

Abstract
INTRODUCTION:
Despite an ever-expanding catalog of noncoding elements that are implicated in the control of mammalian gene expression, how the regulatory input from multiple elements is integrated across a genomic neighborhood has remained largely unclear. This challenge is exemplified at Hox clusters (~100 to 200 kb), which contain genes that specify positional identity along the anterior-posterior axis of the developing embryo. In response to developmental morphogens such as retinoic acid (RA), the HoxA cluster splits into an active (HoxA1–5) and inactive domain (HoxA6–13) at the level of gene expression and chromatin. Although distal enhancers, intracluster transcription factor binding, and topological organization have emerged as the major regulatory modules in establishing this expression pattern, their relative contributions remain elusive.
RATIONALE:
Despite the advent of a vast suite of genome editing tools, it has remained challenging to simultaneously manipulate multiple regulatory elements across large genomic windows to deconvolve their relative contributions. Taking inspiration from the bottom-up approaches of synthetic biology and biochemical reconstitution, we developed “synthetic regulatory reconstitution” as a framework for the study of gene regulation to address this gap. The synthesis of large DNA constructs (>100 kb) permits any combination of complex modifications to be made, at the scale required to probe regulation across a native genomic neighborhood. We fabricated HoxA cluster variants that encode various combinations of the previously identified regulatory modules and integrated them into an ectopic location in the mouse genome. This enabled us to directly test the independent ability of these variant ectopic clusters to reconstitute distinct aspects of HoxA regulation.
RESULTS:
We harnessed the efficient homologous recombination machinery in yeast to construct four rat HoxA variants (130 to 170 kb) and delivered them at single copy to the housekeeping Hprt1 locus of mouse embryonic stem cells. Upon RA-induced differentiation, an ectopic HoxA cluster lacking distal enhancers (SynHoxA) induced both the appropriate subset of HoxA genes and the corresponding chromatin boundary. The presence of distal enhancers (Enhancers+SynHoxA) increased transcription levels, especially at early time points. Further, both SynHoxA and Enhancers+SynHoxA reorganized into active and inactive topological domains upon differentiation, mirroring the endogenous organization. The mutation of just four retinoic acid response elements (RAREs) present in SynHoxA (RAREΔ) almost completely eliminated any response of the ectopic cluster to RA, at the levels of both gene expression and chromatin reorganization. The addition of distal enhancers to RAREΔ could not fully rescue this loss of gene expression phenotype.
CONCLUSION:
Our data suggest that at HoxA, the primary module of active gene and chromatin boundary specification in response to RA is through the presence of internal transcription factor binding sites. Distal enhancers are dispensable for the specification of active genes but synergize with intracluster activator binding to boost the amount of transcription. Therefore, mammalian Hox clusters contain all the regulatory information that is necessary to convert a morphogenetic signal into a stable transcriptional, epigenetic, and topological state. This study showcases the power of synthetic regulatory reconstitution, a generalizable platform for the dissection of gene regulation at other loci in complex genomes.
Graphical Abstract

Bottom-up construction of variant mammalian genomic loci at the >100-kb scale. Making multiple edits on the same allele over large genomic windows has remained challenging in mammalian cells. Variant loci with an arbitrary number of changes can be constructed in yeast using synthetic DNA and site-specifically integrated into the genome of mammalian cells to study their behavior. BAC, bacterial artificial chromosome.
Precise Hox gene expression is crucial for embryonic patterning. Intra-Hox transcription factor binding and distal enhancer elements have emerged as the major regulatory modules controlling Hox gene expression. However, quantifying their relative contributions has remained elusive. Here, we introduce “synthetic regulatory reconstitution,” a conceptual framework for studying gene regulation, and apply it to the HoxA cluster. We synthesized and delivered variant rat HoxA clusters (130 to 170 kilobases) to an ectopic location in the mouse genome. We found that a minimal HoxA cluster recapitulated correct patterns of chromatin remodeling and transcription in response to patterning signals, whereas the addition of distal enhancers was needed for full transcriptional output. Synthetic regulatory reconstitution could provide a generalizable strategy for deciphering the regulatory logic of gene expression in complex genomes.
Mammalian Hox genes encode deeply conserved transcription factors (TFs) that are organized into four dense clusters (HoxA to HoxD) lacking other coding genes (1). In response to developmental morphogens, Hox genes are expressed along the anterior-posterior axis of the developing embryo in a spatial and temporal pattern that mirrors the organization of the genes within the cluster (2–4). The alteration of this “colinear” Hox gene expression pattern results in gross developmental defects or diseases such as cancer (5, 6).
In undifferentiated cells, where no Hox genes are expressed, the entire HoxA cluster (~130 kb) is targeted by Polycomb repressive complex 2 (PRC2) and marked by heterochromatin (7). Patterning signals such as retinoic acid (RA) and Wnt activate transcription through their downstream TFs. In response to RA, activated RA receptors (RARs) are bound to retinoic acid response elements (RAREs) found within the HoxA cluster. This is correlated with the separation of the cluster into two domains that contain transcribed and repressed genes, respectively (8–12) (Fig. 1A). RARE mutations can lead to decreased or abolished expression of the neighboring HoxA genes in the central nervous system, solidifying the notion that RAREs directly control Hox gene expression (9, 13, 14). However, the relationship between RARE activity and chromatin domain formation is not yet well established.
Fig. 1. HoxA regulation relies on the integration of multiple regulatory modules.
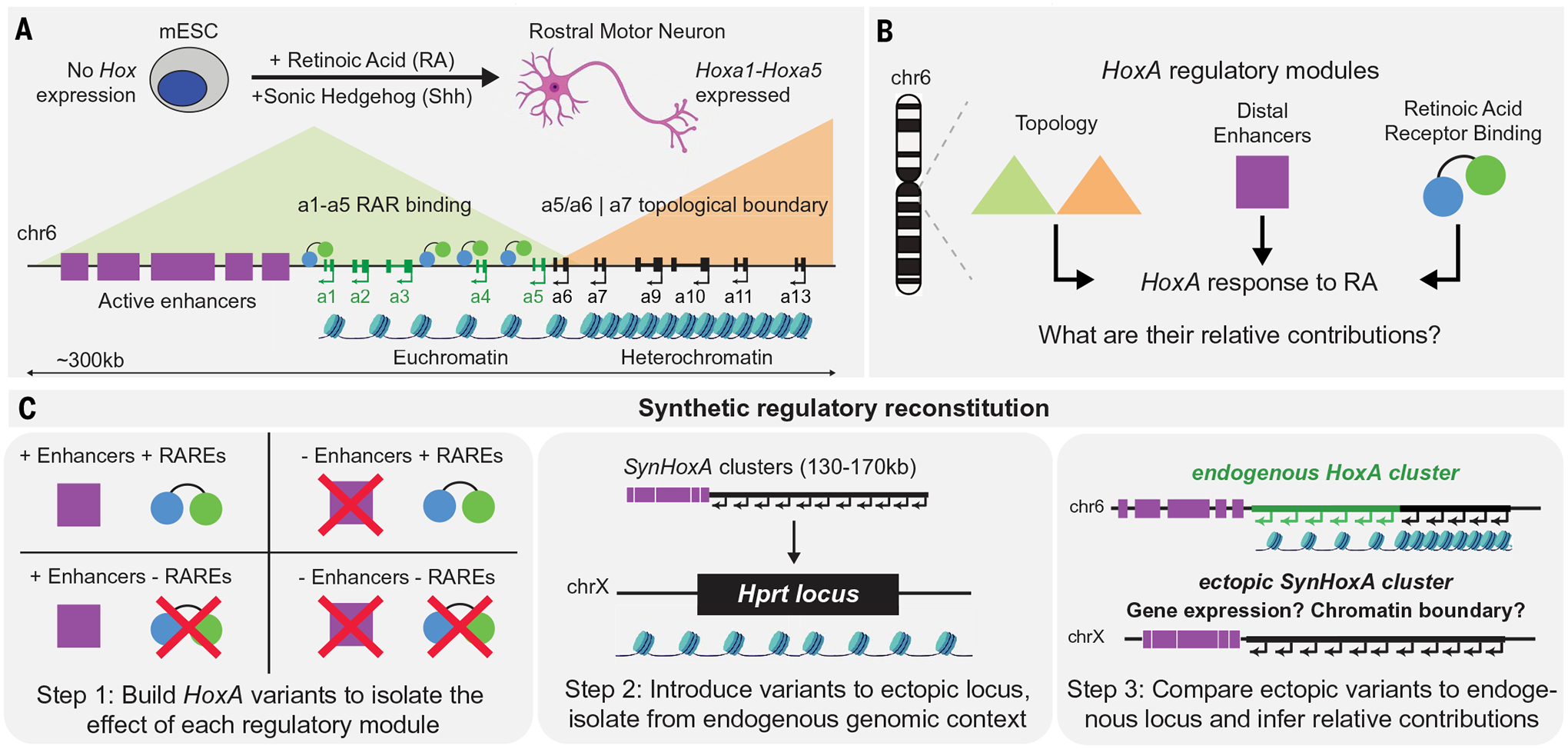
(A) Schematic of HoxA regulation in response to retinoic acid (RA) during in vitro mouse ES cell (mESC)–motor neuron differentiation. (B) Multiple regulatory modules, including enhancers, TF binding, and topology, are integrated to drive HoxA cluster response to RA. (C) Schematic of the synthetic regulatory reconstitution approach. Synthetic HoxA variants encoding various combinations of regulatory modules are built and integrated at an ectopic location in the genome. The response of these synthetic ectopic clusters to RA reveals their sufficiency and relative contribution in driving faithful HoxA expression.
A collection of intricate genetic manipulations has revealed a complex regulatory landscape surrounding the Hox clusters (15–19). For example, the HoxA cluster relies on several RA and Wnt responsive distal regulatory elements (enhancers) located in the gene-poor region between HoxA1 and the next gene, Skap2 (~300 kb away). Deleting some of these enhancers reduces, but never eliminates, HoxA expression in response to RA or Wnt (20–22). Distal enhancer access to the genes in the inactive domain is restricted by the formation of a strong topological boundary at CCCTC-binding factor (CTCF) sites within the cluster upon differentiation. Disruption of this boundary results in the misexpression of posterior HoxA genes in response to RA (15, 23, 24).
Thus, different regulatory modules are integrated to control Hox gene expression: local TF binding, distal enhancers, and topological DNA organization. However, a synergistic model describing their relative contribution and interactions has remained elusive (Fig. 1B).
The relative contribution of a cis-regulatory module could be measured by (i) generating a variant allele that only contains the elements that constitute the module, and (ii) isolating the variant allele from confounding factors, such as the compensatory effect of other regulatory elements in cis. Testing the necessity of individual elements by generating loss-of-function variants has received a lot of attention. However, tests of sufficiency have remained intractable owing to the inability to precisely manipulate DNA at a scale that accurately models the complexity and size of native loci (>100 kb) (25).
Short reporter constructs enable the study of variants only over a small genomic window (<10 kb) and also suffer from a lack of controlled genomic context, as they are largely randomly integrated or reside on episomal plasmids (26). Although they can test the ability of a sequence to drive expression, they lack the scale to study Hox cluster chromatin domain formation (>100 kb). Larger bacterial or yeast artificial chromosomes (BACs/YACs) can provide more genomic context and have been elegantly employed to understand regulation at Hox clusters (27–29). However, they are not easy to manipulate, making it challenging to generate a large number of variants that can be tested in vivo (26, 30, 31). Further, these large constructs are often randomly integrated into the genome, which confounds comparison across different constructs as a result of position effects and the integration of multiple copies. Methods for precise, single-copy integration of large DNA molecules in mammalian cells have not yet been applied to the study of Hox cluster regulation (32, 33). Despite the revolution in genome editing, it is still inefficient and time-consuming to make intricate structural rearrangements or multiple defined edits in cis, phased on a single homolog (34, 35).
We recently described a pipeline that harnesses the endogenous homologous recombination machinery in yeast to de novo assemble ~100-kb regions of mammalian genomes and integrate them into a defined location in mouse embryonic stem cells (mESCs) (36). This bottom-up assembly of genomic loci enables the introduction of an arbitrary number of variants in cis that are independent of any natural template.
Here, we apply and extend this technology to study the relative contributions of genomic context, distal enhancers, and intracluster regulatory elements to HoxA regulation. We constructed variants of the HoxA cluster (ranging from 130 to 170 kb) encoding combinations of the previously identified regulatory modules and integrated them into an ectopic locus, thereby isolating them from the native genomic neighborhood. We then asked whether the variant ectopic clusters were sufficient to reconstitute the transcriptional and epigenetic HoxA response to activating patterning signals (Fig. 1C).
Synthetic HoxA strategy and construction
All HoxA constructs described here were derived from rat (Rattus norvegicus) HoxA sequence, which shares ~90% sequence identity with the mouse HoxA sequence. This facilitated experiments in two distinct genetic backgrounds: (i) cells containing the endogenous HoxA cluster (HoxA+/+), allowing direct comparisons of expression levels to an internal control, and (ii) cells lacking endogenous mouse HoxA to eliminate potential sequence-mapping challenges (HoxA−/−).
We first constructed a 134-kb wild-type minimal rat HoxA cluster (SynHoxA) (Fig. 2 and fig. S1). SynHoxA contains all HoxA coding genes and encompasses the sequence corresponding to the contiguous domain repressed by H3K27me3 (trimethylated histone H3 Lys27) in undifferentiated mESCs. We produced polymerase chain reaction (PCR) amplicons from BACs bearing the rat HoxA cluster, with overlap between adjacent segments to enable homologous recombination. These amplicons were recombined in yeast to produce the 134-kb SynHoxA construct, termed an “assemblon” (fig. S1B). Edits to the assemblon can be made by switching the wild-type amplicons with synthetic DNA bearing the desired changes or by editing the assemblons directly using highly efficient, marker-free CRISPR/Cas9-based engineering in yeast (37). Assemblons were recovered from yeast into bacteria to purify large amounts of DNA (Fig. 2A). To test the contribution of distal enhancers to HoxA regulation, we fused all experimentally verified distal regulatory elements directly upstream of the core SynHoxA assemblon, generating the 170-kb Enhancers+SynHoxA construct (fig. S2).
Fig. 2. Build and delivery of SynHoxA constructs.
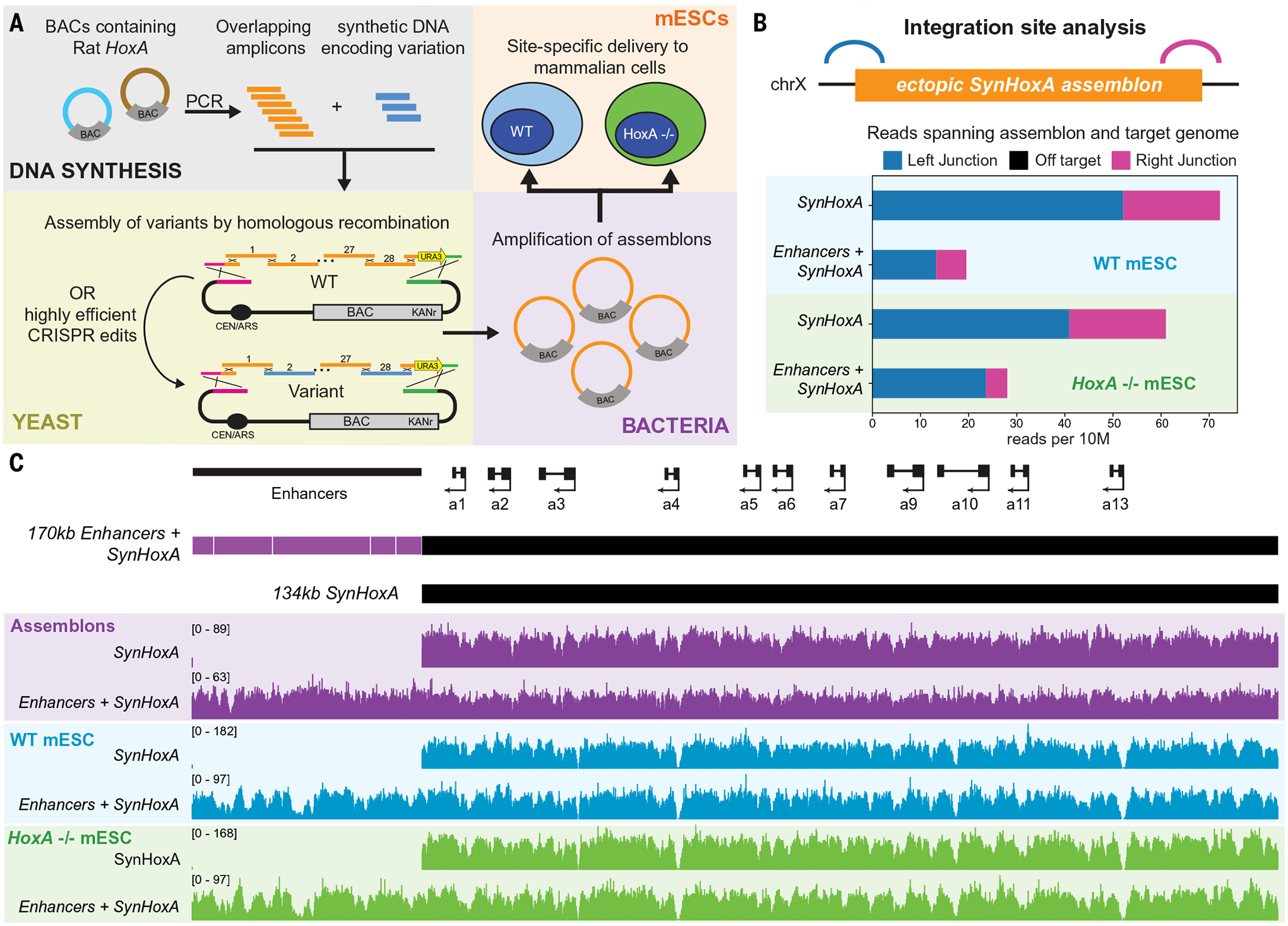
(A) Schematic of the process to generate mESCs bearing ectopic synthetic HoxA clusters via homologous recombination-based assembly in yeast and amplification in bacteria. (B) Analysis of reads spanning the synthetic construct and the host genome by bamintersect revealed only the expected junctions with no off-target integrations. (C) Schematic of the 134-kb SynHoxA and 170-kb Enhancers+SynHoxA constructs. Sequencing data for assemblon DNA isolated from bacteria (purple) and from capture sequencing after integration into wild-type (blue) and HoxA−/− (green) mESCs.
DNA sequencing at each step confirmed assemblon integrity (Fig. 2C and fig. S3). We detected mutations at a frequency of ~1 nucleotide per 6 kb, likely arising from PCR errors (fig. S3). We verified that none of these mutations were likely to affect the function of the clusters in our differentiation system described below (table S1).
All constructs were delivered to the Hprt locus on the X chromosome of both HoxA+/+ and HoxA−/− mESCs using Cre-mediated recombination (38) (figs. S4 and S5). We reasoned that Hprt would be an appropriate site to attempt regulatory reconstitution because it is a housekeeping gene with little regulatory activity in the surrounding regions, and its use as a “safe harbor” site is well documented (39, 40). Furthermore, transposing the HoxA cluster to an open locus would be the most stringent test of its ability to recruit the repressive PRC2 complex in undifferentiated cells. We used capture sequencing to verify that mESC clones contained the entire assemblon, specifically at Hprt and in single copy (41) (Fig. 2B).
Induction of SynHoxA gene expression during motor neuron differentiation
We investigated the response of the ectopic clusters to RA in a widely used differentiation system that recapitulates key aspects of ventral spinal cord development, including the activation of the appropriate HoxA genes (fig. S6A) (42, 43). Endogenous HoxA genes are repressed before exposure to patterning signals, and HoxA1–5 are induced in response to the “anterior” RA signal (10).
We performed RNA-seq on wild-type HoxA+/+ cells containing ectopic SynHoxA variants over the course of the differentiation protocol. Comparison to previously published control datasets by principal components analysis (PCA) revealed that the samples grouped largely according to time since RA treatment (44, 45) (fig. S6B). Cells containing SynHoxA variants down-regulated the expression of pluripotency markers, up-regulated the expression of markers of motor neuron differentiation, and induced HoxA1–5 from the endogenous cluster upon exposure to RA as expected (Fig. 3, A and C, and fig. S6, C and D). Thus, the presence of an ectopic synthetic cluster does not affect the ability of cells to differentiate appropriately after RA treatment.
Fig. 3. SynHoxA variants up-regulate the correct subset of genes in response to RA patterning signal.

(A to D) Fold change (FC) of RNA-seq data for endogenous mouse HoxA [(A) and (C)] and SynHoxA [(B) and (D)] genes during RA differentiation, relative to expression before RA treatment (n = 2). SynHoxA variants induce the correct genes (SynHoxA1–5) in response to RA. (E and F) Ratios of gene expression for SynHoxA genes to endogenous mouse HoxA genes (n = 2). Counts for the endogenous HoxA genes were halved to normalize for two endogenous HoxA versus one ectopic SynHoxA copy.
The ectopic Enhancers+SynHoxA cluster induced SynHoxA1–5 starting 24 hours after RA treatment (Fig. 3B and figs. S7 and S8, A and B), whereas SynHoxA6 to SynHoxA13 remained repressed throughout. Similarly, ectopic SynHoxA specifically induced SynHoxA1–5 (Fig. 3D and figs. S7 and S8C) without misexpression of any posterior genes. Thus, neither endogenous genomic context nor the wide spacing of enhancer elements is strictly required for a Hox cluster to induce the appropriate genes.
We quantified gene induction from the ectopic clusters by comparing each gene to its endogenous mouse HoxA counterpart (Fig. 3, E and F). Both ectopic clusters induced lower SynHoxA1 transcription, whereas SynHoxA2 induction surpassed the endogenous gene. The induction kinetics of SynHoxA3–5 were slower than HoxA3–5, but the mRNA levels became comparable after 96 hours, particularly in the Enhancers+SynHoxA construct. In general, Enhancers+SynHoxA induced higher levels of SynHoxA1–5 expression than SynHoxA, especially at early time points (Fig. 3 and figs. S7 and S8, B and C).
In our differentiation model, temporal colinearity of genes from the endogenous HoxA cluster is exhibited by (i) an early induction of HoxA1 (peaks at 24 hours) and its subsequent down-regulation and (ii) the sequential induction of HoxA2–5 from 24 to 96 hours. At 96 hours, the expression level is HoxA2 < HoxA3 < HoxA4 < HoxA5 (figs. S7 to S9). The minimal SynHoxA cluster does not recapitulate the temporal expression pattern of HoxA1. SynHoxA1 is expressed at low levels throughout but increases steadily with time, mirroring the regulation of SynHoxA2–5 genes. However, this is partially rescued with the addition of enhancers in Enhancers+SynHoxA. SynHoxA1 expression in this context peaks at 48 hours and decreases at 96 hours. The temporal expression of SynHoxA2–5 is retained in both constructs (fig. S9).
The observed bulk differences in gene expression between the ectopic clusters could be attributed either to a change in the number of cells that induce expression or to expression level changes in each cell. To address this, we investigated the response of the synthetic clusters to RA at the single-cell level using(i) HoxA5 antibody staining and (ii) RNA single-molecule fluorescence in situ hybridization for anterior SynHoxA genes. We performed these experiments in the HoxA−/− background. Gene expression from the ectopic clusters was not influenced by the presence or absence of the endogenous HoxA cluster (fig. S10), and the cells differentiated appropriately (fig. S11). In both methods, the observed bulk differences in expression were not due to a difference in the number of cells that express anterior SynHoxA genes, but due to an increase in the amount of expression per cell in the presence of the enhancers (figs. S12 to S15).
We investigated whether these findings could be extended to other patterning signals beyond RA, such as the posteriorizing Wnt signal (10, 22). Both ectopic clusters induced more posterior genes upon treatment with Wnt, up to SynHoxA11 (fig. S16). Intriguingly, the addition of distal enhancers does not seem to consistently modify the Wnt response across SynHoxA genes as much as it did for RA.
Together, these data support a model in which the HoxA cluster contains all the necessary information to decode patterning signals into the appropriate positional identity, independent of distal enhancers and the native genomic architecture. Distal enhancers are not required to specify active genes but have a critical role in modulating transcriptional output in response to RA. Whereas both ectopic clusters induce the correct subset of genes, additional elements may be required to fine-tune expression amplitude and timing.
Chromatin reorganization at SynHoxA clusters through differentiation
In undifferentiated cells, Hox clusters are carpeted with repressive H3K27me3 marks and recruit CTCF to potential boundary positions established in response to extracellular signals. The endogenous HoxA cluster responds to RA by separating into an active and inactive domain, forming a precise chromatin boundary between HoxA5 and HoxA6 (10, 23). We investigated the chromatin dynamics and CTCF recruitment of the relocated HoxA clusters by performing chromatin immunoprecipitation sequencing (ChIP-seq) in the HoxA−/− background to more reliably map reads to the ectopic clusters, thereby enhancing resolution.
Prior to differentiation, both Enhancers+SynHoxA and SynHoxA were entirely covered with high levels of H3K27me3. In addition, CTCF was recruited to the appropriate sites (Fig. 4, A and B). Thus, the ability to recruit all components for correct patterning is intrinsic to the HoxA cluster sequence and is independent of endogenous genomic context or distal enhancers. Upon RA treatment, Enhancers+SynHoxA remodeled chromatin in a manner that is similar to the endogenous cluster. H3K27me3 decreased in the SynHoxA1–5 domain and increased at SynHoxA6–13 (Fig. 4C). An increase in acetylated H3K27 (H3K27ac) complemented H3K27me3 removal from SynHoxA1–5. H3K27me3 was entirely cleared by 48 hours, slightly slower than the endogenous locus, which clears by 24 hours (10). Thus, the endogenous genomic context at HoxA is not required to translate an extracellular signal into an accurate epigenetic chromatin state.
Fig. 4. Distal enhancers are required for full clearance of repressive chromatin and the formation of a sharp chromatin boundary.
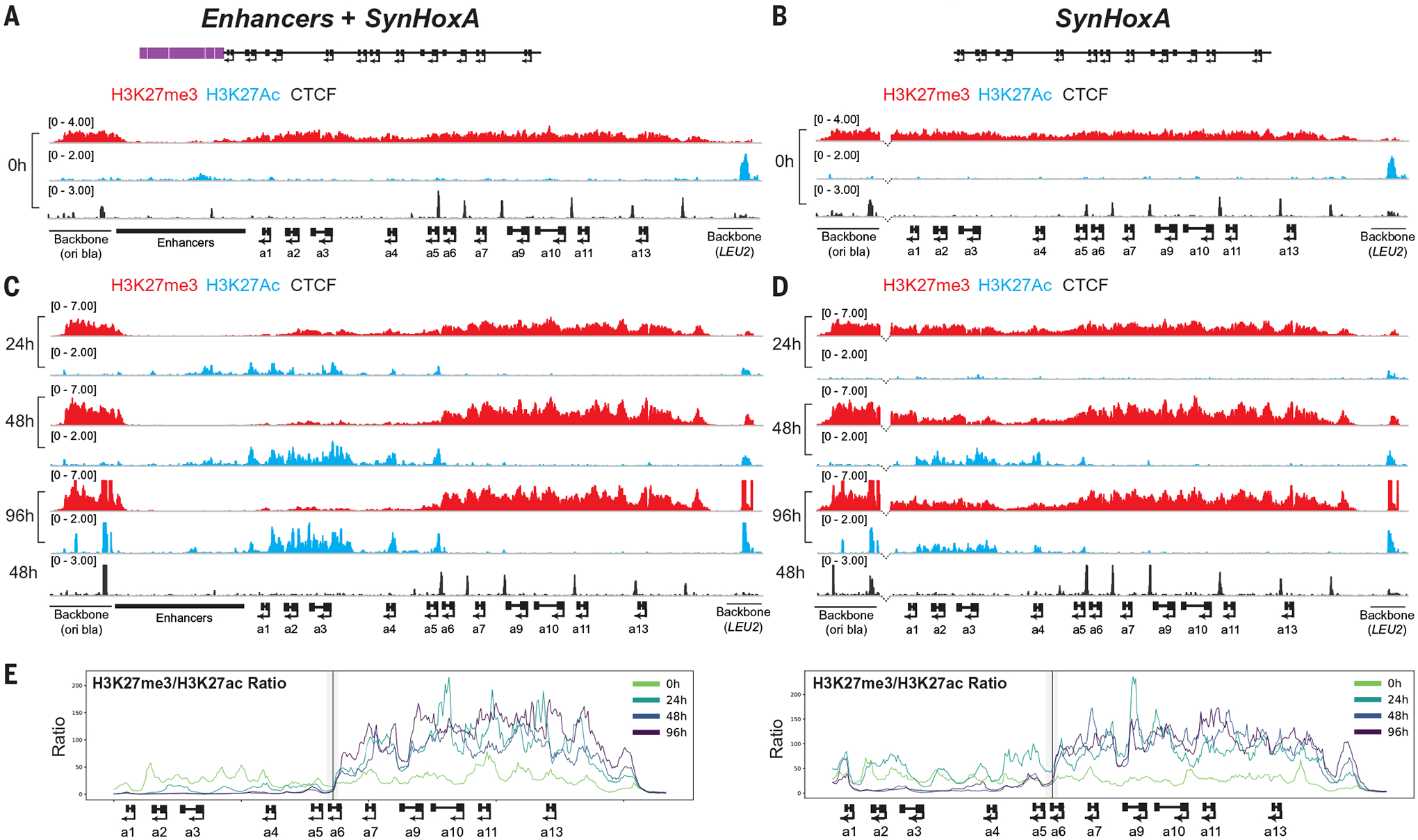
ChIP-seq analysis (n = 2) of activating H3K27ac (blue), repressive H3K27me3 (red), and CTCF (black) at SynHoxA clusters. (A and B) In undifferentiated cells, SynHoxA clusters contained H3K27me3, lacked H3K27ac, and recruited CTCF to correct locations. (C and D) In response to RA, the SynHoxA1–5 domain contained activating H3K27ac and cleared repressive H3K27me3. Dotted lines at the anterior breakpoint between the cluster and the vector sequences in (B) and (D) indicate presence of enhancer sequences in the reference genome to which no reads are mapped. (E) RPKM (reads per kilobase per million mapped reads) normalized ratios of repressive H3K27me3 to active H3K27ac across SynHoxA. The black line marks the HoxA5-HoxA6 CTCF site; gray shading indicates the windows contributing to ChIP-seq signal at the site.
The minimal SynHoxA cluster also recruited H3K27ac to anterior genes upon RA activation (Fig. 4D). Unlike the endogenous HoxA cluster and Enhancers+SynHoxA, H3K27me3 was not entirely lost from the SynHoxA1–5 domain during differentiation (Fig. 4D). Nonetheless, SynHoxA formed the appropriate, albeit weak, chromatin boundary at the SynHoxA5–a6 CTCF binding site (Fig. 4E and fig. S17, A and B). Thus, the minimal SynHoxA cluster has the intrinsic ability to induce dynamic chromatin domains, independent of genomic context or enhancers.
Topological organization of ectopic SynHoxA clusters
The three-dimensional structure of the HoxA cluster changes during motor neuron differentiation, transitioning from a single association domain to two domains containing active or repressed chromatin (15, 23, 24). We investigated the topological organization of the ectopic clusters by performing Hi-C, a technique for detecting genome-wide chromatin interactions, during differentiation (0 and 48 hours after RA treatment). Both clusters formed self-associating domains in undifferentiated cells without generating a de novo topologically associating domain (TAD) boundary (fig. S18). Similar to the endogenous cluster, Enhancers+SynHoxA broke into two domains during differentiation, with enhancers and active genes in one domain and repressed genes in the other (fig. S18A). The minimal SynHoxA similarly transitioned from a compact self-associated state in undifferentiated cells into two domains during differentiation (fig. S18B). We did not observe strong evidence for trans-chromosomal interactions between the ectopic locus and its endogenous enhancers (tables S2 and S3). Thus, the ectopic HoxA clusters have the intrinsic ability to self-organize in three dimensions, mirroring the expression and chromatin changes that occur upon differentiation.
The RARE sites within the HoxA cluster are required for the RA response
The minimal SynHoxA transformed the RA signal into the correct transcriptional and chromatin programs. This behavior could theoretically depend on RAREs located within the HoxA1–5 domain, other sequences within the cluster, or interactions with other regulatory elements at the ectopic locus. To distinguish between these possibilities, we built a third construct lacking both RAREs and enhancers (RAREΔ SynHoxA) and integrated it into wild-type and HoxA−/− mESCs (figs. S19 and S20) (9).
Cells carrying RAREΔSynHoxA differentiated and induced endogenous HoxA genes appropriately (Fig. 5A and fig. S20, D and E). The ectopic cluster was decorated with H3K27me3 in mESCs, indicating that RAREs are not required to recruit the repressive chromatin machinery. However, unlike the previous constructs, RAREΔ SynHoxA failed to up-regulate the expression of SynHoxA1–5 or to form a chromatin boundary in response to RA (Fig. 5, B to D, and figs. S8D and S17C). SynHoxA5 was the only gene with any signal, potentially due to a poorly characterized RAR binding site located between HoxA5 and HoxA6 (9, 10). This null phenotype indicates that the behavior of ectopic SynHoxA clusters reflects their innate regulatory potential and not the effect of novel interactions formed at the Hprt locus.
Fig. 5. Retinoic acid receptor response element (RARE) sites are required for the RA response.
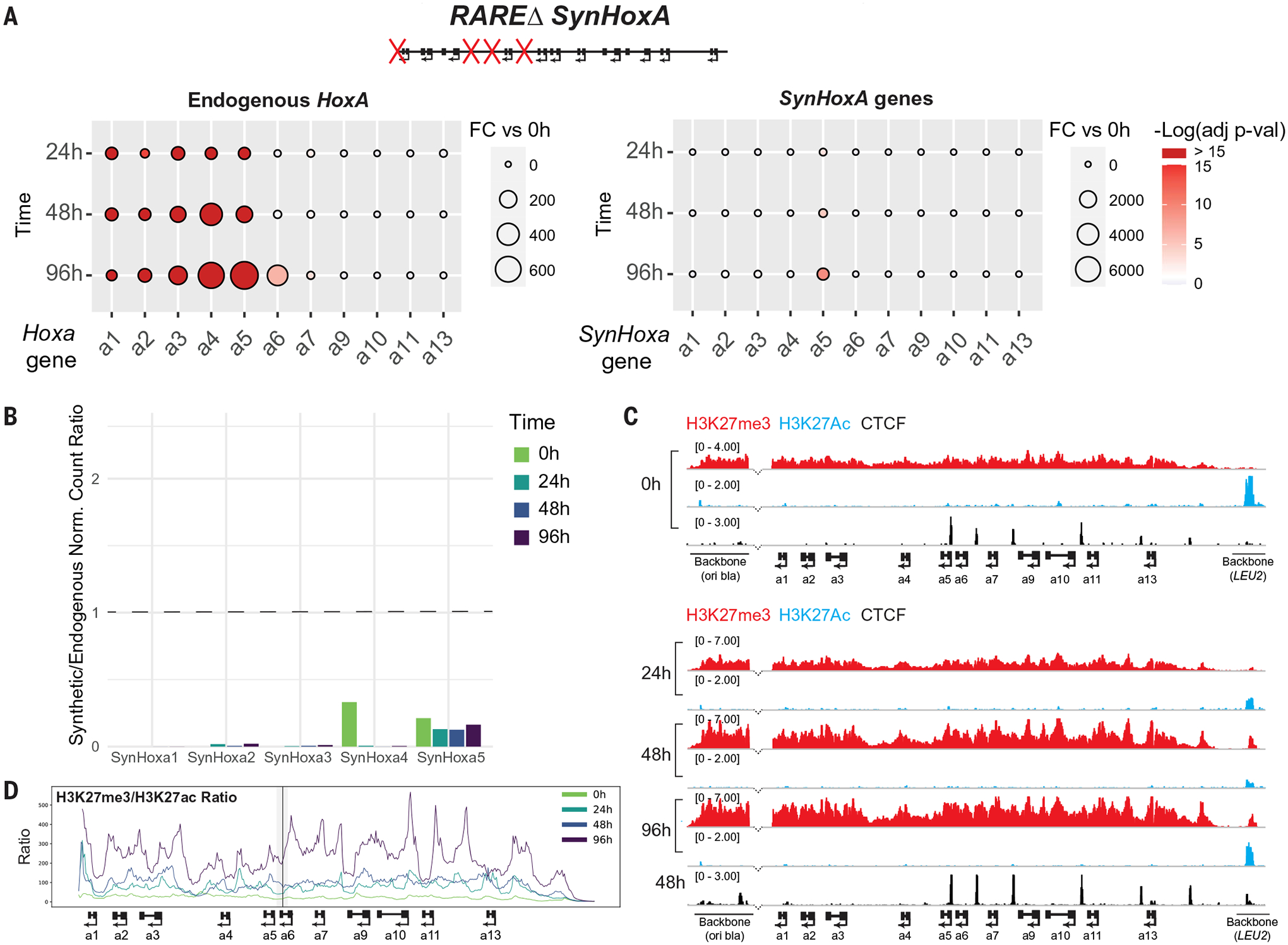
(A) Fold change of RNA-seq data for endogenous HoxA and SynHoxA genes during differentiation (n = 2). (B) Ratios of gene expression for SynHoxA genes to endogenous mouse HoxA genes (n = 2). (C) ChIP-seq revealed no evidence of H3K27me3 (red) clearance and H3K27ac (blue) recruitment at RAREΔ SynHoxA. Dotted lines show the anterior breakpoint between the cluster and the vector sequences in (C) as in Fig. 4. (D) Ratio of repressive H3K27me3 to active H3K27ac chromatin across SynHoxA as in Fig. 4E.
This lack of RA response provided the ideal background to measure the independent contribution of distal enhancers to HoxA gene regulation. To that end, we built and integrated a fourth construct: Enhancers+RAREΔ SynHoxA, with the distal enhancers inserted upstream of RAREΔ SynHoxA (figs. S19 and S20). In Enhancers+RAREΔ SynHoxA, SynHoxA2–5 were induced at low levels in response to RA (Fig. 6, A and B, and fig. S8E). A faint chromatin boundary formed at the appropriate location between SynHoxA5 and SynHoxA6 (Fig. 6, C and D, and fig. S17D). This suggests that the distal enhancers may have a weak ability to activate HoxA gene transcription without the driving force provided by internal RAREs, or that they synergize with the poorly characterized RARE still present in this construct.
Fig. 6. The addition of enhancers to SynHoxA RAREΔ does not rescue gene expression.
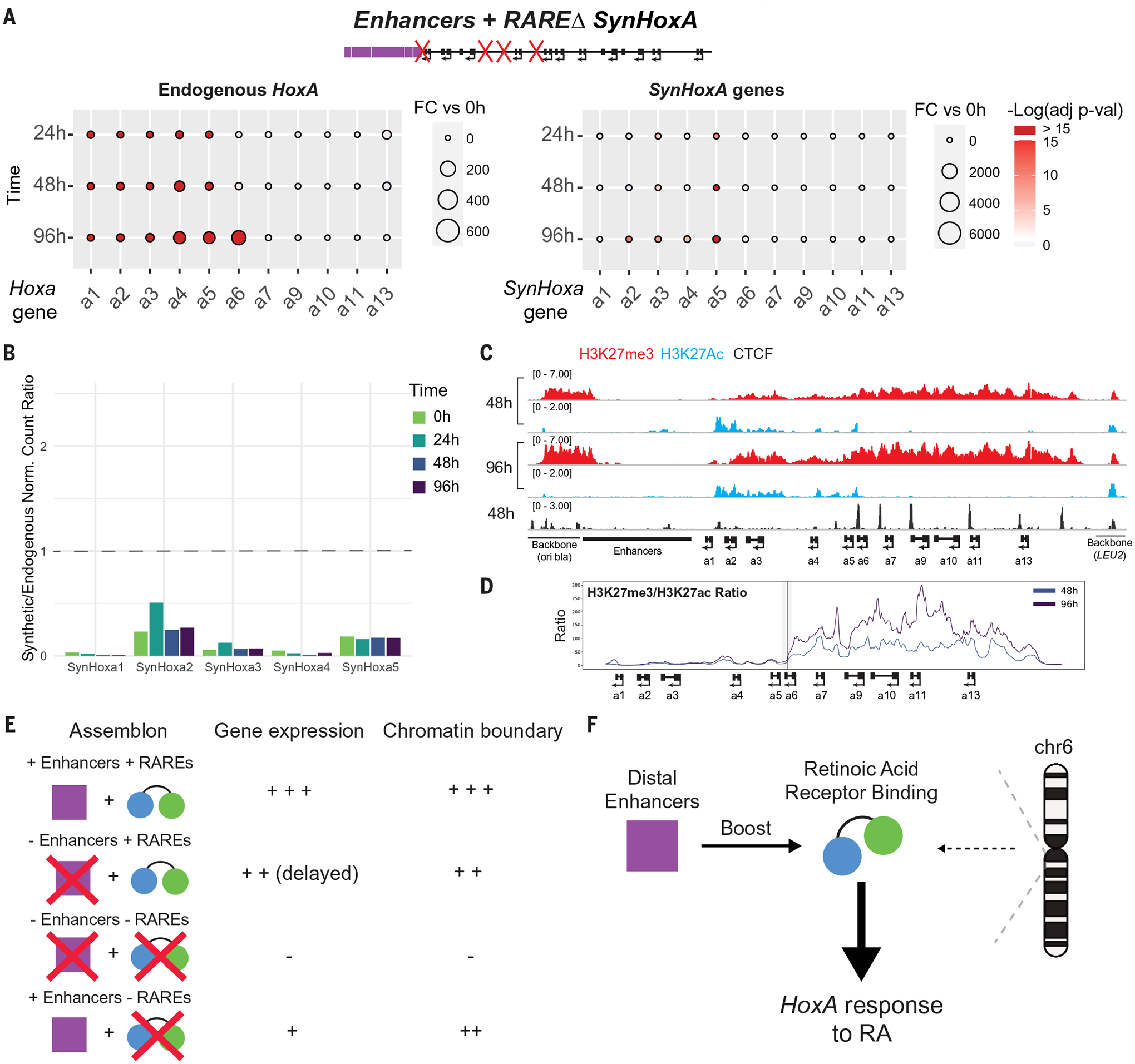
(A) Fold change of RNA-seq data for endogenous HoxA and SynHoxA genes during differentiation (n = 2). (B) Ratios of gene expression for SynHoxA genes to endogenous mouse HoxA genes (n = 2). (C) ChIP-seq revealed the appropriate recruitment of CTCF (black) and the formation of a weak chromatin boundary at Enhancers+RAREΔ SynHoxA upon differentiation (n = 2). (D) Ratio of repressive H3K27me3 to active H3K27ac chromatin across SynHoxA as in Fig. 4E. (E) Summary of gene expression and chromatin boundary phenotypes across all SynHoxA clusters. (F) Model describing relative contributions of distal enhancers, intra-Hox binding, and genomic context to the RA response at HoxA.
Discussion
We developed a “synthetic regulatory reconstitution” approach to characterize the relative contributions of distal enhancers and intracluster TF binding in specifying distinct aspects of HoxA regulation. A minimal ectopic cluster lacking distal enhancers induced the correct subset of genes in response to two developmental signals (RA and Wnt). The presence of distal enhancers increased the transcriptional output from the ectopic cluster in response to RA, especially at earlier time points. These results are consistent with previous studies in which deletion of enhancers from the endogenous locus resulted in lower Hox gene transcription in response to morphogens (20, 22, 46). At the time point we investigated, the ectopic cluster with distal enhancers did not induce higher expression in response to Wnt. This may indicate that some Wnt-responsive enhancers are missing in these constructs, or that the distal enhancers are ineffective in this configuration. Alternatively, earlier time points may reveal differences in transcriptional dynamics or output between these constructs.
The importance of both RAREs and distal enhancers to Hox gene expression was firmly established before this work. However, it was difficult to quantify their relative contributions to the establishment of transcriptional and chromatin domains at the scale of an entire Hox cluster with previous techniques. Our results show that an ectopic cluster lacking all previously described internal RAREs (RAREΔ SynHoxA) failed to respond to RA both at the transcriptional and chromatin levels. This gene expression phenotype was not fully rescued by the addition of distal enhancers in Enhancers+RAREΔ SynHoxA. However, some chromatin remodeling was observed. Together, removal of internal RAREs led to virtually complete loss of gene expression, whereas removal of enhancers led to a reduction in expression at early time points, with almost complete rescue at later time points. Therefore, distal enhancers do not induce high levels of transcription or induce drastic chromatin remodeling in the absence of intracluster RAREs, but synergize with the RAREs to play a critical role in fine-tuning expression levels (Fig. 6, E and F).
All ectopic clusters recruited CTCF and PRC2 in embryonic stem cells, implying that this property is Hox cluster–intrinsic. Therefore, precise CTCF positioning within the cluster does not depend on interactions with other elements at the endogenous TAD boundary. Together, our results imply that Hox clusters are discrete units with an intrinsic ability to respond to patterning signals, strengthening original observations made at the HoxD cluster using random BAC transgenesis (28, 29). This is also congruent with the idea that the evolution of novel distal enhancers is a source of morphological novelty in secondary structures such as limbs (16, 47).
The inability of the minimal SynHoxA to fully clear repressive marks in the anterior domain could have several causes. First, the boost in transcription provided by distal enhancers at early time points might facilitate the clearance of repressive chromatin. Second, the enhancers could serve as platforms to recruit additional chromatin modifiers. Finally, the vector backbone that is introduced as part of the delivery harbors repressive chromatin modifications throughout differentiation (Fig. 4). The spread of repressive chromatin from this region might be more effectively prevented by enhancer sequences that contain fewer CpG islands than the cluster itself. Future experiments using scarless delivery methods will enable us to distinguish between these hypotheses (41). Although we see no strong genetic or topological evidence for trans-chromosomal interactions, we cannot fully exclude the possibility that they may play some part in activating SynHoxA genes. Higher-resolution chromatin conformation data centered on the ectopic clusters may help to address this question.
This study represents a proof of principle for “synthetic regulatory reconstitution.” Targeting large, fully editable constructs to precise genomic locations enables quantitative comparisons between variants and promises to address critical questions in gene regulation and genome organization. Multiple elements required for the finer analysis of constructs through differentiation, such as live-cell imaging of transcription and chromatin mobility, could also be included via bottom-up synthesis (48). Testing different ectopic sites such as those marked with constitutive heterochromatin, cross-species transplants of regulatory landscapes, and phenotyping in richer systems such as living mice and gastruloids are all attractive avenues to explore (49).
Differences from endogenous gene expression dynamics were observed, even for the Enhancers+SynHoxA construct. Thus, even the largest construct does not contain all the regulatory information required for refining gene expression. The great value in pursuing the synthetic regulatory reconstitution strategy is realized in cases where endogenous regulation cannot be fully recapitulated. This points to gaps in knowledge that we can attempt to fill by building successively larger or more intricate ectopic constructs in the future until no differences are observed when compared to the endogenous cluster. Reconstitution is a powerful framework for dissecting complex biochemical processes because it allows for exquisite control over components of the system under study (50, 51). By analogy, our approach allows for the generation of locus-scale variant constructs with any combination of desired changes. We expect synthetic regulatory reconstitution to be a fundamental component of the toolbox for studying transcriptional regulation.
Methods summary
A full description of the methods can be found in the supplementary materials. In brief, SynHoxA constructs were fabricated in yeast and integrated into mESCs as described (36, 41). SynHoxA mESCs were differentiated to motor neurons and characterized by RNA-seq, ChIP-seq, and Hi-C as described (10, 44, 45). Sequencing data were analyzed using custom pipelines.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Mazzoni, Boeke, and Holt labs as well as the Institute for Systems Genetics community for their support, M. Khalfan (Genomics Core Facility at NYU) for making the reform tool publicly available, B. Ragipani for preliminary analysis on motor neuron differentiation markers, N. Zesati and S. Arora for help with preliminary visualization of Hi-C data, the Experimental Pathology core at NYU Langone for help with sectioning, and J. Skok and D. Reinberg for their insights.
Funding:
Supported by NHGRI grant RM1HG009491 (J.B., M.T.M., and E.O.M.), NINDS grant R01NS100897 and NIGMS grant R01GM138876 (E.O.M.), New York State Stem Cell Science predoctoral training grant C322560GG (M.B.), NIH grants R01AG075272 and R01GM127538 and Melanoma Research Foundation Award 687306 (T.L.), and NIH grant F32CA239394 (B.R.K.).
Footnotes
Competing interests: J.D.B. is a founder and director of CDI Labs Inc.; a founder of and consultant to Neochromosome Inc.; a founder of, scientific advisory board member of, and consultant to ReOpen Diagnostics LLC; and past or present scientific advisory board member of Sangamo Inc., Modern Meadow Inc., Rome Therapeutics Inc., Sample6 Inc., Tessera Therapeutics Inc., and the Wyss Institute. The other authors declare no competing interests.
Data and materials availability:
All sequencing data from this study are deposited in NCBI GEO under GSE190906.
REFERENCES AND NOTES
- 1.Duboule D, The rise and fall of Hox gene clusters. Development 134, 2549–2560 (2007). doi: 10.1242/dev.001065; [DOI] [PubMed] [Google Scholar]
- 2.Kmita M, Duboule D, Organizing axes in time and space;25 years of colinear tinkering. Science 301, 331–333 (2003). doi: 10.1126/science.1085753; [DOI] [PubMed] [Google Scholar]
- 3.McGinnis W, Krumlauf R, Homeobox genes and axial patterning. Cell 68, 283–302 (1992). doi: 10.1016/0092-8674(92)90471-N; [DOI] [PubMed] [Google Scholar]
- 4.Duboule D, Morata G, Colinearity and functional hierarchy among genes of the homeotic complexes. Trends Genet 10, 358–364 (1994). doi: 10.1016/0168-9525(94)90132-5; [DOI] [PubMed] [Google Scholar]
- 5.Lewis EB, A gene complex controlling segmentation in Drosophila. Nature 276, 565–570 (1978). doi: 10.1038/276565a0; [DOI] [PubMed] [Google Scholar]
- 6.Shah N, Sukumar S, The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 10, 361–371 (2010). doi: 10.1038/nrc2826; [DOI] [PubMed] [Google Scholar]
- 7.Margueron R, Reinberg D, The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). doi: 10.1038/nature09784; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahony S et al. , Ligand-dependent dynamics of retinoic acid receptor binding during early neurogenesis. Genome Biol 12, R2 (2011). doi: 10.1186/gb-2011-12-1-r2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolte C, De Kumar B, Krumlauf R, Hox genes: Downstream “effectors” of retinoic acid signaling in vertebrate embryogenesis. Genesis 57, e23306 (2019). doi: 10.1002/dvg.23306; [DOI] [PubMed] [Google Scholar]
- 10.Mazzoni EO et al. , Saltatory remodeling of Hox chromatin in response to rostrocaudal patterning signals. Nat. Neurosci 16, 1191–1198 (2013). doi: 10.1038/nn.3490; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noordermeer D et al. , The dynamic architecture of Hox gene clusters. Science 334, 222–225 (2011). doi: 10.1126/science.1207194; [DOI] [PubMed] [Google Scholar]
- 12.Soshnikova N, Duboule D, Epigenetic temporal control of mouse Hox genes in vivo. Science 324, 1320–1323 (2009). doi: 10.1126/science.1171468; [DOI] [PubMed] [Google Scholar]
- 13.Dupé V et al. , In vivo functional analysis of the HoxA-1 3′ retinoic acid response element (3’RARE). Development 124, 399–410 (1997). doi: 10.1242/dev.124.2.399; [DOI] [PubMed] [Google Scholar]
- 14.Frasch M, Chen X, Lufkin T, Evolutionary-conserved enhancers direct region-specific expression of the murine HoxA-1 and HoxA-2 loci in both mice and Drosophila. Development 121, 957–974 (1995). doi: 10.1242/dev.121.4.957; [DOI] [PubMed] [Google Scholar]
- 15.Lonfat N, Montavon T, Darbellay F, Gitto S, Duboule D, Convergent evolution of complex regulatory landscapes and pleiotropy at Hox loci. Science 346, 1004–1006 (2014). doi: 10.1126/science.1257493; [DOI] [PubMed] [Google Scholar]
- 16.Montavon T, Duboule D, Chromatin organization and global regulation of Hox gene clusters. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20120367 (2013). doi: 10.1098/rstb.2012.0367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montavon T, Soshnikova N, Hox gene regulation and timing in embryogenesis. Semin. Cell Dev. Biol 34, 76–84 (2014).doi: 10.1016/j.semcdb.2014.06.005; [DOI] [PubMed] [Google Scholar]
- 18.Montavon T et al. , A regulatory archipelago controls Hox genes transcription in digits. Cell 147, 1132–1145 (2011). doi: 10.1016/j.cell.2011.10.023; [DOI] [PubMed] [Google Scholar]
- 19.Berlivet S et al. , Clustering of tissue-specific sub-TADs accompanies the regulation of HoxA genes in developing limbs. PLOS Genet 9, e1004018 (2013). doi: 10.1371/journal.pgen.1004018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao K et al. , SET1A/COMPASS and shadow enhancers in the regulation of homeotic gene expression. Genes Dev 31, 787–801 (2017). doi: 10.1101/gad.294744.116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Kumar B et al. , Analysis of dynamic changes in retinoid-induced transcription and epigenetic profiles of murine Hox clusters in ES cells. Genome Res 25, 1229–1243 (2015). doi: 10.1101/gr.184978.114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neijts R et al. , Polarized regulatory landscape and Wnt responsiveness underlie Hox activation in embryos. Genes Dev 30, 1937–1942 (2016). doi: 10.1101/gad.285767.116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narendra V et al. , CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021 (2015). doi: 10.1126/science.1262088; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narendra V, Bulajić M, Dekker J, Mazzoni EO, Reinberg D, CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev 30, 2657–2662 (2016). doi: 10.1101/gad.288324.116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrov N et al. , Technological challenges and milestones for writing genomes. Science 366, 310–312 (2019). doi: 10.1126/science.aay0339; [DOI] [PubMed] [Google Scholar]
- 26.Gasperini M, Starita L, Shendure J, The power of multiplexed functional analysis of genetic variants. Nat. Protoc 11, 1782–1787 (2016). doi: 10.1038/nprot.2016.135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehoczky JA, Innis JW, BAC transgenic analysis reveals enhancers sufficient for HoxA13 and neighborhood gene expression in mouse embryonic distal limbs and genital bud. Evol. Dev 10, 421–432 (2008). doi: 10.1111/j.1525-142X.2008.00253.x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spitz F, Gonzalez F, Duboule D, A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell 113, 405–417 (2003). doi: 10.1016/S0092-8674(03)00310-6; [DOI] [PubMed] [Google Scholar]
- 29.Spitz F et al. , Large scale transgenic and cluster deletion analysis of the HoxD complex separate an ancestral regulatory module from evolutionary innovations. Genes Dev 15, 2209–2214 (2001). doi: 10.1101/gad.205701; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peterson KR et al. , Use of yeast artificial chromosomes (YACs) in studies of mammalian development: Production of beta-globin locus YAC mice carrying human globin developmental mutants. Proc. Natl. Acad. Sci. U.S.A 92, 5655–5659 (1995). doi: 10.1073/pnas.92.12.5655; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heintz N, BAC to the future: The use of bac transgenic mice for neuroscience research. Nat. Rev. Neurosci 2, 861–870 (2001). doi: 10.1038/35104049; [DOI] [PubMed] [Google Scholar]
- 32.Liberante FG, Ellis T, From kilobases to megabases: Design and delivery of large DNA constructs into mammalian genomes. Curr. Opin. Syst. Biol 25, 1–10 (2021). doi: 10.1016/j.coisb.2020.11.003 [DOI] [Google Scholar]
- 33.Wallace HA et al. , Manipulating the mouse genome to engineer precise functional syntenic replacements with human sequence. Cell 128, 197–209 (2007). doi: 10.1016/j.cell.2006.11.044; [DOI] [PubMed] [Google Scholar]
- 34.McCarty NS, Graham AE, Studená L, Ledesma-Amaro R, Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun 11, 1281 (2020). doi: 10.1038/s41467-020-15053-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraft K et al. , Deletions, Inversions, Duplications: Engineering of Structural Variants using CRISPR/Cas in Mice. Cell Rep 10, 833–839 (2015). doi: 10.1016/j.celrep.2015.01.016; [DOI] [PubMed] [Google Scholar]
- 36.Mitchell LA et al. , De novo assembly and delivery to mouse cells of a 101 kb functional human gene. Genetics 218, iyab038 (2021). doi: 10.1093/genetics/iyab038; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DiCarlo JE et al. , Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41, 4336–4343 (2013). doi: 10.1093/nar/gkt135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iacovino M et al. , Inducible cassette exchange: A rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cells 29, 1580–1588 (2011). doi: 10.1002/stem.715; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jasin M, Moynahan ME, Richardson C, Targeted transgenesis. Proc. Natl. Acad. Sci. U.S.A 93, 8804–8808 (1996). doi: 10.1073/pnas.93.17.8804; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasperini M et al. , CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet 101, 192–205 (2017). doi: 10.1016/j.ajhg.2017.06.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brosh R et al. , A versatile platform for locus-scale genome rewriting and verification. Proc. Natl. Acad. Sci. U.S.A 118, e2023952118 (2021). doi: 10.1073/pnas.2023952118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wichterle H, Lieberam I, Porter JA, Jessell TM, Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397 (2002). doi: 10.1016/S0092-8674(02)00835-8; [DOI] [PubMed] [Google Scholar]
- 43.Peljto M, Wichterle H, Programming embryonic stem cells to neuronal subtypes. Curr. Opin. Neurobiol 21, 43–51 (2011). doi: 10.1016/j.conb.2010.09.012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aydin B et al. , Proneural factors Ascl1 and Neurog2 contribute to neuronal subtype identities by establishing distinct chromatin landscapes. Nat. Neurosci 22, 897–908 (2019). doi: 10.1038/s41593-019-0399-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bulajić M et al. , Differential abilities to engage inaccessible chromatin diversify vertebrate Hox binding patterns. Development 147, dev.194761 (2020). doi: 10.1242/dev.194761; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su G et al. , CTCF-binding element regulates ESC differentiation via orchestrating long-range chromatin interaction between enhancers and HoxA. J. Biol. Chem 296, 100413 (2021). doi: 10.1016/j.jbc.2021.100413; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freitas R, Gómez-Marín C, Wilson JM, Casares F, Gómez-Skarmeta JL, HoxD13 contribution to the evolution of vertebrate appendages. Dev. Cell 23, 1219–1229 (2012).doi: 10.1016/j.devcel.2012.10.015; [DOI] [PubMed] [Google Scholar]
- 48.Sato H, Das S, Singer RH, Vera M, Imaging of DNA and RNA in Living Eukaryotic Cells to Reveal Spatiotemporal Dynamics of Gene Expression. Annu. Rev. Biochem 89, 159–187 (2020). doi: 10.1146/annurev-biochem-011520-104955; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beccari L et al. , Multi-axial self-organization properties of mouse embryonic stem cells into gastruloids. Nature 562, 272–276 (2018). doi: 10.1038/s41586-018-0578-0; [DOI] [PubMed] [Google Scholar]
- 50.Ganzinger KA, Schwille P, More from less - bottom-up reconstitution of cell biology. J. Cell Sci 132, jcs227488 (2019). doi: 10.1242/jcs.227488; [DOI] [PubMed] [Google Scholar]
- 51.Liu AP, Fletcher DA, Biology under construction: In vitro reconstitution of cellular function. Nat. Rev. Mol. Cell Biol 10, 644–650 (2009). doi: 10.1038/nrm2746; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data from this study are deposited in NCBI GEO under GSE190906.