

Abstract
Ewing's sarcoma tumors (ES) are a rare entity exceptionally localized on the liver. We report a case of an ES of the liver in a 26‐year‐old man who presented with abdominal pain. The diagnosis was confirmed with a histopathological examination of the left hepatectomy specimen and adjuvant chemotherapy was received.
Keywords: Ewing's sarcoma, literature review, liver, MRI, primitive neuroectodermal tumor
Because of their rarity, the diagnosis of Ewing sarcoma is difficult and need a cytogenetic study to be confirmed. Therapeutic strategies and prognosis are not established yet. When the diagnosis is established, patients should be sent to a reference center.
1. INTRODUCTION
Ewing's sarcoma family of tumors (ESFT) is a rare entity of mesenchymal tumors deriving from neural crest tissue with a variable degree of neuroectodermal differentiation and sharing common morphological and cytogenetic aberrations. 1 It includes extraosseous Ewing sarcoma (ES), primitive neuroectodermal tumor (PNET), Askin tumor, and atypical ES. PNETs were recognized for the first time by Arthur Purdy Stout in 1918 2 and constitutes approximately 1% of all sarcomas. ES was first described by James Ewing in 1921. 3 These tumors commonly occur in the young population as the majority of patients are younger than 30 years of age. In adults, ESFT arises in more than 50% of cases in soft tissues (trunk, retroperitoneum, intra‐abdominal tissues, and viscera). The primary involvement of the liver is extremely rare, and only 11 cases have been reported. 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 These tumors are aggressive, with a high tendency to relapse and metastasize especially in the lungs, bone marrow, brain, and lymph nodes. 15 The present work aims to report a new case on primitive hepatic ES and review all cases of primitive hepatic ES/PNET reported in the literature and describe clinical, radiological, histological, cytogenetical, therapeutic, and prognosis features of this singular tumor site in the different cases.
2. CASE REPORT
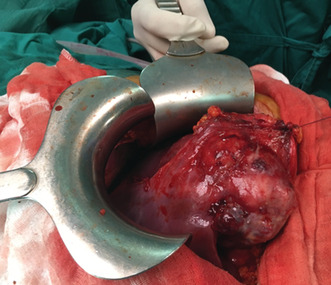
A 26‐year‐old man presented with paroxysmal right upper quadrant pain and progressive distension of his upper abdomen evolving for 1 month prior to admission. On his medical history, he was successfully treated for pulmonary tuberculosis. The physical examination revealed a patient in a good condition with an epigastric mass which was tough, well limited, and mobile with the breathing. There were no significant findings in laboratory investigations particularly liver enzymes were within normal limits. The serum tumor markers, including alpha‐fetoprotein (AFP), carcinoembryonic antigen (CEA), human chorionic gonadotropin (HCG), and Ca19‐9, were negative. Hepatitis B surface antigen and antibody and serum antibodies for hepatitis C virus were negative. The imaging examination, including ultrasonography (US) and magnetic resonance imagery (MRI) of the abdomen, showed a solid mass of the left liver, measuring 8 cm in its largest diameter (Figure 1). These radiological findings were suggestive of mesenchymal neoplasm, the patient underwent exploratory laparotomy. There was a large mass of the left liver with no evidence of hepatic metastasis or peritoneal carcinomatosis (Figure 2). The patient underwent a left hepatectomy. The postoperative course was uneventful. The histopathological examination concluded a small round cell tumor (Figure 3). The immunohistochemical study revealed positive expression for Protein S100 and CD‐99 (Figure 4) and negative immunostaining for pan cytokeratin, EMA, HMB45, CD34, Desmine, B catenin, and DOG1. This immunohistochemical profile was consistent with an Ewing sarcoma of the liver. Cytogenetic analysis was not performed. Adjuvant systemic chemotherapy was then initiated. The patient remains disease‐free at 6 months of follow‐up.
FIGURE 1.
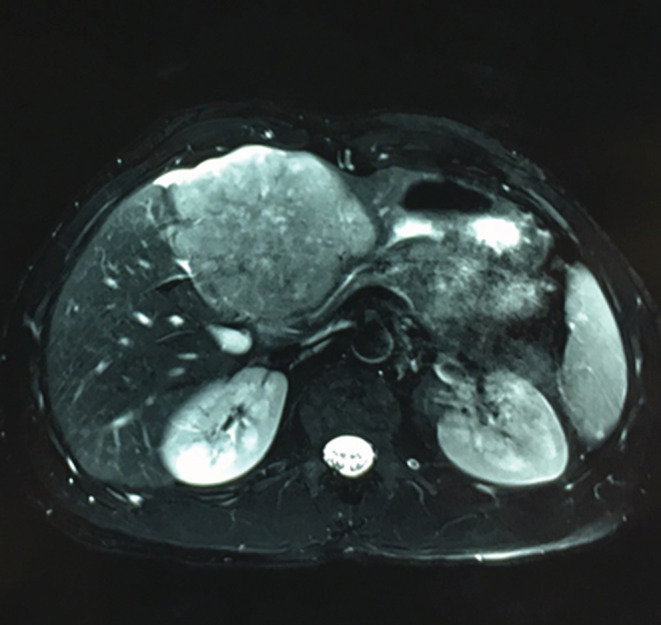
Preoperative MRI showing a solid mass in the left liver
FIGURE 2.
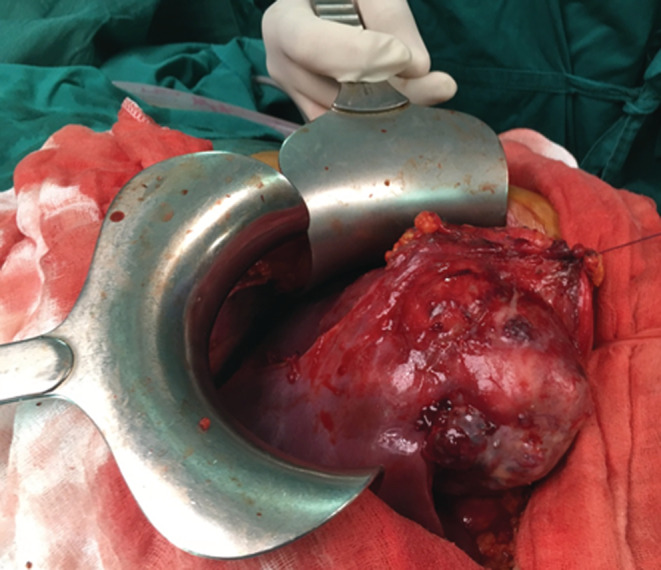
Intraoperative view of the left liver mass
FIGURE 3.
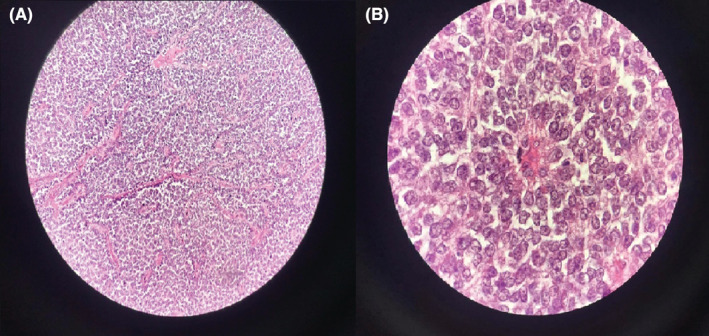
(A) H/E 400x‐Round cell tumor. (B) H/E 1000x‐pseudo rosette aspect
FIGURE 4.
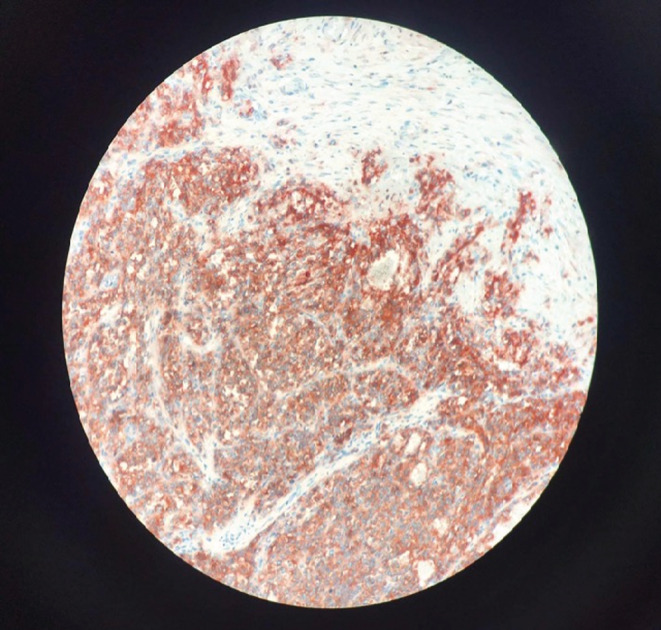
Tumor cells positively stained for CD99 at immunohistochemistry
3. DISCUSSION
An exhaustive literature review was performed after searching the PubMed database. We could find only 11 cases of ES/PNET primarily involving the liver (Table 1). 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 Visceral involvement of Ewing's sarcoma family of tumors has been described in the kidney, gastrointestinal tract (small bowel and rectum), pancreas, gall bladder, vagina, vulva, uterine cervix, breast, ovaries, urinary bladder, ureter, prostate, seminal vesicles, testis, penis, maxillary sinus, trachea, lung, and parotid gland. 16 The liver is a common site of ES/PNET metastasis but it is exceptionally a primary location. ESFT concerns usually young adults, but it also can occur in older patients as the cases reported by Ousadden 4 and Gupta. 13 Following the previous reports and our case PNET is usually diagnosed before the age of 35 years with a slight male preponderance.
TABLE 1.
Primitive Ewing's sarcoma tumors of the liver cases reported in the literature
| Authors | Year | Age | Sex | Type of tumor | Size | Segments | Surgical procedures |
|---|---|---|---|---|---|---|---|
| Ousadden et al. 4 | 2005 | 58 | female | PNET | 10 cm | 7,8 | Right hepatectomy |
| Mani et al. 5 | 2010 | 20 | female | PNET | ‐ | ‐ | No surgery |
| Cambruzzi et al. 6 | 2011 | 18 | male | PNET | 12 cm | 4,5,8 | Central hepatectomy + Cholecystectomy + portal lymphadenectomy |
| Huang et al. 7 | 2011 | 18 | male | ES | 21 cm | 4,5,6,7,8 |
Right hepatectomy with en bloc resection of the right kidney, gallbladder, and partial colectomy (hepatic colon flexure) with primary anastomosis |
| Marquez et al. 8 | 2011 | 19 | male | PNET | 15 cm | 7,8 | ‐ |
| MacGrann et al. 9 | 2013 | 29 | male | ES | 14 cm | 5,6 | Total resection of the tumor + cholecystectomy + hepato‐duodenal lymph node dissection + omentectomy. |
| Ozaki et al. 10 | 2015 | 27 | female | ES | 8 cm | 8 | Right hepatectomy |
| Ates et al. 11 | 2016 | 24 | female | ES | 12 cm | 4,5,6,7,8 | Segmentectomy 4–6 + Cholecystectomy |
| Shah et al. 12 | 2018 | 33 | male | ES | 10 cm | ‐ | ‐ |
| Gupta et al. 13 | 2020 | 56 | male | PNET | 0,2–0,8 cm | Multinodular | No surgery (discovered on autopsy) |
| Lu et al. 14 | 2021 | 27 | female | ES | 10 cm | Caudate lobe | ‐ |
Clinically, these tumors remain silent for a long period of time. Indeed, the mean size at the time of diagnosis is about 12 cm. There were no pathognomonic clinical signs, but most of the patients presented with abdominal pain, mass effect, and compression symptoms.
There was a lack of detailed radiological tumor descriptions in most cases. Radiological imagery information provided by US, CT scan, and MRI were not conclusive for the diagnosis. But they contribute to obtaining a lot of useful information about the tumor as segment involvements, size, the existence of suspicious tumor characteristics for malignancy (irregular thickened septa, nodal walls …), shape, relation with adjacent structures (vessels, bile duct) and the existence or not of metastasis which can guide therapeutic strategy. For soft tissue sarcoma, MRI is known to have the best performance. Preoperative diagnosis is difficult to assess, then biopsy was performed in some of the reported cases. The standard approach to diagnosis consists of multiple core needle biopsies. Fine needle aspiration can also be done, but it is used in some specialized institutions. Nonetheless, the pathway of the biopsy should be carefully planned to minimize contamination and complication. The gross appearance of the tumor varies. In general, it is multilobulated, soft, and friable. It rarely exceeds 10 cm in its largest dimension. Its cut surface has a gray‐yellow or gray‐tan appearance, often with large areas of necrosis, cyst formation, or hemorrhage. Despite the extensive necrosis, calcification is rare. 17 Cytogenetic studies in ES/PNET reveal a specific translocation t (11, 22) (q24; q12) resulting in EWS/FLI‐1 fusion gene in more than 90% of cases. 18 It is detected by the reverse transcriptase polymerase chain reaction (Rt‐Pcr) and is considered to be the most specific feature for diagnosis of ES/PNET. 18 Once the diagnosis is established, evaluation of tumor extension is necessary. Regional lymph node metastases are rare for these tumors. A thoraco‐abdominopelvic CT scan is mandatory. Bone scan, whole‐body MRI, and PET scan are optional. 19 The management of ESFT should be carried out in a reference center for sarcomas. A multidisciplinary approach is required, involving pathologists, radiologists, surgeons, radiation therapists, and medical oncologists. Because of the rarity of these tumors, there is no standard treatment. Surgery alone is considered insufficient, and multimodal treatment with chemotherapy and radiotherapy is frequently performed. There is no consensus on the current role of adjuvant chemotherapy because of study results are conflicting. In this review, all patients with liver ESFT received adjuvant multi‐agent chemotherapy with a relapse‐free survival of 13 months. For metastatic disease, if metastasis is metachronous, resectable and located in the lung, surgery is recommended, otherwise, chemotherapy is recommended. 19 Key prognostic factors that adversely influence the outcome are the presence of metastatic disease at the time of the initial diagnosis, large tumor size, extensive necrosis, and poor response to initial chemotherapy. A large study evaluating more than 2000 ESFTs has shown that localized extraskeletal ESFT have a superior prognosis compared with localized skeletal ESFT. 20 In a study of 57 patients with extraskeletal ESFT, relapse‐free survival was about 35%. 21
4. CONCLUSION
ESFT are rare small round cell tumors that do not have any specific clinical or radiological presentation. That is why they have to be considered especially in young patients with liver mass. Diagnosis requires histopathological, IHC and cytogenetic examinations. There are very few published cases, and data are still insufficient to suggest recommendations for treatment strategies. Therefore, patients should be referred to a specialized center and participate in international trials.
AUTHOR CONTRIBUTIONS
Dr Mohamed Hefdi contributed to manuscript writing. Dr Hakim Zenaidi contributed to study concepts. Dr Imen Ben Ismail helped in data interpretation and manuscript evaluation. Dr Zoghlami Ayoub critically revised the manuscript.
FUNDING INFORMATION
This study was not supported by any institution and company.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVAL
Ethics statement was not required, and patient identifying knowledge was not presented in the report.
CONSENT
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
None.
Hedfi M, Ben Ismail I, Zenaidi H, Bouslama S, Zoghlami A. Primary Ewing sarcoma of the liver: Diagnosis, management, and prognosis: A case report and literature review. Clin Case Rep. 2022;10:e06508. doi: 10.1002/ccr3.6508
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Fletcher CDM, Unni KK, Mertens F. Pathology and genetics of tumours of soft tissue and bone. World Health Organization classification of tumours; 2002:298. [Google Scholar]
- 2. Pinto A, Dickman P, Parham D. Pathobiologic markers of the Ewing sarcoma family of tumors: state of the art and prediction of behaviour. Sarcoma. 2011;2011:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pomara G, Cappello F, Cuttano MG, et al. Primitive neuroectodermal tumor (PNET) of the kidney: a case report. BMC Cancer. 2004;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ousadden A, Mazaz K, Amraoui A, Kettani F, Chefchaouni MC, AitTaleb K. Primary hepatic localization of the PPNET (primitive peripheral neuroectodermal tumors). Case report. Ann Chir. 2005;130:254‐256. [DOI] [PubMed] [Google Scholar]
- 5. Mani S, Dutta D, De BK. Primitive neuroectodermal tumor of the liver: a case report. Jpn J Clin Oncol. 2010;40(3):258‐262. [DOI] [PubMed] [Google Scholar]
- 6. Cambruzzi E, Guerra E, Hilgert HC, et al. Primitive neuroectodermal tumor of the liver: a case report. Case Rep Med. 2011;2011:748194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang SF, Chiang JH, Jan HC, Chou SJ, Chen TK, Chen TH. Intra‐abdomen Ewing's sarcoma. ANZ J Surg. 2011;81(5):377‐378. [DOI] [PubMed] [Google Scholar]
- 8. Marques M, Cardosoa H, Barrocac H, Lopes J, Macedo G. Peripheral primitive neuroectodermal tumour of the liver: a case report and review of the literature. Gastroenterol Hepatol. 2011;34:611‐613. [DOI] [PubMed] [Google Scholar]
- 9. McGrann PF, Pooleman IJ, Wilson CH, Haugk B, Scott J, Charnley RM. Primary hepatic Ewing's sarcoma with cytogenetic confirmation. J Gastrointest Surg. 2014;18(3):635‐637. [DOI] [PubMed] [Google Scholar]
- 10. Ozaki Y, Miura Y, Koganemaru S, et al. Ewing sarcoma of the liver with multilocular cystic mass formation: a case report. BMC Cancer. 2015;15:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ates O, Basel F, Bozdogan N, Aksel B, Oksuzoglu OB. Primary hepatic Ewing sarcoma: a very infrequent case report. IITJM. 2015;10(1):67‐70. [Google Scholar]
- 12. Shah S, Hassan A, Sotiriadis J. Ewing Sarcoma/Primitive Neuroectodermal Tumor of the Liver: A Late Presentation With an Extremely Rare “Large Cell” Variant. Program No. P1597. ACG 2018 Annual Scientific Meeting Abstracts. American College of Gastroenterology.
- 13. Gupta S, Rawat P, Malik S, Marwah N, Singh S. Primitive neuroectodermal tumor in a Cirrhotic liver: an autopsy finding. JMSCR. 2020;08:477‐480. [Google Scholar]
- 14. Lu T, Yang W, Liu X, Yang X, Yang C, Di W. Imaging findings of hepatic Ewing's sarcoma on computed tomography and gadobenate dimeglumine‐enhanced magnetic resonance imaging: a case report and literature review. J Clin Transl Hepatol. 2021;10(3):564‐569. doi: 10.14218/JCTH.2021.00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Helsel JC, Mrak RE, Hanna E, Parham DH, Bardales RH. Peripheral primitive neuroectodermal tumor of the parotid gland region: report of a case with fine‐needle aspiration findings. Diagn Cytopathol. 2000;22(3):161‐166. [DOI] [PubMed] [Google Scholar]
- 16. Koufopoulos N, Kokkali S, Manatakis D, Balalis D, Nasi D, Ardavanis A. Primary peripheral neuroectodermal tumor (PNET) of the adrenal gland: a rare entity. J BUON. 2019;24(2):770‐778. [PubMed] [Google Scholar]
- 17. Vakar‐Lopez F, Ayala AG, Raymond AK, Czerniak B. Epithelial phenotype in Ewing's sarcoma/primitive neuroectodermal tumor. Int J Surg Pathol. 2000;8(1):59‐65. [DOI] [PubMed] [Google Scholar]
- 18. Aurias A, Rimbaut C, Buffe D, et al. Chromosomal translocations in Ewing's sarcoma. N Engl J Med. 1983;309:496‐497. [PubMed] [Google Scholar]
- 19. Casal PG, Abecassis N, Bauer S, et al. Soft tissue and visceral sarcomas: SMO–EURACAN clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2018;29:iv268‐iv269. [DOI] [PubMed] [Google Scholar]
- 20. Applebaum MA, Worch J, Matthay KK, et al. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer. 2011;117:3027‐3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. El Weshi A, Allam A, Ajarim D, et al Extraskeletal Ewing's Sarcoma Family of Tumours in Adults: Analysis of 57 Patients from a Single Institution. Clinical oncology; 2010:374‐381 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.