

Abstract
Propionic acidemia (PA) in the Amish is caused by a homozygous pathogenic variant (c.1606A>G; p.Asn536Asp) in the PCCB gene. Amish patients can have borderline or normal newborn screening (NBS) results and symptoms can present at any time from early childhood to mid-adulthood. Early diagnosis and initiation of treatment for PA in the non-Amish population improves patient outcomes. Here, we present data from a retrospective chart review of Amish patients diagnosed with PA from three different medical centers in order to document its natural history in the Amish and determine the influence of treatment on outcomes in this population. A total of 38 patients with average current age 19.9 years (range 4y-45y), 57.9% males, were enrolled in the study. Fourteen patients (36.8%) were diagnosed with a positive newborn screening (NBS) while 24 patients (63.2%) had negative or inconclusive NBS or had no record of NBS in their charts. These 24 patients were diagnosed by screening after a family member was diagnosed with PA (14; 58.3%), following a hospitalization for metabolic acidosis (5; 20.8%), hospitalization for seizures (3; 12.5%) or via cord blood (2; 8.3%). The majority of patients were prescribed a protein restricted diet (32; 84.2%), including metabolic formula (29; 76.3%). Most were treated with carnitine (35; 92.1%), biotin (2; 76.3%) and/or Coenzyme Q10 (16; 42.1%). However, treatment adherence varied widely among patients, with 7 (24.1%) of the patients prescribed metabolic formula reportedly nonadherent. Cardiomyopathy was the most prevalent finding (22; 63.2%), followed by developmental delay/intellectual disability (15; 39.5%), long QT (14; 36.8%), seizures (12; 31.6%), failure to thrive (4; 10.5%), and basal ganglia strokes (3; 7.9%). No difference in outcome was obvious for those diagnosed by NBS and treated early with dietary and supplement management, especially for cardiomyopathy. However, this is a limited retrospective observational study. A prospective study with strict documentation of treatment adherence and universal screening for cardiomyopathy and long QT should be conducted to better study the impact of early detection and treatment. Additional treatment options such as liver transplantation and future therapies such as mRNA or gene therapy should be explored in this population.
Keywords: Propionic Acidemia, Amish, Newborn screen, Treatment, Cardiomyopathy, Seizures
1. Introduction
The distribution of genetic disorders in the Amish differs from the non-Amish population due to a bottleneck effect arising from the migration of a small subset of Anabaptists in Europe to America in the early 18th century due to religious persecution [1]. Propionic acidemia (PA) is caused by pathogenic variants in the PCCA and PCCB genes encoding the alpha and beta subunits of the mitochondrial enzyme propionyl-CoA carboxylase (PCC), leading to absent or reduced enzyme activity [2]. Certain amino acids including isoleucine, valine, threonine, and methionine, in addition to odd chain fatty acids, are important sources of propionyl CoA, which is metabolized by propionyl-CoA carboxylase to D-methylmalonyl-CoA. Defects in PCC lead to toxic metabolite accumulation that results in episodes of intermittent acidosis. In addition, there appears to be a toxic effect on mitochondrial energy metabolism. In the non-Amish population, most patients with PA are compound heterozygotes for variants in PCCB or PCCA [3]. However, Amish patients with PA are homozygous for a missense variant (c.1606A>G; p.Asn536Asp) in the PCCB gene [2,4].
Elevated levels of propionylcarnitine (C3) identified by tandem mass spectrometry as part of newborn screening (NBS) are characteristic of PA. Non-Amish individuals with PA typically present with symptoms in the neonatal period including lethargy, poor feeding, hypotonia, metabolic acidosis and hyperammonemia, often before newborn screening results are available [[5], [6], [7]]. However, Amish patients generally have a less severe, though variable disease, likely because the mutant enzyme has residual activity [2,8]. Newborn screen results usually identify borderline high or normal C3, and patients rarely present with neonatal symptoms. Rather, symptoms can develop at any time from early childhood to mid-adulthood [7]. Clinical findings in PA include cardiomyopathy, cardiac arrhythmias, metabolic decompensation, seizures, basal ganglia strokes, developmental delay and pancreatitis [3,8]. In the Amish population, cardiomyopathy is frequently the presenting symptom.
Treatment for PA consists of a modified diet focused on protein restriction, often with supplemental metabolic formula with reduced or free propiogenic amino acids, biotin and carnitine supplementation and occasionally antibiotic treatment to reduce intestinal propiogenic bacteria [2,5,[9], [10], [11]]. Liver transplantation has been shown in many cases to reduce episodes of metabolic decompensation, improve cardiac function in patients with cardiomyopathy, and increase protein tolerance [10,12,13].However, long term comparison of transplanted individuals with non-transplanted patients is lacking in the literature, and some reports have shown that transplanted patients can still develop cardiomyopathy or other complications after liver transplant [10,14].Liver transplantation does not provide complete correction of the metabolic disease and therefore these patients should continue to be managed by metabolic specialist after the transplant.
The importance of early diagnosis and treatment of PA for survival has been well documented, but reported long term outcomes have been variable [7,11,15]. Notably, development of cardiomyopathy has been reported in PA patients despite early diagnosis and metabolic management, even with good metabolic control [12,13]. Amish patients with PA in Wisconsin diagnosed and treated early had long-term reduction of neurological sequelae, but no clear change in cardiac complications [7]. The present study is a retrospective examination of medical and laboratory records of Amish patients with PA from Western Pennsylvania, Ohio and Indiana, with a goal of comparing clinical symptoms, progression of disease, and the impact of ongoing management on outcome.
2. Materials and methods
2.1. Study design and setting
This retrospective study involved the review of charts of Amish patients with PA from Western Pennsylvania, Ohio and Indiana. This study was approved by the University of Pittsburgh Institutional Review Board (protocol #STUDY20050254).
2.2. Identification of Amish patients
Informed consent to collect documentation of diagnosis, management, and outcomes was obtained from Amish patients with previously diagnosed PA who are followed at three medical centers that are part of the Plain Community Health Consortium (PCHC): UPMC Children's Hospital of Pittsburgh, New Leaf Center, and the Community Health Clinic in western Pennsylvania, Ohio and Indiana, respectively. Parental informed consent was obtained for underaged participants. For patients outside of the UPMC system, charts were shared as either physical copies or electronically on Ecares, a medical record sharing system.
2.3. Data collection
Information abstracted from patient charts included demographic information, initial presentation and age at diagnosis, clinical characteristics, hospitalizations, treatments and outcomes. Laboratory tests captured included NBS results, plasma acylcarnitine profiles, carnitine levels, ammonia, lactate, plasma amino acids, urine organic acid profiles, EKGs, and clinical imaging such as echocardiograms and MRIs of the brain. Treatment parameters documented included intact protein restriction, use of metabolic formula (most with no or limited amounts of isoleucine, valine, threonine, methionine, and odd chain fatty acids), or medication supplementation, and age at treatment initiation. Prespecified diagnoses recognized as sequelae of PA were assessed including cardiomyopathy, arrhythmias, seizures, metabolic strokes and developmental delays/intellectual disabilities.
2.4. Primary outcome variables
The goals of the study were to identify common characteristics among Amish patients diagnosed with propionic acidemia and determine the associations of treatment with patient outcome as described in the data collection section.
2.5. Analysis
Data analysis included descriptive statistics to summarize demographic information, positive NBS results, initial presentation, average ages at presentation and diagnosis, average lab results and information on special diets and medications. Percentages of patients with previously specified outcomes were also calculated.
3. Results
3.1. Demographics
A total of 38 patients were enrolled in the study. The current age of participants at time of data collection was 19.9 years (range 4y-45y), with 57.9% of participants being male.
3.2. Newborn screening and diagnosis
Of the 38 patients, 22 (57.9%) had a PA NBS reported. Of these, 17 (77.3%) had a presumptive positive result for PA and 5 (22.7%) had negative results. For the 17 patients with presumptive positive NBS, the C3 level on NBS was listed in their chart. The average C3 of these 17 patients was 7.4 ± 1.9 nmol/mL. Normal C3 cutoffs varied depending on the state in which patients received their NBS; for example, the cutoff in Ohio is <5.6 nmol/mL while the cutoff in Wisconsin is <6.92 nmol/mL. One patient had a normal C3/C2 ratio in the setting of a mildly elevated C3 on NBS. A repeat NBS in two patients with slightly elevated C3 was normal. All 3 of these patients were told they likely did not have PA and did not have medical follow up until other family members were diagnosed and they had molecular testing confirmation (Table 1, Fig. 1). Of the 24 patients who were not diagnosed via NBS, 16 participants did not have record of a NBS for PA in their chart, 5 patients had a negative NBS result, and the 3 patients described above were presumed not to have PA despite an initial positive NBS. For these 24 patients, the average age at diagnosis of PA was 5.9 years (3 days to 32 years). The majority (14/24; 58.3%) were diagnosed via testing after a family member was diagnosed. The remaining patients were identified following hospitalization for metabolic acidosis (5/24; 20.8%) or seizures (3/24; 12.5%), or cord blood analysis undertaken because of family history (2/24; 8.3%) (Table 2, Fig. 1). Thirty-three (86.8%) of the 38 participants had record of a molecular study confirming the diagnosis of PA; all were homozygous for the common Amish PCCB (c.1606A>G; p.Asn536Asp) variant (Table 3).
Table 1.
Newborn screening results.
| Newborn Screen for PA completed | |||||
|---|---|---|---|---|---|
| Yes | 22 (57.9%) | Positive Result | 17 (77.3%) | C3 level (reference range N < 5.6 nmol/mL): 7.4 ± 1.9 nmol/mL N = 13 |
Normal C3/C2 ratio and no follow up: 1 (5.9%) |
| Repeat NBS negative: 2 (11.8%) | |||||
| Negative Result | 5 (22.7%) | ||||
| No information available | 16 (42.1%) | ||||
Fig. 1.

Diagram representing the patients included in this study
Table 2.
Diagnostic Features Leading to the Diagnosis in Patients with False Negative NBS or No NBS Result in Their Chart.
| Patients Not Diagnosed by NBS (N = 24) | |
|---|---|
| Average Age at Diagnosis (range) | 5.9 y (3 d – 32 y) |
| Cord Blood⁎ | 2 (8.3%) |
| Hospitalization for Metabolic Acidosis | 5 (20.8%) |
| Hospitalization for Seizure | 3 (12.5%) |
| Relative Diagnosed with PA | 14 (58.3%) |
Two patients had a positive cord blood testing for the homozygous PCCB (c.1606A > G; p.Asn536Asp) variant and negative NBS for PA.
Table 3.
Molecular Study Results for PA.
| Molecular Study (homozygous PCCB c.1606A > G variant) | |
|---|---|
| Yes (positive) | 33 (86.8%) |
| None recorded⁎ | 5 (13.2%) |
Patients diagnosed with NBS and had subsequent abnormal metabolic labs consistent with propionic acidemia.
3.3. Treatment
Dietary modifications such as intact protein restriction and intake of metabolic formula were begun at an average age of 4.2 years with implementation ranging from birth to 17 years of age. Thirty-two patients (84.2%) were instructed to follow a protein restricted diet and 29 (76.3%) patients were prescribed metabolic formula. Of patients prescribed metabolic formula, 7 (24.1%) participants were noted to be non-adherent to treatment (Table 4). Most of the metabolic formulas prescribed were the usual commercially available formulas for use in propionic acidemia. Some patients were prescribed other metabolic formulas restricting isoleucine and valine (MSUD formulas such as MSD Jr. and MSD AA formulas) due to availability, then supplemented with oral Leucine so that it was not restricted. The most common medications prescribed for PA were carnitine (92.1%), followed by biotin (76.3%) and coenzyme Q10 (42.1%). Metronidazole was not prescribed for any of the patients at the time of this chart review; however it was subsequently given to one patient due to a glycine level of ∼3 times the normal range and mild cardiomyopathy, despite protein restriction and formula supplementation (Table 5).
Table 4.
Special Diets.
| Diet | |
|---|---|
| Average Age Started (range) | 4.2 y (birth – 17) |
| Intact Protein Restriction | Yes: 32 (84.2%) |
| N = 38 | No: 6 (15.8%) |
| Metabolic Formula Recommended | Yes: 29 (76.3%) |
| N = 38 | No: 9 (23.7%) |
| Adherence to Metabolic Formula | Unknown: 22 (75.9%) |
| N = 29 | Not adherent: 7 (24.1%) |
Table 5.
PA Medications Prescribed.
| Medications | Percentage of Patients Ever Prescribed (N = 38) |
|---|---|
| Carnitine | 34 (92.1%) |
| Coenzyme Q10 | 15 (42.1%) |
| Biotin | 28 (76.3%) |
| Metronidazole | 0 (0.0%) |
3.4. Outcomes
Cardiomyopathy was the most prevalent symptom in this cohort (63.2% of patients). Mean age at time of diagnosis of cardiomyopathy was 17.5 years (range 3–38 years), with 37.5% of patients symptomatic at the time of diagnosis. Developmental delay/intellectual disability was the next most common finding, present in 39.5% of patients. Forty-seven percent of these patients had both cognitive and motor delays reported, while 26.7% had only motor and 26.7% had only cognitive delays. History of seizures was present in 31.6% of participants, diagnosed at an average of 1 year of age (range 7 months to 2 years). The most common subtype of seizure was with illness or fever (50.0%), followed by generalized tonic-clonic seizures (41.7%), and focal seizures (8.3%). Long QT was present in 36.8% of patients, diagnosed at an average of 11.3 years (range 8 months to 29 years). Basal ganglia strokes were diagnosed in 7.9% of patients at an average age of 17.2 years (range 7 mo–41 y). Failure to thrive was reported in 4 (10.5%) participants (Table 6).
Table 6.
PA-associated outcomes.
| Outcomes (N = 38) | Age at Diagnosis | Presentation | |
|---|---|---|---|
| Cardiomyopathy | 24 (63.2%) | 17.5y (3y - 38y) | Symptomatic: 9 (37.5%) |
| Asymptomatic: 15 (62.5%) | |||
| Basal Ganglia Strokes⁎⁎ | 3 (7.9%) | 17.2y (7mo - 41y) | ND |
| Seizures | 12 (31.6%) | 1.0y (7mo – 2y) | Febrile/With Illness: 6 (50.0%) |
| GTC: 5 (41.7%) | |||
| Focal: 1 (8.3%) | |||
| Long QT | 14 (36.8%) | 11.3y (8mo – 29y) | ND |
| Failure to Thrive | 4 (10.5%) | ND | ND |
| Developmental Delays/Intellectual disability | 15 (42.1%) | ND | Motor: 4 (26.7%) |
| Cognitive: 4 (26.7%) | |||
| Motor and Cognitive: 7 (46.7%) | |||
ND: not described.
GTC: Generalized Tonic-Clonic.
2 of these 3 patients had brain imaging confirming the diagnosis.
Outcomes for patients were grouped based on types of dietary modifications and medications taken by the patients for the overall cohort as well as subgroups based on the age at diagnosis (Fig. 2, Fig. 3, Fig. 4, Fig. 5). All patients diagnosed by NBS were prescribed a protein restricted diet, and the majority were also prescribed metabolic formula and medications. More patients diagnosed by NBS had no disease sequelae at the time of the chart review than the other two groups; however, patients diagnosed via NBS were on average younger. Patients diagnosed after one year of age had the most variability in treatment and included the lowest number of patients without sequelae. There was no clear pattern in outcome relative to treatment.
Fig. 2.

Treatments and outcomes for all patients.
Fig. 3.

Treatments and outcomes for those diagnosed by NBS.
Fig. 4.
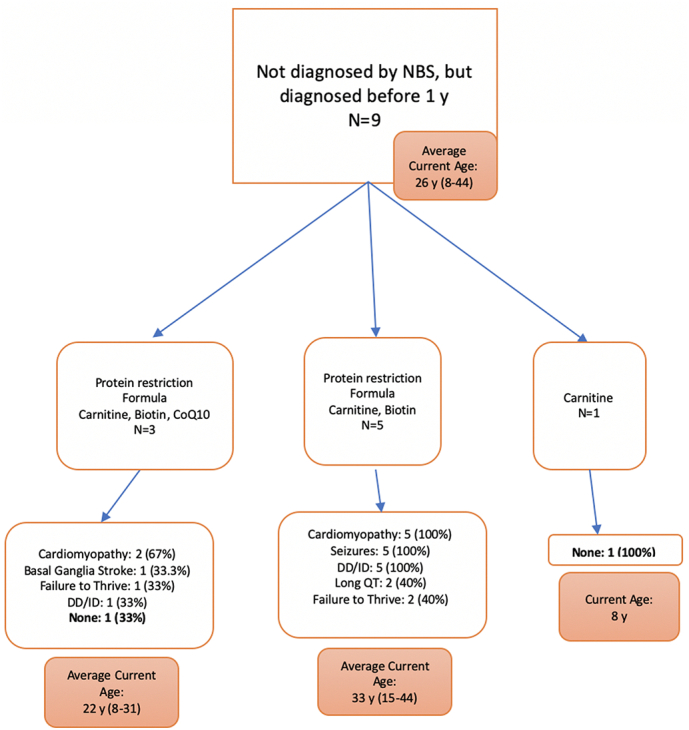
Treatments and outcomes for those diagnosed before the age of 1 year but not via NBS.
Fig. 5.

Treatments and outcomes for those diagnosed after 1 year of age.
3.5. Laboratory results
Twenty-six patients had an acylcarnitine profile recorded in their chart, all showing an elevated C3 level. Ten of twenty-one patients (47.6%) with a recorded ammonia had mild to moderate elevation (ranging from slightly above normal range to ∼4 times the normal range). Four of sixteen patients (25.5%) with a recorded lactate had an elevated level (ranging from slightly above normal range to ∼3 times the normal range). Thirty-two patients had a carnitine battery in their chart; 7 patients (21.9%) had a low free carnitine and 2 (6.3%) had a low total carnitine. Thirty-two patients had plasma amino acids in their chart, and 27 (84.4%) of these patients had an elevated glycine. Twenty-two patients had urine organic acids reported, with 20 (90.9%) noting a metabolite profile consistent with their PA diagnosis (Table 7).
Table 7.
PA-specific Lab Results in Patients (N = number of patients with the specific lab value).
| Lab | Result values |
|---|---|
| Ammonia | High: 10 (47.6%) |
| N = 21 | Normal: 11 (52.4%) |
| Lactate | High: 4 (25.0%) |
| N = 16 | Normal: 12 (75.0%) |
| Free Carnitine | Normal or High: 25 (78.1%) |
| N = 32 | Low: 7 (21.9%) |
| Total Carnitine | Normal or High: 30 (93.8%) |
| N = 32 | Low: 2 (6.3%) |
| C3 Level on Acylcarnitine Profile | High: 26 (100.0%) |
| N = 26 | |
| Glycine on Plasma Amino Acids | High: 27 (84.4%) |
| N = 32 | Normal: 5 (15.6%) |
| Urine Organic Acids | Metabolites excretion consistent with PA diagnosis: 20 (90.9%) |
| N = 22 | Normal: 2 (9.1%) |
4. Discussion
This study reviewed medical records from Amish patients with a known diagnosis of propionic acidemia in order to characterize the disease course, and assess association of age at diagnosis and treatment implementation with patient outcomes (Table 8).
Table 8.
Individual participant outcomes.
| ID | Current Age (years) | Age at Dx/NBS Dx/Cord blood Dx | Age Diet/Formula Started | Diet | Formula | Medications | Outcomes |
|---|---|---|---|---|---|---|---|
| PA-001 | 9 | NBS | 2 y | Protein restricted | Propimex | Carnitine Biotin CoQ10 |
None |
| PA-002 | 7 | NBS | infancy | Protein restricted | MMA-PA Cooler | Carnitine Biotin CoQ10 |
None |
| PA-003 | 5 | NBS | infancy | Protein restricted | Propimex | Carnitine CoQ10 Biotin |
Cardiomyopathy |
| PA-004 | 10 | 5 y | 5 y | Protein restricted | none | Carnitine CoQ10 Biotin |
Cardiomyopathy |
| PA-005 | 31 | 4 mo | 4 mo | Protein restricted | Procitric | Carnitine CoQ10 Biotin |
Cardiomyopathy Developmental delays Basal Ganglia Stroke Failure to Thrive |
| PA-006 | 26 | infancy | 17 y | Protein restricted | Propimex | Carnitine CoQ10 Biotin |
Cardiomyopathy |
| PA-007 | 10 | NBS | infancy | Protein restricted | MMA-PA Cooler | Carnitine Biotin |
Developmental delays Failure to thrive |
| PA-008 | 14 | 10 y | 10 y | Protein restricted | none | Carnitine | Cardiomyopathy Long QT |
| PA-009 | 4 | NBS | infancy | Protein restricted | Propimex | Carnitine | None |
| PA-010 | 8 | Cord Blood | n/a | none | none | Carnitine | None |
| PA-011 | 32 | 2 y | 2 y | Protein restricted | None | Carnitine | None |
| PA-012 | 26 | NBS | infancy | Protein restricted | None | none | None |
| PA-013 | 35 | 13 y | 13 y | none | Propimex | Carnitine CoQ10 Biotin |
Cardiomyopathy |
| PA-014 | 13 | NBS | infancy | Protein restricted | OA-2 | Carnitine Biotin |
Cardiomyopathy Long QT |
| PA-015 | 16 | NBS | infancy | Protein restricted | OA-2 | Carnitine | Long QT |
| PA-016 | 15 | 7 y | 8 y | Protein restricted | none | Carnitine CoQ10 Biotin |
Cardiomyopathy |
| PA-017 | 24 | 22 m | 22 m | none | none | Carnitine Biotin |
Seizure with illness Long QT Cardiomyopathy |
| PA-018 | 28 | 21 y | 21 y | Protein restricted | none | Carnitine CoQ10 |
Long QT |
| PA-019 | 22 | 1 y | 1 y | Protein restricted | Propimex-1 | Carnitine Biotin |
Cardiomyopathy Focal Seizures Failure to thrive Developmental delay Long QT |
| PA-020 | 13 | NBS | infancy | Protein restricted | none | Carnitine | Basal Ganglia Stroke GTC seizures Developmental delay Long QT Cardiomyopathy |
| PA-021 | 15 | 11 m | 11 m | Protein restricted | MSD AA | Carnitine Biotin |
Cardiomyopathy Focal Seizures Failure to thrive Developmental delay |
| PA-022 | 26 | 7 y | 7 y | Protein restricted | MSD AA | Carnitine CoQ10 Biotin |
Cardiomyopathy GTC seizures Cognitive delay |
| PA-023 | 16 | NBS | infancy | Protein restricted | MSD AA | Carnitine Biotin |
Cardiomyopathy Long QT |
| PA-024 | 29 | 12 y | 12 y | Protein restricted | MSD AA | Carnitine Biotin |
Seizure with illness Developmental delay |
| PA-025 | 43 | 1 m | infancy | Protein restricted | S-14 | Carnitine Biotin |
Cardiomyopathy Seizure with illness Developmental delay Long QT |
| PA-026 | 41 | 1 m | infancy | Protein restricted | S-14 | Carnitine Biotin |
Cardiomyopathy Seizure with illness Developmental delay |
| PA-027 | 44 | 7 m | 7 m | Protein restricted | S-14 | Carnitine Biotin |
Cardiomyopathy GTC seizures Developmental delay |
| PA-028 | ⁎44 | 2 y | 2 y | Protein restricted | S-14 | Biotin | Cardiomyopathy Basal Ganglia Stroke GTC seizures Developmental delay |
| PA-029 | 6 | NBS | infancy | Protein restricted | MSD Jr. | Carnitine Biotin |
Developmental delay |
| PA-030 | 14 | 3 y | 7 y | Protein restricted | MSD AA | Carnitine CoQ10 Biotin |
Long QT Cardiomyopathy |
| PA-031 | 13 | 2 y | 4 y | Protein restricted | MSD AA | Carnitine Biotin |
Developmental Delay Long QT Cardiomyopathy |
| PA-032 | 11 | NBS | infancy | Protein restricted | Propimex | Carnitine Biotin |
Seizure with illness Developmental delay Long QT Cardiomyopathy |
| PA-033 | 8 | Cord blood | infancy | Protein restricted | MSD Jr. | Carnitine CoQ10 Biotin |
n/a |
| PA-034 | 24 | 14 y | 14 y | none | MSD AA | Carnitine CoQ10 Biotin |
Cardiomyopathy |
| PA-035 | 42 | 32 y | n/a | none | none | n/a | Cardiomyopathy |
| PA-036 | 15 | 7 y | 7 y | Protein restricted | MSD AA | Carnitine CoQ10 Biotin |
Developmental Delay Long QT |
| PA-037 | 9 | NBS | infancy | Protein restricted | MSD AA | Carnitine CoQ10 Biotin |
Seizures with illness Long QT |
| PA-038 | 7 | NBS | infancy | Protein restricted | MSD Jr. | Carnitine CoQ10 Biotin |
n/a |
Deceased at age listed.
PA is identified by NBS through an elevated C3 level on acylcarnitine profile. In non-Amish patients with neonatal-onset disease, symptoms can arise before NBS results are available [2,16], but Amish individuals typically have milder disease. Thus, the NBS is often normal, or identifies a biochemical abnormality long before symptoms arise [7]. It has been suggested that lowering the C3 cutoff for an abnormal NBS would decrease the number of false negative results [7]. However, this would likely result in an increased number of false-positive results, leading to unnecessary testing, healthcare costs and psychosocial burden for families [17]. Instead, additional targeted screening for individuals of Amish descent has been proposed using molecular testing for the PCCB common Amish variant [7,18].
Over half of the patients in our study had record of a NBS for PA and the majority were positive. For patients without NBS or with a negative NBS, over half were diagnosed with PA after a family member was found to have the condition. A smaller subset of patients had more severe presentations in early childhood such as metabolic decompensation or seizures resulting in hospitalization. This is consistent with other studies showing that PA in the Amish has a variable presentation and is less likely to cause metabolic decompensation in the neonatal period when compared to PA in the non-Amish population [7,19].
Early diagnosis and treatment of inborn errors of metabolism has been shown to have beneficial outcomes for patients [20]. Treatment for PA in the form of an intact protein restricted diet with supplemental metabolic formulas decreases exposure to propiogenic precursors, decreasing episodes of metabolic decompensation and other negative outcomes [2]. In addition, early diagnosis allows care providers, patients, and their families to better prepare for and manage severe metabolic symptoms during periods of otherwise minor illness [21]. Early diagnosis can also facilitate better understanding of the natural history of the disorder, leading to optimization of interventions.
Cardiomyopathy has been recognized as a common complication of PA. The age at diagnosis of PA, degree of metabolic control, or degree of residual enzymatic activity do not seem to modify the risk for cardiomyopathy, and cardiomyopathy can occur in patients who have mild to moderate forms of well-controlled PA [12]. Cardiomyopathy can spontaneously resolve or progress to cardiac failure, and lead to sudden death [22]. Cardiac arrhythmias are frequent, including prolonged QTc interval associated with syncope [9,23], and cardiac arrest [24]. The post-partum period also seems to be a high-risk time for women who have PA, with some presenting with cardiomyopathy only after pregnancy. Although there was no difference found in this study for risk for cardiomyopathy relative to age at diagnosis or degree of metabolic control, earlier diagnosis of PA could lead to earlier screening for cardiac dysfunction and subsequent intervention. Per the 2021 management guidelines for PA, EKG and echocardiogram are suggested for monitoring in PA patients. The recommended frequency for echocardiogram is not provided, but suggest for EKGs every 12 months [10]. We suggest for the PA Amish patients a baseline echocardiogram at time of diagnosis and every year thereafter, unless the patient has symptoms of cardiomyopathy. Further follow- up if cardiomyopathy develops will depend on the cardiologist's recommendations. Additionally, adding N-terminal pro–B-type natriuretic peptide (NT-proBNP) to the routine metabolic follow up labs during clinical evaluations could be useful in these patients as it can add value for the prediction of heart failure/ cardiomyopathy [[25], [26], [27]]. Fibroblast growth factor (FGF21) and 2-methylcitrate monitoring also has the potential to predict long term outcomes as seen in previous studies [10].
Liver transplantation has been shown to improve or slow progression of cardiomyopathy secondary to PA, including in individuals with severe heart failure [12,13]. The exact mechanism is not known but could be due to decreased accumulation of toxic metabolites in the heart. In addition, successful liver transplant appears to achieve metabolic stabilization, resulting in fewer hospitalizations, less dietary restriction, and improved linear growth [28,29]. There have, however, been reports of new or recurrent cardiomyopathy following liver transplantation for PA [30,31], though it is unclear if cardiomyopathy is secondary to the presence of a second genetic condition in these individuals or if it is a result of immunosuppressant medications. This is especially important to keep in mind in the Amish since other genetic causes of cardiomyopathy and arrythmia are well described in them [32]. Therefore, gene panel testing for cardiomyopathy/arrythmia in Amish patients diagnosed with PA is highly recommended upon diagnosis with propionic acidemia. In fact, one of the patients in this study had worsening heart failure despite PA management and was being evaluated for liver transplant. He underwent gene panel testing for cardiomyopathy and was found to have a truncating heterozygous pathogenic variant in the TTN gene (c.59693G>A; p.Trp1989*), encoding the sarcomeric protein titin. This changed his management plan with evaluation for heart instead of the liver transplant.
Early diagnosis and treatment in patients with neonatal-onset PA decrease risk of metabolic decompensations and basal ganglia strokes. Although Amish individuals with PA are less likely to present with severe metabolic decompensations early in life, patients in this study were diagnosed with episodic metabolic acidosis, seizures, developmental delays/intellectual disabilities, and metabolic strokes. Prediction of which patients will have recurrent metabolic decompensations and associated sequelae is not possible, therefore it is important to diagnose and begin treatment for all patients as early as possible. Without consistent dietary modifications/adherence and adherence to medication regimen, it is unclear if current conservative treatment significantly improves outcomes in this population, especially cardiomyopathy and arrythmias. A prospective research study including strict dietary control, medication monitoring, close monitoring of metabolic labs and predictive biomarkers, follow up echocardiograms and EKGs throughout the course of the disease and structural neurodevelopmental assessment for all patients, in addition to investigating other secondary genetic factors, is required to better understand the natural history of PA disease in patients with the Amish variant. Additional existing therapies such as liver transplantation or new emerging therapies such as mRNA or gene therapy could be considered in this population.
Limitations to this study are inherent to the nature of chart reviews. The amount and type of information present in patient charts varied widely depending on how often the patient utilized the healthcare system. Although the majority of patients were prescribed some sort of dietary modification such as protein restriction or metabolic formula, adherence to these recommendations often was not recorded in charts. In addition, adherence to other therapeutic recommendations, especially screening for cardiac complications, was inconsistent, with many apparently receiving no screening. Given that nearly half of the patients with cardiomyopathy were asymptomatic at time of diagnosis, it is possible that consistent monitoring and screening would identify a larger proportion of patients with cardiomyopathy. Lastly, screening Amish couples for the common Amish PA variant is recommended given the possibility of missing PA diagnosis on the NBS in this population.
Author disclosures
The authors have no competing interests to declare.
Funding
LGG is funded in part by the National Human Genome Research Institute (NHGRI) grant #1K08 HG010490, a component of the National Institutes of Health (NIH). The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official view of NHGRI/NIH.
CRediT authorship contribution statement
Sarah Ehrenberg: Data curation, Writing - original draft. Catherine Walsh Vockley: Data curation, Project administration, Supervision, Writing - review & editing. Paige Heiman: Data curation. Zineb Ammous: Writing – review & editing. Olivia Wenger: Writing – review & editing. Jerry Vockley: Writing – review & editing. Lina Ghaloul-Gonzalez: Conceptualization, Methodology, Supervision, Writing - review & editing.
Acknowledgements
The authors are grateful to the patients and their families who agreed to participate in this research study.
Data availability
The data that has been used is confidential.
References
- 1.Strauss K.A., Puffenberger E.G. Genetics, medicine, and the plain people. Annu. Rev. Genomics Hum. Genet. 2009;10:513–536. doi: 10.1146/annurev-genom-082908-150040. [DOI] [PubMed] [Google Scholar]
- 2.Shchelochkov OA, Carrillo N, Venditti C. Propionic Acidemia. In: Adam MP, Ardinger HH, Pagon RA, et al., GeneReviews(®). Seattle (WA): University of Washington, Seattle.
- 3.Pena L., Franks J., Chapman K.A., et al. Natural history of propionic acidemia. Mol. Genet. Metab. 2012;105(1):5–9. doi: 10.1016/j.ymgme.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Gravel R.A., Akerman B.R., Lamhonwah A.M., Loyer M., Léon-del-Rio A., Italiano I. Mutations participating in interallelic complementation in propionic acidemia. Am. J. Hum. Genet. 1994;55(1):51–58. [PMC free article] [PubMed] [Google Scholar]
- 5.Grünert S.C., Müllerleile S., de Silva L., et al. Propionic acidemia: neonatal versus selective metabolic screening. J. Inherit. Metab. Dis. 2012;35(1):41–49. doi: 10.1007/s10545-011-9419-0. [DOI] [PubMed] [Google Scholar]
- 6.Grünert S.C., Müllerleile S., De Silva L., et al. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet Journal of Rare Diseases. 2013;8(1):6. doi: 10.1186/1750-1172-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwoerer J.S., Candadai S.C., Held P.K. Long-term outcomes in Amish patients diagnosed with propionic acidemia. Mol Genet Metab Rep. 2018;16:36–38. doi: 10.1016/j.ymgmr.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pena L., Burton B.K. Survey of health status and complications among propionic acidemia patients. Am. J. Med. Genet. A. 2012;158a(7):1641–1646. doi: 10.1002/ajmg.a.35387. [DOI] [PubMed] [Google Scholar]
- 9.Baumgartner M.R., Hörster F., Dionisi-Vici C., et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet Journal of Rare Diseases. 2014;9(1):130. doi: 10.1186/s13023-014-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forny P., Horster F., Ballhausen D., et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: first revision. J. Inherit. Metab. Dis. 2021;44(3):566–592. doi: 10.1002/jimd.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jurecki E., Ueda K., Frazier D., et al. Nutrition management guideline for propionic acidemia: an evidence- and consensus-based approach. Mol. Genet. Metab. 2019;126(4):341–354. doi: 10.1016/j.ymgme.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Romano S., Valayannopoulos V., Touati G., et al. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J. Pediatr. 2010;156(1):128–134. doi: 10.1016/j.jpeds.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Ameloot K., Vlasselaers D., Dupont M., et al. Left ventricular assist device as bridge to liver transplantation in a patient with propionic acidemia and cardiogenic shock. J. Pediatr. 2011;158(5):866–867. doi: 10.1016/j.jpeds.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 14.Yap S., Vara R., Morais A. Post-transplantation outcomes in patients with PA or MMA: a review of the literature. Adv. Ther. 2020;37(5):1866–1896. doi: 10.1007/s12325-020-01305-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nizon M., Ottolenghi C., Valayannopoulos V., et al. Long-term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J Rare Dis. 2013;8:148. doi: 10.1186/1750-1172-8-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malvagia S., Forni G., Ombrone D., la Marca G. Development of strategies to decrease false positive results in newborn screening. International Journal of Neonatal Screening. 2020;6(4):84. doi: 10.3390/ijns6040084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haijes H.A., Molema F., Langeveld M., et al. Retrospective evaluation of the Dutch pre-newborn screening cohort for propionic acidemia and isolated methylmalonic acidemia: what to aim, expect, and evaluate from newborn screening? J. Inherit. Metab. Dis. 2020;43(3):424–437. doi: 10.1002/jimd.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hannah W.B., Dempsey K.J., Schillaci L.-A.P., et al. Life-threatening presentations of propionic acidemia due to the Amish PCCB founder variant. Molecular genetics and metabolism reports. 2019;21:100537. doi: 10.1016/j.ymgmr.2019.100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenger O., Brown M., Smith B., et al. Biochemical phenotype and its relationship to treatment in 16 individuals with PCCB c.1606A > G (p.Asn536Asp) variant propionic acidemia. Mol. Genet. Metab. 2020;131(3):316–324. doi: 10.1016/j.ymgme.2020.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Vernon H.J., Manoli I. Milestones in treatments for inborn errors of metabolism: reflections on where chemistry and medicine meet. Am. J. Med. Genet. A. 2021;185(11):3350–3358. doi: 10.1002/ajmg.a.62385. [DOI] [PubMed] [Google Scholar]
- 21.Hannah W.B., Dempsey K.J., Schillaci L.-A.P., et al. Life-threatening presentations of propionic acidemia due to the Amish PCCB founder variant. Molecular Genetics and Metabolism Reports. 2019;21 doi: 10.1016/j.ymgmr.2019.100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dionisi-Vici C., Deodato F., Röschinger W., Rhead W., Wilcken B. ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J. Inherit. Metab. Dis. 2006;29(2–3):383–389. doi: 10.1007/s10545-006-0278-z. [DOI] [PubMed] [Google Scholar]
- 23.Kakavand B., Schroeder V.A., Di Sessa T.G. Coincidence of long QT syndrome and propionic acidemia. Pediatr. Cardiol. 2006;27(1):160–161. doi: 10.1007/s00246-005-1129-7. [DOI] [PubMed] [Google Scholar]
- 24.Jameson E., Walter J. Cardiac arrest secondary to long QT(C)in a child with propionic acidemia. Pediatr. Cardiol. 2008;29(5):969–970. doi: 10.1007/s00246-007-9160-5. [DOI] [PubMed] [Google Scholar]
- 25.Iacob D., Butnariu A., Leucuta D.C., Samasca G., Deleanu D., Lupan I. Evaluation of NT-proBNP in children with heart failure younger than 3 years old. Rom. J. Intern. Med. 2017;55(2):69–74. doi: 10.1515/rjim-2017-0002. [DOI] [PubMed] [Google Scholar]
- 26.Rusconi P.G., Ludwig D.A., Ratnasamy C., et al. Serial measurements of serum NT-proBNP as markers of left ventricular systolic function and remodeling in children with heart failure. Am. Heart J. 2010;160(4):776–783. doi: 10.1016/j.ahj.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratnasamy C., Kinnamon D.D., Lipshultz S.E., Rusconi P. Associations between neurohormonal and inflammatory activation and heart failure in children. Am. Heart J. 2008;155(3):527–533. doi: 10.1016/j.ahj.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Mazariegos G., Shneider B., Burton B., et al. Liver transplantation for pediatric metabolic disease. Mol. Genet. Metab. 2014;111(4):418–427. doi: 10.1016/j.ymgme.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Pillai N.R., Stroup B.M., Poliner A., et al. Liver transplantation in propionic and methylmalonic acidemia: a single center study with literature review. Mol. Genet. Metab. 2019;128(4):431–443. doi: 10.1016/j.ymgme.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hejazi Y., Hijazi Z.M., Al-saloos H., Omran T.B. The re-occurrence of dilated cardiomyopathy in propionic acidemia after liver transplantation requiring heart transplant, first case from Middle East. Cardiol. Young. 2022;1-4 doi: 10.1017/S104795112200035X. [DOI] [PubMed] [Google Scholar]
- 31.Berry G.T., Blume E.D., Wessel A., et al. The re-occurrence of cardiomyopathy in propionic acidemia after liver transplantation. JIMD Rep. 2020;54(1):3–8. doi: 10.1002/jmd2.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Streeten E.A., See V.Y., Jeng L.B.J., et al. KCNQ1 and long QT syndrome in 1/45 Amish: the road from identification to implementation of culturally appropriate precision medicine. Circ Genom Precis Med. 2020;13(6) doi: 10.1161/CIRCGEN.120.003133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that has been used is confidential.