

Abstract
Background:
Intellectual disability syndromes with or without developmental delays affect up to 3% of the world population. We sought to clinically and genetically characterize a novel intellectual disability syndrome segregating in five unrelated consanguineous families.
Methods:
Clinical analyses were performed for eight patients with intellectual disability. Whole-exome sequencing for selected participants followed by Sanger sequencing for all available family members was completed. Identity-by-decent (IBD) mapping was carried out for patients in two Egyptian families harboring an identical variant. RNA was extracted from blood cells of Turkish participants, followed by cDNA synthesis and real-time PCR for TTC5.
Results:
Phenotype comparisons of patients revealed shared clinical features of moderate-to-severe intellectual disability, corpus callosum agenesis, mild ventriculomegaly, simplified gyral pattern, cerebral atrophy, delayed motor and verbal milestones, and hypotonia, presenting with an intellectual disability syndrome. Four novel homozygous variants in TTC5: c.629A>G;p.(Tyr210Cys), c.692C>T;p.(Ala231Val), c.787C>T;p.(Arg263Ter), and c.1883C>T;p.(Arg395Ter) were identified in the eight patients from participating families. IBD mapping revealed that c.787C>T;p.(Arg263Ter) is a founder variant in Egypt. Missense variants c.629A>G;p.(Tyr210Cys) and c.692C>T;p.(Ala231Val) disrupt highly conserved residues of TTC5 within the fifth and sixth TPR motifs which are required for p300 interaction, while the nonsense variants are predicted to decrease TTC5 expression. Functional analysis of variant c.1883C>T;p.(Arg395Ter) showed reduced TTC5 transcript levels in accordance with nonsense-mediated decay.
Conclusion:
Combining our clinical and molecular data with a recent case report, we identify the core and variable clinical features associated with TTC5 loss-of-function variants and reveal the requirement for TTC5 in human brain development and health.
Keywords: STRAP, tetratricopeptide, whole-exome sequencing, delayed milestones, facial dysmorphism, developmental delay, speech
INTRODUCTION
Intellectual disability syndromes (IDSs) are a widespread group of neurological disorders impacting 1–3% of the world-wide population.[1] An individual is diagnosed with intellectual disability (ID) when they exhibit significant limitations of intellectual functioning and adaptive behaviors prior to 18 years of age.[2] Hundreds of genomic variants have been associated with IDSs, yet nearly 60% of cases have unknown molecular etiology.[3] The recent use of next-generation sequencing technologies in the clinic have enabled the discovery of novel candidate genes and variants for this genetically complex and heterogeneous disorder.[4] For example, heterozygous variants in EP300, encoding p300, a co-activator of p53, are observed in Rubinstein-Taybi syndrome type 2 (RSTS [MIM: 613684]) in roughly 3% of patients. RSTS2 patients present with intellectual disability, pronounced microcephaly, broad thumbs and halluces, and characteristic dysmorphic facial features that include high arched eyebrows, mild downslanting of palpebral fissures, and retrognathia.[5] Most of the reported variants are small, frameshifting insertions or deletions within the EP300 coding sequence, suggesting the mechanism of disease may be haploinsufficiency.[6–10] Only recently, genotyping of two female patients with Menke-Hennekam syndrome 2 (MKHK2 [MIM: 618333]) uncovered de novo missense and in-frame indel heterozygous variants in EP300.[11] Both individuals had low IQ, feeding difficulties, recurrent upper airway infections, hearing impairment, hypermetropia, and facial features consisting of square flat face with upslanting, short palpebral fissures.[11, 12] Together, these genetic and clinical findings suggest the functional impact and position of variants in EP300, and likely components of the p300 co-activator complex, can lead to diverse clinical phenotypes and distinct IDSs.
p300 directly interacts with TTC5 (also known as STRAP, stress-responsive activator of p300) and JMY (junction mediating and regulatory protein, p53 cofactor) in the nucleus to form the p300 co-activator complex that mediates the p53 transcriptional response to cell stress.[13, 14] TTC5 encodes a 440 amino acid protein containing six tetratricopeptide repeat (TPR) domains and an oligonucleotide-binding (OB) fold that functions as a scaffold to stabilize p300-JMY interactions. This interaction prevents MDM2-dependent degradation of p53 to promote apoptosis.[13] In addition to this nuclear role, TTC5 has two known functions in the cytoplasm. TTC5 inhibits actin nucleation and autophagosome formation in the cytoplasm during cell stress,[15] as well as associates with the active ribosome and nascent tubulin polypeptides during protein synthesis to control tubulin autoregulation.[16] TTC5 is ubiquitously expressed in human tissues, with the highest levels found in the brain.[13]
Based on the expression pattern of TTC5 and biological association with JMY and p300, we hypothesized that variants in TTC5 might underlie an intellectual disability syndrome. A recent report identified TTC5 as a candidate gene for ID among 183 other genes profiled in 404 families by whole genome sequencing.[17] The authors reported two variants (c.51+1G>A and c.599delC;p.(Pro200LeufsTer29)) in three patients from two families with ID and discordant clinical features.[17] Here, we expand on these findings by using whole-exome sequencing to identify four novel bi-allelic TTC5 variants in eight individuals from five unrelated families with an intellectual disability syndrome.
MATERIALS AND METHODS
Patient Recruitment
This research was conducted after obtaining approval from Institutional Review Board of School of Biological Sciences, University of the Punjab, Lahore Pakistan (IRB No. 00005281), the Regional Ethics Committee of Harran University, Sanliurfa, Turkey, and the University of California, San Diego (140028). All participants or legal guardians signed written informed consent, including permission to publish photographs of the affected children. Medical histories were obtained by questioning. Collaboration was enabled by GeneMatcher.[18] For inclusion in the study, patients needed to be diagnosed with a moderate to severe intellectual disability with no known etiology.
Sampling
Peripheral blood was obtained from all available family members. DNA was extracted using a standard sucrose lysis, proteinase K digestion and salting out method (RDHM-05) or the MagNA Pure Compact Nucleic Acid Isolation Kit (CW07) (Roche, Switzerland) or as previously described.[19]
Whole-exome Sequencing
Whole-exome sequencing (WES)[20] was performed on DNA from all affected individuals in family RDHM-05, or individual CW07-A, 5543-A, 5377-A, or 6772-B at 100X resolution using Agilent V4 enrichment kit (Agilent Technologies, Santa Clara, CA, USA) or Nextera Rapid Capture Exome (Illumina, San Diego, CA USA), using the Illumina platform.[19, 21] ANNOVAR (http://wannovar.usc.edu/)[22] was used to annotate the variants. 97.41% of target regions had a coverage depth of more than 10-fold.
Variant Prioritization and amino acid conservation analyses
Rare homozygous exonic and splice variants with a population allele frequency of less than 1% in public databases (1000 genome database (http://www.internationalgenome.org/), Exome Aggregation Consortium (ExAC) (www.exac.broadinstitute.org/),[23] Exome Sequencing Project ESP (http://evs.gs.washington.edu/EVS/), genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/)[24] were filtered for further consideration. Missense variants were prioritized by the pathogenicity scores predicted from CADD (Combined Annotation Dependent Depletion),[25] SIFT (Scale-Invariant Feature Transform),[26] REVEL (Rare Exome Variant Ensemble Learner)[27] and ClinPred (https://omictools.com/clinpred-tool).[28] Each affected individual was analyzed independently for disease candidate variants; however, if multiple affected family members were sequenced, shared variants were prioritized as potentially pathogenic. Variants segregation was tested by PCR amplification and Sanger sequencing using Big Dye Terminator v 3.1 with Seqman (Lasergene, DNAstar) or SeqScape Software 3 (Life Technologies Corp.) analyses. In addition, the allele frequency of the TTC5 missense and nonsense variants were validated using DNA from ethnically matched controls. Protein sequence alignments from the UCSC genome browser (https://genome.ucsc.edu/) and homologene (https://www.ncbi.nlm.nih.gov/homologene) were used to assess amino acid conservation among diverse species. TTC5 protein orthologue sequences were obtained from UniProtKB (https://www.uniprot.org/help/uniprotkb) and amino acid conservation was observed following Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) alignment.
Identity-by-descent mapping
Homozygous intervals were detected from whole -exome sequencing data of the affected individuals in family 5543 and 5377 using HomozygosityMapper.[29] Variant calls within the homozygous interval surrounding TTC5 were aligned by chromosomal position and tabulated to identify a shared haplotype.
TTC5 qRT-PCR
Peripheral blood mononuclear cells were isolated from patients and family matched controls for family CW07 that consented to blood resampling using Lymphoprep (Stemcell Technologies, Inc.) according to manufacturer recommendations. The cells were lysed and the total RNA was extracted using TriZOL LS (Thermo Fisher Scientific). The cDNA libraries were prepared with SuperScript Vilo with ezDNase (Invitrogen). qRT-PCR was performed for TTC5 and GAPDH (loading control) expression using equal amounts of cDNA input per individual, Power SYBR Green Master Mix (Thermo Fisher Scientific), and primers: (TTC5 fwd) 5ʹ– TCA TCC TGG GAA AGG TGG TA – 3ʹ, (TTC5 rev) 5ʹ – GCT TCC CAT TCA CCA CTA GC – 3ʹ, or (GAPDH fwd) 5ʹ – GAG GCA GGG ATG ATG TTC TG – 3ʹ, (GAPDH rev) 5ʹ – CTG CAC CAC CAA CTG CTT AG – 3ʹ in triplicate. Triplicate Ct values were averaged and normalized to the loading control (GAPDH) for each individual. Normalized expression of TTC5 in unaffected relatives was set to 1, and the ΔCt calculated to determine fold change of TTC5 in affected patients.
RESULTS
Clinical Analyses:
Family 1, (RDHM-05, figure 1A, table 1) with two affected siblings from a first cousin marriage was ascertained from Lahore, Pakistan. Male patient RDHM-05-A, now age 8, was born through C-section and had an undescended testicle (HP:0000028), which was repaired through surgery. He suffered from lactose intolerance (HP:0004789) during early infancy. All assessed verbal (HP:0000750) and motor milestones (HP:0002194) were developmentally delayed (table 1) and he was diagnosed with moderate intellectual disability (HP:0002342). Muscular hypotonia (HP:0001252), aggression (HP:0000718) and recurrent chest infections (HP:0002205) were observed. Brain MR imaging revealed cerebral atrophy (HP:0002059) (data not shown). Facial dysmorphisms of pointed chin (HP:0000307), thin upper lip (HP:0000219), low-set ears (HP:0000369), and prominent midface were noted. Female patient RDHM-05-B, age 3, was born pre-term (HP:0001622) at 34 weeks gestation by C-section. Similar to her brother, her developmental milestones were delayed (HP:0001263). To date, she has shown mild, but identical age-matched features to her brother but with the addition of hirsutism (HP:0001007). Facial dysmorphisms include thin upper lip (HP:0000219) and low-set ears (HP:0000369). (figure 1B, table 1).
Figure 1. Pedigree and clinical data from families RDHM-05, CW07, 5543 and 5377.
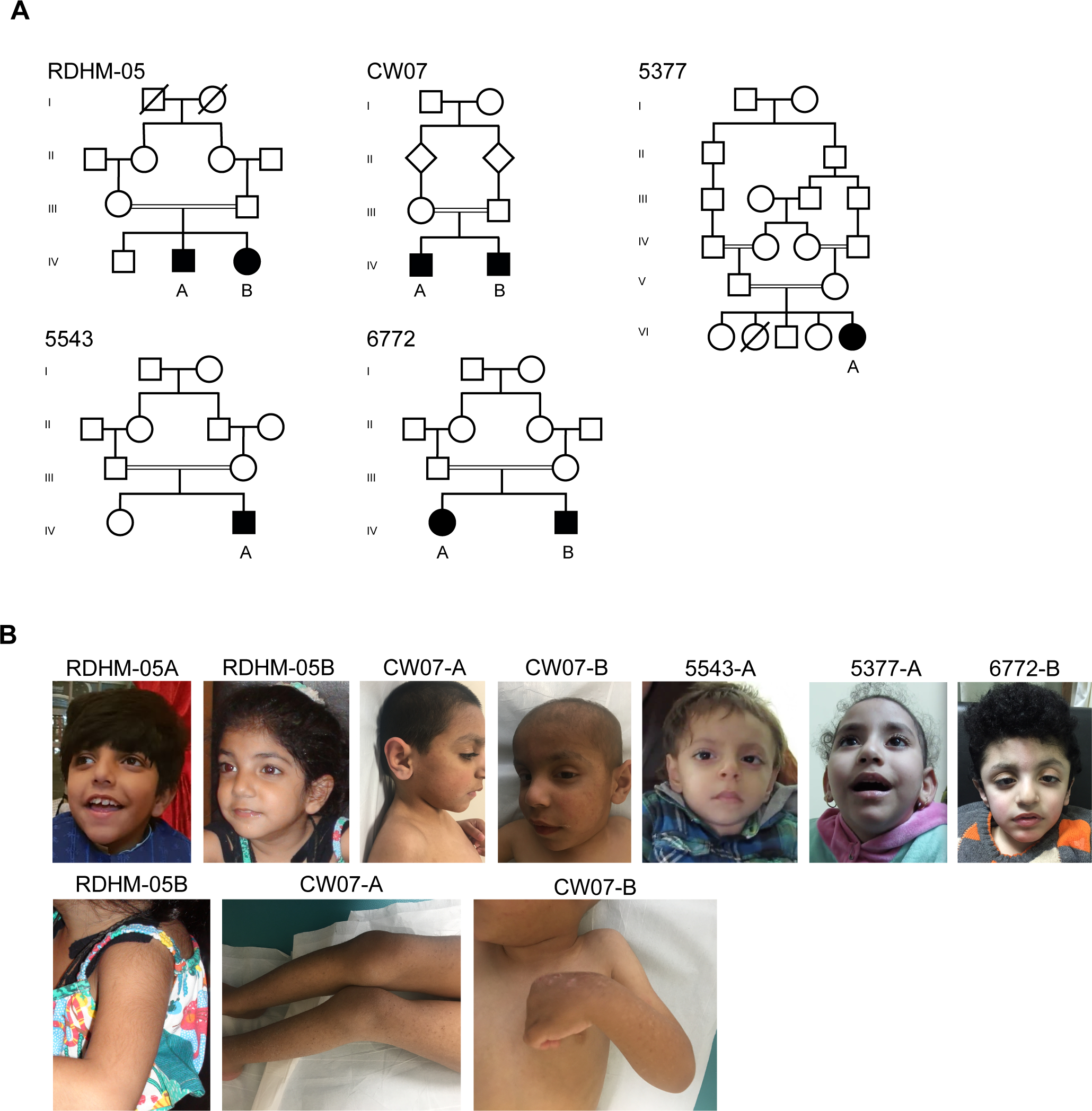
A. Family RDHM-05, CW07, 5543, 5377 and 6772. Parental consanguinity, double bar; square, male; circle, female; diamond, sex unknown; filled, affected; unfilled, unaffected; backslash, deceased.
B. Photographs of patients from family CW07 showing dry, acneiform lesions of the skin, RDHM-05, 5543, 5377 and 6772 showing thin upper lips.
Table 1.
Clinical features of individuals with bi-allelic TTC5 variants
| Family | RDHM-05 | CW07 | 5543 | 5377 | Summary of features | ||
|---|---|---|---|---|---|---|---|
| Subject | A | B | A | B | A | A | |
| TTC5 Variant | c.629A>G p.Tyr210Cys |
c.629A>G p.Tyr210Cys |
c.1183 C>T p.Arg395Ter |
c.1183 C>T p.Arg395Ter |
c.787C>T p.Arg263Ter |
c.787C>T p.Arg263Ter |
|
| Sex | Male | Female | Male | Male | Male | Female | 2f / 4m |
| Gestational age at delivery (weeks) | 37 | 34 | 35 | 34 | 39 | 40 | 34–40 weeks |
| Head Size (cm) | BPD: 8.8 | BPD: 8.5 | BPD: 8.7 | NA | HC: 32 | HC: 31 | 0.1–50.8 percentile |
| Onset of disorder | At birth | At birth | At birth | At birth | At birth | At birth | At birth |
| Birth weight (kg) | 2.5 | 1.8 | 2.0 | 2.4 | 2.75 | 3 | 3.2–56.9 percentile |
| Present weight (kg) | 21 | 10 | 18.5 | 16.1 | 6.8 | 10 | 0.1–12.71 percentile |
| Current age (years) | 8 | 3 | 7 | 5 | 5 | 6 | 3–8 years |
| Present height (cm.) | 106.68 | 86.36 | 115 | 110 | 92 | 82 | 0.1–57.1 percentile |
| Age at unassisted sitting (months) | 12 | NA | After 1 year | After 1 year | Not Yet | Not Yet | 3/6 1–6+ years |
| Age at unassisted standing (months) | 36 | 12 | After 3 years | After 2 years | Not Yet | Not Yet | 4/6 1–3+ years |
| Age at first word (months) | 24 | None | None | None | None | None | 6/6 24+ months |
| Present speech ability | Equals 2 years and 3 months | No speech | No speech | No speech | No speech | No speech | 5/6 no speech |
| Intellectual disability | Moderate | Mild | Severe | Moderate | Severe | Severe | 6/6 |
| Abnormal brain MRI | + | NA | + | + | + | + | 5/5 |
| Cerebral atrophy | + | NA | + | + | + | + | 5/5 |
| Enlarged lateral ventricles | − | NA | + | + | + | + | 4/5 |
| Corpus callosum atrophy | − | NA | + | + | + | + | 4/5 |
| Simplified gyral pattern | − | NA | + | + | + | + | 4/5 |
| Other brain abnormality | − | NA | + distended straight sinus, gliosis |
+ distended straight sinus, enlarged third ventricle, bilateral silvian fissures observed |
+ | + central atrophy |
4/5 |
| Hypotonia | − | − | + | + | + | + | 4/6 |
| Facial Dysmorphism | − | − | + abnormal outer ear, wide forehead, flared eyebrows and thin upper lip (v-shaped) |
+ abnormal outer ear, wide forehead, flared eyebrows, v-shaped upper lip |
+ V-shaped upper lip, high arched palate, long philtrum, prominent eyes |
+ Thin upper lip, high arched palate |
4/6 Thin v-shaped upper lip |
| Microcephaly | − | − | − | − | − | + | 1/6 |
| Skin Anomaly | − | + Hirsutism |
+ Acneiform lesions, dry skin |
+ Acneiform lesions, dry skin |
− | − | 3/6 |
| Paranasal sinus volume increased | − | NA | + | + | − | − | 2/5 |
| Autistic behavior | − | − | + | + | − | − | 2/6 |
| Aggression | + | − | − | + | − | − | 2/6 |
| Genitourinary Abnormalities | + Undescended testes |
− | − | − | + Bilateral undescended testes |
− | 2/6 |
| Sleep Disturbance | − | − | − | − | + | + | 2/6 |
| Scoliosis or kyphosis | − | − | − | − | − | + Mild kyphosis |
1/6 |
| Seizures | − | − | − | − | − | + Left centro- parieto- temporal epileptogenic discharge |
1/6 |
| Internipple distance | NA | NA | − | + wide |
− | − | 1/6 |
NA, not available; HC, head circumference; BPD, biparietal diameter; +, present; −, absent.
Family 2, (CW07, figure 1A, table 1) with two affected brothers, aged 7 and 5, from a first cousin marriage was ascertained from Sanliurfa, Turkey after approval by the Regional Ethics Committee of Harran University, Sanliurfa, Turkey. Patient CW07-A was born pre-term at 35 weeks gestation (HP:0001622). He presented with autistic behaviors (HP:0000729), severe intellectual disability (HP:0010864), delayed motor milestones (HP:0002194), lack of speech (HP:0001344), and muscular hypotonia (HP:0001252) (table 1). MRI revealed several brain abnormalities, including prominent anterior ventricle horns (HP:0002119), dilated paranasal sinus (HP:0000245), distended straight sinus, atrophy of the corpus callosum (HP:0007371), simplified gyral pattern (HP:0009879), and gliosis (HP:0002171) (figure 2). Moreover, he had dermatological findings of acneiform lesions with dry skin (HP:0000958) (figure 1B) together with minor dysmorphic features of abnormal outer ear (HP:0000356), wide forehead (HP:0000337), flared eyebrows (HP:0011229), and v-shaped, thin upper lip (HP:0000219). CW07-B presented with autistic (HP:0000729) and aggressive behavioral pattern (HP:0000718), moderate intellectual disability (HP:0002342), delayed motor milestones (HP:0001270), lack of speech (HP:0001344), muscular hypotonia (HP:0001252), and severe brain abnormalities, including enlarged paranasal (HP:00000245) and distended straight sinus, cerebral atrophy (HP:0002059) with simplified gyral pattern (HP:0009879), ventriculomegaly of the third and lateral ventricles (HP:0002119), atrophy of the corpus callosum (HP:0007371) and prominent sylvian fissures (HP:0100952) (figure 2). He shared the dermatological and dysmorphic findings observed in his brother, as well as increased internipple distance (HP:0006610) (table 1, figure 1B).
Figure 2. Magnetic resonance images (MRI) of patients from families CW07, 5543, 5377, and 6772.
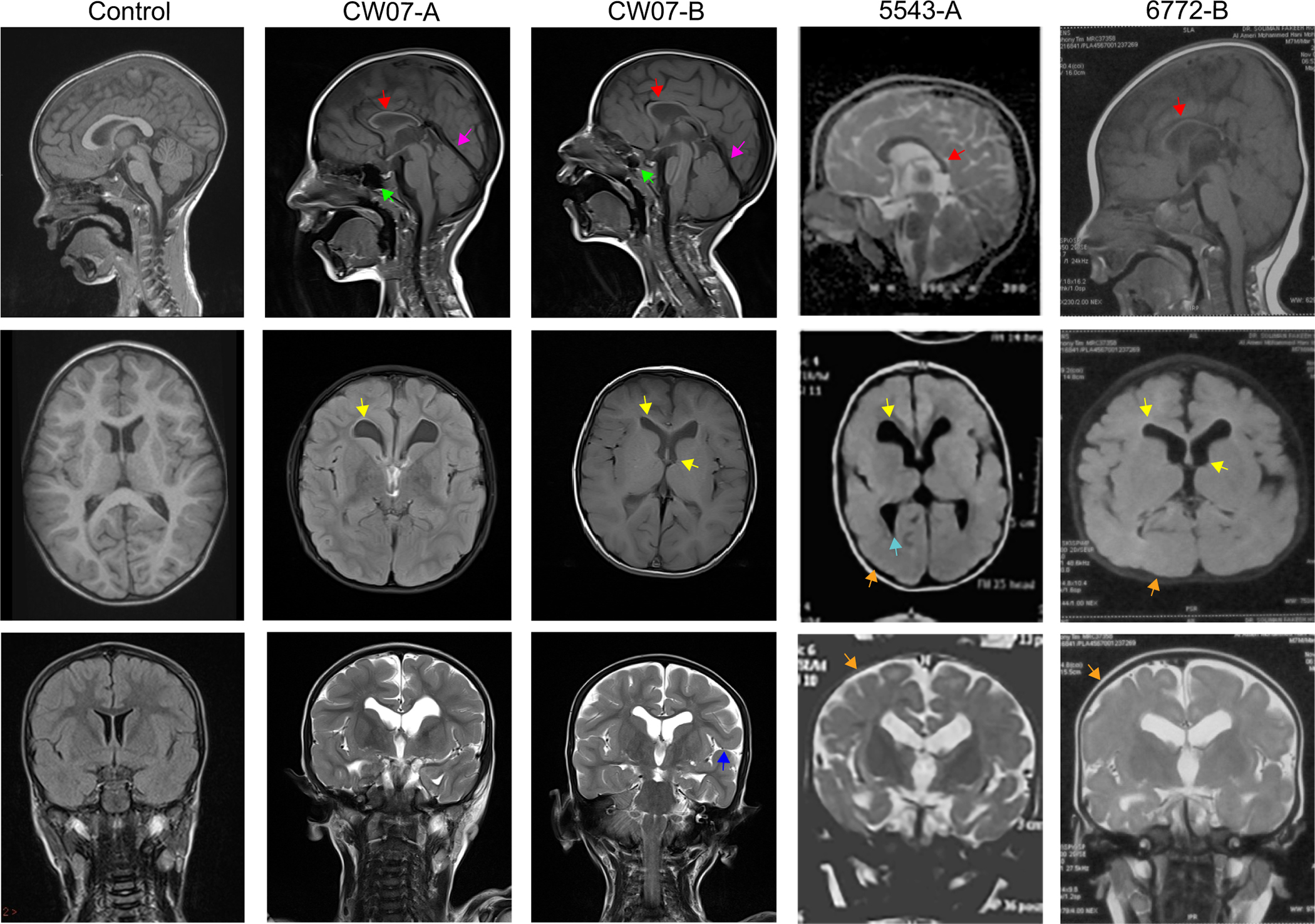
MRIs from a healthy control individual (left column) and affected individuals. Midline sagittal images (top row) show corpus callosum atrophy (red arrow), increased paranasal sinus area (green arrow), and distended straight sinus (pink arrow). Axial (middle row) images depict dilated ventricles (yellow arrow), white matter changes at the occipital horn (blue arrow), and abnormal gyri (orange arrow). Coronal (bottom row) images show prominent sylvian fissures in patient CWO7-B (purple arrow) and abnormal gyri in patient 5543-A and 6772-B (orange arrow). Control cases courtesy of Dr. Derek Smith, Radiopaedia.org, rID:53692
Family 3 (5543, figure 1, table 1), with an affected male from a first cousin marriage, was ascertained from Cairo, Egypt. Patient 5543-A, now 5 years of age, was born full term at 39 weeks gestation with bilateral undescended testes (HP:0000028). He presented with severe intellectual disability (HP:0010864), delayed motor milestones (HP:0001270), lack of speech (HP:0001344), muscular hypotonia (HP:0001252), and facial dysmorphisms of v-shaped upper lip (HP:0010804), high arched palate (HP:0002705), long philtrum (HP:0000343), and prominent eyes (HP:0000520) (table 1, figure 1B). MR imaging revealed cerebral atrophy (HP:0002059) with simplified gyral pattern (HP:0009879), hypoplastic corpus callosum (HP:0002079), white matter abnormalities around the occipital horn (HP:0007052), and dilated lateral ventricles (HP:0002119) (figure 2).
Family 4, (5377, figure 1, table 1), with an affected female of noted parental consanguinity, was ascertained from Cairo, Egypt. Patient 5377-A, now 6 years of age, was born by vaginal delivery at 40 weeks gestation. She presented with severe intellectual disability (HP:0010864), microcephaly (HP:0011451), hypotonia (HP:0001290), delayed motor milestones (HP:0001270), no speech (HP:0001344), mild kyphosis (HP:0002808), and minor facial dysmorphisms of thin upper lip (HP:0000219), curly hair (HP:000212) and high arched palate (HP:0002705) (table 1, figure 1B). At the age of 2, she developed severe epilepsy resulting in roughly four tonic (HP:0010818) and two myoclonic (HP:0002123) seizures per day with cyanosis (HP:0000961) that is now controlled with antiepileptic medications (table 1). She has severe developmental brain abnormalities including abnormal gyri (HP:0009879), dilated lateral ventricles (HP:0002119), a thin corpus callosum (HP:0002079), and central (HP:0002506) and cortical atrophy (HP:0002120) (data not shown).
Family 5, (6772, figure 1, table 1), with two affected siblings from a first cousin marriage, was ascertained from Cairo, Egypt. Female patient 6772-A, now age 12, was born by vaginal delivery at 40 weeks gestation. She presented with moderate intellectual disability (HP:0010864), microcephaly (HP:0011451), hypotonia (HP:0001290), delayed verbal (HP:0000750) and motor (HP:0001270) milestones, increased internipple distance (HP:0006610), and minor facial dysmorphisms of long face (HP:0000276), high-arched eyebrows (HP:0002553), prominent nose (HP:0000448), short philtrum (HP:0000322), and large low-set ears (HP:0000369). MR imaging revealed severe structural brain defects including abnormal gyri (HP:0009879), dilated lateral ventricles (HP:0002119), a thin corpus callosum (HP:0002079), and cortical atrophy (HP:0002120) (data not shown). Male patient 6772-B, now age 5, was born by vaginal delivery at 38 weeks gestation. He was diagnosed with severe intellectual disability and presented with delayed motor milestones (HP:0001270), lack of speech (HP:0001344), muscular hypotonia (HP:0001252), increased internipple distance (HP:0006610), and facial dysmorphisms of long face (HP:0000276), brachycephaly (HP:0000248), low anterior hairline (HP:0000294), woolly scalp hair (HP:0040149), mild sagging eyelid (HP:0001488), prominent nose (HP:0000448) and nasal tip (HP:0005274), v-shaped upper lip (HP:0010804), short philtrum (HP:0000322), and low-set protruding ears (HP:0000369) (table 1, figure 1B). MR imaging revealed cerebral atrophy (HP:0002059) with simplified gyral pattern (HP:0009879), hypoplastic corpus callosum (HP:0002079), and dilated lateral and third ventricles (HP:0002119) (figure 2).
Molecular Analyses
Affected individuals from RDHM-05 had multiple homozygous variants present (Supplementary table 1), however, only a single variant in TTC5: NM_13837:g.20298807T>C; c.629A>G; p.(Tyr210Cys), passed segregation analysis according to a strict recessive mode of inheritance (figure 3A, B, C). The variant was not previously described in public databases, including Exome Aggregation Consortium (ExAC), HGMD Professional 2018.3, 1000 genomes, gnomAD, the NHLBI GO Exome Sequencing Project (ESP), or observed in 200 chromosomes from ethnically matched controls. Likewise, exome sequencing of affected individual 6772-A in family 6772 detected a single homozygous missense variant in TTC5: NM_13837:g.20296394G>A; c.692C>T; p.(Ala231Val) which segregated with the phenotype (figure 3A, B, C). It was observed only once as heterozygous in ExAC (1/251420).
Figure 3. Molecular analysis of variants in TTC5.
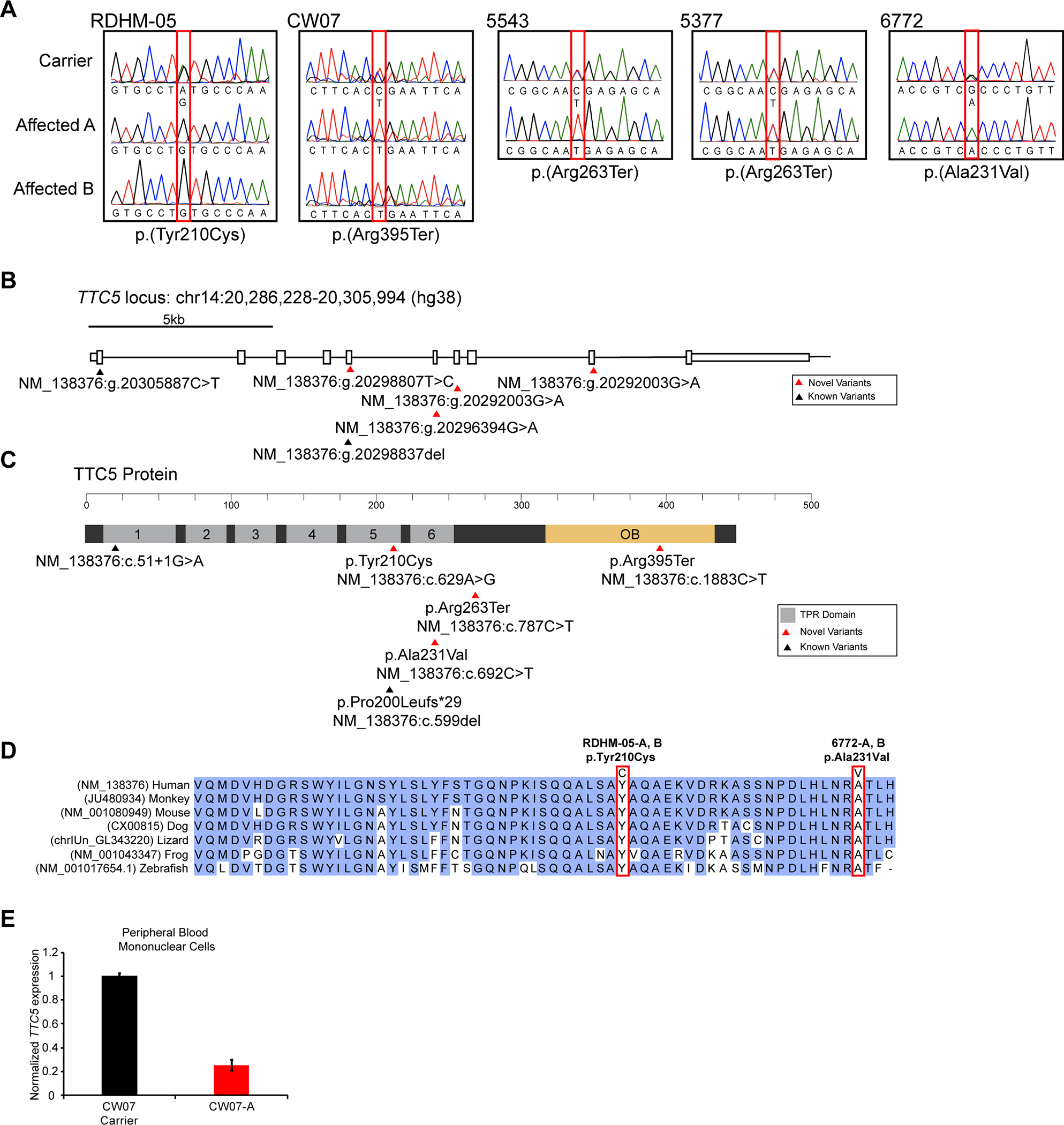
A. Partial Sanger sequence traces for TTC5 exons 5, 6, 7, and 9, encompassing c.629A>G;p.(Tyr210Cys), c.1883C>T;p.(Arg395Ter), c.787C>T;p.(Arg263Ter), and c.692C>T;p.(Ala231Val) variants, respectively. Affected individuals are homozygous for the variant, while unaffected carriers are heterozygous.
B. TTC5, located on chromosome 14 on the minus strand has ten exons (shown by rectangles). Novel TTC5 variants occur at hg38 position 20298807 in exon 5 (family RDHM-05), 20296394 in exon 6 (family 6772), 20292003 in exon 7 (families 5543 and 5377), and 20292003 in exon 9 (family CW07).
C. TTC5 protein domain structure. TPR motif domains are shown in grey. Previously reported variants are annotated with black triangles and novel variants with red triangles. The cDNA and protein positions are shown. TPR; Tetratricopeptide repeat.
D. Clustal-Omega alignment of TTC5 protein orthologues in vertebrate species showing complete evolutionary conservation of p.Tyr210 and p.Ala231.
E. Quantitative RT-PCR for TTC5 from peripheral blood mononuclear cells isolated from an unaffected carrier or affected individual in family CW07 showing variant p.(Arg395Ter) decreases TTC5 expression.
The TTC5 missense variants c.629A>G; p.(Tyr210Cys) and c.692C>T; p.(Ala231Val) had high CADD (p.(Tyr210Cys), 19.99; p.(Ala231Val), 32.00),[25] Grantham (p.(Tyr210Cys), 194; p.(Ala231Val),64),[30] and GERP (p.(Tyr210Cys), 4.65; p.(Ala231Val), 5.10)[31] scores indicating high nucleotide and amino acid residue conservation at both positions. Indeed, Clustal-Omega alignment of TTC5 orthologues revealed the p.Tyr210 and p.Ala231 residues are evolutionarily conserved among all vertebrate species examined (figure 3D) as well as in ciliates and plants.[16] In addition, in-silico tools predict the variants to be damaging or disease causing: PolyPhen,[32] probably damaging; SIFT,[26] deleterious; PROVEAN,[33] deleterious; and MutationTaster,[34] disease causing.
Exome sequencing of affected individuals CW07-A, 5477-A and 5543-A revealed two novel homozygous nonsense variants in TTC5: NM_138376:g.20292003G>A; c.1883C>T; p.(Arg395Ter) in family CW07, and NM_138376:g.20295764G>A; c.787C>T; p.(Arg263Ter) in families 5477 and 5543, respectively, that segregated with the phenotypes in the respective families (figure 3A, B, C). Nonsense variant p.(Arg395Ter) was not present in 120 chromosomes from ethnically matched control individuals but was observed twice in ExAC and four times in gnomAD (4/235016), while, nonsense variant p.(Arg263Ter) was reported once in ExAC (1/120536) and twice in gnomAD (1/246074); all as heterozygous. Both nonsense variants had not been described in 1000 genomes or in the NHLBI GO Exome Sequencing Project (ESP), and are predicted to be disease causing by in-silico prediction tools.
To functionally determine whether the nonsense variant p.(Arg395Ter) in TTC5 would elicit nonsense-mediated decay and thereby reduce transcript levels, we investigated mRNA extracted from peripheral blood mononuclear cells from healthy and affected individuals in family CW07 that consented to resampling. Quantitative RT-PCR measurement of TTC5 transcript levels in cells isolated from the father of family CW07 (monoallelic for p.(Arg395Ter)) or CW07-A (bi-allelic for p.(Arg395Ter)) found a 4-fold decrease in expression in the affected individual (figure 3E). Taken together, the high nucleotide and amino acid conservation scores of the reference TTC5 alleles, the in-silico variant predictions, and real-time PCR results suggest that the novel TTC5 variants are deleterious and may be pathogenic.
Since patients 5377-A and 5543-A share an identical homozygous nonsense variant p.(Arg263Ter) in TTC5, and both families originated from Egypt, we sought to determine if families 5377 and 5543 share a homozygous haplotype block. Identity-by-descent mapping[29] of exome sequencing data from both affected individuals uncovered a 6,301,093 base pair region spanning the TTC5 locus, encompassing 59 single nucleotide polymorphisms (SNPs) rs199831317-rs4983176 (Supplementary table 2), indicating p.(Arg263Ter) is likely a founder mutation.
DISCUSSION
TTC5 is a member of a large family of paralogous proteins in humans, which contain a TPR motif (https://www.genenames.org/data/genegroup/#!/group/769). Variants in TTC family proteins have been reported to be causative for various neurological disorders. TTC3, TTC8 or TTC15 variants are linked to familial Alzheimer’s disease,[35] Bardet-Biedl Syndrome,[36] or encephalopathy,[37] respectively. TTC19 mutations are reported in patients with mitochondrial complex III deficiency, nuclear type 2 [MIM: 615157],[38] in which individuals have developmental delay, hypotonia, ataxia, dysarthria, dystonia, spasticity and cognitive impairment.
Protein domain interaction studies for TTC5, p300, JMY, and p53 have demonstrated that the six TPR protein-binding motifs and OB fold of TTC5 are critical for JMY and p300 associations, respectively. Loss of TPR protein-binding motifs 1–3 or 3–5 abolishes association with JMY, whereas TPR protein-binding motifs 5, 6, and the OB fold are required for p300 interaction.[13, 39] The p.(Tyr210Cys) and p.(Ala231Val) missense variants identified in this study occur within the tetratricopeptide-like helical structure of the TPR protein-binding motif 5 and 6, respectively, which are important for protein-protein interaction via the formation of anti–parallel α–helices.[40] These helices together form a super helix structure that constitutes a ligand binding pocket.[41] TPR protein-binding domain 5 is comprised of two alpha-helices of 17 residues each and harbors two phosphorylation sites that are essential for intramolecular interaction of TRP protein-binding motifs 5 and 6.[39] Phosphorylation of serine 203 by ataxia telangiectasia (ATM) and serine 221 by checkpoint kinase 2 (CHK2) stabilizes TTC5 in the nucleus to increase levels and histone acetylation activity of the p300 co-activator complex.[42, 43] Since amino acids Tyr210 and Ala231 are highly conserved and adjacent to these phosphorylation sites, we hypothesize that these residues may play a critical role in TTC5 function (figure 3D). Perhaps, p.(Tyr210Cys) or p.(Ala231Val) impairs TTC5-JMY/p300 interactions or TTC5 protein folding and stability. Western blot and co-immunoprecipitation analyses from patient derived cell lines, if available, could distinguish between these possibilities.
Premature termination codons resulting from nonsense mutations typically lead to transcript degradation by the cellular process of nonsense-mediated decay (NMD), thereby reducing overall abundance of the target mRNA.[44] However, various factors influence the efficiency of NMD, including the location of the premature termination codon within long or ultimate exons and the proximity of the variant to an intron.[45, 46] Since the homozygous nonsense variant p.(Arg263Ter) is located in TTC5 exon 7 of 10, we predict the variant will likely elicit nonsense-mediated decay. In contrast, nonsense variant p.(Arg395Ter) is located in TTC5 exon 9 of 10, 18 base pairs from the terminal intron (figure 3B). Experimental evidence has shown premature termination codons that occur less than 50–55 base pairs from the penultimate exon splice donor site might not cause NMD,[46, 47] therefore, p.(Arg395Ter) could result in a truncated TTC5 protein lacking the complete OB fold domain. However, we found a marked reduction in TTC5 expression in patients with p.(Arg395Ter) suggesting this variant diminishes levels of TTC5, consistent with NMD.
Thus, following genetic and molecular analysis, all affected patients from the families described herein were diagnosed with TTC5-associated intellectual disability syndrome, an autosomal recessive syndrome caused by loss-of-function variants in TTC5. The four novel homozygous variants in TTC5 lead to delayed developmental milestones and intellectual disability with or without microcephaly. It is challenging to distinguish intellectual disability syndromes from one another, since many share overlapping and non-specific symptoms. Our work, together with a prior study,[17] enables us to define to-date, the cornerstone clinical features of TTC5-associated intellectual disability syndrome as severe speech impairment, cerebral atrophy and hypotonia that can be used to prioritize this syndrome for diagnosis. We find patients with bi-allelic variants present with moderate to severe intellectual disability, delayed motor and verbal milestones, hypotonia, and a thin, often v-shaped, upper lip. Shared radiological findings observed in more than one family include corpus callosum agenesis, mild ventriculomegaly, abnormal gyri, and cerebral atrophy. Variable clinical features include autistic and aggressive behavior, microcephaly, wide internipple distance, and dysmorphic facial features including flared or arched eyebrows, and low-set ears with abnormalities. Variable radiological findings of gliosis, white matter changes, and mild leukodystrophy may be present. While core clinical features of the disorder are highly consistent, phenotypes unique to families or individual patients, such as autistic behavior and dry acneiform lesions (CW07-A, B), genitourinary abnormalities (RDHM-05A and 5543-A), epilepsy (5377-A), kyphosis (5377-A), and dilated paranasal and straight sinus areas (CW07-A, B), are likely attributed to pathogenic variants in other genes. However, we cannot rule out the possibility that TTC5 variants may contribute to these phenotypes.
Patients with TTC5-associated IDS share clinical features with Rubinstein-Taybi syndrome type 2 affected individuals harboring EP300 variants, indicating the same molecular mechanism may be driving these similarities, namely reduced p300 co-activator complex activity. In our cohort of TTC5-associated IDS patients, microcephaly is present 50% of the time, while in the EP300-associated Rubinstein-Taybi syndrome type 2, this rate varies between 87–100%.[48–51] In addition to this feature, low hanging columella (HP:0009765) (92–100%), broad thumbs (69–87.5%) long eyelashes (HP:0000527) (90%), a large beaked nose (HP:0003683) (81%), and distinct facial grimacing’ (HP:0000273) (47%) are observed in patients with Rubinstein-Taybi syndrome type 2. %.[48–51] We did not find these features in our TTC5-associated IDS cohort. Furthermore, while normal IQ levels can be observed in Rubinstein-Taybi syndrome type 2, all patients with TTC5-associated IDS have moderate to severe ID. [5] Lastly, TTC5-associated IDS is inherited in an autosomal recessive pattern, with parents as carriers. Rubinstein-Taybi syndrome type 2 shows an autosomal dominant inheritance pattern and all patients identified to date have de novo variants, except one individual. [48–51]
The phenotypic differences between patients with Rubinstein-Taybi syndrome type 2 and TTC5-associated IDS may be due to a limited requirement for TTC5 during developmental periods of cell stress, while EP300 appears less restricted.[13, 14, 52, 53] Alternatively, the clinical variation observed among these different IDSs could be attributed to the additional functions of TTC5 in autophagy regulation and tubulin autoregulation in the cytoplasm, a cellular process unassociated with p300.[15],[16] Taken together, the diverse clinical features observed in TTC5-associated intellectual disability syndrome patients involve multiple organ systems and suggest a critical role of TTC5 for disparate cellular processes. Our findings establish TTC5 as a disease associated gene and delineate recurrent and variable features of this novel intellectual disability syndrome.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the family members for their participation.
FUNDING
The study was funded by family RDHM-05, Higher Education Commission, Pakistan #2877 to S.N., the National Institutes of Health R00HD082337 to A.E.S., R01NS048453, R01NS09800 and QNRF NPRP 6–1463-3–351 to J.G.G., SFARI 51486313 to J.G.G., UM1HG008900/Broad Institute Center for Mendelian Genomics U54HG006504/Yale Center for Mendelian Disorders, N01 268201700006I-0–26800029 to CIDR for genotyping. J.G.G. is an investigator with the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
The authors declare that they have no conflict of interest
PATIENT CONSENT FOR PUBLICATION
Obtained
ETHICS APPROVAL
Institutional Review Board of School of Biological Sciences, University of the Punjab, Lahore Pakistan (IRB No. 00005281), the Regional Ethics Committee of Harran University, Sanliurfa; Turkey, and the University of California, San Diego (140028).
SUPPLEMENTAL DATA
Supplemental Data includes two tables.
DATA AVAILABILITY STATEMENT
All data is available under reasonable request from the corresponding authors. The accession numbers for the TTC5 variants reported in this paper are: LOVD:0000602976 LOVD:0000604184 and dbGap:phs000288.v1.p1
REFERENCES
- 1.McKenzie K, Milton M, Smith G, Ouellette-Kuntz H. Systematic Review of the Prevalence and Incidence of Intellectual Disabilities: Current Trends and Issues. Current Developmental Disorders Reports 2016;3:1–12. [Google Scholar]
- 2.Schalock RL, Luckasson R, Tassé MJ. The contemporary view of intellectual and developmental disabilities: Implications for psychologists. Psicothema 2019;31:223–8. [DOI] [PubMed] [Google Scholar]
- 3.Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, Zenker M, Hüffmeier U, Thiel C, Rüschendorf F, Nurnberg P, Reis A, Trautmann U. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A 2006;140:2063–74. [DOI] [PubMed] [Google Scholar]
- 4.de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BBA, Brunner HG, Veltman JA, Vissers LELM. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N Engl J Med 2012;367:1921–9. [DOI] [PubMed] [Google Scholar]
- 5.Bartsch O, Labonté J, Albrecht B, Wieczorek D, Lechno S, Zechner U, Haaf T. Two patients with EP300 mutations and facial dysmorphism different from the classic Rubinstein-Taybi syndrome. Am J Med Genet 2009;152A:181–4. [DOI] [PubMed] [Google Scholar]
- 6.Roelfsema JH, White SJ, Ariyürek Y, Bartholdi D, Niedrist D, Papadia F, Bacino CA, Dunnen den JT, van Ommen G-JB, Breuning MH, Hennekam RC, Peters DJM. Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Mutations in Both the CBP and EP300 Genes Cause Disease. The American Journal of Human Genetics 2005;76:572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartholdi D, Roelfsema JH, Papadia F, Breuning MH, Niedrist D, Hennekam RC, Schinzel A, Peters DJM. Genetic heterogeneity in Rubinstein-Taybi syndrome: delineation of the phenotype of the first patients carrying mutations in EP300. Journal of Medical Genetics 2007;44:327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zimmermann N, Acosta AMBF, Kohlhase J, Bartsch O. Confirmation of EP300 gene mutations as a rare cause of Rubinstein–Taybi syndrome. Eur J Hum Genet 2007;15:837–42. [DOI] [PubMed] [Google Scholar]
- 9.Foley P, Bunyan D, Stratton J, Dillon M, Lynch SA. Further case of Rubinstein-Taybi syndrome due to a deletion in EP300. Am J Med Genet 2009;149A:997–1000. [DOI] [PubMed] [Google Scholar]
- 10.Hamilton MJ, Newbury-Ecob R, Holder-Espinasse M, Yau S, Lillis S, Hurst JA, Clement E, Reardon W, Joss S, Hobson E, Blyth M, Al-Shehhi M, Lynch SA, Suri M. Rubinstein-Taybi syndrome type 2: report of nine new cases that extend the phenotypic and genotypic spectrum. Clin Dysmorphol 2016;25:135–45. [DOI] [PubMed] [Google Scholar]
- 11.Menke LA, The DDD study, Gardeitchik T, Hammond P, Heimdal KR, Houge G, Hufnagel SB, Ji J, Johansson S, Kant SG, Kinning E, Leon EL, Newbury-Ecob R, Paolacci S, Pfundt R, Ragge NK, Rinne T, Ruivenkamp C, Saitta SC, Sun Y, Tartaglia M, Terhal PA, van Essen AJ, Vigeland MD, Xiao B, Hennekam RC. Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am J Med Genet 2018;176:862–76. [DOI] [PubMed] [Google Scholar]
- 12.Menke LA, van Belzen MJ, Alders M, Cristofoli F, Ehmke N, Fergelot P, Foster A, Gerkes EH, Hoffer MJV, Horn D, Kant SG, Lacombe D, Leon E, Maas SM, Melis D, Muto V, Park S-M, Peeters H, Peters DJM, Pfundt R, van Ravenswaaij-Arts CMA, Tartaglia M, Hennekam RCM. CREBBP mutations in individuals without Rubinstein-Taybi syndrome phenotype. Am J Med Genet A 2016;170:2681–93. [DOI] [PubMed] [Google Scholar]
- 13.Demonacos C, Krstic-Demonacos M, La Thangue NB. A TPR motif cofactor contributes to p300 activity in the p53 response. Mol Cell 2001;8:71–84. [DOI] [PubMed] [Google Scholar]
- 14.Shikama N, Lee C-W, France S, Delavaine L, Lyon J, Krstic-Demonacos M, La Thangue NB. A Novel Cofactor for p300 that Regulates the p53 Response. Mol Cell 1999;4:365–76. [DOI] [PubMed] [Google Scholar]
- 15.Hu X, Mullins RD. LC3 and STRAP regulate actin filament assembly by JMY during autophagosome formation. The Journal of Cell Biology 2019;218:251–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Z, Gasic I, Chandrasekaran V, Peters N, Shao S, Mitchison TJ, Hegde RS. TTC5 mediates autoregulation of tubulin via mRNA degradation. Science 2019;:eaaz4352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu H, Kahrizi K, Musante L, Fattahi Z, Herwig R, Hosseini M, Oppitz C, Abedini SS, Suckow V, Larti F, Beheshtian M, Lipkowitz B, Akhtarkhavari T, Mehvari S, Otto S, Mohseni M, Arzhangi S, Jamali P, Mojahedi F, Taghdiri M, Papari E, Soltani Banavandi MJ, Akbari S, Tonekaboni SH, Dehghani H, Ebrahimpour MR, Bader I, Davarnia B, Cohen M, Khodaei H, Albrecht B, Azimi S, Zirn B, Bastami M, Wieczorek D, Bahrami G, Keleman K, Vahid LN, Tzschach A, Gärtner J, Gillessen-Kaesbach G, Varaghchi JR, Timmermann B, Pourfatemi F, Jankhah A, Chen W, Nikuei P, Kalscheuer VM, Oladnabi M, Wienker TF, Ropers H-H, Najmabadi H. Genetics of intellectual disability in consanguineous families. Molecular Psychiatry 2018;24:1027–39. [DOI] [PubMed] [Google Scholar]
- 18.Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, Brunner HG, Buske OJ, Carey K, Doll C, Dumitriu S, Dyke SOM, Dunnen den JT, Firth HV, Gibbs RA, Girdea M, Gonzalez M, Haendel MA, Hamosh A, Holm IA, Huang L, Hurles ME, Hutton B, Krier JB, Misyura A, Mungall CJ, Paschall J, Paten B, Robinson PN, Schiettecatte F, Sobreira NL, Swaminathan GJ, Taschner PE, Terry SF, Washington NL, Züchner S, Boycott KM, Rehm HL. The Matchmaker Exchange: A Platform for Rare Disease Gene Discovery. Human Mutation 2015;36:915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaffer AE, Breuss MW, Caglayan AO, Al-Sanaa N, Al-Abdulwahed HY, Kaymakçalan H, Yılmaz C, Zaki MS, Rosti RO, Copeland B, Baek ST, Musaev D, Scott EC, Ben-Omran T, Kariminejad A, Kayserili H, Mojahedi F, Kara M, Cai N, Silhavy JL, Elsharif S, Fenercioglu E, Barshop BA, Kara B, Wang R, Stanley V, James KN, Nachnani R, Kalur A, Megahed H, Incecik F, Danda S, Alanay Y, Faqeih E, Melikishvili G, Mansour L, Miller I, Sukhudyan B, Chelly J, Dobyns WB, Bilguvar K, Jamra RA, Günel M, Gleeson JG. Biallelic loss of human CTNNA2, encoding αN-catenin, leads to ARP2/3 complex overactivity and disordered cortical neuronal migration. Nature Genetics 2018;50:1093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane SM, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA 2009;106:19096–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yüksel Z, Yazol M, Gümüş E. Pathogenic homozygous variations in ACTL6B cause DECAM syndrome: Developmental delay, Epileptic encephalopathy, Cerebral Atrophy, and abnormal Myelination. Am J Med Genet A 2019;179:1603–8. [DOI] [PubMed] [Google Scholar]
- 22.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, The Genome Aggregation Database Consortium, Neale BM, Daly MJ, MacArthur DG. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019;49:806–44. [Google Scholar]
- 25.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics 2014;46:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc 2015;11:1–9. [DOI] [PubMed] [Google Scholar]
- 27.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh C-L, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS, Sieh W. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet 2016;99:877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alirezaie N, Kernohan KD, Hartley T, Majewski J, Hocking TD. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am J Hum Genet 2018;103:474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seelow D, Schuelke M, Hildebrandt F, Nurnberg P. HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic Acids Research 2009;37:W593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grantham R Amino acid difference formula to help explain protein evolution. Science 1974;185:862–4. [DOI] [PubMed] [Google Scholar]
- 31.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP. PLoS Comput Biol 2010;6:e1001025–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015;31:2745–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature Methods 2010;7:575–6. [DOI] [PubMed] [Google Scholar]
- 35.Kohli MA, Cukier HN, Hamilton-Nelson KL, Rolati S, Kunkle BW, Whitehead PL, Züchner SL, Farrer LA, Martin ER, Beecham GW, Haines JL, Vance JM, Cuccaro ML, Gilbert JR, Schellenberg GD, Carney RM, Pericak-Vance MA. Segregation of a rare TTC3 variant in an extended family with late-onset Alzheimer disease. Neurol Genet 2016;2:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goyal S, Jäger M, Robinson PN, Vanita V. Confirmation of TTC8 as a disease gene for nonsyndromic autosomal recessive retinitis pigmentosa (RP51). Clin Genet 2016;89:454–60. [DOI] [PubMed] [Google Scholar]
- 37.Milev MP, Grout ME, Saint-Dic D, Cheng Y-HH, Glass IA, Hale CJ, Hanna DS, Dorschner MO, Prematilake K, Shaag A, Elpeleg O, Sacher M, Doherty D, Edvardson S. Mutations in TRAPPC12 Manifest in Progressive Childhood Encephalopathy and Golgi Dysfunction. Am J Hum Genet 2017;101:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, Benna C, D’Adamo P, Diodato D, Costa R, Mariotti C, Uziel G, Smiderle C, Zeviani M. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nature Genetics 2011;43:259–63. [DOI] [PubMed] [Google Scholar]
- 39.Adams CJ, Pike ACW, Maniam S, Sharpe TD, Coutts AS, Knapp S, La Thangue NB, Bullock AN. The p53 cofactor Strap exhibits an unexpected TPR motif and oligonucleotide-binding (OB)–fold structure. PNAS 2012;109:3778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 2000;101:199–210. [DOI] [PubMed] [Google Scholar]
- 41.Zeytuni N, Zarivach R. Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 2012;20:397–405. [DOI] [PubMed] [Google Scholar]
- 42.Adams CJ, Graham AL, Jansson M, Coutts AS, Edelmann M, Smith L, Kessler B, La Thangue NB. ATM and Chk2 kinase target the p53 cofactor Strap. EMBO reports 2008;9:1222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demonacos C, Krstic-Demonacos M, Smith L, Xu D, O’Connor DP, Jansson M, La Thangue NB. A new effector pathway links ATM kinase with the DNA damage response. Nature Cell Biology 2004;6:968–76. [DOI] [PubMed] [Google Scholar]
- 44.Kurosaki T, Maquat LE. Nonsense-mediated mRNA decay in humans at a glance. Journal of Cell Science 2016;129:461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindeboom RGH, Supek F, Lehner B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nature Genetics 2016;48:1112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoek TA, Khuperkar D, Lindeboom RGH, Sonneveld S, Verhagen BMP, Boersma S, Vermeulen M, Tanenbaum ME. Single-Molecule Imaging Uncovers Rules Governing Nonsense-Mediated mRNA Decay. Molecular Cell 2019;75:324–339.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dostie J, Dreyfuss G. Translation is required to remove Y14 from mRNAs in the cytoplasm. Current Biology 2002;12:1060–7. [DOI] [PubMed] [Google Scholar]
- 48.López M, Seidel V, Santibáñez P, Cervera-Acedo C, Castro-de Castro P, Domínguez-Garrido E. First case report of inherited Rubinstein-Taybi syndrome associated with a novel EP300 variant. BMC Med Genet 2016;17:97–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fergelot P, Van Belzen M, van Gils J, Afenjar A, Armour CM, Arveiler B, Beets L, Burglen L, Busa T, Collet M, Deforges J, de Vries BBA, Domínguez-Garrido E, Dorison N, Dupont J, Francannet C, García-Miñaur S, Gabau Vila E, Gebre-Medhin S, Gener Querol B, Genevieve D, Gérard M, Gervasini CG, Goldenberg A, Josifova D, Lachlan K, Maas S, Maranda B, Moilanen JS, Nordgren A, Parent P, Rankin J, Reardon W, Rio M, Roume J, Shaw A, Smigiel R, Sojo A, Solomon B, Stembalska A, Stumpel C, Suarez F, Terhal P, Thomas S, Touraine R, Verloes A, Vincent-Delorme C, Wincent J, Peters DJM, Bartsch O, Larizza L, Lacombe D, Hennekam RC. Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused by EP300 mutations. Am J Med Genet A 2016;170:3069–82. [DOI] [PubMed] [Google Scholar]
- 50.López M, García-Oguiza A, Armstrong J, García-Cobaleda I, García-Miñaur S, Santos-Simarro F, Seidel V, Domínguez-Garrido E. Rubinstein-Taybi 2 associated to novel EP300 mutations: deepening the clinical and genetic spectrum. BMC Med Genet 2018;19:36–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar S, Suthar R, Panigrahi I, Marwaha RK. Rubinstein-Taybi syndrome: Clinical profile of 11 patients and review of literature. Indian J Hum Genet 2012;18:161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The Transcriptional Coactivators p300 and CBP Are Histone Acetyltransferases. Cell 1996;87:953–9. [DOI] [PubMed] [Google Scholar]
- 53.Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM. Molecular cloning and functional analysis of the adenovirus E1A-assoclated 300-kD protein.(p300) reveals a protein with properties of a transcriptional adaptor. Genes And Development 1994;8:869–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available under reasonable request from the corresponding authors. The accession numbers for the TTC5 variants reported in this paper are: LOVD:0000602976 LOVD:0000604184 and dbGap:phs000288.v1.p1