differentiation (Crotty, 2011). In addition, rescued T cells not only phenotypically resembled TFH cells through their expression of CXCR5, PD-1, Bcl-6, and ICOS (Figures 2B and 2C) but they were functional because they induced GC B cell formation which reflects TFH/B cell crosstalk. Furthermore, the spleen of infected “rescue” mice, but not CD28−/− mice, featured organized GCs containing GL7+ B cells and CD4+ T cells (Figure 2E) demonstrating the restoration of another hallmark defect found in CD28−/− mice (Ferguson et al., 1996). Finally, we analyzed TH1 responses and found that transgenic miR-17∼92 restored the defect in TH1 differentiation observed in CD28−/− mice (Figures 2F and 2G). We noticed that fewer CD28−/− cells expressed Tbx21 compared to wt cells (Figure 2F). However, some of those cells that did express Tbx21 co-expressed IFNγ, even in CD28−/− cells. Analyzing the ratio of Tbx21+IFNγ+/Tbx21+ T cells confirmed that the missing costimulatory signal mainly resulted in defective Tbx21 induction rather than IFNγ production (Figure 2H). These results suggest that the CD28−/− defect acts during T cell activation, i.e. before TH1 differentiation. Accordingly, the miR-17∼92 transgene appears to restore T cell activation signals.
Figure 2.

Transgenic miR-17∼92 enables T cell activation of CD28-deficient T cells in vivo
6–8 week old mice were infected with LCMV Armstrong; spleens were analyzed at d8 post-infection. wt (black), CD28−/− (purple), rescue (dark blue).
(A–D) represent data from 4 independent experiments with 4 mice per group, pre-gating on viable CD4+CD3+ or viable CD19+B220+ cells. Representative FACS plots and quantification of relative numbers of (A) CD44 expression, (B) Bcl6+ICOS+ population (TFH), (C) CXCR5+PD-1+ population (TFH) and (D) Fas+GL7+ population (GC B cells).
(E) Cryosections of spleens stained for GL-7 (green), CD4 (red) and CD19 (blue). Scale bar 40 μm.
(F–H) Splenocytes were re-stimulated with GP-64 and BFA for 4 h and investigated for TH1 phenotype, pre-gated on viable CD3+CD4+ cells. Shown are 3 independent experiments with 4 biological replicates per group. Representative FACS plots (F) and quantification (G) of relative numbers of Tbx21+IFNγ+ CD3+CD4+ cells. (H) Ratio of Tbx21+IFNγ+ to total Tbx21+ cells. Error bars represent mean with SD, Dunn’s multiple comparison test; p values: ns = not significant, ∗<0.05, ∗∗<0.002, ∗∗∗<0.0002, ∗∗∗∗<0.0001.
T1792tg/tg T cells displayed increased proliferation and IL-2 secretion compared to wt cells (Figures S1A–S1C) and intracellular IL-2 was not only restored but even higher in “rescue” T cells than wt T cells (Figure 1G), suggesting that the effect of miR-17∼92 depended on its abundance. Therefore, we investigated the effect of a single copy of the miR-17∼92 transgene in CD28−/− cells. In addition, we directly compared the effect of the miR-17∼92 transgene in CD28-deficient and CD28-sufficient T cells in vivo to test if CD28 triggering and the miR-17∼92 transgene were additive. The effect of various genotypes on miR-17 expression is shown in Figure S1D. We infected CD28−/−, T1792Δ/Δ, wt, het rescue, rescue, and T1792tg/tg mice with LCMV Armstrong. “het rescue” were CD28−/− that only carried one copy of the miR-17∼92 Tg while “rescue” mice were the rescue mice used above (Figures 1 and 2) with two copies of the miR-17∼92 transgene. The relative number of CD44+ T cells was lowest in CD28−/− and highest in T1792tg/tg mice (Figure S2A). Moreover, CD28−/− and T1792Δ/Δ mice exhibited similar defects compared to wt mice. In contrast, even one copy of the miR-17∼92 Tg was sufficient to rescue the CD28−/− phenotype of TFH and GC B cell formation (Figures S2B–S2D).
In summary, we found an unexpectedly complete the restoration of CD28 costimulatory function and T cell activation exerted by transgenic miR-17∼92 expression in vitro as well as in vivo.
Restoration of T cell activation of CD28-deficient T cells by miR-17∼92 is cell intrinsic
Although CD28’s main function is on T cells, we sought to formally test whether the miR-17∼92-mediated rescue effect was cell intrinsic. We crossed MHC class II-restricted CD4+ TCR transgenic mice specific for LCMV (SMARTA; Vα2+Vβ8.3+) to wt, CD28−/− and “rescue” mice. We adoptively transferred (AT) naive CD4+ T cells to CD28−/− host mice followed by acute LCMV infection. Eight days post-infection we isolated spleen, mesenteric, and peripheral lymph nodes (LN). In all three organs the frequency and absolute number of Vα2+Vβ8.3+ CD28−/− cells was strongly reduced compared to Vα2+Vβ8.3+ CD28 w/w cells. In contrast, the miR-17∼92 transgene restored relative and absolute numbers of Vα2+Vβ8.3+ CD28−/− T cells (Figures 3A, S3A, and S3B). Furthermore, among Vα2+Vβ8.3+ T cells, fewer CD28−/− cells upregulated CD44 than in wt cells, a defect that was entirely restored in rescue cells (Figures 3B, S3C, and S3D). Thus, these data unequivocally demonstrate that transgenic miR-17∼92 cell intrinsically compensated for CD28 deficiency.
Figure 3.

Restoration of T cell activation of CD28-deficient T cells by miR-17∼92 is cell intrinsic
Adoptive transfer of naive SMARTA+ CD4+ T cells into CD28−/− hosts, subsequent LCMV Armstrong infection, and analysis of organs at d8 post-infection. Donor genotypes wt (black), CD28−/− (purple), rescue (dark blue). Dotted line indicates recipient’s intrinsic Vα2+Vβ8.3+ population measured in a non-transferred control host.
(A and B) (A) Vα2+Vβ8.3+ cells from peripheral LN pre-gated on viable, CD3+CD4+ cells (B) CD44 expression in Vα2+Vβ8.3+ population from peripheral LN. 2 independent experiments, 4 recipients per group. Error bars represent mean ± SD, Dunn’s multiple comparison test, p values: ns = not significant, ∗<0.05, ∗∗<0.002, ∗∗∗<0.0002. See also Figure S3 for spleen and mesenteric LN.
miR-17∼92 shapes the transcriptome after CD4+ T cell activation
To unravel the molecular mechanism underlying miR-17∼92-mediated function during T cell activation we performed RNA-sequencing on naive and in vitro activated (24 and 48 h) CD4+ T cells from T1792Δ/Δ, wt, and T1792tg/tg mice. Principal component analysis (PCA) revealed that the transcriptomes of naive T cells from all three genotypes were very similar (Figure 4A, 0 h). T cell activation induced major changes in gene expression (PC1, 56.7% of variance explained and PC2, 14% of variance explained) and also made the genotypes separate at 24 h and even more clearly at 48 h after activation (Figures 4A and S4A) (PC1 and PC3, 7.2% of variance explained). Since miRNAs often repress individual genes only mildly (Bartel, 2018), we compared the most extreme genotypes, i.e. T1792Δ/Δ to T1792tg/tg to increase the power of differential gene expression analysis, at each time point. At a false discovery rate (FDR) of 1%, the number of differentially expressed genes (DEG) increased over time (830 genes up-regulated and 789 genes down-regulated at 0 h, 2,493 up and 2,370 down at 24 h, and 3,173 up and 3,242 down at 48 h). Unsupervised hierarchical clustering of DEG 24 h after activation revealed a nuanced pattern of gene clusters (Figure 4B). As expected from the PCA (Figure 4A), gene expression across genotypes was very similar in naive T cells and the magnitude of expression differences increased after activation (Figure 4B). According to their expression profile, we highlighted 4 different groups of genes (Figure 4B): cluster I genes were induced over time and enhanced in T1792tg/tg compared to wt but reduced or delayed in T1792Δ/Δ. Cluster II genes decreased with time and miR-17∼92 supported their repression. Overall cluster III gene expression increased after activation but expression per time point inversely correlated with the genotype (T1792Δ/Δ > T1792tg/tg). Thus, miR-17∼92 limited the maximal expression of genes in this group after induction. Finally, genes displaying the most obvious inverse correlation with the genotype were grouped in clusters IVa and IVb.
Figure 4.

miR-17∼92 shapes the transcriptome after CD4+ T cell activation
Naive CD4+ T cells from T1792Δ/Δ (gray), wt (black) and T1792tg/tg (light blue) were activated with plate-bound anti-CD28 and anti-CD3 for 0, 24, and 48 h. Total RNA was extracted for bulk RNA sequencing.
(A) PCA (PC1 vs. PC2) based on the 25% most variable genes.
(B) Hierarchical clustering of the set of genes selected with abs(log2FC) > 1 & adj.P.Val<0.001 in the T1792tg/tg vs. T1792Δ/Δ comparison at 24 h. The heatmap displays the centered log of counts per million, with blue indicating low and red indicating high expression. Annotations: “DE” indicates the fold change direction, “DE intron” indicates if significant changes are observed in EISA analysis, “TS” indicates presence (gray) or absence (blank) of a seed match and its location and “AHC” indicates the 3′UTR signal intensity in HITS-CLIP data. Boxes I–IVb designate gene clusters.
(C) Examples of genes in cluster I.
(D and E) Genome-wide transcriptome analysis presented as the log2 value of the gene-expression ratio for each gene versus the cumulative fraction of all log2 ratios in naive (D) and 24 h activated (E). Shown is the miR-17 seed family for T1792tg/tg vs. wt and T1792Δ/Δ vs. wt comparisons. Black curve: all genes without a seed match and ≤5 AHC reads; green: subset of genes with a seed sequence for the seed family and >5 reads in the AHC.
(F) Examples of genes defined as empirically validated miR-17∼92 targets. log2 RNA expression level in activated CD4+ T cells, numbers correspond to FDR<0.05. Boxplot representing values distribution over minimum and maximum values, median, 25th and 75th percentiles.
To disentangle the gene regulation modalities of the different clusters, we used exon-intron split analysis (EISA) to discriminate between transcriptional versus posttranscriptional regulation (Gaidatzis et al., 2015a). In addition, we employed computational target gene predictions for miR-17∼92 from the Targetscan (“TS”) database (Agarwal et al., 2015) and used a dataset of biochemically detected direct miRNA:mRNA interactions in T cells defined by Argonaute 2 high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (“AHC”) (Gagnon et al., 2019). For each gene, we quantified the read coverage on TS seed matches for each of the miR-17∼92 cluster seed families. These predictors of transcriptional (“DE intron,” for differential expression pattern detected at the nascent transcripts) and posttranscriptional regulation (“TS,” “AHC”), annotated on the heatmap (Figure 4B) revealed different patterns of regulation for the 4 groups of genes highlighted above. Cluster I genes were enriched for transcriptional regulation (both DE and DE intron) and displayed few TS sites or AHC reads (Figure 4B, box I). Thus, they were mainly induced by increased gene transcription, and miR-17∼92 promoted this transcriptional activity. Examples include Cd44, IL-21, Tbx21, and IFNγ (Figure 4C). In contrast, genes from clusters IVa and IVb (Figure 4B, boxes IVa, IVb), which displayed a colinear reduction of expression levels across genotypes, contained few transcriptionally regulated genes but were enriched for TS sites (p value = 0.001037; Fisher test) and experimentally determined AHC reads (p value = 0.003598; Fisher test) (Figure 4B). Thus, these clusters were mainly regulated posttranscriptionally and were likely enriched for direct miR-17∼92 target genes.
To further characterize direct miR-17∼92 target genes we focused on naive T cells and the 24 h time point since indirect effects likely increased after this time point. To visualize the effect of individual miR-17∼92 cluster miRNAs on their target genes we compared the expression of genes identified by TS and with >5 AHC reads for each miRNA seed family to all genes without any seed match for that family. As illustrated by the miR-17 seed family, the miR-17∼92 transgene repressed miR-17 target genes in naive T cells (Figure 4D, left panel) but the absence of miR-17∼92 had no effect on the expression of miR-17 target genes (Figure 4D, right panel). In contrast, after T cell activation miR-17 target genes were repressed in T1792tg/tg T cells (Figure 4E, left panel) and derepressed in T1792Δ/Δ T cells (Figure 4E, right panel). Similar effects were observed for all seed families although to a lesser extent for the miR-18 seed family (Figures 4B and 4C). We defined genes as empirically validated miR-17∼92 targets if they fulfilled the following criteria: i) significant derepression in T1792Δ/Δ vs wt and significant repression in T1792tg/tg vs wt at 24 h ii) predicted TS match iii) > 5 AHC reads and iv) posttranscriptional regulation based on EISA. Applying these criteria across the 4 seed families from miR-17∼92 defined a set of 68 empirically supported direct miR-17∼92 target genes (Table S1). These genes included previously described miR-17∼92 targets validated in T cells, such as Phlpp2 (Kang et al., 2013) and Cyld, validated in B cells (Jin et al., 2013) (Figure 4F). In addition, this approach identified many less studied genes (e.g. Rcan3, Nrbp1, Rnf38, and Rnf167) (Figure 4F). Notably, several of the target genes negatively regulate pathways important for T cell activation. For instance, Phlpp2 is a PI3K inhibitor (Kang et al., 2013) and Cyld is an NF-κB inhibitor (Reiley et al., 2007). In addition, we identified the putative calcineurin inhibitor Rcan3 (Mulero et al., 2007) as a new miR-17∼92 target and validated its RNA-Seq data by qPCR on independent biologic replicates (Figure S4D). Its 3′UTR contains a conserved miR-17-5p 8mer binding site and AHC confirmed a single discrete peak at this site, evidence for a direct interaction of a miRNA with the Rcan3 3′UTR in primary T cells (Figure S4E and (Loeb et al., 2012; Gagnon et al., 2019)). Together, these data empirically validate Rcan3 as a miR-17 target in T cells.
Thus, using a combination of experimentally validated differential gene expression (T1792Δ/Δ, wt, and T1792tg/tg T cells), evidence of posttranscriptional gene regulation and biochemical detection of miRNA binding we defined a high confidence set of miR-17∼92 target genes in T cells. miR-17∼92 became functionally relevant after T cell activation and shaped the transcriptome in intricate ways. Our data demonstrate that miR-17∼92 can promote and inhibit gene expression, through posttranscriptional gene regulation, enhancing gene silencing or dampening expression of induced genes. Thus, miR-17∼92-mediated gene repression is important to shape the T cell transcriptome during T cell activation.
miR-17∼92 promotes the calcineurin/NFAT pathway in CD4+ T cells
To identify which molecular pathways were regulated by miR-17∼92, we analyzed curated gene sets enriched for differentially expressed genes between T1792tg/tg and T1792Δ/Δ. At 24 h, the gene sets with the highest statistical significance and largest average fold change were related to cytokines, inflammation, and T cell differentiation (Figure S5A) while at 48 h many metabolic pathways were altered (Figure S5B). Next, we performed enrichment analysis on DoRothEA regulons (Garcia-Alonso et al., 2019) to identify TF activity that could explain the regulated pathways. Regulons were defined using any existing documented interaction with a particular TF. At 24 h, the five most significantly enriched TF regulons with the highest fold change contained two NFAT members (NFATC2, NFATC3) as well as RELA, NF-κB1, and GATA3 (Figure 5A). Since NFAT TFs are important for T cell activation and differentiation (including TFH differentiation (Martinez et al., 2016)) but miR-17∼92 is not known to promote NFAT activity in T cells, we focused on the calcineurin/NFAT axis. Genes belonging to regulons NFATC2 and NFATC3 include many T cell lineage-defining TFs, cytokines, and cytokine receptors and most of them – including IL-21, Tbx21, and IFNγ (Figure 4C) – were regulated by miR-17∼92 (Figure 5B). This confirmed that miR-17∼92 constitutes a central regulator of T cell activation and suggested that transgenic miR-17∼92 enhanced canonical pathways that resulted in the functional substitution of CD28 for the differentiation of TFH and TH1 in vivo (Figure 2). Since miR-17∼92 promoted the expression of signature TF and cytokines defining multiple T cell subsets, we tested if miR-17∼92 more generally could replace CD28. To this end, we differentiated TH1, TH17, and iTreg cells in vitro. We confirmed that also in this setup transgenic miR-17∼92 was sufficient to compensate for the absence of CD28 and functionally corrected the defects of CD28−/− T cells to differentiate into all 3 subsets (Figure S6).
Figure 5.

miR-17∼92 promotes the NFAT and NF-κB pathways in CD4+ T cells
(A and B) Dataset from Figure 4. Naive CD4+ T cells from T1792Δ/Δ (gray), wt (black), and T1792tg/tg (light blue) were activated with plate-bound anti-CD28 and anti-CD3 for 0, 24, and 48 h. Total RNA was extracted for bulk RNA sequencing. (A) Volcano plot showing the absolute log2 fold change and -log10 p value from regulon analysis. A threshold of 1% FDR was applied. Dot size indicates the number of genes within each regulon and colors indicate fold change direction. (B) Heatmap of genes under NFATC2_D and NFATC3_D regulons plus several known activated genes in CD4+ T cells (Il10, Il12a, Il6, Rorc, Il23a). Hierarchical clustering was applied on genes. Centered log of counts per million is displayed, with blue indicating low and red indicating high expression.
(C) CD4+ wt (black), CD28−/− (purple), and rescue (dark blue) cells were activated for 48 h in the presence of increasing concentrations of cyclosporin A (CsA) as indicated and stained for CD25 and CD69. Shown are 2 independent experiments, error bars represent means ± SD. Tukey’s multiple comparison, p values: ∗∗<0.002, ∗∗∗∗<0.0001 refer to the difference between CD28−/− and wt.
(D) representative FACS plots of CD25/CD69 expression in viable CD4+ T cells activated for 48 h with no or 6.25 ng/mL CsA.
(E) influence of 6.25 ng/mL CsA on blasting (FSC-A of the lymphocyte gate) of viable CD4+ cells.
(F) Imagestream analysis of CD4+ T cells, activated for 48 h in presence of 6.25 ng/mL CsA; DAPI (blue) and NFATc2 (red) staining. Examples for cytoplasmic (top) and nuclear (bottom) NFATc2 in a CD28−/− sample.
(G) histograms of the similarity dilate indicative of the co-localization of NFATc2 and DAPI signals; gates indicate the nuclear (high similarity dilate) and the cytoplasmic population (low similarity dilate).
See also Figures S5 and S6.
While miR-17∼92 predominantly promoted inflammatory pathways at 24 h, at 48 h several metabolic pathways were regulated by miR-17∼92 (Figures S5A and S5B). Thus, we hypothesized that initial transcriptional changes could lead to altered metabolism. It was recently shown that cell cycle entry of quiescent T cells was controlled by store-operated Ca2+ entry (SOCE) and calcineurin/NFAT through control of glycolysis and oxidative phosphorylation (Vaeth et al., 2017). Therefore, we analyzed T cell metabolism in naive and activated T1792Δ/Δ, wt, and T1792tg/tg T cells. In line with the transcriptome results (Figure 4), metabolic flux analysis demonstrated comparable glycolytic and respiratory activity of naive T cells in T1792Δ/Δ, wt, and T1792tg/tg T cells (Figures S5C and S5D). In contrast, 48 h after activation, glycolytic and respiratory activity positively correlated with the miR-17∼92 genotype (Figure S5E and S5F). Furthermore, genes associated with the TCA cycle and respiratory electron transport positively correlated with miR-17∼92 at 48 h but not before, supporting the notion that miR-17∼92 indirectly promoted this metabolic activity (Figure S5G).
Since transcriptome analysis identified NFAT as a key pathway promoted by miR-17∼92 (Figures 5A and 5B), we designed experiments to functionally validate this increased activity. We activated wt, CD28−/− and “rescue” T cells in the presence of cyclosporin A (CsA), a drug known to inhibit calcineurin/NFAT, and quantified CD69/CD25 expression. Compared to wt cells, CD28−/− cells were 4-fold more sensitive to CsA (Figure 5C). At a CsA concentration that did not affect wt cells (arrow in Figure 5C), T cell activation of CD28−/− T cells was clearly inhibited (Figures 5C and 5D). Thus, compared to wt cells CD28−/− T cells are hypersensitive to CsA. In contrast, CD28−/− T cells with forced miR-17∼92 expression (rescue cells) were normally activated (Figures 5C and 5D). In fact, at higher CsA concentrations the “rescue” T cells were even more resistant to CsA inhibition than wt T cells (Figure 5C). These findings were further corroborated by the analysis of cell size which revealed that in the presence of low dose CsA the blasting defect of CD28−/− T cells was compensated for by transgenic miR-17∼92 (Figure 5E). Finally, we assessed the nuclear translocation of activated NFATC2 as a direct readout of calcineurin activity. ImageStream analysis of T cells activated in the presence of 6.25 ng/mL CsA showed T cells with cytoplasmic (top row) or nuclear (lower row) NFATC2 (Figure 5F). Quantification of this data demonstrated that the presence of a low CsA concentration reduced nuclear NFATC2 translocation in CD28−/− T cells but was restored in “rescue” cells (Figure 5G). Thus, the miR-17∼92 transgene replaced CD28-enhanced NFAT signals for blasting, CD69/CD25 upregulation, and nuclear translocation of NFATC2.
miR-17∼92 is necessary for the repression of a subset of genes during T cell activation
Next, we examined whether miR-17∼92 was physiologically required to shape the molecular program triggered by CD28 engagement. We repeated transcriptome analysis with RNA-seq on naive and activated (24 h) CD4+ T cells from T1792Δ/Δ, wt, and T1792tg/tg mice but added CD28−/− and “rescue” T cells. The correlation of miR-17∼92 target gene expression levels with the first RNA-seq experiment (Figure 4) was very high (Figure S7A). A PCA showed that the transcriptomes of naive T cells of all 5 genotypes were closely related (Figure 6A). However, after T cell activation (PC1, 67.6% of variance) the 5 genotypes formed 5 distinct groups that were separated on PC2 (5.6% of variance). Notably, the “rescue” samples were located between CD28−/− and T1792Δ/Δ, wt, and T1792tg/tg T cells (Figure 6A). This implied that transgenic miR-17∼92 partially restored the genetic networks dysregulated by CD28 deficiency, supporting the notion that the phenotypic rescue (Figure 1, Figure 2, Figure 3) was not a transgene artifact. Moreover, compared to all genes without a miR-17 seed match, the miR-17 target gene set based on the same criteria specified earlier but using a second dataset differential gene expression analysis was overall derepressed in CD28−/− T cells compared to wt cells (Figures 6B and S7B). It is important to note that these cells have an untouched miR-17∼92 locus and therefore should repress miR-17∼92 target genes similar to wt cells. However, the absence of CD28 resulted in increased expression of those genes. This result demonstrated that CD28-dependent effects normally rely on miR-17∼92 to repress dozens of genes during T cell activation. Remarkably, the expression of target genes was corrected in “rescue” cells (Figures 6C and S7B). Thus, transgenic miR-17∼92 repressed the physiologic targets that were derepressed in CD28−/− T cells.
Figure 6.

miR-17∼92 is necessary for repression of a subset of genes during T cell activation
CD4+ T cells from T1792Δ/Δ (gray), wt (black), T1792tg/tg (light blue), CD28−/− (purple) and rescue (dark blue) mice were activated for 24 h. Total RNA was extracted for sequencing.
(A) PCA (PC1 vs. PC2) based on the 25% most variable genes.
(B and C) (B) Genome-wide transcriptome analysis, presented as the log2 value of the gene-expression ratio for each gene versus the cumulative fraction of all log2 ratios. Shown are the contrasts between activated samples separated by the miR-17 seed family for the comparison CD28−/− vs. wt (B) and rescue vs. wt (C). Black curve: genes without a seed match, ≤5 AHC reads, and no differential expression in the second RNA sequencing. Green: genes with a conserved binding site for the miR-17 seed family (TS) and >5 reads in the AHC, red: genes with a conserved binding site for the miR-17 seed family (TS), >5 reads in the AHC and differential expression in the second RNA sequencing dataset.
(D) Hierarchical clustering of the set of genes selected with abs(log2FC) > 1 & adj.P.Val<0.001 in the T1792tg/tg vs. T1792Δ/Δ comparison at 24 h. The heatmap displays the centered log of counts per million, with blue indicating low and red indicating high expression.
(E) Examples of transcripts that were restored in “rescue” compared to CD28−/− T cells, i.e. contained in the box (D).
(F) Examples of direct miR-17∼92 targets. E, F: log2 mRNA expression (counts per million) in activated CD4+ T cells; numbers correspond to FDR<0.05. Boxplot representing values distribution over minimum and maximum values, median, 25th and 75th percentiles.
See also Figure S7.
However, the molecular rescue effect on the entire transcriptome was incomplete (Figure 6A). We, therefore, analyzed which transcripts were restored and whether they were regulated transcriptionally or posttranscriptionally. Similarly to Figure 4B, we selected genes differentially expressed in the T1792Δ/Δ to T1792tg/tg comparison at 24 h, and performed unsupervised hierarchical clustering using their gene expression profiles across all 5 genotypes. At 24 h, one gene cluster stood out in which “rescue” T cells were more similar to wt and T1792tg/tg cells than CD28−/− and T1792Δ/Δ cells (Figure 6D, box). Genes in this cluster contained several NFAT-dependent transcripts including IL-4, IL-12a, IL12rb2, IL-21 and IFNγ (Table S2, Figure 5B). Importantly, Cd44, IL-21, Tbx-21, and IFNγ transcripts, also contained in this cluster, were reduced in CD28−/− compared to wt cells (Figure 6E). In contrast, transgenic miR-17∼92 partially or completely restored these transcripts in “rescue” cells to wt levels. Furthermore, T1792tg/tg T cells expressed supraphysiologic mRNA levels (Figure 6E). Thus, the phenotypic rescue of CD44 and IFNγ in CD28−/− T cells (Figure 1, Figure 2, Figure 3) could at least partially be attributed to increased expression of these genes driven by the miR-17∼92 transgene in “rescue” cells.
Conversely, as illustrated above (Figure 6B) many direct targets were not only derepressed in T1792Δ/Δ but also in CD28−/− cells. Several genes (e.g. Rcan3, Nrbp1, Rnf38, and Rnf167) were similarly derepressed in CD28−/− and T1792Δ/Δ cells, expression was restored in “rescue” cells to levels similar to wt and even further repressed in T1792tg/tg cells (Figure 6F). Others, such as Phlpp2 and Cyld were clearly regulated by miR-17∼92 but their expression differed in CD28−/− and T1792Δ/Δ cells (Figure 6F). This suggests that these targets are sensitive to regulation by miR-17∼92 and in multiple cases, their derepression observed in CD28−/− T cells is largely attributed to reduced repression by miR-17∼92. However, functional validation of candidate targets will be necessary.
miR-17∼92 target genes regulate discrete functions
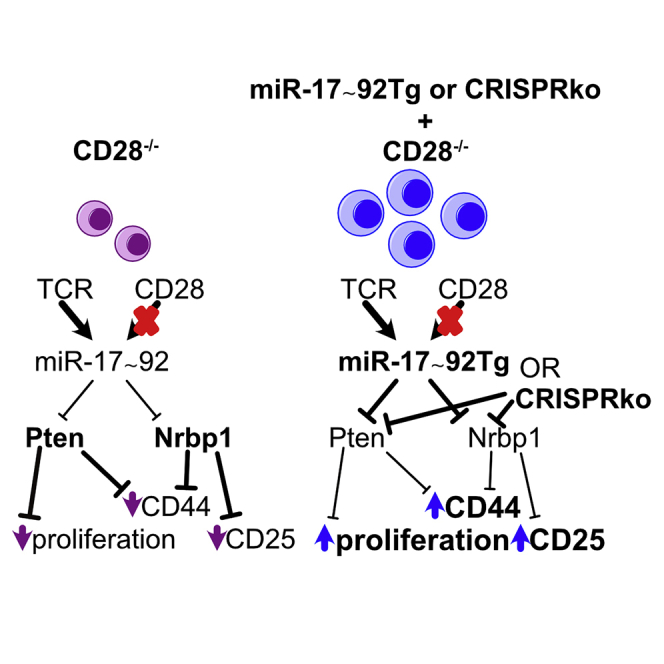
To understand the functional relevance of the empirically validated miR-17∼92 targets (Table S1) we reasoned that genetically inactivating targets should have phenotypically similar effect as miR-17∼92-mediated repression. We therefore chose to ablate candidate target genes in CD28−/− T cells using CRISPR/Cas9. In order to be able to analyze a candidate gene’s role during T cell activation we sought to delete candidate genes in naive T cells, i.e. before activation. To this end, we adapted a protocol to use CRISPR/Cas9 in naive CD4+ T cells (Seki and Rutz, 2018) (Figure 7A). We used a non-targeting control (NTC) crRNA as a negative control, included the well-validated miR-17∼92 target Pten as a positive control and chose Phlpp2, Cyld, Rcan3, Nrbp1, Rnf38, and Rnf167 (Figures 4F and 6F) to test our hypothesis. As readouts for potential phenotypic rescue we analyzed proliferation, CD25, CD44, and ICOS since these parameters were clearly restored in “rescue” T cells (Figures 1D–1F and 2B). In line with a previous report that genetic Pten-ablation in T cells removed the requirement for CD28 costimulation for proliferation (Buckler et al., 2006), we observed increased proliferation in cells electroporated with a Pten-targeting gRNA but not any of the other gRNAs (Figure 7B). This effect was clearly evident at 48 h but could no longer be detected at 72 h. Ablation of nuclear receptor binding protein 1 (Nrbp1) resulted in increased expression of CD25 after 72 h while the ablation of the other genes did not affect CD25 expression (Figure 7C). Furthermore, CD44 expression was increased by Pten-ablation at 48 and 72 h and by Nrbp1-ablation at 72 h. Finally, contrary to expectations, Pten-ablation resulted in decreased ICOS expression at 72 h. Thus, CRISPR/Cas-mediated ablation of individual target genes resulted in distinct phenotypic consequences. Deleting two individual miR-17∼92 target genes in naive CD28−/− CD4+ T cells increased 3 of the 4 tested parameters that were also rescued by transgenic miR-17∼92 expression, suggesting that their repression by transgenic miR-17∼92 in “rescue” T cells contributed to the phenotypic rescue observed. In contrast, the deletion of Pten resulted in two expected phenotypic consequences and one opposite to expectations. This suggests that ICOS upregulation observed in “rescue” T cells is not mediated by repression of Pten but rather an as yet unidentified miR-17∼92 target gene. Together, these results support CRISPR/Cas-based gene deletion as a means to functionally validate candidate miRNA target genes and strongly suggest that miR-17∼92-mediated repression of these targets is functionally relevant during T cell activation.
Figure 7.

miR-17∼92 target genes regulate discrete functions
(A) Schematic representation of Cas9 RNP electroporation experiments. Naive CD28−/− CD4+ T cells were cultured with IL-7 to preserve a naive state for 5 days post-electroporation and activated for 48 or 72 h with plate-bound anti-CD3/anti-CD28.
(B–E) proliferation index (B), MFI of CD25 (C), CD44 (D) and ICOS (E) after 48 and 72 h of activation. Data from 3 independent experiments. Error bars represent mean ± SD, Dunnett’s multiple comparison test; p values: ns = not significant, ∗<0.05 ∗∗< 0.002 ∗∗∗<0.0002 ∗∗∗∗<0.0001.
Discussion
T cell activation depends on TCR and CD28 engagement to trigger complex molecular mechanisms including the activation of the PI3K, NF-κB, and calcineurin/NFAT pathways. Intense research in the past decades uncovered many molecules that transmit TCR and CD28 signals (Esensten et al., 2016; Tian et al., 2015; Liu et al., 2018). Most studies focused on proteins as signaling intermediates but we and others previously reported that the combined engagement of TCR and CD28 also alters the expression of non-coding RNAs. Most miRNAs are downregulated after T cell activation but a few, including miR-17∼92, remain relatively constant or are slightly induced (Bronevetsky et al., 2013; de Kouchkovsky et al., 2013). This suggests that miR-17∼92 might be functionally relevant during T cell activation. We previously found that miR-17∼92 was induced after combined stimulation of TCR and CD28 in vitro but not after TCR stimulation alone (de Kouchkovsky et al., 2013). Here, we found that TCR stimulation alone does induce miR-17 but that costimulation through CD28 synergistically further increased miR-17 expression. Even adding low CD28 costimulation resulted in higher miR-17 induction than high TCR stimulation alone. These findings are consistent with experiments using stimulation by the natural ligands CD80/CD86. Heterozygosity or absence of CD80/CD86 on B cells resulted in a dose-dependent reduction in miR-17 induction in co-cultured T cells while deficiency or blockade of CTLA-4 resulted in increased miR-17 expression (Wang et al., 2015). Collectively, these studies demonstrate that TCR stimulation and costimulation/coinhibition by CD28 or CTLA-4 are intimately linked to miR-17∼92 expression. Furthermore, stimulating T cells with antibodies directed to CD3 is sufficient to induce proliferation in T1792tg/tg T cells suggesting that transgenic miR-17∼92 renders T cells CD28 costimulation independent (Xiao et al., 2008). Therefore, we set out to formally investigate if and how transgenic miR-17∼92 was sufficient to substitute for the absence of CD28 and whether miR-17∼92 was required for CD28-mediated T cell activation. We found an unexpectedly potent rescue effect both in vitro and in vivo. Many defects of CD28−/− T cells were functionally compensated for by the miR-17∼92 transgene. This is notable for two reasons. First, miR-17∼92 is a non-coding RNA and second, miRNAs are negative regulators. Thus, overexpression of an inhibitory non-coding RNA was sufficient to enable T cell activation in the absence of CD28 costimulation.
To analyze the molecular mechanism by which miR-17∼92 enabled T cell activation of CD28−/− CD4+ T cells we first defined miR-17∼92 target genes in CD28-sufficient CD4+ T cells before and after activation. RNA-seq analysis of T cells with miR-17∼92 loss- or gain of function and sampled over a time course revealed that miR-17∼92 mainly influenced the transcriptome after T cell activation, a finding consistent with the phenotypic analysis of ex vivo characterized naive T cells. There is an emerging notion that evidence of miRNA binding combined with differential gene expression in primary cells constitutes a precise approach to empirically define miRNA:target relationships (Gagnon et al., 2019; Hsin et al., 2018). We, therefore, employed this approach but refined it by combining EISA with AHC to discriminate whether differentially expressed gene clusters were primarily regulated transcriptionally or posttranscriptionally. This analysis provides a highly granular view of miRNA-mediated gene regulation. Specifically, pathway and regulon enrichment analysis revealed that miR-17∼92 enhanced the calcineurin/NFAT pathway. Consistent with this, a gene cluster enriched for genes that positively correlated with miR-17∼92 was mainly transcriptionally regulated and contained many genes known to be regulated by NFAT TF including genes for various CD4+ T cell subsets such as TFH (IL-21) and TH1 (Tbx-21 and IFNγ). Experiments with pharmacologic CNIs, e.g. CsA, confirmed that miR-17∼92 functionally promoted the calcineurin/NFAT pathway. Importantly, we demonstrated that the expression of genes we validated empirically as miR-17∼92 targets were elevated in stimulated CD28−/− T cells. In contrast, transgenic miR-17∼92 partially restored the molecular program that was defective in CD28−/− T cells. In particular, the expression of key NFAT-regulated genes found to be driven by miR-17∼92 in CD28-sufficient T cells were restored by the transgene in CD28−/− T cells. We conclude that during T cell activation miR-17∼92 is required to repress this set of genes.
With the list of stringently defined miR-17∼92 target genes, we set out to investigate the functional relevance of identified miRNA target genes. This is generally challenging because miRNAs bind to and regulate many genes and because the per gene repression is mostly modest (Baumjohann and Ansel, 2013). We previously addressed this using siRNA-mediated gene repression (Pua et al., 2016). Here, we reasoned that the disruption of genes that physiologically are to be repressed by miR-17∼92 during T cell activation could partially restore defects of CD28-deficient T cells resulting in the gain of function (rescue) phenotypes. Indeed, some parameters that were rescued by transgenic miR-17∼92 were also restored by the disruption of a single miR-17∼92 target gene (Pten: proliferation; Nrbp1: CD25; see model in Figure S8) while others were restored by the disruption of more than one target (Pten, Nrbp1: CD44). For yet other targets we could not detect any functional relevance for the measured parameters (Phlpp2, Cyld, Rcan3, Rnf38, Rnf167). Importantly, due to the known dependency on the cellular context, the absence of a change in the assessed parameters does not exclude that repression of these genes may be relevant in a different context, timepoint, or for a function not assessed here (Lu et al., 2015; Hsin et al., 2018). For instance, it’s interesting to note that CRISPR/Cas9-mediated disruption of Pten restored proliferation at the earlier time point but the effect waned with time. Similarly, we previously found that removing one copy of Pten was able to restore the defect observed in T1792Δ/Δ cells during early TFH differentiation but not during later stages (Baumjohann et al., 2013). In addition, the disruption of Pten resulted in the opposite effect (decrease) than miR-17∼92 transgene expression (increase) for ICOS expression. Therefore, although Pten is an important miR-17∼92 target, others must be functionally relevant and remain to be identified. For instance, our limited CRISPR/Cas9 validation revealed that disrupting Nrbp1 increased CD25 and CD44 expression suggesting that it regulates these important proteins. Interestingly, Nrbp1 is a known tumor suppressor and Nrbp1-disruption increases CD44 and c-myc in the intestine of conditional KO mice (Wilson et al., 2012). These results suggest that Nrbp1 is another negative regulator of T cell activation. Together, these results clearly demonstrate that a single target is very unlikely to explain the phenotypic rescue observed in CD28−/− T cells expressing transgenic miR-17∼92. Rather, we propose that miR-17∼92 represses a network of genes fulfilling distinct functions. A scaled up, targeted CRISPR/Cas9 screen could be used to identify regulators of T cell function. For instance, our data suggest that ICOS expression may be restrained by yet unknown miR-17∼92 targets. In addition, not all target genes have binding sites for each of the miR-17∼92 cluster’s miRNAs. Therefore, such a screen could help to identify which miRNA of the cluster affects which function. As an example, we identify Nrbp1 as a functionally relevant miR-17∼92 target. Since it contains binding sites for miR-17 and miR-19 it is likely that one or both of these miRNAs are functionally relevant for its repression and consequently CD25 and CD44 regulation. However, investigation of the relevance of the respective binding sites needs separate validation experiments. Thus, our empirically validated target list provides a blueprint for an arrayed CRISPR/Cas9 screening approach to reveal new biologic insight. Expanding such a screen to additional readouts, e.g. IL-2 or Tbx21, might reveal the regulation of distinct functions by unexpected genes. Moreover, CRISPR/Cas9 screening in naive T cells could be useful beyond miRNA research, particularly in CD28−/− T cells, e.g. to identify T cell negative regulators.
Our results are in line with limited experimental evidence that gene deletion can partially rescue CD28 deficiency. For instance, genetic deletion of Casitas B lymphoma-b protein (Cbl-b) or TRAF6 is sufficient to restore IL-2 production and proliferation in CD28−/− T cells (Bachmaier et al., 2000; Chiang et al., 2000; King et al., 2006). However, neither IFNγ, IL-4 or ICOS expression nor GC formation was restored by Cbl-b deletion (Bachmaier et al., 2000; Chiang et al., 2000). Thus, while loss of Cbl-b uncoupled T cells from the strict requirement for CD28 costimulation, it could not restore all aspects of CD28-mediated costimulation suggesting that additional genes restrain T cell activation. As a group of genes, negative regulators impose a requirement for a costimulatory signal to actively remove these brakes to allow a productive T cell response. CD28 costimulation, therefore, serves a dual purpose by enhancing TCR signaling and simultaneously overcoming T cell repression (Paolino and Penninger, 2010; Buckler et al., 2006; Martinez-Llordella et al., 2013). Using Pten-deficient T cells it was previously shown that PTEN imposes a requirement for CD28 costimulation by setting a threshold for activation (Buckler et al., 2006). Here, we show that CRISPR/Cas-mediated Pten disruption can partially overcome defective proliferation and CD44 upregulation of CD28−/− CD4+ T cells but negatively affects ICOS expression. In addition, we identify the tumor suppressor Nrbp1 as an additional gene whose removal enables the the expression of important proteins in CD28−/− CD4+ T cells. Moreover, our list of empirically validated miR-17∼92 target genes contains several other known or suspected tumor suppressor genes. The growing list (e.g. Pten, Phlpp2, Cyld, Nrbp1) of tumor suppressors/negative regulators whose expression is controlled by miR-17∼92 is noteworthy and suggests that these could all contribute to setting a threshold for T cell activation. Although it’s remarkable that removing individual genes in CD28−/− T cells is sufficient to restore specific functions, it’s important to note that during T cell activation the inhibitors are not completely eliminated. Therefore, although experimental deletion can demonstrate that a given gene has the potential to act as a powerful negative regulator, its physiologic function is more delicate to uncover and complete deletion likely overestimates the contribution of the gene under investigation. To date, no single gene or pathway can explain the function of T cell activation and/or CD28 costimulation. More likely, multiple pathways need to be induced and multiple negative regulators need to be removed. It is conceivable that precise control of genetic programs is particularly important for an exponential process like clonal T cell expansion. Even minor changes in setting the T cell activation threshold or the rate of proliferation or survival could result in major consequences for the host organism. Since miRNAs fine-tune expression of many genes they are ideally suited to mediate controlled release from multiple restraining proteins preventing T cell activation (Bartel, 2018). Our findings greatly expand the list of high-confidence miR-17∼92 targets with a suspected function in T cell activation and illustrate that miR-17∼92 can exert complex regulation of the T cell transcriptome through direct and indirect gene regulation.
In summary, we propose a model (graphical abstract) in which miR-17∼92 acts as a downstream mediator, modulator, and/or amplifier of the molecular program triggered by combined TCR/CD28 engagement serving as a gatekeeper of a network of restrainers of T cell activation and function. In this model, non-coding RNA-mediated direct repression of inhibitors indirectly supports the transcriptional induction of genes necessary for further T cell differentiation and function (e.g. CD44, IL-21, Tbx21, IFNγ). Importantly, we not only show that transgenic miR-17∼92 is largely sufficient to substitute for CD28-deficiency but that miR-17∼92 is physiologically required to shape the transcriptome after CD28 costimulation and that miR-17∼92 overexpression in CD28-sufficient T cells results in “super-costimulation.” In support of the proposed model, constitutive transgenic miR-17∼92 expression leads to a lupus-like systemic autoimmune syndrome (Xiao et al., 2008), not unlike the ones observed in mice deficient in Cbl-b, PTEN, TRAF6, or Cyld (Bachmaier et al., 2000; Chiang et al., 2000; Suzuki et al., 2001; King et al., 2006; Reiley et al., 2007). Our results support the consideration of miR-17∼92 or its target genes for pharmacologic intervention or for its integration into engineered cellular therapies.
Limitations of the study
Our study is limited in scope and by technical constraints concerning the mechanistic link between i) CD28, miR-17∼92 and T cell activation, ii) miR-17∼92 target validation as well as iii) relevance for other cells. We cannot definitively discern the relative functional relevance of miR-17∼92 induced by TCR stimulation alone versus combined TCR and CD28 costimulation. This study did not address the molecular mechanism of miR-17∼92 regulation in T cells. Although the presence of myc binding sites is suggestive of transcriptional regulation (O'Donnell et al., 2005), miRNA biogenesis can be controlled by extracellular cues and maturation of miR-17∼92 is known to be regulated posttranscriptionally (Du et al., 2015). It will be important to identify the individual miRNAs of the cluster that regulate specific functions and to determine whether the expression of miR-17 (which we measured) corresponds to other miRNAs of the cluster, such as miR-18a, miR-19a, and miR-19b-1 that reportedly have an important function in T cells (Montoya et al., 2017; Simpson et al., 2014; Jiang et al., 2011). Together, future studies addressing these open questions will untangle the molecular connection between TCR stimulation/CD28 costimulation/T cell activation and miR-17∼92 expression/function. For instance, such studies could address whether miR-17∼92-deficient T cells receiving both TCR and costimulatory signal become anergic similar to costimulation-deficient T cells. With regards to miR-17∼92 target validation the study did not address whether miR-17∼92 increased calcineurin/NFAT activity through repression of a direct calcineurin inhibitor, e.g. Rcan3, or indirectly, e.g. through repression of a PI3K inhibitor, e.g. Pten or Phlpp2. Furthermore, a more comprehensive analysis of the repression of single or combinations of targets will be required but this will remain challenging since our results suggest a context and time-dependent regulation of multiple genes as observed for other miRNAs. Technically, the deletion efficiencies of the CRISPR/Cas9 approach varied, ranging from 13.4 to 28% (Phlpp2) to 65.5–84% (Rnf167) but 30–49% was sufficient to see phenotypic effects (Nrbp1) in this experiment. Interpretation of the results further needs to take into account that the inhibitors are not completely eliminated by miRNAs during T cell activation. Therefore, complete deletion likely overestimates the contribution of the gene under investigation. Finally, future studies need to determine whether T cell subsets that rely less on CD28 than naive T cells (e.g. memory and CD8+ T cells) display differential sensitivity to miR-17∼92-mediated gene regulation or express more miR-17∼92.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-mouse CD4 clone GK1.5 or RM4-5 | Biolegend | Cat#100423, 100532 |
| Rat anti-mouse/human CD44 clone IM7 | Biolegend | Cat#103032, 103008 |
| Rat anti-mouse CD62L clone MEL-14 | Biolegend | Cat#104412 |
| Armenian Hamster anti-mouse CD69 clone H1.2F3 | Biolegend | Cat#104537 |
| Rat anti-mouse CD25 clone PC61 | Biolegend | Cat#102016 |
| Mouse anti-mouse Tbet clone 4B10 | Biolegend | Cat#644810 |
| Rat anti-mouse IFNγ clone XMG1.2 | Biolegend | Cat#505813 |
| Rat anti-mouse IL-17a clone TC11-18H10.1 | Biolegend | Cat#506904 |
| Mouse anti-mouse Rorγt clone Q31-378 | BD BioScience | Cat#562684 |
| Rat anti-mouse Foxp3 clone FJK-16s | eBioscience | Cat#17-5773-82 |
| Armenian Hamster anti-mouse Fas (CD95) clone Jo2 | BD BioScience | Cat#554257 |
| Rat anti-mouse anti-GL-7 clone GL7 | Biolegend | Cat#144612, 144610 |
| Mouse anti-mouse anti-Bcl-6 clone K112-91 | BD BioScience | Cat#561522 |
| Armenian Hamster anti-mouse anti-ICOS (CD278) clone C398.4A | Biolegend | Cat#313525 |
| Rat anti-mouse CXCR5 (CD185) clone SPRCL5 | Biolegend | Cat#145513 |
| Rat anti-mouse PD-1 (CD279) clone 29F.1A12 | Biolegend | Cat#135216 |
| Rat anti-mouse B220 clone RA3-6B2 | Biolegend, Tonbo | Cat#103229, 20-0452-U100 |
| Rat anti-mouse CD19 clone 1D3 | Biolegend | Cat#115555, 115522 |
| Armenian Hamster anti-mouse CD3e clone 145-2C11 | Biolegend | Cat#100319, 100309 |
| Rat anti-mouse anti-IL-2 clone JES6-5H4 | Biolegend | Cat#503808 |
| Rat anti-mouse Vα2 clone B20.1 | Biolegend, | Cat#127816 |
| Armenian Hamster anti-mouse anti-Vβ8.3 clone 1B3.3 | BD BioScience | Cat#553664 |
| Armenian Hamster anti-mouse CD3 clone 2C11 (for coating) | BioXcell | Cat#BP0001-1 |
| Armenian Hamster anti-mouse CD28 clone PV-1 (for coating) | BioXcell | Cat#BE0015-5 |
| Rat anti-mouse CD16/CD32 clone 2.4G2 (for blocking) | BioXcell | Cat#BE0307 |
| anti-IL4 clone 11B11 | UCSF | AM039-PURE-B25 |
| anti-IFNγ clone XMG1.2 | UCSF | AM034-PURE-B25 |
| anti-IL12/23 clone C17.8 | UCSF | AM037-PURE-B25 |
| Goat anti-mouse IgG1 | ThermoFisher | A21240 |
| Mouse anti-mouse NFATC2 clone 25A10.D6.D2 | ThermoFisher | MA1-025 |
| Bacterial and virus strains | ||
| LCMV Armstrong | Annaise Jauch | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| FCS for medium | Atlanta | S11150 |
| HEPES | SIGMA | H0887 |
| Sodium pyruvate | Gibco | 11360-039 |
| non-essential amino acids | Gibco | 11140-050 |
| Glutamax | Gibco | 35050-038 |
| 0.1% 2-Mercaptoethanol | Gibco | 31350-010 |
| FCS for FACS buffer | Milian | S0750 |
| NaN3 | SIGMA | S2002-5G |
| NH4Cl | SIGMA | A9434-500G |
| NaHCO3 | SIGMA | S5761-500G |
| Proteinase K | Promega | V3021 |
| TE buffer | SIGMA | 93283-100ML |
| Fixation/Permeabilization Concentrate | invitrogen | 00-5123-43 |
| Fixation/Permeabilization Diluent | invitrogen | 00-5223-5 |
| Permeabilization Buffer 10X | eBioscience | 00-8333-56 |
| OneComp eBeads | invitrogen | 01-111-42 |
| TRI Reagent | Sigma-Aldrich | T9424-200ML |
| 1-Bromo-3-chloropropane | Sigma-Aldrich | B9673-200ML |
| M-MLV Reverse Transcriptase | Sigma-Aldrich | M1302-40KU |
| TaqMan™ MicroRNA Reverse Transcription Kit | ThermoFisher | 4366597 |
| TaqMan™ Fast Universal PCR Master Mix (2X), no AmpErase™ UNG | ThermoFisher | 4364103 |
| TaqMan Gene Expression Assays RCAN3 | ThermoFisher | 4351372 |
| TaqMan Gene Expression Assays 18S | ThermoFisher | 4331182 |
| MicroAmp™ Fast Optical 96-Well Reaction Plate, 0.1 mL | ThermoFisher | 4346907 |
| EasySep naïve CD4+ T cell Isolation Kit | STEMCELL | 19765A |
| CellTAK adhesive | Corning | 354241 |
| Seahorse XFe96/XF96 FluxPak (18 cartr.) | SeahorseBiotech | PN 102416-100 |
| Glucose-free unbuffered RPMI | SIGMA | R6504-10X1L |
| Oligomycin | SIGMA | 495445 |
| FCCP | SIGMA | C2920 |
| Rotenone | SIGMA | R8875 |
| D-(+)-Glucose | Sigma-Aldrich | G7021 |
| Fixable Viability Dye eF780 | eBioscience | 65-0865-14 |
| Cell Trace Violet (CTV) | ThermoFisher | C34557 |
| Phorbol 12-Myristate 13-Acetate (PMA) | Sigma | P1585 |
| Ionomycin | Sigma | 10634-1MG |
| Brefeldin A (BFA) | Sigma | B7651-5MG |
| GP-64 | Neosystem | SP991567B |
| IL-2 | R&D | 202-IL |
| Retinoic acid | Sigma | R2625 |
| rhTGFβ | R&D | P01137 |
| IL-6 | R&D | 406ML |
| IL-7 | R&D | 407-ML-005 |
| Cyclosporin A (CsA) | Sigma | 30024 |
| DAPI | Sigma | 10236276001 |
| IL-12 | R&D | 419ML |
| TMB | BD | 555214 |
| Cas9 nuclease | QB3 MacroLab, UC Berkeley | N/A |
| Sodium deoxycholate | Thermo Scientific | 89904 |
| TRIS | Sigma, | T6791 |
| TCEP | Sigma | 646547-10x1ml |
| PR-Sulfonate Cartridges | PreOmics | N/A |
| Microplate BCA Protein Assay Kit | Thermo-Pierce | 23252 |
| Chloroacetamide | Sigma | C0267-100G |
| TFA | Pierce/Thermo | 28904 |
| Lysin-C | Wako | 125-05061 |
| Porcine Trypsin | Promega | V5113 |
| 2-Propanol | Sigma-Aldrich | I9516-500ML |
| Ethanol | Sigma-Aldrich | 51976-500ML-F |
| Ammonium hydroxide solution | Sigma-Aldrich | 17093-1L |
| HPLC water | Chemie Brunswick | W/0106/17 |
| Acetonitrile | Thermo Scientific | A955-212 |
| Formic acid | Sigma | 94318-50ml |
| Poly-L-glutamic acid sodium salt (PGA) | Sigma | P4761 |
| QuickExtract | Lucigen | QE09050 |
| Critical commercial assays | ||
| ELISA MAX mouse IL-2 set | BioLegend | 431002 |
| Nunc-Immuno™ MicroWell™ 96 well solid plates | SIGMA | M9410-1CS |
| Zymol Directzol kit | ZymoResearch | R2060 |
| Deposited data | ||
| Raw and analyzed data | This paper | N/A |
| Mouse reference genome | UCSC | MM10 |
| Generated data GEO accession number | This paper | GSE140568 |
| Experimental models: Organisms/strains | ||
| Mir17-92<tm1.1Tyj>/J (crossed to CD4cre, T1792Δ/Δ) | Dr. Bluestone, UCSF | N/A |
| C57Bl/6-Gt(ROSA)26Sor<tm3(CAG-MIRN17-92,-EGFP)Rsky>/J (crossed to CD4cre, T1792tg/tg) | The Jackson Laboratory | stock number 008517 |
| B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ | The Jackson Laboratory | stock number 022071 |
| B6.129S2-Cd28tm1Mak/J | SWIMR | N/A |
| B6-Tg(TCRGP13)1 Brl | SWIMR | N/A |
| Oligonucleotides | ||
| Rcan3 crRNA GAGAAATACGAACTGCACGC |
IDT | N/A |
| Cyld crRNA GTTGGCAATTACCAACTGTG |
IDT | N/A |
| Pten crRNA CCTCCAATTCAGGACCCACG |
IDT | N/A |
| Phlpp2 crRNA TTGTACACTGCAGCAGACGA |
IDT | N/A |
| Nrbp1 crRNA AAACTGCTTAAGACTCCCAG |
IDT | N/A |
| Rnf38 crRNA CTCCTACACGGTAACTACGG |
IDT | N/A |
| Rnf167 crRNA GGAATGAGGTAATAGCCCAA |
IDT | N/A |
| Negative control crRNA#2 | IDT | 1072545 |
| tracrRNA | IDT | 1072532 |
| fwd primer Rcan3 TCAGCCACTGAATGACGTAGTAG | Microsynth | N/A |
| rev primer Rcan3 GCGTCTGCGTGATTTTCTGT | Microsynth | N/A |
| Fwd primer Cyld CCTGTGGAGCCAAGAGAAAGT | Microsynth | N/A |
| Rev primer Cyld TCCTGGGGTCTATCCTGAGAA | Microsynth | N/A |
| Fwd primer Pten AGAAGTCCTTACATGGGTTGGT | Microsynth | N/A |
| Rev primer Pten GCTTTAAGCAAAAGGTCTGTGGT | Microsynth | N/A |
| Fwd primer Phlpp2 GTGTCTGCCCTGGCTATGGA | Microsynth | N/A |
| Rev primer Phlpp2 GTGGTCCAGGAAGGAGTAACAG | Microsynth | N/A |
| Fwd primer Nrbp1 GTTCTCACTGTGCTCTTAAGCC | Microsynth | N/A |
| Rev primer Nrbp1 CCCACTAGTCCTTGCTATCCC | Microsynth | N/A |
| Fwd primer Rnf38 TACTTGAGCTACCTCCCCCG | Microsynth | N/A |
| Rev primer Rnf38 TCAGTTGGAAGTGAAGCTACAAA | Microsynth | N/A |
| Fwd primer Rnf167 AAGCGCAGAGTACCTACGAG | Microsynth | N/A |
| Rev primer Rnf167 TCCCCTCTACCGATCCAGTC | Microsynth | N/A |
| Software and algorithms | ||
| FlowJo 10.6.0 | BD | N/A |
| Prism 8.2.0 | GraphPad | N/A |
| IDEAS software v6.2 | Merck | N/A |
| TIDE | Brinkman et al. (2014) | https://tide.nki.nl/ |
| Fragment Analyzer | Advanced Analytical | N/A |
| FastQC tool version 0.11.5 | Andrews (2010) | N/A |
| STAR version 2.5.2a | Dobin et al. (2013) | N/A |
| R software version 3.5 | R Core Team 2015 | http://www.R-project.org/ |
| R Bioconductor package QuasR version 1.18 | Gaidatzis et al., 2015b | N/A |
| R Bioconductor package edgeR version 3.24.3 | Robinson et al. (2010) | N/A |
| ImageJ | Schneider et al. (2012) | https://imagej.nih.gov/ij/ |
| Other | ||
| Sequence data, analyses, and resources related to the RNA-sequencing of T1792Δ/Δ, wt, T1792tg/tg | This paper | N/A |
| Sequence data, analyses, and resources related to the RNA-sequencing of CD28−/−, rescue, T1792Δ/Δ, wt, T1792tg/tg | This paper | N/A |
| Curated gene set collection of the Molecular Signature Database (MSigDB v6.0) |
Subramanian et al. (2005) | N/A |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Lukas Jeker (lukas.jeker@unibas.ch).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
All animal work was performed in accordance with the federal and cantonal laws of Switzerland. Protocols were approved by the Animal Research Commission of the Canton of Basel-Stadt, Switzerland. Most of the mouse lines were imported from the JAX laboratory as indicated in the key resources table. T1792Δ/Δ mice were imported from the laboratory of Dr. Bluestone (UCSF). Rescue SM+ mice were obtained by crossing the rescue strain to other transgene lines in house so that their precise genetic origin is difficult to determine. As for the transfer cells, we used SM CD45.1+ wt cells imported from SWIMR. All other mouse lines were crossed to CD45.2 expression. Cre negative littermates (from T1792tg/tg or T1792Δ/Δ) were used as wt controls, and cre negative littermates from the rescue strain were used as CD28ko. 6–8 week old females and males were used for all experiments.
Method details
Organ isolation
Organs were obtained after CO2 euthanization and kept on ice until processing. Mesenteric lymph nodes (LN), peripheral LN (inguinal, axillary, brachial, six cervical) and spleen were taken for most of the experiments. Spleens were injected with 0.5 mL ACK lysis buffer (0.155 M NH4Cl, 200 μL 0.5 M EDTA pH = 8.0, 0.012 M NaHCO3 pH 7.2) for erythrolysis before processing. The organs were meshed with 0.4 μm filters to obtain single cell suspensions which were then washed with FACS buffer (2% heat-inactivated FCS in PBS, for stainings add 0.02% NaN3). Cells were centrifuged at 4°C, 5 min at a speed of 370 g for most washing procedures.
Naïve CD4+ T cell isolation
Naïve CD4+ T cells were isolated from cell suspensions with pooled lymph nodes and spleen. Isolation was performed with StemCell mouse naïve CD4+ T cell isolation kit according to manufacturer’s instructions. In brief, the cell suspensions were incubated with rat serum and CD4+ isolation antibody for 7.5 minutes, then with memory depletion antibody for 2.5 minutes, and in the end with magnetic beads for another 2.5 minutes before incubating with the isolation magnet for 2.5 minutes. The resulting untouched naïve CD4+ T cells were then washed with FACS buffer, and purity was routinely checked with a staining for CD4+, CD44− and viability.
Plate-bound CD4+ T cell activation
Plates were coated over night with 0.2 μg anti-CD28 and 0.5 μg anti-CD3 per mL PBS for most of the experiments (low stimulation as according to (Baumjohann et al., 2013)). We used 1 mL/well for 24 well plates and 0.2 mL/well for 96 well plates. Before plating of the cells, plates were washed with PBS. We plated 2∗105 naive CD4+ T cells per well in 96 well flat-bottom in 200 μL medium. For 24 well plates, 2∗106 naïve CD4+ T cells per mL medium in 1 mL medium were plated. Complete T cell medium (RPMI-1640 Medium, 10% FCS, 1% HEPES, 1% non-essential amino acids, 1% Glutamax, 0.1% 2-Mercaptoethanol) was supplemented with 50U IL-2/mL. Cells were cultured at 37°C, 5% CO2 for 24 h or longer depending on the purpose of the experiment as indicated in figure legends.
Proliferation assay with cell trace violet (CTV)
Freshly isolated naïve CD4+ T cells were washed with PBS. 1 μL of Cell Trace stock solution (dissolved in DMSO according to the manufacturer’s instructions) was then used per mL PBS for 10∗106 cells. Cells were incubated at 37°C for 20 minutes, then 5x the original staining volume of normal T cell culture medium was added for 5 minutes to remove residual dye. Cells were washed and plated in complete culture medium supplied with 50U IL-2 per mL for 48 h.
Enzyme-linked immunosorbent assay (ELISA)
IL-2 secretion was addressed in 48 h culture supernatants from cells that were plated in pre-coated wells in complete T cell medium without IL-2. IL-2 ELISA was performed with the BioLegend ELISA MAX mouse IL-2 set according to the manufacturer’s instructions. After the last washing step, TMB substrate was added for the readout. Absorbance was measured with an ELISA plate reader (Synergy H1 Hybrid Reader, BioTek) at 450 nm as well as 570 nm wavelength, and normalized to wild type control samples that were run in the same experiment.
FACS staining
Generally, cells were stained for viability with viability dye eFluor780 in PBS for 20 min at 4°C and then washed with PBS or FACS buffer. Non-specific binding was blocked with anti-CD16/anti-CD32 0.5 mg/mL on ice for 10 minutes. Surface staining was performed in FACS buffer for 20–30 minutes at 4°C. In intracellular staining or LCMV experiments, cell fixation was performed with Fix-Perm for 20 minutes on 4°C (1 h for LCMV experiments). Intracellular staining was done in permeabilization buffer for 1 h at 4°C. Activation status of the cells was assessed by staining and gating for singlets/lymphocytes/viable cells/CD4+ and early activation marker CD25/CD69 as well as CD44/CD62L expression. Blasting of lymphocytes was addressed by pre-gating on singlets/viable cells. For cytokine staining after in vitro differentiation or ex vivo e.g., for IL-2 staining, cells were stimulated with 50 ng/mL PMA, 500 ng/mL Ionomycin and 10 μg/mL Brefeldin A (BFA) for 3 h at 37°C before staining. Data was acquired with an LSR Fortessa (BD) and analyzed with FlowJo.
LCMV Armstrong infection model
Mice were infected with 2∗105 PFU LCMV-Armstrong strain i.p. with U-100 insulin syringes (0.30 mm (30G) x 8 mm). Eight days post infection the spleens were harvested for staining. We gated for singlets/lymphocytes/viable cells/CD3+CD4+ to look at CD44 expression, and moreover we addressed TFH cells by the co-expression of key markers Bcl-6, PD-1, ICOS and CXCR5. We gated for singlets/lymphocytes/viable cells/CD19+B220+ to look at GC B cells expressing Fas and GL-7. Re-stimulation of splenocytes was performed in flat bottom 96 well plates with 1 μg/mL LCMV-specific peptide GP-64 in comparison to polyclonal 50 ng/mL PMA, 500 ng/mL Ionomycin stimulation for one hour, then 10 μg/mL Brefeldin A was added for another three hours before staining. We then gated for TH1 cells using singlets/lymphocytes/viable cells/CD3+CD4+ and finally looking at Tbx21/IFNγ expression. All in vivo experiments were performed blinded with assignment of animal numbers after data analysis.
Histology
Spleens were embedded in cryo embedding medium and frozen on dry ice before storage at −80°C. Sections were cut at a thickness of 6 μm and dried on air. Single sections were then fixed with acetone for 5 minutes and circled with PAP pen. Staining for CD19, CD4 and GL-7 was performed in FACS buffer with anti-CD16/anti-CD32 in a wet chamber at 4°C overnight. Slides were then washed with PBS on a shaker for 15 minutes before drying and mounting. Imaging was performed with a 20x objective on a Nikon Ti2 microscope and analyzed with ImageJ version 2.0.0.
Adoptive transfer (AT) with subsequent LCMV infection
Naïve SMARTA+ CD4+ T cells from wt, CD28−/− and rescue mice were isolated transferred into CD28−/− recipients. Each recipient received 1∗105 cells in 100 μL PBS i.v. The recipients were infected with 2∗105 PFU LCMV Armstrong i.p. two days after cell transfer. Eight days after infection, the mesenteric and peripheral LN as well as the spleen were analyzed separately for the presence of Vα2+Vβ8.3+ T cells (pre-gated on singlets/lymphocytes/viable cells/CD4+), and these cells were further characterized for their CD44 expression. One CD28−/− mouse that did not receive donor cells was used as a negative control in each experiment to display the recipient-intrinsic Vα2+Vβ8.3+ population.
RNA sequencing
For any experiment involving RNA, cells were washed with PBS before counting and the RNA was kept on ice during the experiments, storage at −80°C. All pipetting was performed with filter-tips and RNAse-free tubes. All procedures for the extractions were performed at the facilities with materials, protocols and supervision of the facility experts. For RNA sequencing, 2.5∗105 cells were washed with PBS and resuspend in 200 μL TRI Reagent. RNA was extracted from Trizol-samples with a Zymo Direct-zol kit which includes DNAse treatment. Quality control was run with a Bioanalyzer. RNA quality was assessed with a Fragment Analyzer (Advanced Analytical) and RNA-seq library preparation was performed using Illumina Truseq stranded kit. Sequencing was performed on an Illumina NexSeq 500 machine to produce single-end 76-mers reads. All steps were performed at the Genomics Facility Basel (ETH Zurich).
Read quality was assessed with the FastQC tool (version 0.11.5). Reads were mapped to the mouse genome (UCSC version mm10) with STAR (version 2.5.2a) (Dobin et al., 2013) with default parameters, except filtering out reads mapping to more than 10 genomic locations (outFilterMultimapNmax = 10), reporting only one hit in the final alignment for multimappers (outSAMmultNmax = 1) and filtering reads without evidence in the spliced junction table (outFilterType = "BySJout").
All subsequent gene expression data analysis was performed using the R software (version 3.5). Read alignment quality was evaluated using the qQCReport function of the R Bioconductor package QuasR (version 1.18). Gene expression was quantified using the qCount function of QuasR (Gaidatzis et al., 2015b) as the number of reads (5′ends) overlapping with the exons of each gene assuming an exon union model (using the UCSC knownGenes annotation downloaded on 2015-12-18). To quantify intronic expression levels, exonic coordinates were extended by 10 bp on each side of the exons, and for each gene the resulting read count was subtracted to the read count obtained on the whole gene (extended by 10 bp on each side).
The R Bioconductor package edgeR (version 3.28) (Robinson et al., 2010) was used for differential gene expression analysis. Between samples normalization was done using the TMM method (Robinson and Oshlack, 2010). Only genes with CPM (counts per million reads mapped) values more than 1 in at least 4 samples (the number of biological replicates) were retained. Principal component analysis (PCA) was performed on the log-transformed normalized CPM values. An generalized linear model including a genotype effect, an activation effect, and a replicate effect (nested within genotype) was fitted to the raw counts (function glmFit), and differential expression was tested using likelihood ratio tests (function glmLRT). p-values were adjusted by controlling the false-discovery rate (Benjamini-Hochberg method) and genes with a FDR lower than 1% were considered differentially expressed.
To select a restricted list of most likely miRNA targets for each seed family, we restricted the genes to those with a conserved 3′UTR seed match (using TargetScan (Friedman et al., 2009)), and a coverage of more than 5 AHC reads. We additionally restricted the list by using differential expression criteria from the first RNA-seq dataset: genes should be significantly differentially expressed between T1792tg/tg and wt, as well as between T1792Δ/Δ and wt, with fold-changes in the expected directions; using intronic read counts, genes should not be significantly differentially expressed in the same comparison, at an increased FDR threshold of 10%.
Gene set enrichment analysis was performed with the function camera (Wu and Smyth, 2012) from the limma package (version 3.42; using the default parameter value of 0.01 for the correlations of genes within gene sets) using gene sets from the curated gene set collection (C2) of the Molecular Signature Database (MSigDB v7.0) (Subramanian et al., 2005) with a special focus on gene sets from pathway databases, including KEGG (Kanehisa et al., 2017), Biocarta (http://cgap.nci.nih.gov/Pathways/BioCarta_Pathways), PID (Schaefer et al., 2009) and Reactome (Fabregat et al., 2018). We tested a total of 1,576 gene sets containing more than 10 genes and those with a false discovery rate (FDR) lower than 1% were considered significant.
Regulon enrichment analysis
Regulon enrichment analysis was performed similarly to pathway enrichment analysis using camera from the limma package. DoRothEA v2 regulons (Fabregat et al., 2018) were downloaded from https://github.com/saezlab/DoRothEA.
More specifically, we used human TOP10score regulons in VIPER format, containing the targets with the highest quality score possible (and at least 10 genes) per transcription factor. Mouse regulons were obtained by orthology: mouse genes with a 1-to-1 or Many-to-1 relationship to human genes of each DoRothEA regulon were obtained using Ensembl Compara (release 97); only orthology relationships annotated to taxonomic levels "Boreoeutheria", "Eutheria", "Euarchontoglires", "Theria", or "Amniota" were retained. We tested a total of 1,307 regulons including at least 10 mouse genes and those with a false discovery rate (FDR) lower than 1% were considered significant.
AGO2 HITS-CLIP (AHC) data
Sample preparation and read depths on seed regions of genes targeted by different miRNA seed families were obtained as previously described (Loeb et al., 2012; Gagnon et al., 2019). Briefly, libraries were constructed using CD4+ T cells where nuclei were eliminated. AGO2 immunoprecipitation was performed using a monoclonal antibody (Wako; clone 2D4). Libraries were multiplexed and sequenced on an Illumina HiSeq 2500 system. Reads were binned based on individual barcodes and aligned to the mm10 reference genome using Bowtie (Langmead et al., 2009). Maximum read depths across mature miRNAs and miRNA targets were generated using the samtools package (Li et al., 2009).
In vitro differentiation
TH1 differentiation conditions were generated with 50U IL-2, 5 ng/mL IL-12 and 10 μg/mL anti-IL-4 per mL T cell medium (Vigne et al., 2011). iTregs were differentiated with retinoic acid (0.9 mM), 250U IL-2, 0.75 ng/mL TGFβ, 10 μg/mL anti-IFNγ and 10 μg/mL anti-IL-4 (Barrat et al., 2002). TH17 were generated with 50 ng/mL IL-6, 3 ng/mL TGFβ, 5 μg/mL anti-IFNγ and 10 μg/mL anti-IL-4 per mL T cell medium (Chen et al., 2003). For the differentiation, 2∗105 naïve CD4+ T cells were plated on a pre-coated 96-well flat bottom plate (coated over night with 0.2 μg anti-CD28, 0.5 μg anti-CD3 per mL PBS) and harvested at 24 h, 48 h or 72 h after plating.
Seahorse
Calibration plates were coated over night with 200 μL calibrant. Cell plates were coated with 18 μL 0.1 M NaHCO3 pH8.0 6.67% CellTak (Seahorse XF96 flux pack, Bucher Biotech, CH). The following day, cell plates were washed with H2O and left for drying during cell preparation. Compounds were prepared for a final in-well concentration of 1 μM for Oligomycin, 2 μM for FCCP and 11 μM for Rotenone. CD4+ T cells (naïve or activated) were harvested with glucose-free, unbuffered RPMI, washed and counted multiple times before plating 3∗105 cells per well. Mitochondrial perturbation was performed by sequential injection of glucose (80 mM stock), oligomycin, FCCP and rotenone. Measurements of oxygen consumption rate (OCR, pMoles/min) and extracellular acidification rate (ECAR; mpH/min) were performed with a Seahorse XF96 flux analyzer (Seahorse Bioscience, USA). Data analysis was performed using Prism (Version7.0d), mitochondrial parameters were calculated as described by Gubser et al. (2013).
Cyclosporin A (CsA) titration
The sensitivity of CD28−/− and rescue cells to compounds interfering with Ca2+ signaling was tested. Increasing amounts of CsA were added to the cultures during activation: 2∗105 naïve CD4+ T cells were plated in 100 μL complete T cell medium in pre-coated 96 well plates. 100 μL of a serial dilution of CsA were then added to result in in-well concentrations of 100 ng/mL, 50 ng/mL, 25 ng/mL, 12.5 ng/mL, 6.5 ng/mL, 3.125 ng/mL, 1.5625 ng/mL or 0 ng/mL. Cells were then activated in the presence of these CsA concentrations for 48 h before harvesting and staining for viability and activation markers.
ImageStream
Naïve CD4+ T cells were isolated and activated for 48 h in the presence of 6.25 ng/mL CsA. They were then harvested and washed with PBS before fixation for 20 min at 4°C. Intracellular staining for NFATc2 was performed in a two-step staining with first 1 h at room temperature with anti-NFATc2 in permeabilization buffer, and subsequently 1 h at room temperature with goat anti-mouse IgG1 in permeabilization buffer. Nuclei were stained with DAPI in the last 5 min of incubation. Acquisition was run on an ImageStreamX Mark2 Imaging flow cytometer (Amnis), and data analysis was performed with the IDEAS software (v6.2).
Reverse transcription (RT) and quantitative PCR (qPCR)
5∗105 cells were washed resuspend in 400 μL TRIreagent. RNA isolation was then performed according to the isolation protocol from TRIreagent supplier (SIGMA). In brief, 0.1 mL of 1-bromo-3-chloropropane per mL of TRI Reagent was added, samples were mixed by vigorous shaking, incubated for 15 min at room temperature and then centrifuged at 12’000 g for 15 min at 4°C for phase separation. The aqueous phase was then mixed with 0.5 mL of isopropanol per mL of TRI Reagent used, again centrifuged for 10 min for RNA precipitation. RNA was then washed with 70% Ethanol and finally resuspend in RNAse-free water. RNA concentration and purity was determined with a Nanodrop2000 Spectrophotometer (ThermoScientific).
For Rcan3 qPCR, reverse transcription was performed using the SIGMA MMLV kit on 1 μg RNA according to the manufacturer’s instructions. qPCR was run with TaqMan FAST Universal PCR master mix on an Applied Biosystems® Real-Time PCR System. 18S was used as a reference gene.
For miRNA17 qPCR, reverse transcription was performed using the TaqMan MicroRNA Reverse Transcription Kit (ABI) and a miRNA-specific reverse stem-loop primer according to the manufacturer’s instructions (Moltzahn et al., 2011). qPCR was run with TaqMan FAST Universal PCR master mix on an Applied Biosystems® Real-Time PCR System. SNO234 was used as a reference gene.
CRISPR/Cas9 editing of naïve CD4+ T cells
Protospacer sequences were taken from the Brie library (Doench et al., 2016) and ordered as CRISPR RNAs (crRNAs) from IDT. Cas9 ribonucleoproteins (RNPs) were prepared as described (Dolz et al., 2021). During preparation, 1.44 μL of 100 mg/mL of poly-L-glutamic acid (PGA) solution (Sigma) was added to gRNAs to improve cutting efficiency (Nguyen et al., 2020).
Naïve CD4+ T cells were isolated from CD28−/− mice as described above, labelled with CTV and cultured for 24 h in 24-well plates at a density of 3∗106/mL in T cell medium supplemented with 5 ng/mL IL-7. Prior to electroporation, 1.8–2∗106 cells/electroporation were washed with PBS, counted and resuspended in 20 μL P4 buffer (Lonza). Cells were electroporated with Lonza Nucleofector 4D in 16-well strips using pulse code DS137. After electroporation, cells were cultured in IL-7 supplemented medium for 5 days and activated with plate-bound anti-CD3/anti-CD28 as described above for low stimulation, i.e. 0.2 μg anti-CD28, 0.5 μg anti-CD3 per mL PBS. Cells were harvested and stained for flow cytometry after 48 and 72 hours of activation. Leftover naïve cells were washed with PBS, resuspended in QuickExtract solution (Lucigen), vortexed for 1 min, incubated at 60°C for 6 minutes, vortexed for 1 min and incubated at 98°C for 10 minutes to extract genomic DNA. Genomic DNA was used for PCRs that were sequenced to measure editing efficiency using TIDE (https://tide.nki.nl/).
Quantification and statistical analysis
Statistical analysis was performed with GraphPad Prism (v7.0d, Graphpad Software). Normal distribution was not assumed, and non-parametric tests were chosen individually depending on the type of experiment as indicated in the figure legends. The overall statistical significance was set to 5% (α = 0.05), and error bars represent standard deviation (SD). N corresponds to number of biological replicates. We used Kruskal-Wallis tests for most of the experiments with more than two unpaired groups and one factor (e.g., %Fas+GL7+ population in three genotypes), followed by Dunn’s multiple comparison test or Dunnet’s multiple comparison test for CRISPR experiments. We used two-way Anova for experiments including two factors (e.g., %CD44hiCD62lo population in three genotypes activated with or without anti-CD28), followed by Tukey’s multiple comparison test.
Acknowledgments
We would like to thank the current and former lab members for scientific discussions; Daniel Pinschewer and Weldy Bonilla Pinschewer for advice on LCMV, reagents (GP61 peptide), and scientific discussions; Annaïse Jauch for providing LCMV Armstrong; Mihaela Zavolan and Ed Palmer for deep scientific discussions and critical comments on the article; members of the NCCR RNA & Disease for discussions; the University of Basel, the Basel University Hospital and Department of Biomedicine (DBM) for institutional support; the team from the DBM animal facility for animal husbandry; the DBM flow cytometry core team; Robert Ivanek from the DBM bioinformatics core; Philippe Demougin from the D-BSSE/DBM Genomics Facility Basel; Alexander Schmidt from the University of Basel Biozentrum Proteomics Core Facility for support and Marco Amsler, Caroline Schwenzel, Hélène Rossez, Giuseppina Capoferri and Corinne Engdahl-Démollière for technical assistance. Calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing center at the University of Basel. This work was supported by grants of the Swiss National Science Foundation (SNSF Professorship PP00P3_144860 to LTJ), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health, USA, under Award Number R56/R01AI106923 and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 818806) (to LTJ), and the National Heart, Lung, and Blood Institute of the National Institutes of Health, USA, under Award Numbers R01HL109102 and P01HL107202 (to KMA). The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
M.D. performed and analyzed most experiments; M.H. performed all CRISPR experiments, G.B. and C.H. helped to design and interpret metabolic experiments; R.M. performed qPCR experiments, provided scientific input, key experimental and infrastructure support; M.K. provided scientific input and experimental support; R.K. generated HITS-CLIP data; D.S. and J.R. performed the bioinformatic analysis with the help of J.G.; K.M.A. provided key scientific input and access to HITS-CLIP data; M.D. and L.T.J. designed the experiments, interpreted the data and discussed results; L.T.J. designed the study, obtained funding, supervised all experiments and wrote the article (MS) with the help of M.D. and M.H.; all authors approved the final MS.
Declaration of interests
L.T.J. is a co-founder and board member of, holds equity in, and has a sponsored research agreement with Cimeio Therapeutics AG. M.D. and L.T.J. are inventors on a patent application related to the findings reported here.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105372.
Supplemental information
List of genes that fulfilled the following criteria: i) significant derepression in T1792Δ/Δ vs wt and significant repression in T1792tg/tg vs wt at 24 h ii) predicted TS match iii) >5 AHC reads and iv) posttranscriptional regulation based on EISA. Applying these criteria across the 4 seed families from miR-17∼92 cluster defined a set of 68 empirically supported direct miR-17∼92 target genes. Individual seed families are reported.
Data and code availability
Data generated in this study are available at GEO under accession number GEO:GSE140568.
Code is accessible at this location: https://gitlab.com/JekerLab/mir17-92-rescues-cd28-deficient-cd4-t-cells. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Acuto O., Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat. Rev. Immunol. 2003;3:939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- Agarwal V., Bell G.W., Nam J.W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Bachmaier K., Krawczyk C., Kozieradzki I., Kong Y.Y., Sasaki T., Oliveira-Dos-Santos A., Mariathasan S., Bouchard D., Wakeham A., Itie A., et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- Barrat F.J., Cua D.J., Boonstra A., Richards D.F., Crain C., Savelkoul H.F., De Waal-Malefyt R., Coffman R.L., Hawrylowicz C.M., O'garra A. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J. Exp. Med. 2002;195:603–616. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D.P. Metazoan microRNAs. Cell. 2018;173:20–51. doi: 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumjohann D., Ansel K.M. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat. Rev. Immunol. 2013;13:666–678. doi: 10.1038/nri3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumjohann D., Kageyama R., Clingan J.M., Morar M.M., Patel S., De Kouchkovsky D., Bannard O., Bluestone J.A., Matloubian M., Ansel K.M., Jeker L.T. The microRNA cluster miR-17 approximately 92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat. Immunol. 2013;14:840–848. doi: 10.1038/ni.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman E.K., Chen T., Amendola M., Van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronevetsky Y., Villarino A.V., Eisley C.J., Barbeau R., Barczak A.J., Heinz G.A., Kremmer E., Heissmeyer V., Mcmanus M.T., Erle D.J., et al. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. J. Exp. Med. 2013;210:417–432. doi: 10.1084/jem.20111717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler J.L., Walsh P.T., Porrett P.M., Choi Y., Turka L.A. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J. Immunol. 2006;177:4262–4266. doi: 10.4049/jimmunol.177.7.4262. [DOI] [PubMed] [Google Scholar]
- Chen W., Jin W., Hardegen N., Lei K.J., Li L., Marinos N., Mcgrady G., Wahl S.M. Conversion of peripheral CD4(+)CD25(-) naive T cells to CD4(+)CD25(+) regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang Y.J., Kole H.K., Brown K., Naramura M., Fukuhara S., Hu R.J., Jang I.K., Gutkind J.S., Shevach E., Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- Crotty S. Follicular helper CD4 T cells (TFH) Annu. Rev. Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- De Kouchkovsky D., Esensten J.H., Rosenthal W.L., Morar M.M., Bluestone J.A., Jeker L.T. microRNA-17-92 regulates IL-10 production by regulatory T cells and control of experimental autoimmune encephalomyelitis. J. Immunol. 2013;191:1594–1605. doi: 10.4049/jimmunol.1203567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolz M., Marone R., Jeker L.T. Plasmid- or ribonucleoprotein-mediated CRISPR/Cas gene editing in primary murine T cells. Methods Mol. Biol. 2021;2285:255–264. doi: 10.1007/978-1-0716-1311-5_20. [DOI] [PubMed] [Google Scholar]
- Du P., Wang L., Sliz P., Gregory R.I. A biogenesis step upstream of microprocessor controls miR-17 approximately 92 expression. Cell. 2015;162:885–899. doi: 10.1016/j.cell.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edner N.M., Carlesso G., Rush J.S., Walker L.S.K. Targeting co-stimulatory molecules in autoimmune disease. Nat. Rev. Drug Discov. 2020;20:82. doi: 10.1038/s41573-020-00116-x. [DOI] [PubMed] [Google Scholar]
- Esensten J.H., Helou Y.A., Chopra G., Weiss A., Bluestone J.A. CD28 costimulation: from mechanism to therapy. Immunity. 2016;44:973–988. doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46:D649–D655. doi: 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S.E., Han S., Kelsoe G., Thompson C.B. CD28 is required for germinal center formation. J. Immunol. 1996;156:4576–4581. [PubMed] [Google Scholar]
- Friedman R.C., Farh K.K.H., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon J.D., Kageyama R., Shehata H.M., Fassett M.S., Mar D.J., Wigton E.J., Johansson K., Litterman A.J., Odorizzi P., Simeonov D., et al. miR-15/16 restrain memory T cell differentiation, cell cycle, and survival. Cell Rep. 2019;28:2169–2181.e4. doi: 10.1016/j.celrep.2019.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D., Burger L., Florescu M., Stadler M.B. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat. Biotechnol. 2015;33:722–729. doi: 10.1038/nbt.3269. [DOI] [PubMed] [Google Scholar]
- Gaidatzis D., Lerch A., Hahne F., Stadler M.B. QuasR: quantification and annotation of short reads in R. Bioinformatics. 2015;31:1130–1132. doi: 10.1093/bioinformatics/btu781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alonso L., Holland C.H., Ibrahim M.M., Turei D., Saez-Rodriguez J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019;29:1363–1375. doi: 10.1101/gr.240663.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross G., Eshhar Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu. Rev. Pharmacol. Toxicol. 2016;56:59–83. doi: 10.1146/annurev-pharmtox-010814-124844. [DOI] [PubMed] [Google Scholar]
- Gubser P.M., Bantug G.R., Razik L., Fischer M., Dimeloe S., Hoenger G., Durovic B., Jauch A., Hess C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 2013;14:1064–1072. doi: 10.1038/ni.2687. [DOI] [PubMed] [Google Scholar]
- Homet Moreno B., Zaretsky J.M., Garcia-Diaz A., Tsoi J., Parisi G., Robert L., Meeth K., Ndoye A., Bosenberg M., Weeraratna A.T., et al. Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol. Res. 2016;4:845–857. doi: 10.1158/2326-6066.CIR-16-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin J.P., Lu Y., Loeb G.B., Leslie C.S., Rudensky A.Y. The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat. Immunol. 2018;19:1137–1145. doi: 10.1038/s41590-018-0208-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E., Cheung J., Zhu J., Su X., Taylor M.J., Wallweber H.A., Sasmal D.K., Huang J., Kim J.M., Mellman I., Vale R.D. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeker L.T., Bluestone J.A. MicroRNA regulation of T-cell differentiation and function. Immunol. Rev. 2013;253:65–81. doi: 10.1111/imr.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S., Li C., Olive V., Lykken E., Feng F., Sevilla J., Wan Y., He L., Li Q.J. Molecular dissection of the miR-17-92 cluster's critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood. 2011;118:5487–5497. doi: 10.1182/blood-2011-05-355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H.Y., Oda H., Lai M., Skalsky R.L., Bethel K., Shepherd J., Kang S.G., Liu W.H., Sabouri-Ghomi M., Cullen B.R., et al. MicroRNA-17∼92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J. 2013;32:2377–2391. doi: 10.1038/emboj.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst A.O., Wieland A., Nasti T., Yang S., Zhang R., Barber D.L., Konieczny B.T., Daugherty C.Z., Koenig L., Yu K., et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.G., Liu W.H., Lu P., Jin H.Y., Lim H.W., Shepherd J., Fremgen D., Verdin E., Oldstone M.B.A., Qi H., et al. MicroRNAs of the miR-17 approximately 92 family are critical regulators of T(FH) differentiation. Nat. Immunol. 2013;14:849–857. doi: 10.1038/ni.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C.G., Kobayashi T., Cejas P.J., Kim T., Yoon K., Kim G.K., Chiffoleau E., Hickman S.P., Walsh P.T., Turka L.A., Choi Y. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat. Med. 2006;12:1088–1092. doi: 10.1038/nm1449. [DOI] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach D.R., Krummel M.F., Allison J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linterman M.A., Denton A.E., Divekar D.P., Zvetkova I., Kane L., Ferreira C., Veldhoen M., Clare S., Dougan G., Espéli M., Smith K.G. CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. Elife. 2014;3:e03180. doi: 10.7554/eLife.03180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linterman M.A., Rigby R.J., Wong R., Silva D., Withers D., Anderson G., Verma N.K., Brink R., Hutloff A., Goodnow C.C., Vinuesa C.G. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes CD28 and ICOS. Immunity. 2009;30:228–241. doi: 10.1016/j.immuni.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Liu Q., Zheng J., Sun W., Huo Y., Zhang L., Hao P., Wang H., Zhuang M. A proximity-tagging system to identify membrane protein-protein interactions. Nat. Methods. 2018;15:715–722. doi: 10.1038/s41592-018-0100-5. [DOI] [PubMed] [Google Scholar]
- Loeb G.B., Khan A.A., Canner D., Hiatt J.B., Shendure J., Darnell R.B., Leslie C.S., Rudensky A.Y. Transcriptome-wide miR-155 binding map reveals widespread noncanonical MicroRNA targeting. Mol. Cell. 2012;48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L.F., Gasteiger G., Yu I.S., Chaudhry A., Hsin J.P., Lu Y., Bos P.D., Lin L.L., Zawislak C.L., Cho S., et al. A single miRNA-mRNA interaction affects the immune response in a context- and cell-type-specific manner. Immunity. 2015;43:52–64. doi: 10.1016/j.immuni.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G.J., Hu J.K., Pereira R.M., Crampton J.S., Togher S., Bild N., Crotty S., Rao A. Cutting edge: NFAT transcription factors promote the generation of follicular helper T cells in response to acute viral infection. J. Immunol. 2016;196:2015–2019. doi: 10.4049/jimmunol.1501841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Llordella M., Esensten J.H., Bailey-Bucktrout S.L., Lipsky R.H., Marini A., Chen J., Mughal M., Mattson M.P., Taub D.D., Bluestone J.A. CD28-inducible transcription factor DEC1 is required for efficient autoreactive CD4+ T cell response. J. Exp. Med. 2013;210:1603–1619. doi: 10.1084/jem.20122387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moltzahn F., Olshen A.B., Baehner L., Peek A., Fong L., Stöppler H., Simko J., Hilton J.F., Carroll P., Blelloch R. Microfluidic-based multiplex qRT-PCR identifies diagnostic and prognostic microRNA signatures in the sera of prostate cancer patients. Cancer Res. 2011;71:550–560. doi: 10.1158/0008-5472.CAN-10-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya M.M., Maul J., Singh P.B., Pua H.H., Dahlström F., Wu N., Huang X., Ansel K.M., Baumjohann D. A distinct inhibitory function for miR-18a in Th17 cell differentiation. J. Immunol. 2017;199:559–569. doi: 10.4049/jimmunol.1700170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulero M.C., Aubareda A., Schlüter A., Pérez-Riba M. RCAN3, a novel calcineurin inhibitor that down-regulates NFAT-dependent cytokine gene expression. Biochim. Biophys. Acta. 2007;1773:330–341. doi: 10.1016/j.bbamcr.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Nguyen D.N., Roth T.L., Li P.J., Chen P.A., Apathy R., Mamedov M.R., Vo L.T., Tobin V.R., Goodman D., Shifrut E., et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020;38:44–49. doi: 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'connell R.M., Rao D.S., Chaudhuri A.A., Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- O'donnell K.A., Wentzel E.A., Zeller K.I., Dang C.V., Mendell J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Paolino M., Penninger J.M. Cbl-b in T-cell activation. Semin. Immunopathol. 2010;32:137–148. doi: 10.1007/s00281-010-0197-9. [DOI] [PubMed] [Google Scholar]
- Pua H., Steiner D., Patel S., Gonzalez J., Ortiz-Carpena J., Kageyama R., Chiou N.T., Gallman A., De Kouchkovsky D., Jeker L., et al. MicroRNAs 24 and 27 suppress allergic inflammation and target a network of regulators of T helper 2 cell-associated cytokine production. Immunity. 2016;44:821–832. doi: 10.1016/j.immuni.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley W.W., Jin W., Lee A.J., Wright A., Wu X., Tewalt E.F., Leonard T.O., Norbury C.C., Fitzpatrick L., Zhang M., Sun S.C. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J. Exp. Med. 2007;204:1475–1485. doi: 10.1084/jem.20062694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riha P., Rudd C.E. CD28 co-signaling in the adaptive immune response. Self. Nonself. 2010;1:231–240. doi: 10.4161/self.1.3.12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., Mccarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R., Neilson J.R., Sarma A., Sharp P.A., Burge C.B. Proliferating cells express mRNAs with shortened 3' untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom D.M., Walker L.S.K. CD28 costimulation: walking the immunological tightrope. Eur. J. Immunol. 2013;43:42–45. doi: 10.1002/eji.201243211. [DOI] [PubMed] [Google Scholar]
- Schaefer C.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay T., Buetow K.H. PID: the pathway interaction database. Nucleic Acids Res. 2009;37:D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki A., Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 2018;215:985–997. doi: 10.1084/jem.20171626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinian A., Pfeffer K., Lee K.P., Kündig T.M., Kishihara K., Wakeham A., Kawai K., Ohashi P.S., Thompson C.B., Mak T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- Shang W., Jiang Y., Boettcher M., Ding K., Mollenauer M., Liu Z., Wen X., Liu C., Hao P., Zhao S., et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc. Natl. Acad. Sci. USA. 2018;115:E4051–E4060. doi: 10.1073/pnas.1801340115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson L.J., Patel S., Bhakta N.R., Choy D.F., Brightbill H.D., Ren X., Wang Y., Pua H.H., Baumjohann D., Montoya M.M., et al. A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nat. Immunol. 2014;15:1162–1170. doi: 10.1038/ni.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner D.F., Thomas M.F., Hu J.K., Yang Z., Babiarz J.E., Allen C.D.C., Matloubian M., Blelloch R., Ansel K.M. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity. 2011;35:169–181. doi: 10.1016/j.immuni.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Yamaguchi M.T., Ohteki T., Sasaki T., Kaisho T., Kimura Y., Yoshida R., Wakeham A., Higuchi T., Fukumoto M., et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14:523–534. doi: 10.1016/s1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- Tian R., Wang H., Gish G.D., Petsalaki E., Pasculescu A., Shi Y., Mollenauer M., Bagshaw R.D., Yosef N., Hunter T., et al. Combinatorial proteomic analysis of intercellular signaling applied to the CD28 T-cell costimulatory receptor. Proc. Natl. Acad. Sci. USA. 2015;112:E1594–E1603. doi: 10.1073/pnas.1503286112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M., Maus M., Klein-Hessling S., Freinkman E., Yang J., Eckstein M., Cameron S., Turvey S.E., Serfling E., Berberich-Siebelt F., et al. Store-operated Ca(2+) entry controls clonal expansion of T cells through metabolic reprogramming. Immunity. 2017;47:664–679.e6. doi: 10.1016/j.immuni.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne S., Palmer G., Lamacchia C., Martin P., Talabot-Ayer D., Rodriguez E., Ronchi F., Sallusto F., Dinh H., Sims J.E., Gabay C. IL-36R ligands are potent regulators of dendritic and T cells. Blood. 2011;118:5813–5823. doi: 10.1182/blood-2011-05-356873. [DOI] [PubMed] [Google Scholar]
- Walker L.S., Gulbranson-Judge A., Flynn S., Brocker T., Raykundalia C., Goodall M., Förster R., Lipp M., Lane P. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J. Exp. Med. 1999;190:1115–1122. doi: 10.1084/jem.190.8.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.J., Heuts F., Ovcinnikovs V., Wardzinski L., Bowers C., Schmidt E.M., Kogimtzis A., Kenefeck R., Sansom D.M., Walker L.S.K. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc. Natl. Acad. Sci. USA. 2015;112:524–529. doi: 10.1073/pnas.1414576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C.H., Crombie C., Van Der Weyden L., Poulogiannis G., Rust A.G., Pardo M., Gracia T., Yu L., Choudhary J., Poulin G.B., et al. Nuclear receptor binding protein 1 regulates intestinal progenitor cell homeostasis and tumour formation. EMBO J. 2012;31:2486–2497. doi: 10.1038/emboj.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Smyth G.K. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012;40:e133. doi: 10.1093/nar/gks461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C., Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26–36. doi: 10.1016/j.cell.2008.12.027. [DOI] [PubMed] [Google Scholar]
- Xiao C., Srinivasan L., Calado D.P., Patterson H.C., Zhang B., Wang J., Henderson J.M., Kutok J.L., Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of genes that fulfilled the following criteria: i) significant derepression in T1792Δ/Δ vs wt and significant repression in T1792tg/tg vs wt at 24 h ii) predicted TS match iii) >5 AHC reads and iv) posttranscriptional regulation based on EISA. Applying these criteria across the 4 seed families from miR-17∼92 cluster defined a set of 68 empirically supported direct miR-17∼92 target genes. Individual seed families are reported.
Data Availability Statement
Data generated in this study are available at GEO under accession number GEO:GSE140568.
Code is accessible at this location: https://gitlab.com/JekerLab/mir17-92-rescues-cd28-deficient-cd4-t-cells. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.