

Summary
Transmembrane protein 16A (TMEM16A) localizes at plasma membrane and controls chloride influx in various type of cells. We here showed an intracellular localization pattern of TMEM16A molecules. In myoblasts, TMEM16A was primarily localized to the cytosolic compartment and partially co-localized with intracellular organelles. The global deletion of TMEM16A led to severe skeletal muscle developmental defect. In vitro observation showed that the proliferation of Tmem16a−/− myoblasts was significantly promoted along with activated ERK1/2 and Cyclin D expression; the myogenic differentiation was impaired accompanied by the enhanced caspase 12/3 activation, implying enhanced endoplasmic reticulum (ER) stress. Interestingly, the bradykinin-induced Ca2+ release from ER calcium store was significantly enhanced after TMEM16A deletion. This suggested a suppressing role of intracellular TMEM16A in ER calcium release whereby regulating the flux of chloride ion across the ER membrane. Our findings reveal a unique location pattern of TMEM16A in undifferentiated myoblasts and its role in myogenesis.
Subject areas: Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
TMEM16A locates to the intracellular organelles of undifferentiated myoblast
-
•
Intracellular TMEM16A modulates IP3R calcium channels-mediated Ca2+ release
-
•
Intracellular TMEM16A is necessary for skeletal muscle development
Molecular biology; Cell biology
Introduction
TMEM16A (also called anoctamin 1, ANO1) is a member of the classical Ca2+-activated chloride (Cl−) channel (CaCC) family (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008). It is broadly expressed in many types of cells, localized to the plasma membrane, and contributes to a myriad of biological functions (Duvvuri et al., 2012; Gomez-Pinilla et al., 2009; Jin et al., 2013; Kunzelmann et al., 2009; Tian et al., 2011). TMEM16A molecules adopt a homodimer architecture in which the Cl− _selective pores are surrounded. Each subunit of TMEM16A is composed of ten transmembrane segments and binds two Ca2+ ions (Dang et al., 2017; Paulino et al., 2017). Activation of TMEM16A can be triggered by G-protein-coupled receptor signaling pathways that activate phospholipase C for inositol 1,4,5-trisphosphate (IP3) production and/or Ca2+ release from intracellular stores mediated by the binding of IP3 to IP3 receptors (IP3Rs) (Dayal et al., 2019; Schredelseker et al., 2010; Wang et al., 2018). The resultant TMEM16A activation facilitates the passive flow of Cl− across the plasma membrane, whereby regulates diverse functions such as smooth muscle contraction, nociception, neuronal excitability, insulin secretion, cell proliferation, and migration (Duvvuri et al., 2012; Gomez-Pinilla et al., 2009; Jin et al., 2013; Kunzelmann et al., 2009; Tian et al., 2011).
In zebrafish, TMEM16A expressed in mature skeletal musculature locates at plasma membrane near the sarcoplasmic reticulum (SR) and plays a potential role in the excitation-contraction coupling process (Dayal et al., 2019). There are reports showing that TMEM16A also localizes at the plasma membrane of the tether part of endoplasmic reticulum (ER)/SR membrane and facilitates TMEM16A activation by the calcium released from ER/SR (Cabrita et al., 2017; Dayal et al., 2019; Jin et al., 2013; Wang et al., 2020). Interestingly, we here found that TMEM16A could alternatively localize at intracellular organelles of skeletal muscle precursor cells (myoblasts).
During embryonic and fetal developmental processes, myoblasts differentiate into mononucleotide myocytes, and then fuse to form multinucleated myotubes and myofibers (Chal and Pourquié, 2017; Kablar and Rudnicki, 2000; Magli and Perlingeiro, 2017; Tran et al., 2013). After birth, muscle myofibers undergo massive growth, fiber type specialization, hypertrophy, and maturation of musculature. In mouse, skeletal muscle development originates at approximately embryonic day 10.5 (E10.5) and is completed at adult (Chal and Pourquié, 2017). Within this process, several transcription factors including muscle regulatory factors (MRFs) with b-HLH domains (e.g., myogenin), and ERK1/2 signaling modules, cyclin D1, caspases, and microRNAs are involved in the regulation of myogenesis (Adi et al., 2002; Holstein et al., 2020; Kablar and Rudnicki, 2000; Li et al., 2012; Skapek et al., 1995; Zhang et al., 1999). As a second messenger, calcium released from ER takes an essential role in myogenesis (Nakanishi et al., 2005, 2007, 2015). ER is the main intracellular calcium store and heavily regulates calcium metabolism through IP3R and RyR systems. IP3Rs can directly mediate Ca2+ release from the ER lumen upon IP3 binding (Bare et al., 2005; Wu et al., 2006), and the released Ca2+ then regulates several biological functions including proliferation and differentiation. However, when the ER calcium homeostasis is impaired, ER stress and apoptosis will be induced (Bare et al., 2005; Santulli et al., 2017; Wu et al., 2006). In addition, the released Ca2+ in cytosol also activates Ca2+-related signaling pathways such as extracellular signal-regulated kinase 1/2 (ERK1/2), thereby interfering the process of cellular proliferation. We here found that intracellular TMEM16A could essentially regulate the IP3R-mediated calcium release from ER and thereby functions in myogenesis. The deletion of TMEM16A led to overproduction of cytosolic calcium induced by augmented release of Ca2+ from ER through IP3R calcium channels. As a consequence, the myoblast displayed enhanced proliferation and inhibited differentiation capacities. Consistently, the TMEM16A knockout mice (KO) exhibited impaired skeletal muscle development. Our findings reveal a novel expression and localization pattern of TMEM16A and its biological relevance in skeletal muscle.
Results
Intracellular expression of TMEM16A in developing skeletal muscles
To examine the expression pattern of TMEM16A in skeletal muscle, we firstly measured TMEM16A protein and mRNA of adult skeletal muscles. The results showed that both TMEM16A mRNA and protein were abundantly expressed in the muscles including tongue muscle, tibialis anterior muscle, quadriceps muscle, and soleus muscle (Figures 1A–1C). It was noted that both mRNA and protein levels of TMEM16A in the tongue muscles were much higher than those in other muscles. We next measured TMEM16A in embryonic muscles. The limb muscles at embryonic day (E) 11.5–17.5 expressed TMEM16A protein abundantly, but the expression level decreased apparently by postnatal day 1 (P1) (Figure 1D). To test if this expression pattern paralleled to muscular differentiation process, we used an in vitro model of muscle differentiation by culturing C2C12 myoblast with differentiation medium (DM) and then measured the dynamic expression of TMEM16A. When the myoblasts were cultured with DM, TMEM16A protein level increased immediately and peaked around the third day after the switch. The expression level was reduced afterward (Figure 1E). This observation suggested that the dynamic expression of TMEM16A was associated with skeletal muscle development. We next determined the spatial expression of TMEM16A in skeletal muscle. To our surprise, the immunofluorescence staining of the transverse cross sections showed that, in the immature soleus muscle of 3-week-old mice, most TMEM16A protein was expressed randomly in the cytosol of myofibers rather than the sarcolemma (Figure 1F). In the 8-week-old mice, however, TMEM16A localized exclusively in the sarcolemma of mature soleus myofibers in a similar manner of laminin. To further resolve the TMEM16A expression within myofibers, we isolated individual myofibers from the extensor digitorum longus (EDL) muscle of 8-week-old mice and subjected to immunofluorescence staining. TMEM16A located at the sarcolemma of mature sarcomeres in a very good order, and it did not overlap with either DHPRα1 (dihydropyridine receptor, T-tubule marker protein) or RyRs (ryanodine receptors, SR marker protein) (Figures 1G and 1H). Thus, we concluded that TMEM16A did not reside in the T-tubule or SR of mature myofibers. Results from co-staining of the myofibers with anti-TMEM16A and actinin antibodies consistently indicated that TMEM16A did not locate to the Z-discs corresponding regions of the sarcolemma (Figure S1). In immature myofibers isolated from 3-week-old mice, part of TMEM16A molecules showed similar location as mature myofibers. However, the other TMEM16A molecules were diffusely distributed in the cytosol of cells. Combined with the results from the cross-section staining, our observations indicated an intracellular localization of TMEM16A in the developing skeletal muscle cells. We here did not study the biological relevance of TMEM16A localization in mature myofibrils.
Figure 1.

Tmem16a expression in skeletal muscle cells
(A) q-RT PCR analysis of the mRNA levels of Tmem16a expressed in skeletal muscle tissues of adult mice. GAPDH was used as an internal control.
(B and C) Western blot analysis of TMEM16A expression in skeletal muscle tissues of adult mice.
(D) Western blot analysis of TMEM16A/Myogenin expression in limb muscles during development.
(E) Western blot analysis of TMEM16A/myosin heavy chain (MyHC) expression in C2C12 cells during myogenic differentiation.
(F) Immunofluorescence images of the cross sections of immature (3-week-old) and mature (8-week-old) soleus muscles co-stained with TMEM16A and Laminin antibodies. Scale bars, 10 μm.
(G) Immunofluorescence images of mature EDL single myofibers co-stained with TMEM16A/RYRs and TMEM16A/DHPR antibodies. Scale bars, 5 μm.
(H) Line scan analysis of the images in G panel.
Partial of TMEM16A molecules may locate at ER of undifferentiated myoblast
The intracellular localization of TMEM16A in immature myofibers allowed us to predict that TMEM16A might exist in subcellular organelles. As there was evidence showing that TMEM16A had capacity to bind IP3R (Cabrita et al., 2017), we guess that TMEM16A may locate at ER or SR of myoblasts. To validate this point, we firstly transfected the vector-expressing GFP fusing with full length of TMEM16A (GFP-TMEM16A) to primary cultured myoblasts. GFP signals were detected abundant in the cytosol and modest in the plasma membrane (Figure 2A). This expressing pattern was unlikely attributable to the abnormality of the recombinant fusing protein because typical plasma membrane localization of this protein could be measured in Chinese hamster ovary cells (CHOs) transfected with the same plasmid (Figure S2). When the proliferative myoblasts underwent myogenic differentiation for 4 days (DM4), a large partial of the fluorescence signals moved to the plasma membrane of the newly formed myotubes. Interestingly, the CaCC activity in myoblast was almost undetectable in primary cultured myoblasts but apparent in myotubes (Figure S3), verified the distinguished localization patterns of TMEM16A in the skeletal muscle cells at different developmental stages. To explore the intracellular localization of TMEM16A in myoblast, we co-transfected the primary cultured myoblasts with expressive vectors of recombinant GFP-TMEM16A and mCherry-Sec61β, a subunit of the Sec61 translocation channel on the ER membrane (Gemmer and Förster, 2020). Interestingly, the GFP-TMEM16A molecules were overlapped with mCherry-Sec61β signals (Figure 2B), whereas almost no co-localized signals were detected in myotubes after 4 days of myogenic differentiation induction. We next co-stained the primary cultured myoblasts of mice with anti-TMEM16A and IP3Rs (IP3RI/II/III) antibodies. The results showed that only a part of TMEM16A signals overlapped with those of IP3Rs (Figures 2C and 2D). This result indicated that a part of intracellular TMEM16A might locate at ER and others might locate at other intracellular organelles.
Figure 2.

Intracellular localization of TMEM16A in myoblast
(A) Localization of transduced GFP-TMEM16A proteins in mouse primary myoblast and myotube (DM4) was determined by confocal microscopy. The myoblasts transfected with GFP were used as a control. Scale bars, 20 μm.
(B) Confocal microscopy images show the co-localization of transduced GFP-TMEM16A and mCherry-Sec61β in the cytosol of mouse primary myoblast, note the surface membrane location of TMEM16A in myotube (DM4). Scale bars, 20 μm.
(C) Co-staining of primary myoblast with TMEM16A and IP3Rs antibodies. Scale bars, 20 μm. (D) Line scan analysis of part region of myoblast in C panel indicated by white arrow. Black arrows indicate the co-localization of TMEM16A and IP3Rs signals.
TMEM16A inhibits calcium release from intracellular calcium stores through IP3R Ca2+ channels
As ER is the primary intracellular calcium store, we speculate a role of TMEM16A in calcium release from ER. To test this hypothesis, we isolated myoblast cells from wild-type control (CTR) and TMEM16A knockout mice (KO) and cultured them in vitro, then stimulated the primary cells with bradykinin, an agonist of IP3Rs Ca2+ channels (Tarroni et al., 1997). To our surprise, upon stimulation with 100 nM bradykinin, the mutant myoblast cells displayed stronger cytosolic calcium signals in contrast to the control (Figure 3A). Quantitation analysis showed that both the signal amplitude and the area under the signal curve of the myoblast cells were significantly increased in the mutant myoblast (Figures 3B and 3C). No apparent intracellular calcium concentration elevations were observed in both the mutant and control myoblasts against stimulation with 20 mM caffeine (a RyR agonist) (Meissner, 2017) (Figures 3D–3F), indicating the undifferentiated state of the myoblasts. It was notable that both the bradykinin and caffeine-induced Ca2+ signals in the mutant myotubes were much less than the controls (Figures 3G–3L). Interestingly, the protein expression level of IP3Rs was slightly declined in the KO myoblast compared with WT control (Figure S4), suggesting that the elevated calcium release capacity of IP3Rs may attribute to the functional alteration of the channel. In addition, the expression of RyRs was also decreased in KO myotubes, consistent with the downregulated calcium response against caffeine. This result suggested that TMEM16A was not involved in the modulation of calcium release through RyR calcium channels. Our above observation clearly indicated that TMEM16A served to inhibit IP3R-mediated release of Ca2+ from intracellular stores of myoblasts. However, no physical interaction was detected between endogenous TMEM16A and IP3Rs molecules by immunoprecipitation (Figure S5).
Figure 3.

Deletion of TMEM16A enhances IP3Rs-mediated calcium release in myoblast
(A) Fluo-4 direct staining shows the bradykinin-induced global Ca2+ transients in the KO myoblast versus CTR.
(B and C) The amplitude (B) and the area under the curve (C) of the bradykinin-induced global Ca2+ transients in panel A were calculated. n = 3. Data are mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
(D) Fluo-4 direct staining shows the caffeine induced global Ca2+ transients in the KO myoblast versus CTR.
(E and F) The amplitude (E) and the area under the curve (F) of the caffeine-induced global Ca2+ transients in (D) were determined. n = 3. Data are mean ± SD.
(G) Fluo-4 direct staining shows the bradykinin-induced global Ca2+ transients in the KO myotube (DM4) versus CTR.
(H and I) The amplitude (H) and the area under the curve (I) of the bradykinin-induced global Ca2+ transients in (G) were determined. n = 3. Data are mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
(J) Fluo-4 direct staining shows the caffeine-induced global Ca2+ transients in the KO myotubes (DM4) versus CTR.
(K and L) The amplitude (K) and the area under the curve (L) of the caffeine-induced global Ca2+ transients in (J) were determined. n = 3. Data are mean ± SD. ∗p < 0.05.
TMEM16A is required for skeletal muscle development
To determine the role of TMEM16A in skeletal muscle development in vivo, we analyzed the muscular phenotypes of the mice with global deletion of Tmem16a gene. The deletion efficiency in skeletal muscle was confirmed by immunoblotting, real-time quantitative RT-PCR, and immunofluorescence staining (Figures 4A–4C). Accordingly, the CaCC activity was significantly declined in the myotubes-deleted TMEM16A (Figure S6). Consistent with previous observations of another group (Rock et al., 2008), we found that the KO neonates (P1) were seemingly healthy and had comparable body weights to the control littermates (Tmem16a+/+). However, the KO mice were visibly distinguishable from the control littermates with lower body weights as the age increased (Figures 4D and 4E). Moreover, the skeletal muscle mass was also smaller in the KO mice (Figures 4F and 4G). Further examination showed that all the mutant muscles were smaller than the control muscles. Figure 4G represents the gross morphologies of mutant and control tongue muscles and limb muscles of 5-day-old mice. These data together indicated that the reduced muscle mass was a primary cause for the lower body mass in the KO mice. Moreover, histological analysis showed that the limb muscles of neonatal KO mice contained sparse and disorganized myofibers embedded within an amorphous mass of extracellular matrix. Similarly, the tongue muscle of the KO mice was smaller than the control, and the myofibers were shorter and disorganized (Figure 4H). We also examined the skeleton of P10 mice and observed apparent abnormality in the mutant mice including the shorter bones and kyphosis of cervical vertebrae (Figure 4I). Since TMEM16A is not expressed in the chondrogenic mesenchyme at any time during mouse development (Rock et al., 2008), this abnormality was likely secondary to the defect of skeletal muscle. Given the body weight of KO mice was comparable to the WT mice at perinatal stage, we also performed the histological analysis of the muscles. The results showed that the gross morphology of the KO limb muscles was almost normal. However, the muscles were smaller, and the sparse and disorganized myofibers embedded within an amorphous mass of enlarged extracellular matrix were also observed in the KO muscles, similar to the elder mice (Figure S7). Above observations collectively showed that the ablation of TMEM16A caused an apparent defect of skeletal muscle development in mice both during embryonic stage and postnatal growth.
Figure 4.

TMEM16A knockout mice display developmental defects in skeletal muscles
(A) Western blot analysis shows loss of TMEM16A protein expression in the KO skeletal muscle of neonatal mice.
(B) q-RT PCR analysis shows loss of Tmem16a mRNA expression in the KO hind limb muscle of neonatal mice. GAPDH was used as an internal control. n = 3. Data are mean ± SD. ∗∗∗p < 0.001.
(C) Immunofluorescence images show the deletion of TMEM16A protein in limb muscle of neonatal KO mice. Scale bars, 20 μm.
(D) Representative images of P1-, P5-, P10-, and P20-old KO mice and CTR littermates.
(E) Body weight curve of the CTR and KO pups (CTR: n = 15; KO: n = 15). ∗p < 0.05.
(F) 20-day-old CTR and TMEM16A-KO mice were skinned to reveal the lack of muscle in the hind limbs.
(G) Gross morphological analysis of the hind limb and tongue muscles in 5-day-old CTR and KO mice determined by hematoxylin and eosin (H/E) staining of the transverse-sections. Scale bars, 500 μm for hind limb, 100 μm for tongue.
(H) H&E staining shows the nonuniform and unregularly patterned myofibers in neonatal KO mice versus CTR littermates. Scale bars, 20 μm. (I) The skeleton of 10-day-old KO and CTR mice.
TMEM16A is necessary for the coordination of proliferation and myogenic differentiation of myoblast
The developmental defect of the Tmem16a−/− skeletal muscle implies impaired proliferation and/or differentiation of muscular precursors. As Ca2+ release from intracellular stores has diverse roles in regulating tremendous physiological processes including cell proliferation and differentiation (Nakanishi et al., 2005, 2007, 2015; Wu et al., 2006), we firstly assessed the effect of TMEM16A on primary cultured myoblasts expansion with 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay. The result showed that the proliferation capability was apparently enhanced in the mutant myoblasts isolated from KO mice compared with the control cells (Figure 5A). We then examined the expression of phosphorylated ERK1/2 (p-ERK1/2) and CyclinD1, the key signal modules of cell proliferation. The results showed that the levels of p-ERK1/2 and CyclinD1 were significantly upregulated in Tmem16a−/− myoblasts (Figures 5B–5E). These observations indicated that the augmented elevation of cytosolic Ca2+ by TMEM16A deletion led to enhancement of myoblast proliferation. We next investigated the effects of TMEM16A deletion on the myogenic differentiation. The differentiation process was inhibited in the KO myoblast as evidenced by the smaller size of the formed myotubes and the decreased fusion index (Figures 5F and 5G), and reduced myogenin and myosin heavy chain protein (MyHC) (Figures 5H–5K).
Figure 5.

Enhanced proliferation but inhibited myogenic differentiation capacities in TMEM16A deleted myoblasts
(A) EdU staining shows the proliferation capacity of primary myoblasts isolated from the KO and CTR mice. n = 3. Data are mean ± SD. ∗p < 0.05.
(B and C) The increase in expression levels of p-ERK1/2 in the KO versus CTR myoblasts determined by Western blotting. n = 3. Data are mean ± SD. ∗∗∗p < 0.001.
(D and E) The increase in expression levels of Cyclin D in the KO versus CTR myoblasts determined by Western blotting. n = 3. Data are mean ± SD. ∗∗p < 0.01.
(F) Immunostaining with MyHC antibody shows the new formed KO versus CTR myotubes (DM6) differentiated from primary myoblasts. Scale bars, 10 μm.
(G) The fusion index (the percentage of nuclei contained in myosin-positive myotubes) analysis in (F). n = 3. Data are mean ± SD. ∗∗∗p < 0.001.
(H and I) Western blotting shows the expression of MyHC in KO versus CTR myotubes differentiated from the primary myoblasts. n = 3. Data are mean ± SD. ∗p < 0.05.
(J and K) Western blotting shows the expression of Myogenin in KO versus CTR myotubes differentiated from the primary myoblasts. n = 3. Data are mean ± SD. ∗p < 0.05.
Proper apoptosis of myoblasts was essential for myogenic differentiation (Hochreiter-Hufford et al., 2013; Ikeda et al., 2009). We then examined the apoptosis-associated molecules during myogenesis progression. As shown in Figures 6A and 6B, the activation of caspase 3 (cleaved caspase 3) was significantly heightened in the myoblasts cultured from the KO mice at DM1 and DM2 compared with control. The impaired myogenic differentiation of the KO myoblasts was significantly improved by the administration of Z-VAD-FMK, a pan-caspase inhibitor (Figures 6C and 6D). Moreover, as ER stress is the main initial event attributed to myogenic differentiation-induced apoptosis that is characterized by the expression and activation of the initiator, capase12, and the downstream effector, caspase 3 (Nakagawa et al., 2000; Nakanishi et al., 2005, 2007), we then measured these proteins in the differentiating myoblasts. As expected, both the expression levels of pro-caspase 12 and the activated caspase 12 (cleaved caspase 12) were significantly elevated at the early stage of myogenesis (DM1) in KO myoblasts, which was in parallel to the elevated caspase 3 activation (Figures 6E and 6F). These observations suggested that the excessive ER stress and apoptosis caused by TMEM16A deletion underlies the impaired differentiation of TMEM16A KO skeletal muscle. Collectively, above findings indicated that Tmem16A deletion altered the pathways of cell fate commitment and ER stress, and the latter might be more prone to cell death secondary to inhibition of differentiation.
Figure 6.

TMEM16A deletion leads to enhanced apoptosis and ER stress in myoblasts
(A and B) Western blotting shows the expression of cleaved caspase 3 and caspase 3 protein levels in the KO versus CTR myoblasts during myogenic differentiation. n = 3. Data are mean ± SD. ∗∗∗p < 0.001.
(C) The impaired myogenic differentiation in the KO myoblasts was improved using pan-caspase inhibitor Z-VAD-FMK. Scale bars, 20 μm.
(D) The fusion index analysis in (C). n = 3. Data are mean ± SD. ∗p < 0.05.
(E and F) Western blotting shows the expression of cleaved caspase 12 and pro-caspase 12 protein levels in the KO versus CTR myoblasts during myogenic differentiation. n = 3. Data are mean ± SD. ∗p < 0.05.
Discussion
In this report, we demonstrated that TMEM16A could localize to the intracellular organelles of undifferentiated myoblast, revealing a novel expression pattern of TMEM16A. Biochemical analysis showed that the deletion of TMEM16A led to an enhanced elevation of cytosolic calcium after agonistic activation of IP3R, the resultant calcium promoted myoblast proliferation, and the excessive ER stress inhibited myoblast differentiation as well. We thus proposed a role of such intracellular TMEM16A was to control ER calcium homeostasis through the IP3R calcium channels-mediated Ca2+ release, and such an elaborative modulation was particularly important for coordination of myogenic processes. These findings revealed an alternative regulatory scheme for skeletal muscle development.
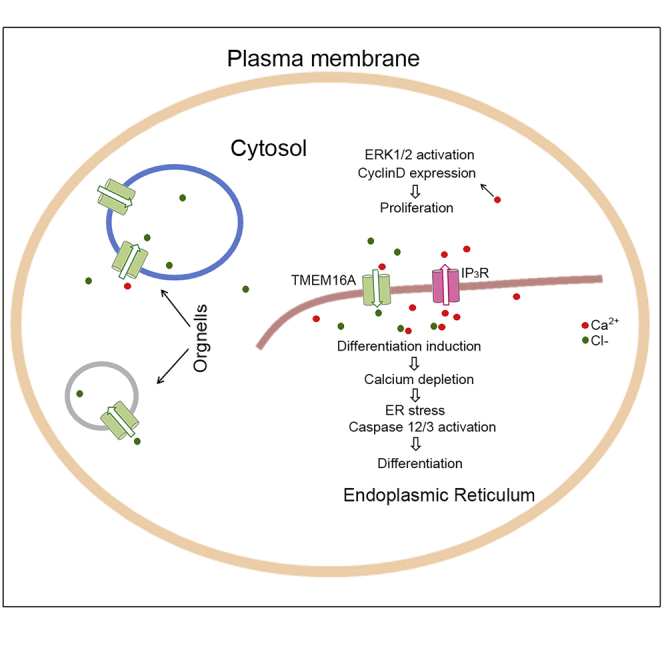
According to current knowledge and the evidence provided by this report, a working model for this scheme may be proposed (Figure 7). In this model, partial of intracellular TMEM16A is expressed in the ER of myoblast cells and functions to maintain intracellular calcium homeostasis through facilitating the influx of chloride, thereby coordinating proliferation and differentiation processes of skeletal muscle precursor cells. After these processes have been completed, the intracellular TMEM16A protein amount was reduced and then exclusively expressed at sarcolemma membrane of myofiber, to regulate the process of muscle contraction (Dayal et al., 2019). When the intracellular TMEM16A is inhibited, the influx of chloride to ER is blocked, resulting in over release of ER calcium. The elevated cytosolic calcium then promotes muscular precursors proliferation via upregulation of cell cycle-related signals/factors (e.g., p-ERK1/2 and CyclinD1). Simultaneously, the imbalance of calcium homeostasis in ER caused excessive ER stress showed by overproduction/activation of caspase 12/caspase 3, resulting in inhibition of myogenic differentiation. Among these processes, the intracellular TMEM16A serves as a negative regulator of IP3Rs-mediated calcium release that is necessary for coordinating the processes of proliferation and differentiation.
Figure 7.

Working scheme of the modulatory roles of TMEM16A in myoblast
Intracellular TMEM16A in myoblast modulates the Ca2+ release from ER through IP3R calcium channels to coordinate the processes of proliferation and myogenic differentiation. Cl− effects as counter ion by binding Ca2+ and modulates the calcium homeostasis in ER and global calcium transients. TMEM16A deletion causes the disturbance of ER calcium homeostasis and global calcium.
Previously, it was reported that inhibition of caspase 3 inhibited the formation of myotubes (Fernando et al., 2002; Hochreiter-Hufford et al., 2013). We here show that intracellular-resident TMEM16A functions to suppress calcium release from ER lumen thus inhibit the activation of caspase 3. The discrepancy between our study and previous study might be explained by the reasons below. Although the caspase 3 activation is required for the myogenesis progression, the caspase 3 activation level should be maintained at a proper level. Too strong caspase 3 activity led to cell death and inhibition of myogenic differentiation. We did not observe inhibition effects of the pan-caspase inhibitor (Z-VAD-FMK) on the differentiation in control wild-type myoblasts as other labs. The reason may be related to the degree of inhibition effect of lower concentration of the inhibitor than other labs. Megeney lab used caspase-3-specific inhibitor, Z-DEVD.FMK at concentration of 20 μM, while Ravichandran lab used the same agent as ours, Z-VAD-FMK at 50 μM (Fernando et al., 2002; Hochreiter-Hufford et al., 2013), both of which had much stronger inhibitory functions than that of our current study (Z-VAD-FMK at 10 μM). It seems that the caspase 3 functions in a dose-dependent manner.
Within this working model, how does the TMEM16A molecule inhibit IP3Rs-mediated calcium release is an interesting issue. In the plasma membrane of neurons, the featured localization of TMEM16A at the proximal region of the ER membrane through physical interaction with IP3R allows TMEM16A to be activated more efficiently by IP3R-mediated calcium (Jin et al., 2013). However, no physical interaction between TMEM16A and IP3Rs was detected in myoblast in this study. We thus proposed the ER-resident TMEM16A to be activated by IP3R-mediated calcium release. The resultant activation of TMEM16A allows chloride to move into ER lumen and bind Ca2+, and hence the ER calcium release is inhibited. As a matter of fact, similar effect was observed in the case of intracellular CFTR (cystic fibrosis transmembrane conductance regulator) (Benedetto et al., 2017; Divangahi et al., 2009; Huang et al., 2020).The intracellular CFTR promotes Ca2+ influx to ER through facilitating the flux of chloride and its binding to Ca2+ in the ER in smooth muscle cells. As CFTR is also expressed in skeletal muscle cells (Divangahi et al., 2009), whether it is involved in the role of TMEM16A in ER calcium release remains to be determined in the future.
As a second messenger, cytosolic calcium functions in diverse physiological processes. Undoubtedly, elaborative control and homeostasis of this messenger is extremely important for various physiological activities. In this study, we show that intracellular TMEM16A is involved in the modulation of calcium release from intracellular ER through IP3Rs in a chloride channel activity-dependent manner. It would be interesting to extend this mechanism to other type of cells.
In summary, we here revealed a unique expression pattern of TMEM16A in skeletal muscle precursors and defined a role of intracellular TMEM16A in myogenesis. The deletion of TMEM16A caused overproduction of cytosolic calcium and excessive ER stress through abolishment of the chloride channel activity of TMEM16A, thereby enhancing cell growth and inhibiting differentiation.
Limitations of the study
In this study, we observed a unique localization of TMEM16A molecules in the cytosolic organelles of undifferentiated myoblast. However, we did not delineate the individual organelles targeting TMEM16A by organelle fraction analysis. It would be interesting to address this limitation in the future studies. Another limitation of this study is lacking measurement of chloride concentration in the ER of differentiating myoblast cells due to our shortage of measurement method, which would weaken our proposal that the intracellular TMEM16A modulates the chloride flux across ER membrane.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TMEM16A | Abcam Rabbit pAb | Cat# ab53212; RRID: AB_883075 |
| L-type Ca ++ CP α1S | Santa Cruz, Mouse mAb | Cat# sc-514685; RRID: AB_2923287 |
| Alpha-Actinin | Boster Biological Technology, Mouse mAb | Cat# BM0003; RRID: AB_2861430 |
| IP3R-I/II/III | Santa Cruz, Mouse mAb | Cat# sc-377518; RRID: AB_2637028 |
| Myosin heavy chain | DSHB, Mouse mAb | Cat# MF20; RRID: AB_2147781 |
| Laminin | Sigma Aldrich, Rabbit pAb | Cat# L9393; RRID: AB_477163 |
| Myogenin | DSHB, Mouse mAb | Cat# F5D; RRID: AB_2146602 |
| RyR | Santa Cruz, Mouse mAb | Cat# sc-376507; RRID: AB_11149759 |
| Cyclin D1 | CST, Rabbit pAb | Cat#2978; RRID: AB_2259616 |
| Caspase 3 | CST, Rabbit pAb | Cat#9662; RRID: AB_331439 |
| Cleaved Caspase 3 | CST, Rabbit pAb | Cat#9661; RRID: AB_2341188 |
| Caspase 12 | Santa Cruz, Rat mAb | Cat#sc-21747; RRID: AB_626799 |
| GAPDH | Applied Biological Materials, Mouse mAb | Cat#G041; RRID: AB_2813868 |
| Chemicals, peptides, and recombinant proteins | ||
| Bradykinin | MCE | HY-P0206 |
| Caffeine | Calbiochem | 58-08-2 |
| LipoMax DNA Transfection Reagent | Sudgen, China | 32011 |
| EdU assay kit | Beyotime, China | ST067 |
| PrimeScript RT reagent kit | Takara, Japan | RR037B |
| HiScript II One Step qRT-PCR SYBR Green Kit | Vazyme, China | Q221-01 |
| Fluo-4 Direct™ Calcium Assay Kit | Thermofisher | F10471 |
| Experimental models: Cell lines | ||
| C2C12 myoblasts | ATCC | CVCL_0188 |
| CHO-K1 | Institute of Cell Biology, Chinese Academy of Science | https://doi.org/10.1016/j.bbalip.2020.158640 |
| primary myoblasts | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mice: C57BL/6JGpt | Gem Pharmatech | N000013 |
| Mice: Tmem16a conventional knockout mice | This paper | https://doi.org/10.1016/j.jaci.2017.05.053 |
| Oligonucleotides | ||
|
Tmem16a –forward: 5′-CTGGCCCCTCTCCTCATTTTCCACAGACAT-3′ |
Sango Biotech, China | N/A |
|
Tmem16a –reverse: 5′-AGAATTACCCCCTTGGTCTACTATGGCTT-3′ |
Sango Biotech, China | N/A |
| Myogenin –forward: 5′-CATCCAGTACATTGAGCGCCTA-3′ |
Sango Biotech, China | N/A |
| Myogenin –reverse: 5′-GAGCAAATGATCTCCTGGGTTG-3′ |
Sango Biotech, China | N/A |
| GAPDH –forward: 5′-AGGTCGGTGTGAACGGATTTG-3′ | Sango Biotech, China | N/A |
| GAPDH–reverse: 5′-TGTAGACCATGTAGTTGAGGTCA-3′ | Sango Biotech, China | N/A |
| Recombinant DNA | ||
| Plasmid: GFP-Tmem16a | Dr. Uhtaek Oh from Seoul National University of Korea | N/A |
| Plasmid: mCherry-Sec61β | Dr. Haijia Yu from Nanjing Normal University | N/A |
| Software and algorithms | ||
| Adobe Photoshop | https://www.adobe.com | RRID:SCR_014199 |
| ImageJ | https://imagej.net/ | RRID:SCR_003070 |
| GraphPad Prism | www.graphpad.com | RRID:SCR_002798 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Huaqun Chen (chenhuaqun@njnu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Ethical approval
The animal experiments were approved by the Experimental Committee of Nanjing Normal University (No: IACYUC-1903023; Approve date: 1 March 2019).
Cell lines
This study used C2C12 myoblast cell line and CHO-K1 hamster ovary epithelial cell line.
Method details
Animals
3-week and 8-week-old male C57BL/6 (B6) mice were purchased from Gem Pharmatech (Nanjing, China). Tmem16a conventional knockout mice were generated as previously reported (Zhang et al., 2016). The mice were maintained in the specific pathogen free (SPF)-grade animal facility at Nanjing Normal University. All animal procedures were performed in accordance with the guidelines of the Animal Care and Use Committee of Nanjing Normal University. The animal experiments were approved by the Experimental Committee of Nanjing Normal University. Deletion of TMEM16A was evaluated by qRT-PCR, Western blotting, and immunofluorescence staining.
Gross phenotypic analysis of neonatal TMEM16A KO mice
Since the TMEM16A KO mice were neonatal lethal, we performed all the analysis in one month. The body weight mass was measured at 1, 2, 5-, 10-, 20- and 26-days old age. The muscle tissues were dissected and fixed, transverse cross sections or cryosections were prepared, followed by the histological or immunofluorescence staining analysis with specific antibodies. The pups were fixed in 95% ethanol for 8–12 h, the skin and all the tissues were removed. Then the skeletons were fixed in 95% ethanol for 3 days, defat in acetone for 1 day, washed with tap water for 8 h, stained with Alcian blue (Sigma Aldrich #TMS-010C) and Alizarin red (Solarbio #G1450, China) for 6 days, washed in tap water for 30 min, decolorized in 2% potassium hydroxide for 12 h. The skeleton images were captured under a stereoscope (Motic, China).
Histology and immunofluorescence staining
For preparation of cryosections, the fresh tissues were flash frozen in isopentane soaked in liquid nitrogen, embedded in OCT Compound (Sakura, USA). The samples were sectioned at 11 μm thick by a freezing microtome (Leica CM1950). For paraffin sections, the fresh tissues were fixed with 4% PFA (paraformaldehyde) (Biosharp #BL539A, China) and processed for routine paraffin histology. Paraffin sections (7 μm) were stained with hematoxylin-eosin (Beyotime #C0105S, China) using routine procedures. Immunofluorescence staining was performed by fixation of the cryosections or cells with 4% PFA, permeabilization with 0.2% Triton X-100 in PBS, blocking with 2.5% normal goat serum (Thermo Fisher #R37624), incubation with a primary antibody at 4°C overnight, and incubation with the corresponding Alexa Fluor-conjugated secondary antibodies (Thermo Fisher #A-11008, A-21422; CST #4413, #4408). The slides were then incubated with DAPI (Sigma Aldrich #D9542) to visualize the cell nuclei. The primary antibodies used were specific for TMEM16A (Abcam #ab53212), myosin heavy chain (MyHC) (DSHB #MF20) and Laminin (Sigma Aldrich #L9393). The slides were washed and mounted with ProLong Gold Anti-fade Mountant (Thermo Fisher #P10144). Images were acquired under a confocal microscope (Nikon A1). Image processing was performed by using Adobe Photoshop software.
Preparation of whole muscle homogenates
Whole muscle homogenates were prepared from the soleus, gastrocnemius, tibialis anterior and hind limb muscles. Aliquots taken for the whole muscle homogenates were combined with SDS sample buffer and incubated at 100°C for 5 min, with two rounds of vigorous vortex during the incubation. The homogenates were centrifuged at 12 000 rpm for 20 min, and the supernatants were used as the whole muscle homogenate samples.
Isolation of single myofibers
The single myofibers were prepared according to previous method (Pasut et al., 2013). Briefly, EDL muscles were digested with an enzyme mixture (0.05% collagenase D (Sigma Aldrich #11088858001), 0.25 μM CaCl2 and 0.5U/ml Dispase (Sigma Aldrich #SCM133)) in high-glucose DMEM (Thermo Fisher #11965092) at 37°C for about 30 min. The released single myofibers were rinsed with DMEM, followed by recovered in the incubator at 37°C for 30 min.
Culture of myoblasts and induction of myogenic differentiation
Skeletal muscle tissues were removed from the limbs of neonatal mice. The tissues were cut into small pieces and digested in the enzyme mixture (same as above) at 37°C for 0.5–1 h. 10 mL of DMEM was added to the digested mixture and triturated prior to centrifugation at 1500 × g for 10 min at room temperature. The pellet was resuspended in Ham’s F-10 nutrient mixture (Thermo Fisher #41550088) containing 20% FBS, bFGF (R&D #3339-FB) (5 ng/mL) and 1% penicillin/streptomycin (Thermo Fisher #15140122) (growth medium, GM) and plated in a 10 cm culture dish for 1 h. The unattached cells were harvested and cultured in Matrigel-coated 24-well culture plates. When the cell confluence reached approximately 80%, the cells were switched to high-glucose DMEM containing 2% horse serum (Sangon #ES10006-0100, China) and 1% penicillin/streptomycin (differentiation medium, DM) to induce myogenesis.
C2C12 cells were cultured in GM, and myogenic differentiation was induced in DM.
Transfection of the recombinant plasmids
The GFP-Tmem16a recombinant plasmid or mCherry-Sec61β recombinant plasmid was transfected into cultured primary myoblasts using LipoMax transfection reagent (Sudgen # 32011, China) according to the manufacturer’s instructions. Cells were collected for subsequent analysis after 48 h of transfection. The transfected myoblasts were switched to DM for another 4 days to induce myogenic differentiation.
Ethynyl-2ʹ-deoxyuridine (EdU) incorporation assay
The primary cultured myoblasts (the 2nd passage) were seeded into 24-well plates at 1×105 cells per well and incubated for 24 h. The 5-ethynyl-20-deoxyuridine (EdU) incorporation assay was performed with an EdU assay kit (Beyotime #C0071S, China) according to the manufacturer’s manual. Briefly, EdU (10 μM) was added into the wells and incubated for 2 h, followed by counterstaining of the cell nuclei with Hoechst 33342 (Beyotime #C1025, China). Images were captured under a fluorescence microscope (Leica). The percentage of EdU-positive cells was calculated in five fields per well.
Western blotting
Protein samples from tissues or cells were subjected to electrophoresis on 4%, 8% or 12% (wt/vol) polyacrylamide gels and transferred onto a PVDF membranes (Millipore # IPVH00010, USA). Membranes were blocked with 5% (wt/vol) skim milk and sequentially reacted with specific primary antibodies and the appropriate horseradish peroxidase–conjugated secondary antibody (Santa Cruz #sc-2004, sc-2005). The signals were visualized by incubation in Clarity Western ECL Substrate (Beyotime P0018S, China) prior to scanning (Tanon 4500). The primary antibodies used were specific for TMEM16A (Abcam# ab53212), myosin heavy chain (DSHB #MF20), myogenin (DSHB #F5D), RyR (Santa Cruz #sc-376507), IP3RI/II/III (Santa Cruz #sc-377518), Cyclin D1 (CST #2978), caspase 3/cleaved caspase 3 (CST #9662) and caspase 12 (Santa Cruz #sc-21747). Band intensities were determined by ImageJ software.
Quantitative RT-PCR
To assess mRNA expression of the genes, total RNA from tissues or cells was extracted with TRIzol Reagent (Invitrogen). cDNA was synthesized by using a PrimeScript RT reagent kit (Takara #RR037B), and the transcription was quantified by quantitative PCR using a SYBR green kit (Vazyme #Q221-01, China) in a StepOnePlus thermal cycler (Applied Biosystems, USA) according to the manufacturer’s instructions. The specific PCR primers are listed in Table S1.
Calcium measurements
Ca2+ signals were measured by using the Ca2+ indicator dye Fluo-4 (Fluo-4 Direct Calcium Assay Kit, Thermo Fisher) according to the manual and previous study (Spoida et al., 2014). Briefly, undifferentiated or differentiated myoblasts (myotubes) grown in a 96-well black plate (clear bottom with lid, Corning) were incubated with 50 μL Fluo-4 calcium indicator dye in a CO2 incubator for 45 min at 37°C. Baseline fluorescence was detected every 2 s for 20 s on a luminescence spectrometer (BioTek Synergy H1 393176 microplate reader) with excitation at 490 nm and emission at 525 nm at room temperature. The agonists were added to the wells and the fluorescence signals were recorded every 2 s for 300 s. For each condition, fluorescence counts were normalized to the basal fluorescence signal. The elevation of intracellular calcium was indicated by the increase in fluorescence counts.
Co-immunoprecipitation
Cells were rinsed twice with ice-cold PBS and protein samples were prepared according to our previous study (Chen et al., 2006). Proteins (500 μg) were immunoprecipitated for 2 h with IP3R antibody (1 μg). The precleared Protein A/G agarose beads (Thermo Fisher #20423) were incubated with immunocomplexes for another 2 h and washed four times with the lysis buffer. The samples were separated by SDS-PAGE, transferred to a PVDF membrane, and detected by Western blot analysis.
Quantification and statistical analysis
Statistical analyses of the data were performed using GraphPad Prism software. Western blotting and Co-IP data are representative of three independent experiments. All biological replicates and group sizes used for statistical analyses are indicated in the figure legends. The results are presented as the mean ± SD values. Student’s t-test (for paired or unpaired samples) was used to determine the significance of differences between two groups; a p value less than 0.05 was considered statistically significant.
Acknowledgments
We are grateful to Professor Uhtaek Oh (from Seoul National University of Korea), Professor Haijia Yu and Professor Cheng Deng (from Nanjing Normal University of China) for kindly providing GFP-Tmem16a recombinant plasmids, mCherry-Sec61recombinant plasmids, and the Fluo-4 Direct Calcium Assay Kit. This work was supported by the National Key Research and Development Program of China (Grant No. 2022YFF0710800 to M.S.Z.), the National Natural Science of China (No. 31371356 to H.Q.C. and No. 9184910039 to M.S.Z.) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (to H.Q.C.).
Author contributions
Methodology, Investigation, Analyses, Writing, & Editing, W.Y., C.C.C., J.L., Y.H.X., C.E.F., Y.C.C., H.W.F., B.X.H., M.Y.S., Z.Y.S., T.X.M., P.W., and Z.Z.; Resources, Writing and Review, M.S.Z.; Conceptualization, Writing, Review, Supervision, Project Administration, H.Q.C.
Declaration of interests
The authors declare that they have no conflict of interest.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105446.
Supplemental information
Data and code availability
-
•
The data reported in this paper is available from the lead contact upon request.
-
•
The paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adi S., Bin-Abbas B., Wu N.Y., Rosenthal S.M. Early stimulation and late inhibition of extracellular signal-regulated kinase 1/2 phosphorylation by IGF-I: a potential mechanism mediating the switch in IGF-I action on skeletal muscle cell differentiation. Endocrinology. 2002;143:511–516. doi: 10.1210/endo.143.2.8648. [DOI] [PubMed] [Google Scholar]
- Bare D.J., Kettlun C.S., Liang M., Bers D.M., Mignery G.A. Cardiac type 2 inositol 1,4,5-trisphosphate receptor: interaction and modulation by calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 2005;280:15912–15920. doi: 10.1074/jbc.M414212200. [DOI] [PubMed] [Google Scholar]
- Benedetto R., Ousingsawat J., Wanitchakool P., Zhang Y., Holtzman M.J., Amaral M., Rock J.R., Schreiber R., Kunzelmann K. Epithelial chloride transport by CFTR requires TMEM16A. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-10910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita I., Benedetto R., Fonseca A., Wanitchakool P., Sirianant L., Skryabin B.V., Schenk L.K., Pavenstädt H., Schreiber R., Kunzelmann K. Differential effects of anoctamins on intracellular calcium signals. FASEB. J. 2017;31:2123–2134. doi: 10.1096/fj.201600797RR. [DOI] [PubMed] [Google Scholar]
- Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., Galietta L.J.V. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- Chal J., Pourquié O. Making muscle: skeletal myogenesis in vivo and in vitro. Development. 2017;144:2104–2122. doi: 10.1242/dev.151035. [DOI] [PubMed] [Google Scholar]
- Chen H., Wu Y., Zhang Y., Jin L., Luo L., Xue B., Lu C., Zhang X., Yin Z. Hsp70 inhibits lipopolysaccharide-induced NF-kappaB activation by interacting with TRAF6 and inhibiting its ubiquitination. FEBS Lett. 2006;580:3145–3152. doi: 10.1016/j.febslet.2006.04.066. [DOI] [PubMed] [Google Scholar]
- Dang S., Feng S., Tien J., Peters C.J., Bulkley D., Lolicato M., Zhao J., Zuberbühler K., Ye W., Qi L., et al. Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature. 2017;552:426–429. doi: 10.1038/nature25024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal A., Ng S.F.J., Grabner M. Ca(2+)-activated Cl(-) channel TMEM16A/ANO1 identified in zebrafish skeletal muscle is crucial for action potential acceleration. Nat. Commun. 2019;10:115. doi: 10.1038/s41467-018-07918-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M., Balghi H., Danialou G., Comtois A.S., Demoule A., Ernest S., Haston C., Robert R., Hanrahan J.W., Radzioch D., Petrof B.J. Lack of CFTR in skeletal muscle predisposes to muscle wasting and diaphragm muscle pump failure in cystic fibrosis mice. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvvuri U., Shiwarski D.J., Xiao D., Bertrand C., Huang X., Edinger R.S., Rock J.R., Harfe B.D., Henson B.J., Kunzelmann K., et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72:3270–3281. doi: 10.1158/0008-5472.Can-12-0475-t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando P., Kelly J.F., Balazsi K., Slack R.S., Megeney L.A. Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA. 2002;99:11025–11030. doi: 10.1073/pnas.162172899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmer M., Förster F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020;133:jcs231340. doi: 10.1242/jcs.231340. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla P.J., Gibbons S.J., Bardsley M.R., Lorincz A., Pozo M.J., Pasricha P.J., Van de Rijn M., West R.B., Sarr M.G., Kendrick M.L., et al. Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G1370–G1381. doi: 10.1152/ajpgi.00074.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter-Hufford A.E., Lee C.S., Kinchen J.M., Sokolowski J.D., Arandjelovic S., Call J.A., Klibanov A.L., Yan Z., Mandell J.W., Ravichandran K.S. Phosphatidylserine receptor Bai1 and apoptotic cells as new promoters of myoblast fusion. Nature. 2013;497:263–267. doi: 10.1038/nature12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein I., Singh A.K., Pohl F., Misiak D., Braun J., Leitner L., Hüttelmaier S., Posern G. Post-transcriptional regulation of MRTF-A by miRNAs during myogenic differentiation of myoblasts. Nucleic Acids Res. 2020;48:8927–8942. doi: 10.1093/nar/gkaa596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Lam H., Koziol-White C., Limjunyawong N., Kim D., Kim N., Karmacharya N., Rajkumar P., Firer D., Dalesio N.M., et al. The odorant receptor OR2W3 on airway smooth muscle evokes bronchodilation via a cooperative chemosensory tradeoff between TMEM16A and CFTR. Proc. Natl. Acad. Sci. USA. 2020;117:28485–28495. doi: 10.1073/pnas.2003111117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T., Kanazawa T., Otsuka S., Ichii O., Hashimoto Y., Kon Y. Expression of caspase family and muscle- and apoptosis-specific genes during skeletal myogenesis in mouse embryo. J. Vet. Med. Sci. 2009;71:1161–1168. doi: 10.1292/jvms.71.1161. [DOI] [PubMed] [Google Scholar]
- Jin X., Shah S., Liu Y., Zhang H., Lees M., Fu Z., Lippiat J.D., Beech D.J., Sivaprasadarao A., Baldwin S.A., et al. Activation of the Cl- channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci. Signal. 2013;6:ra73. doi: 10.1126/scisignal.2004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kablar B., Rudnicki M.A. Skeletal muscle development in the mouse embryo. Histol. Histopathol. 2000;15:649–656. doi: 10.14670/hh-15.649. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Kongsuphol P., Aldehni F., Tian Y., Ousingsawat J., Warth R., Schreiber R. Bestrophin and TMEM16-Ca(2+) activated Cl(-) channels with different functions. Cell Calcium. 2009;46:233–241. doi: 10.1016/j.ceca.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Li X., Wang X., Zhang P., Zhu L., Zhao T., Liu S., Wu Y., Chen X., Fan M. Extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway is involved in inhibition of myogenic differentiation of myoblasts by hypoxia. Exp. Physiol. 2012;97:257–264. doi: 10.1113/expphysiol.2011.061382. [DOI] [PubMed] [Google Scholar]
- Magli A., Perlingeiro R.R.C. Myogenic progenitor specification from pluripotent stem cells. Semin. Cell Dev. Biol. 2017;72:87–98. doi: 10.1016/j.semcdb.2017.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner G. The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 2017;149:1065–1089. doi: 10.1085/jgp.201711878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B.A., Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nakanishi K., Dohmae N., Morishima N. Endoplasmic reticulum stress increases myofiber formation in vitro. FASEB. J. 2007;21:2994–3003. doi: 10.1096/fj.06-6408com. [DOI] [PubMed] [Google Scholar]
- Nakanishi K., Kakiguchi K., Yonemura S., Nakano A., Morishima N. Transient Ca2+ depletion from the endoplasmic reticulum is critical for skeletal myoblast differentiation. FASEB. J. 2015;29:2137–2149. doi: 10.1096/fj.14-261529. [DOI] [PubMed] [Google Scholar]
- Nakanishi K., Sudo T., Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J. Cell Biol. 2005;169:555–560. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasut A., Jones A.E., Rudnicki M.A. Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J. Vis. Exp. 2013;22:e50074. doi: 10.3791/50074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulino C., Kalienkova V., Lam A.K.M., Neldner Y., Dutzler R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature. 2017;552:421–425. doi: 10.1038/nature24652. [DOI] [PubMed] [Google Scholar]
- Rock J.R., Futtner C.R., Harfe B.D. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev. Biol. 2008;321:141–149. doi: 10.1038/nature24652. [DOI] [PubMed] [Google Scholar]
- Santulli G., Nakashima R., Yuan Q., Marks A.R. Intracellular calcium release channels: an update. J. Physiol. 2017;595:3041–3051. doi: 10.1113/jp272781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schredelseker J., Shrivastav M., Dayal A., Grabner M. Non-Ca2+-conducting Ca2+ channels in fish skeletal muscle excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 2010;107:5658–5663. doi: 10.1073/pnas.0912153107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder B.C., Cheng T., Jan Y.N., Jan L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapek S.X., Rhee J., Spicer D.B., Lassar A.B. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science. 1995;267:1022–1024. doi: 10.1126/science.7863328. [DOI] [PubMed] [Google Scholar]
- Spoida K., Masseck O.A., Deneris E.S., Herlitze S. Gq/5-HT2c receptor signals activate a local GABAergic inhibitory feedback circuit to modulate serotonergic firing and anxiety in mice. Proc. Natl. Acad. Sci. USA. 2014;111:6479–6484. doi: 10.1073/pnas.1321576111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarroni P., Rossi D., Conti A., Sorrentino V. Expression of the ryanodine receptor type 3 calcium release channel during development and differentiation of mammalian skeletal muscle cells. J. Biol. Chem. 1997;272:19808–19813. doi: 10.1074/jbc.272.32.19808. [DOI] [PubMed] [Google Scholar]
- Tian Y., Kongsuphol P., Hug M., Ousingsawat J., Witzgall R., Schreiber R., Kunzelmann K. Calmodulin-dependent activation of the epithelial calcium-dependent chloride channel TMEM16A. FASEB. J. 2011;25:1058–1068. doi: 10.1096/fj.10-166884. [DOI] [PubMed] [Google Scholar]
- Tran T., Andersen R., Sherman S.P., Pyle A.D. Insights into skeletal muscle development and applications in regenerative medicine. Int. Rev. Cell Mol. Biol. 2013;300:51–83. doi: 10.1016/b978-0-12-405210-9.00002-3. [DOI] [PubMed] [Google Scholar]
- Wang P., Zhao W., Sun J., Tao T., Chen X., Zheng Y.Y., Zhang C.H., Chen Z., Gao Y.Q., She F., et al. Inflammatory mediators mediate airway smooth muscle contraction through a G protein-coupled receptor-transmembrane protein 16A-voltage-dependent Ca(2+) channel axis and contribute to bronchial hyperresponsiveness in asthma. J. Allergy Clin. Immunol. 2018;141:1259–1268.e11. doi: 10.1016/j.jaci.2017.05.053. [DOI] [PubMed] [Google Scholar]
- Wang Q., Bai L., Luo S., Wang T., Yang F., Xia J., Wang H., Ma K., Liu M., Wu S., et al. TMEM16A Ca(2+)-activated Cl(-) channel inhibition ameliorates acute pancreatitis via the IP(3)R/Ca(2+)/NFκB/IL-6 signaling pathway. J. Adv. Res. 2020;23:25–35. doi: 10.1016/j.jare.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Zhang T., Bossuyt J., Li X., McKinsey T.A., Dedman J.R., Olson E.N., Chen J., Brown J.H., Bers D.M. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J. Clin. Invest. 2006;116:675–682. doi: 10.1172/jci27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.D., Cho H., Koo J.Y., Tak M.H., Cho Y., Shim W.S., Park S.P., Lee J., Lee B., Kim B.M., et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- Zhang C.H., Wang P., Liu D.H., Chen C.P., Zhao W., Chen X., Chen C., He W.Q., Qiao Y.N., Tao T., et al. The molecular basis of the genesis of basal tone in internal anal sphincter. Nat. Commun. 2016;7 doi: 10.1038/ncomms11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.M., Wei Q., Zhao X., Paterson B.M. Coupling of the cell cycle and myogenesis through the cyclin D1-dependent interaction of MyoD with cdk4. EMBO J. 1999;18:926–933. doi: 10.1093/emboj/18.4.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The data reported in this paper is available from the lead contact upon request.
-
•
The paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.