

SUMMARY
The fields of proteomics and machine learning are both large disciplines, each producing well over 5,000 publications per year. However, studies combining both fields are still relatively rare, with only about 2% of recent proteomics papers including machine learning. This review, which focuses on the intersection of the fields, is intended to inspire proteomics researchers to develop skills and knowledge in the application of machine learning. A brief tutorial introduction to machine learning is provided, and research advances that rely on both fields, particularly as they relate to proteomics tools development and biomarker discovery, are highlighted. Key knowledge gaps and opportunities for scientific advancement are also enumerated.
INTRODUCTION
The term machine learning was introduced in 1959 by Arthur Samuel, who “taught” a computer to learn the game of checkers; the machine could beat Samuel after 8–10 h of learning the game through successive playing.1 Samuel’s seminal paper and ideas sparked imagination and amazement among the masses. Today, machine learning assists society daily, when people ask Siri to tell a joke or interact with chatbots that set up medical appointments.
Within the scope of biomedical science, machine learning is driving discoveries in fields that have found few successes based on human-derived knowledge. For example, while effective medications for treating or preventing Alzheimer’s disease have been sought for decades with little success, a recent study combining machine learning and thousands of medical records suggests that the erectile dysfunction drug, sildenafil, may provide substantial protection against memory decline.2
Capturing the power and potential of machine learning is not only exciting but necessary. It can enable discoveries that affect the world on a timescale that maximizes the usefulness of the knowledge gained. This review focuses on the intersection of machine learning and proteomics, and the goal is to catalyze the application of these mathematical tools by proteomics researchers so that new discoveries in proteomics can be imagined by humans and enabled by the assistance of machines. While we provide a brief introduction to proteomics, we assume familiarity with the field and its methods on the part of the reader.
PROTEOMICS IN BRIEF
Proteomics, in one sentence, is a large-scale comparison of the expression of the proteins in a set of biologically derived samples, typically from a particular cell type or biofluid. Since this review highlights examples of machine learning on clinical proteomics samples, a typical work flow for those types of experiments is briefly described next. For complete newcomers to the proteomics field, a more in-depth treatment of the topic will be necessary but is beyond the scope of this piece: two well-known reviews are recommended.3,4 Existing practitioners may, instead, enjoy reading a modern perspective on the field and its future, written by industrial experts.5
Figure 1 describes a typical bottom-up proteomics experiment using isobaric mass tagging. Quantitative information is obtained about each protein detected, and this overall approach is commonly implemented for clinical proteomics experiments that include a machine learning component. Samples from both a control group and a test group are first subjected to a variety of processing steps, typically including depletion of the highly abundant proteins, proteolytic digestion, and isotopic labeling. The digestion step usually involves first reducing the disulfide bonds and alkylating the resulting free cysteines, followed by treatment with one or more proteases, where use of trypsin is most common. Isotopic labeling can be done in a variety of ways, but the use of a tandem mass tag (TMT) reagent is a common choice. The labeled peptide samples are then combined into batches such that each sample can still be uniquely identified by its encoded label. These batches are then subjected to chromatographic separation, and the eluent is directed into the mass spectrometer where high-resolution mass spectrometry (MS) data and tandem MS (MS/MS) data are acquired. Both these MS data types are used, in combination with proteomics software, which leverages human proteome databases and predicted fragmentation patterns of the resulting peptides that would come from those databases to assign the acquired mass spectra to known peptides and, by extension, the proteins from which they originate. The peptides are quantified for all the individual samples within each batch in an isotope-encoded experiment based on the characteristic, mass-encoded product ions that are generated during the MS/MS or MS3 experiments. Peptide-level information is also frequently agglomerated and reported at the protein level. After each batch has been characterized, the data from all the batches are recombined so that the full dataset can be leveraged to identify the underlying protein expression changes. The combination step can involve batch normalization steps and/or filtering out proteins that were detected in only a small minority of the samples.
Figure 1. Example of a quantitative proteomics workflow.

Samples undergo treatment prior to labeling and combining into batches. Each batch separately undergoes liquid chromatography-tandem mass spectrometry (LC-MS/MS) and data analysis. Individual datasets are then processed and recombined to a single dataset of samples and features. Data from each person in the dataset occupy a unique row, while the abundances of each of the proteins quantified are in columns.
While the work flow shown in Figure 1 is one common way to acquire quantitative proteomics data for machine learning experiments, it is by no means the sole approach used in the proteomics field. For example, the MS/MS data, needed for assigning the peptides, can be collected in either a data-dependent or a data-independent fashion. In the former approach, the most abundant peaks in any given high-resolution MS scan can be targeted, one at a time, for MS/MS studies. This data-dependent approach is often used with TMT tagging, as shown in Figure 1. Alternatively, larger swaths of the mass range can be selected sequentially for collisions-induced dissociation (CID) experiments, and all the precursors within the range are fragmented simultaneously. This approach, known as data-independent acquisition (DIA), produces fragmentation data for more peptides, particularly for less-abundant species, but the caveat is that the data are harder to assign, since multiple precursors are fragmented together. Further information on DIA can be found in a recent review.6 Another alternative work flow is to forego the proteolytic digestion step and analyze intact proteins. This paradigm, top-down proteomics, enables studies of protein modifications occurring in concert, and the field has its own hurdles and opportunities.7
Two places where machine learning combines with proteomics are in the peptide analysis step and at the end of the workflow, where the protein expression changes can be used to predict a disease state on a set of patient samples. Significant developments in both subfields are described herein, after an introduction to machine learning.
MACHINE LEARNING DEMYSTIFIED
Just as proteomics is an umbrella term, encompassing a variety of workflows and concepts, the phrase machine learning has a similarly broad meaning. This review focuses on a subset of the machine learning field, supervised classification; its application to proteomics could be described as leveraging generalized mathematical tools that use data from a set of known sample types to make predictions about samples of unknown type. Examples of sample types include a disease state versus a healthy one, or peptides with a particular sequence or characteristic (e.g., a retention time). One important distinction for newcomers to appreciate is how machine learning is different from software development strategies that proteomics researchers have historically used to assign their mass spectral data.
In the expert-writes-software-based approach, which dominated early peptide assignment algorithms, researchers would use their knowledge to write rules that guide the assignment of new data. Consider the task of determining whether ions undergoing MS/MS are glycosylated peptides. Using an expert-based approach, rule writers may specify which ions need to be present for an MS/MS spectrum to be considered a glycopeptide; the abundances of the key ions are also likely included as part of the algorithm that assigns the spectrum to either being glycosylated or not. Those rules would be based on the development teams’ expectations and knowledge of the diagnostic ions that appear in MS/MS data of glycopeptides.
By contrast, in a machine learning paradigm, no pre-set rules specific to the task of interest (determining whether or not the precursor is a glycopeptide) are required; rather, what are needed are existing data and a generalized math tool, the machine learning algorithm, that could be used to match the known data to the given outcomes. In this hypothetical case, the known data would be derived from the MS/MS data of precursors that are known to be glycosylated or not. The outcome would be the answer to whether or not each example in the dataset was from a glycosylated peptide. The rules for assigning new data to the appropriate class (glycosylated or not) would be generated by the machine learning algorithm, based on the data provided in the training set. Importantly, the same machine learning algorithm could also be used in a vast array of other applications; for example, to determine the likelihood that an online shopper would purchase a specific product or to identify the political candidate that an individual is likely to vote for. In these alternate cases, different types of related examples of existing data are needed, but the math algorithm could stay the same.
The salient point is that machine learning algorithms used in proteomics have no MS-specific components or expert-defined rules in them, and, as a result, they can be applied to solve complex problems where the approach to optimally assigning new data is not obvious. However, the heart of these algorithms is not some Franken-steinian computers-learning-to-be-smarter-than-humans sci-fi magic, as it is sometimes depicted on the Internet. Rather, the algorithms are, in their essence, a set of mathematical manipulations. Machine learning could in principle be done by paper and pencil, if anyone had the patience and the exacting precision to accurately perform millions of simple calculations by hand.
The most common type of machine learning associated with the applications of interest herein, namely, proteomics tools development and biomarker discovery problems, is supervised classification; sometimes an up-front feature selection step is also included. The key steps and options for these workflows are briefly described here. An in-depth example of a feature selection study using proteomics data can be found in Dakna et al.,8 and high-quality publications with more detail on supervised classification can also be found.9,10
Figure 2 shows the two basic components needed for supervised classification studies: existing datasets and math algorithms. Let us first consider the requirements for the datasets. In some types of studies, where proteomics and machine learning are combined, developing the set of known data to train the algorithm is straightforward. For example, if the goal is to use a clinical proteomics dataset, such as the one described in Figure 1, to build a model that could predict the disease state of new patients, researchers could directly use the matrix containing the (normalized) protein abundances from each sample that was produced at the end of the work flow in Figure 1. The basic requirements for any dataset are that the features (for example, the protein abundances) are useful in predicting the class (whether or not the person is healthy, for example). The final, normalized datasets from proteomics experiments are, therefore, already ready for machine learning. In other applications, the question of what goes into the dataset is more perplexing. When machine learning is used as part of a work flow where the goal is to generate a proteomics tool, more thought needs to go into considering the best data to use that will give an accurate answer to the question at hand. The machine learning algorithm will typically give researchers a yes/no type answer (e.g., do I have cancer? Yes or no. Is this peptide correctly assigned? Yes or no), although multiple-choice answers (e.g., choosing A, B, C, D, etc.) are also possible. In cases where machine learning is incorporated to build or improve a proteomics software tool, choosing both the question that machine learning can answer and the data that will provide that answer takes more skill. Questions such as “Is the precursor ion a glycosylated peptide?,” for example, would require careful planning to generate an optimal set of features to include for each sample in the dataset. These may simply be the product ions and their abundances, but the features could be more complex than that; they could be neutral losses from the precursor, ratios of specific peaks, etc.
Figure 2. Required components and example work flow for supervised classification.
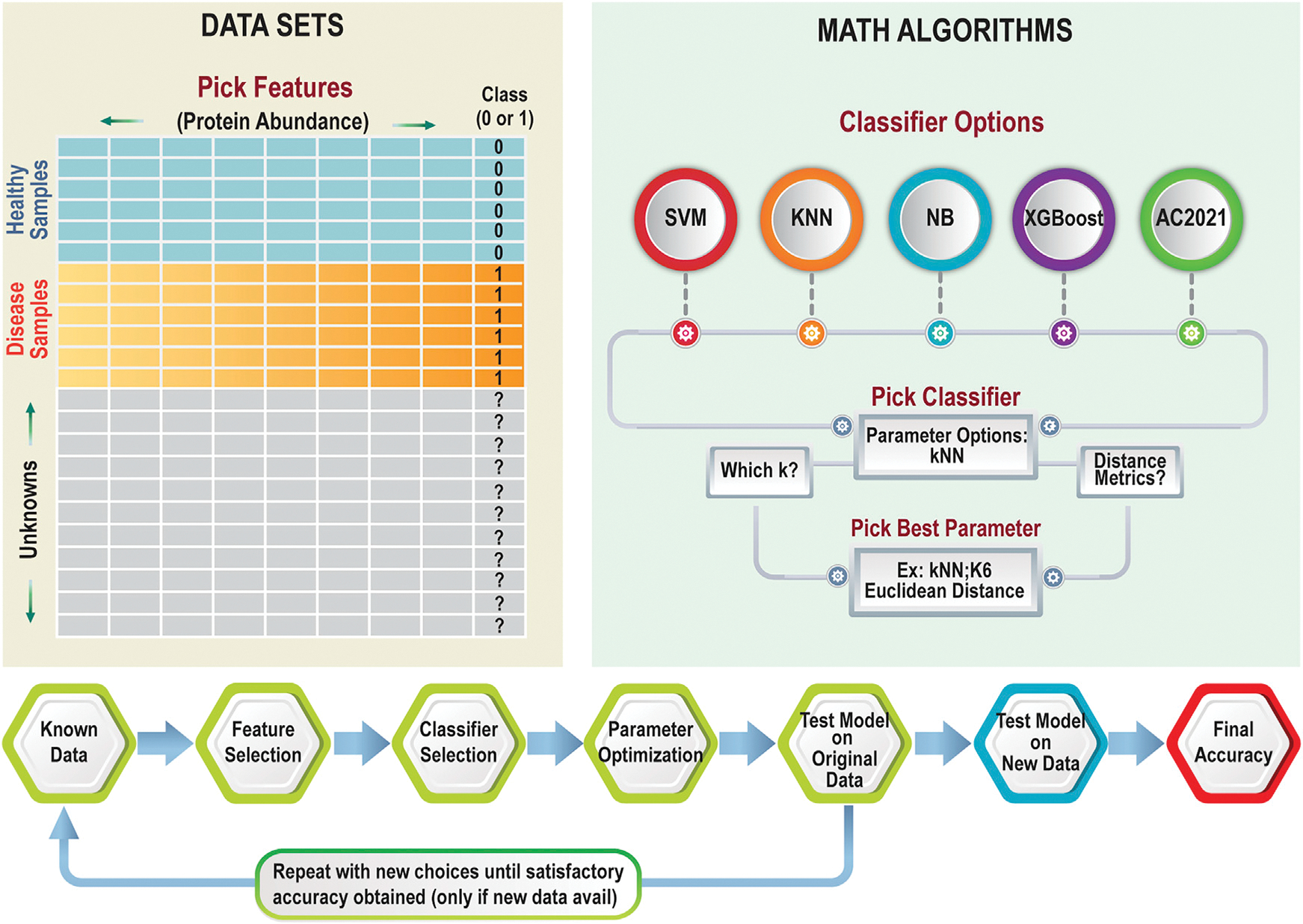
Data input requirements (left) include a data table of samples with known outcomes (health status, for example) and features that could be used to determine the status in unknown cases. Several standard classifier options are shown on the right; once the classifier is selected, decisions about its hyperparameters are also made. The work flow (bottom) shows one logical way in which supervised machine learning can be done: one first makes decisions about the features to use, then tests a classifier, then adjusts hyperparameters, using cross-validation to make decisions (along the way) about which options are optimal. Finally, after a satisfactory model is built, it must be tested on new data to determine the accuracy reliably.
One way to improve the chances of supervised classification generating a highly predictive model is to use math strategies that identify the most useful data in the dataset to keep and to remove the non-useful information. For example, proteomics researchers may want to down-select their protein set from an initial list of 12,000 proteins to a biomarker panel of the four proteins whose abundances are, together, most predictive of the disease state. This down-selection process is known by the general term feature selection. Three feature selection studies are cited as examples of applying this technique.8,11,12 Feature selection methods can be an important part of the workflow, when the ultimate goal of the experiment is to develop a biomarker panel with just a few proteins that may be monitored in clinical laboratories without doing a proteomics experiment.
After one decides on the existing samples and features to use, the math tool(s) for classification need to be selected and applied (Figure 2). Some classifiers to consider, and examples where they have been used, include k-nearest neighbors (kNNs),9,12 support vector machine (SVM),11,13 extreme gradient boosting (XGBoost),14,15 naive Bayes (NB),12,16 or the Aristotle Classifier (AC.2021).13,17 While an in-depth mathematical discussion of each of these methods is beyond the scope of this review, newcomers should understand that the methods have different underlying principles used to optimize their models, and these different principles result in some methods having strengths or weaknesses in certain domains. For example, both XGBoost and AC.2021 can classify datasets with missing values, a common problem in proteomics datasets. The other methods (kNN, SVM, and NB) require researchers to provide only datasets with no missing values or to find ways to provide reasonable approximations for those unreported values. (This estimation process, called imputation, has its own subdomain of machine learning research.)
Deep learning strategies are also heavily used in supervised classification;18 however, to really benefit from the additional complexity that deep learning brings to bear on a classification problem, datasets with very large numbers of samples and few features are optimal. In the referenced example, the dataset contained 1,000 samples and 34 features.18 Comparisons with larger feature sets show that deep learning does not necessarily give better results than those obtained by simpler classifiers.19 A recent review of deep learning in proteomics covers this field in more depth.20
In addition to selecting a classifier, a machine learning expert would typically tune the classifier to optimally perform with the specific type of data in hand. This tuning process involves adjusting hyperparameters. One can think of this process as “bending the rules” for the given classifier until an optimal result is obtained. Tuning is typically done using cross-validation, and the concept is discussed in more depth elsewhere.9,10
Depending on the problem to be solved, different test metrics can be used to assess the final model. Sometimes overall accuracy is most important, but not always. For example, if the goal is to identify patients at risk of a rare disease, a model that identifies everyone as not at risk might be highly accurate for the general population but completely useless for diagnostic purposes. In a case like this, a metric other than overall accuracy would need to be used.
Because there are so many ways to optimize the results that come out of a machine learning work flow—by doing feature selection, classifier selection, and tuning hyperparameters—researchers should always strive to test their solution on new data that have never been any part of the machine learning workflow. If this is not possible, because the data simply do not exist, then obtaining an estimate of accuracy by cross-validation is typically used in lieu of a test set.
The use of cross-validation to estimate accuracy, instead of using a test set, is problematic in some cases, and feature selection is one clear-cut example where completely new test data are needed to assess the accuracy of the method.21,22 In one radical demonstration of this point, a dataset of simulated proteomics data, which was generated using only random numbers, was used to develop a machine learning-based model for classifying mock patient samples as healthy or diseased. The authors used all the (randomly generated) data from all the samples for feature selection, then built a classification model and tested it using cross-validation. This approach resulted in a model that was, alarmingly, >90% accurate, even though datasets of random numbers should generate a model with ~50% accuracy.21 This demonstration study emphasizes that unrealistically high accuracies are obtained when no naive test data are available to assess the utility of the feature selection steps. A similar, earlier study, geared toward genomics researchers, reaches the same conclusion.22 In cases where the sample numbers are small, and no secondary set of test samples are available, supervised classification can be applied without implementing feature selection steps, and, in this case, cross-validation provides a more accurate estimate of the model’s true performance.21 This approach has been used in several published studies; two example are provided.13,23
MACHINE LEARNING INCORPORATED INTO PROTEOMICS TOOLS
Researchers have benefitted from the marriage of machine learning and proteomics for decades, perhaps without realizing it; some of the proteomics field’s early essential tools, PeptideProphet24 and Percolator,25 leverage these capabilities. PeptideProphet was the first widely successful automated method of determining which software-generated peptide assignments, made by matching MS/MS data to database candidates, were likely to be correct and which were likely to be incorrect.24 To build this tool, developers first generated a reliable ground-truth dataset containing thousands of well-characterized CID spectra of known peptides from 18 proteins; they used these data to determine which measurable parameters (i.e., mass error of the precursor) were most important in distinguishing correct from incorrect assignments using Bayesian statistics.24 They then used this information to generate a single test metric that provided a very reliable probability-of-correctness score for each peptide assignment. The development of a robust, automated approach to determine which software-generated assignments were likely correct provided the rigor and rapid analytics necessary to catalyze growth in the burgeoning field of proteomics.
Similar in ultimate objective, but different in method and approach, is Percolator. This software script increases peptide identifications after analysis by peptide assignment algorithms such as Sequest,25 but it does so without ever having generated an expert-curated ground-truth dataset. Instead, a decoy database is used to generate the known incorrect assignments, and a fraction of the highest-scoring spectra are used to generate a set of the ground-truth correct assignments. Also different from PeptideProphet is the underlying machine learning approach. The algorithm driving Percolator uses an SVM to build a model that separates good assignments from poor ones. The developers demonstrated that applying their approach can increase the number of assigned peptides by 50%.25,26 Both these early tools demonstrate the immense value of using math algorithms and ground-truth data to drive broad advancements in the development of the field of proteomics. They also demonstrate that, while a set of known data and a classifier are the two essential components needed for machine learning, the careful selection of each of these is the art behind the science.
Twenty years after the development of PeptideProphet, the question of how to best assign proteomics data in an automated fashion is still being answered. One of today’s biggest challenges is assessing MS/MS data that combine multiple possible precursors; this data type is ubiquitous in DIA datasets, where precursor ions are not individually selected for collisional activation but rather activated in sets. DIA-NN is an example of a widely known proteomics tool that attacks this challenge.27 DIA-NN relies on a spectral library as its known dataset and employs a neural network to build its classification strategy.27 Neural networks are examples of deep learning, which, in the broadest sense, is an approach that better captures trends in the data when the features contribute to the overall outcome through complex relationships.
The transition from shallow learning methods, such as SVM used in Percolator, to deep learning methods, used 15 years later in DIA-NN, is not entirely unpredictable. Computational power has come a long way in the last 15 years, enabling some deep learning studies to be executable on laptops. Furthermore, while deep learning methods require high sample numbers in their training datasets, these datasets are relatively easy to generate for unmodified peptides. Consequently, the field is now frequently turning to deep learning strategies to assist in the assignment of unmodified and simply modified peptides. Additional examples of deep learning-based tools include pDeep, which is a notable early tool from 2017 that assigns CID data to peptide compositions.28 In another example, Prosit uses deep learning and a dataset with over half a million spectra to predict fragmentation spectra for peptides; the tool can be used to assign peptide compositions to experimental data with high accuracy.29 In parallel with these publications, several more algorithms leveraging machine learning, and specifically deep learning, are emerging for assisting in the task of assigning MS/MS data to their correct peptide sequences. These approaches typically show performance enhancements over the older algorithms.
Proteomics analyses encompasses more complexity than simply identifying all the unmodified peptides in the proteome; one key example is the need to identify peptide modifications; where complexity can be found, machine learning can contribute. A relatively old example in this field, from 2013, demonstrated that proteins modified by ubiquitination could be identified more readily by UbiProber, a tool that relies on supervised classification with SVM.30 A host of other tools are now present to assist in identifying other modifications, including phosphorylation,31 nitration,32 glutarylation,33 malonylation,34 threonine isoforms,35 and O-GlcNAc modification.36 All of these rely on supervised classification as at least one key step in the algorithm. These first-generation machine learning methods analyzing post-translational modifications (PTMs) could likely be replaced by even better products as both the machine learning field and the proteomics field mature.
The above-mentioned peptide modifications have one shared element of simplicity: the modification is a fixed mass. When researchers need to identify multitudes of modifications on a peptide (or protein), as in top-down proteomics studies, or when the modification can have a highly variable mass, such as for glycosylated peptides, then the analysis problem, and the best tool to solve it, become more complicated. An algorithm that leveraged machine learning to analyze N-linked glycosylation sites, called Sweetheart, was published in 2013,37 but the glycoproteomics field still relies predominantly on tools built using rules inferred by human learning, with Byonic, a commercial product, being the field leader currently.38 Newer automated methods are emerging, both those with human-learning-inspired algorithms and those that include machine learning; while the verdict is still out on which approach will ultimately reign superior, the authors’ money is being bet on the machines.
Like glycoproteomics, the field of top-down proteomics can make a strong case that its data analysis challenges are supremely complex. Best practices for analysis of proteins using top-down strategies have recently been reviewed;7 machine learning is beginning to make inroads,39 but it is not yet contributing heavily in this field, and numerous analysis challenges remain. The Ge lab has recently shown progress on one of the problems, data deconvolution, which is an (automated) process of grouping the MS peaks into their isotopic envelopes. They demonstrate that a machine learning strategy, using data deconvolution results from multiple algorithms as input, does a better job than any single algorithm.40 Data deconvolution may be “low-hanging fruit” in the orchard of top-down proteomics data analysis, but starting by incorporating machine learning into this component of the work flow makes sense; it exemplifies a long-held computing principle that a complex problem can be solved by finding creative ways to break the seemingly impossible task into sets of smaller problems, each of which is solvable.
Moving forward, machine learning will certainly continue to improve proteomics data analysis, and second-generation PTM analysis tools will likely emerge. We expect, though, that machine learning may offer the most advantage to fields with high-complexity analyses, such as glycoproteomics and top-down proteomics, as shown in Figure 3, where the appropriate analysis rules cannot be easily inferred. The biggest barriers to overcome are the creative development of the question that machine learning can optimally answer and the generation of the datasets that will allow a generalized math algorithm to untangle the complexity of the problem to find an optimal solution.
Figure 3. Potential impact of machine learning on proteomics.
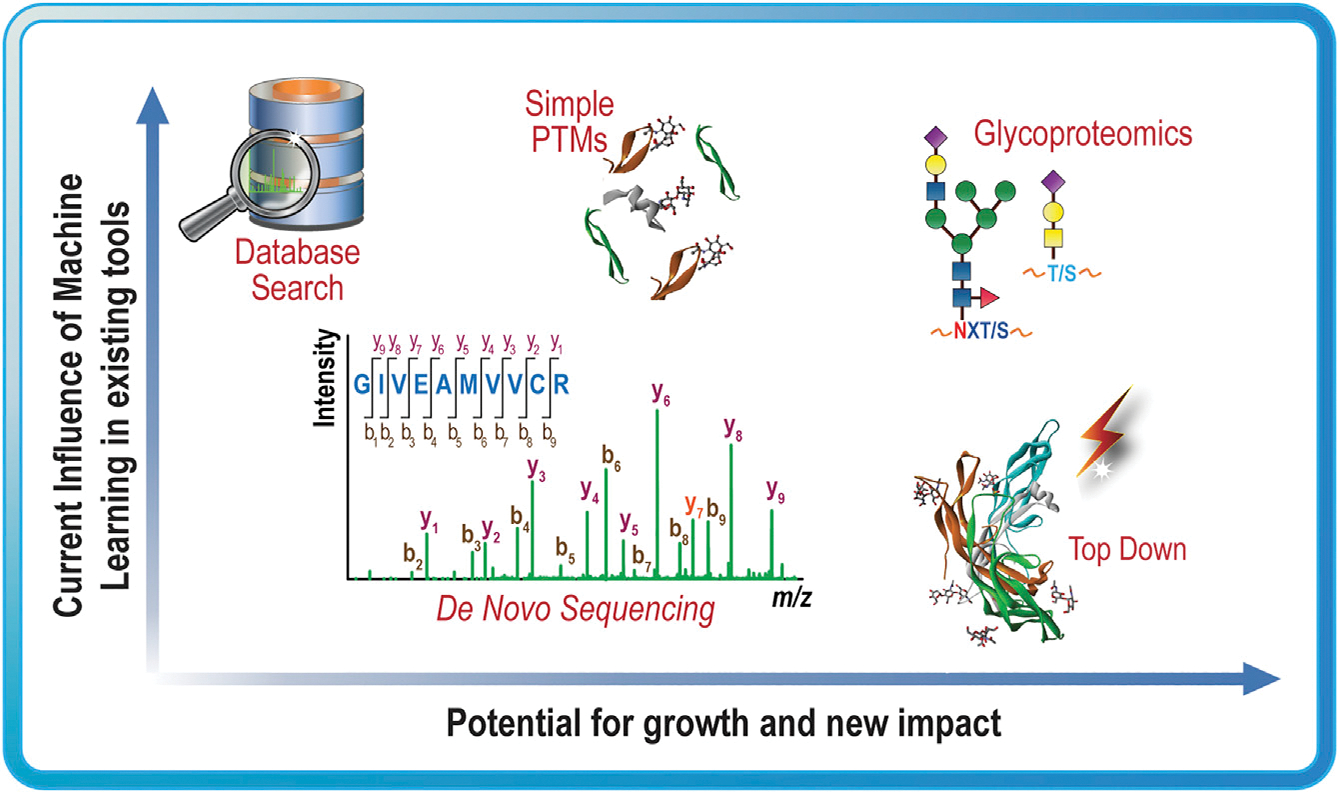
Examples of proteomics analysis problems that have incorporated machine learning in the past and the authors’ predictions about the potential for new impact in the future if these fields were to more heavily leverage machine learning.
MACHINE LEARNING IN BIOMARKER STUDIES
The grandest challenge of combining machine learning and proteomics may be to diagnose disease at its earliest stages and to identify the optimal treatment path for complex diseases. About 20 years ago, several exciting studies demonstrated the ability to diagnose various cancers with high accuracy, including stage I ovarian cancer and prostate cancer, using SELDI-TOF of serum proteins and machine learning.41 While such findings would truly revolutionize medicine, questions arose about the reliability and reproducibility of the work,41 and, because of the inability of these and other studies to deliver on their promises of improving patient outcomes, some insurance providers currently deem the entire field of disease diagnosis by proteomics to be considered investigational and not medically necessary for all indications (see https://provider.healthybluenc.com/dam/medpolicies/healthybluenc/active/policies/mp_pw_a049883). We note that several proteomics-derived biomarker panels have received US Food and Drug Administration (FDA) approval.42,43 The path to successful translation of discoveries that combine machine learning and proteomics into routinely used diagnostics is still an uncertain one. Demonstrating the potential to translate these research findings into routine tests that improve human health should be a top priority for the field.
The two main challenges in translating a promising biomarker candidate into a clinical assay are the need to validate the findings in independent tests and to demonstrate clinical utility.44 Both these metrics depend on the panel’s accuracy in assessing independent test data, and one weakness of many recent biomarker studies is lack of a completely independent test set for initially establishing accuracy.21,22,45 Data in a true test set are those that are not used in any way to build the feature set or model. Instead of including a completely independent test set, many researchers have allowed their test data to leak into their feature selection step, invalidating the final accuracies.21,22,45
One recent high-quality biomarker study is a project aimed at predicting the type of ovarian cancer, so treatment could be optimized;46 the input was MS imaging data of excised ovarian tissue. Notably, the authors carefully split samples into training, validation, and test sets in a way that ensured all the spectra in the test set were from patients who did not contribute any data to the training or validation sets. This is an essential component for any study, where multiple spectra are acquired from individual donors; ultimately, the model needs to distinguish samples based only on the disease state for new individuals, and two samples from the same individual will be similar for reasons other than the disease state, unfairly biasing the results toward higher accuracies. A respectable measure of accuracy, of 75%, was obtained for distinguishing four closely related cancer types.46 Considering the small size of the training sample set, these results are very promising. They could perhaps be considered Biomarker Version 1.0,” and better accuracy may be obtained by building a model with more samples, more representative samples, better features, or perhaps better machine learning approaches. In essence, each aspect of the biomarker work flow could be considered as a point for further improvement for this or any existing biomarker study.
IMPROVING THE ACCURACY OF BIOMARKER PANELS GENERATED BY MACHINE LEARNING METHODS
To get to the clinic, many Biomarker Version 1.0 panels need to increase their accuracy on test data. The best place to start optimization is with the earliest part of the workflow, the collection of samples. The optimal number of samples to analyze is debatable. In one study, researchers found that 16 samples were enough to get statistically meaningful data from a proteomics study with 1,000 proteins.47 Others point out that, in fields with similar analysis challenges and dataset sizes, the target sample size would be 10 times the number of features.48 (This would imply that 10,000 patient samples would be needed for that proteomics study with 1,000 proteins.) While obtaining and analyzing 10,000 proteomics samples is typically not feasible, machine learning practitioners who work with proteomics data know that bigger sample sets are always better: more samples allow for more sophisticated types of learning (i.e., deep learning, feature selection) and much better estimates of accuracy by retaining more samples for a test set.
In some cases, the problem with the quality of the biomarker panel may not be that not enough samples were studied initially but that not enough of the right samples were studied; in other words, the sample set was not representative enough of the population that the test was designed for. For example, in the field of Alzheimer’s disease, many of the sample sets studied to date overrepresent non-Hispanic white adults and underrepresent other racial and ethnic populations within the US.17 Racial bias is a well-studied problem in the field of machine learning, and, in general, models trained on samples from people of predominantly one racial background do not work as well on participants that were underrepresented in the training set.49 This general principle applies to proteomics studies. In one example, we demonstrated that the racial composition of proteomics datasets of brain samples dramatically influenced the utility of heat-shock protein beta 1 (HSPB1) for indicating Alzheimer’s disease.17 While this marker was considerably less effective than the well-known marker, amyloid precursor protein, for indicating the disease state in non-Hispanic white adults, HSPB1’s ability to discriminate the disease state in African American/black adults was significantly better across multiple datasets; see Figure 4.
Figure 4. Racial bias in datasets.
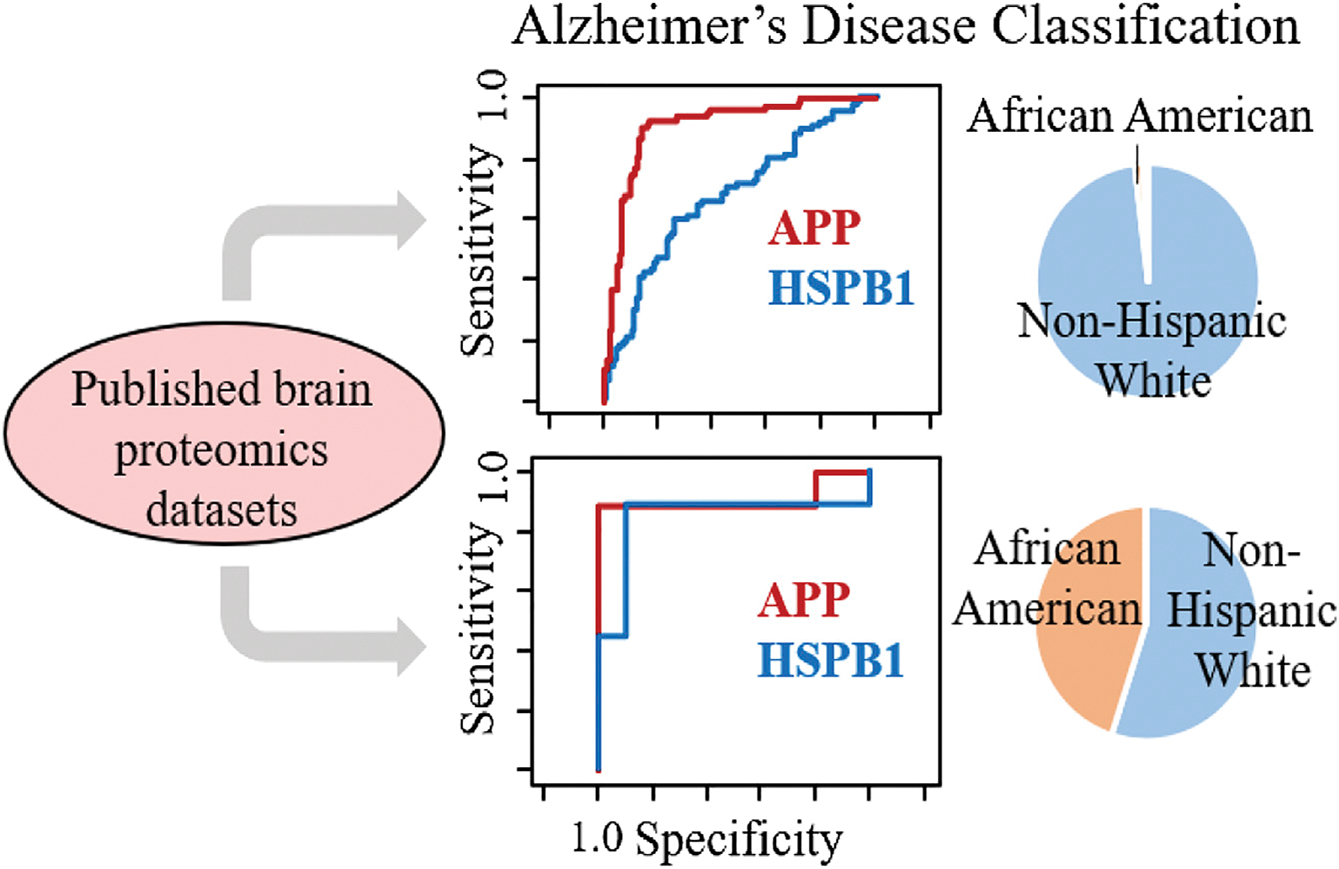
Demographic data (right) and receiver operating characteristic (ROC) curves (middle) from two brain proteomics datasets with differing representation of African American/black and non-Hispanic white participants in two different Alzheimer’s disease studies. In the dataset containing mostly white adults (top), amyloid precursor protein (APP) is a much better marker than heat-shock protein beta 1 (HSPB1), as shown by its larger area under the ROC curve in the top panel. In a more racially diverse dataset (bottom), the utility of HSPB1 as a potential biomarker for Alzheimer’s disease becomes more obvious. Reprinted with permission from Desaire et al.17 Copyright 2022, American Chemical Society.
In a different example, a biomarker panel for predicting Alzheimer’s disease, developed from the study of plasma samples from white patients either with or without Alzheimer’s disease, was >90% effective when applied to a second dataset of samples from white participants, yet the same biomarker panel was no better than a coin toss for predicting the Alzheimer’s disease status of African Americans in two separate sample sets.50 These studies, and the large body of literature assessing racial bias in machine learning, support the view that carefully considering the impacts of racial diversity during study design and validation will be necessary for developing a biomarker that will be highly accurate in a diverse population when relying on proteomics and machine learning. (We note that this issue, of requiring appropriately diverse samples, is not a caveat of machine learning in particular; it is broadly applicable to any clinical study.) Limitations in the number and diversity of samples are possibly the single biggest barrier to dramatically improved, validated biomarker panels. Any advance that addresses better access to samples and/or better throughput for proteomics studies would benefit the scientific community and, ultimately, human health.
Once the samples are collected and proteomics experiments are performed, biomarker panels can be improved by carefully choosing the data that are included about each sample.51 The benefits and risks of restricting the list of proteins to include in the machine learning work flow using feature selection methods were described above. Beyond paring down the feature set, another strategy is increasing it, selectively. A biomarker panel that contains only proteomics data can be expanded to include non-protein features also known to influence the disease state. As an example, in the field of Alzheimer’s disease, combining plasma protein concentrations with the ApoE genotype, age, sex, and years of education are useful in generating a panel with higher predictive accuracy for disease status, compared with the protein-only panel for non-Hispanic white participants, although the inclusion of these features does not improve the panel for African Americans, whose disease status may be less strongly associated with these variables.50 Opportunities exist in the machine learning field to identify optimal ways of including different types of features (i.e., proteins and demographic data) into models to generate the best possible predictions.
Other ways to improve upon an initial biomarker panel are also possible. In some cases, the problem is getting the highest-quality ground-truth dataset. This need is particularly important in imaging applications where the tissue to be imaged is heterogenous.52 Researchers have demonstrated that careful histological annotations were essential for generating an accurate model for predicting certain types of cancer from biopsy slides that had been subjected to MS imaging. In these cases, accuracies of 75%–97% were reported on independent test data.52
Identifying better ways to account for signal variability in the MS data is another means of improving the outcomes of machine learning. Researchers have developed creative normalization methods that reduce instrumental variability, and applying these methods leads to better differentiation of the disease state. One example of a useful tool in this domain is EigenMS; it was first successfully applied to lipidomics data53 and has also been used on proteomics data.54 Normalization for proteomics data is an ongoing challenge, and testing multiple methods is usually necessary to identify an approach that works best on the dataset at hand. The Elo group recently tested different normalization methods on a few proteomics datasets,54 and their strong study could be used as a model for evaluating different methods on new datasets.
Beyond normalization, we showed that signal variability can be effectively accounted for by using a learning strategy called a local-balanced model.23 A local-balanced model combines three concepts: selecting a unique subset of training samples for each test sample by choosing samples that have the most similar instrument response (a local model), optimizing the size of both classes of training set samples (balanced classes), and then finally applying a supervised classification algorithm, such as SVM, to make the final assignments. This approach shows enhanced accuracy on independent test data for various types of instrument-derived data, including samples of different bacterial types undergoing MALDI-MS.23
While the delivery of biomarkers into the clinic, based on workflows combining proteomics and machine learning, is still far from fulfilling its promise, the potential for medical revolutions has always underpinned the excitement in the field. A careful analysis of past failures provides immense guidance for the future.44,48,55 By following this guidance and taking every available opportunity to optimize the accuracy of a biomarker panel, researchers can be well positioned to cross the critical threshold of delivering new medical diagnostics that become routine—not exploratory—and essential for improving patient outcomes.
Researchers are now pursing many diverse and interesting problems that can be addressed by machine learning and proteomics. This technology could be pivotal for understanding the impact of head collisions on National Football League (NFL) players,56 for assessing periodontal disease through proteomic analysis of saliva,57 for using amniotic fluid to assess preterm delivery,58 and in attempting to diagnose early-stage Parkinson’s disease.59 The combination of proteomics and MS imaging has particular promise for typing cancers so that treatments can be optimized,46,60,61 and proteomic analysis of liquid biopsies from prostate cancer patients may also direct optimal treatment options.62 Finally, doctors may be able to better understand and predict disease progression in diabetes63 and heart disease64 with panels that are developed by machine learning and proteomics. In the next 5 years, as more proteomics laboratories include machine learning into their toolbox, the potential impact of combining these technologies will increase even further. We implore researchers in this area to continually work toward making the treacherous transition from potentially impactful biomarker candidates to clinically important, validated biomarkers.
CONCLUSIONS AND OUTLOOK
Machine learning has much to offer proteomics researchers; both tools developers and those interested in using proteomics data to understand biology can leverage this capability to enhance their research and drive new developments. Looking forward, machine learning will significantly enhance the field of proteomics tools development in instances where researchers who understand the unique capabilities of machine learning can design creative strategies to leverage this technology in software designed to automate the analysis of complex MS data. By contrast, in the biomarker field, the biggest barrier to advancement is not creativity on the data analysis side but, rather, on the data collection end of the equation. Obtaining optimal datasets that contain large and diverse sample numbers and are collected on a biofluid that ultimately contains useful information for the diagnostic task at hand is the biggest challenge at this point; once large, diverse, and useful sample sets are present, the machine learning field has many tricks in its bag already to pull out the meaningful markers. The barrier today, though, is that, when markers are identified on small sample sets, or those that are not representative of the full population for which the marker is intended, validation failure is high. Whether a researcher’s primary expertise is in proteomics or machine learning, developing a functional understanding of both fields will give researchers a competitive edge in imagining and implementing innovations that drive science forward and improve health and the human condition.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health, grants R35GM130354 and RF1AG072760 to H.D.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Samuel AL (1959). Some studies in machine learning using the game of checkers. IBM J. Res. Dev. 3, 210–229. 10.1147/rd.33.0210. [DOI] [Google Scholar]
- 2.Fang J, Zhang P, Zhou Y, Chiang C-W, Tan J, Hou Y, Stauffer S, Li L, Pieper AA, Cummings J, and Cheng F (2021). Endophenotype-based in silico network medicine discovery combined with insurance record data mining identifies sildenafil as a candidate drug for Alzheimer’s disease. Nat. Aging 1, 1175–1188. 10.1038/s43587-021-00138-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bantscheff M, Schirle M, Sweetman G, Rick J, and Kuster B (2007). Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031. 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 4.Domon B, and Aebersold R (2006). Review - mass spectrometry and protein analysis. Science 312, 212–217. 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 5.Lill JR, Mathews WR, Rose CM, and Schirle M (2021). Proteomics in the pharmaceutical and biotechnology industry: a look to the next decade. Expert Rev. Proteomics 18, 503–526. 10.1080/14789450.2021.1962300. [DOI] [PubMed] [Google Scholar]
- 6.Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, and Aebersold R (2018). Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 14, e8126. 10.15252/msb.20178126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaffer LV, Millikin RJ, Miller RM, Anderson LC, Fellers RT, Ge Y, Kelleher NL, LeDuc RD, Liu X, Payne SH, et al. (2019). Identification and quantification of proteoforms by mass spectrometry. Proteomics 19, 1800361. 10.1002/pmic.201800361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dakna M, Harris K, Kalousis A, Carpentier S, Kolch W, Schanstra JP, Haubitz M, Vlahou A, Mischak H, and Girolami M (2010). Addressing the challenge of defining valid proteomic biomarkers and classifiers. BMC Bioinf. 11, 594. 10.1186/1471-2105-11-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chicco D (2017). Ten quick tips for machine learning in computational biology. BioData Min. 10, 35. 10.1186/s13040-017-0155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teschendorff AE (2019). Avoiding common pitfalls in machine learning omic data science. Nat. Mater. 18, 422–427. 10.1038/s41563-018-0241-z. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Lu X, Shi Q, Xu XQ, Leung HCE, Harris LN, Iglehart JD, Miron A, Liu JS, and Wong WH (2006). Recursive SVM feature selection and sample classification for mass-spectrometry and microarray data. BMC Bioinf. 7, 197. 10.1186/1471-2105-7-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Q, Sung AH, Qiao M, Chen Z, Yang JY, Yang MQ, Huang X, and Deng Y (2009). Comparison of feature selection and classification for MALDI-MS data. BMC Genom. 10, S3. 10.1186/1471-2164-10-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hua D, and Desaire H (2021). Improved discrimination of disease states using proteomics data with the updated Aristotle classifier. J. Proteome Res. 20, 2823–2829. 10.1021/acs.jproteome.1c00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carnielli CM, Macedo CCS, De Rossi T, Granato DC, Rivera C, Domingues RR, Pauletti BA, Yokoo S, Heberle H, Busso-Lopes AF, et al. (2018). Combining discovery and targeted proteomics reveals a prognostic signature in oral cancer. Nat. Commun. 9, 3598. 10.1038/s41467-018-05696-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villarreal AE, O’Bryant SE, Edwards M, Grajales S, and Britton GB; Panama Aging Research Initiative; Panama Aging Research Initiative (2016). Serum-based protein profiles of Alzheimer’s disease and mild cognitive impairment in elderly Hispanics. Neurodegener. Dis. Manag. 6, 203–213. 10.2217/nmt-2015-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long Y, Wang L, and Sun M (2019). Structure extension of tree-augmented naive Bayes. Entropy 21, 721. 10.3390/e21080721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desaire H, Stepler KE, and Robinson RAS (2022). Exposing the brain proteomic signatures of Alzheimer’s disease in diverse racial groups: leveraging multiple datasets and machine learning. J. Proteome Res. 21, 1095–1104. 10.1021/acs.jproteome.1c00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim H, Kim Y, Han B, Jang JY, and Kim Y (2019). Clinically applicable deep learning algorithm using quantitative proteomic data. J. Proteome Res. 18, 3195–3202. 10.1021/acs.jproteome.9b00268. [DOI] [PubMed] [Google Scholar]
- 19.Smith AM, Walsh JR, Long J, Davis CB, Henstock P, Hodge MR, Maciejewski M, Mu XJ, Ra S, Zhao S, et al. (2020). Standard machine learning approaches outperform deep representation learning on phenotype prediction from transcriptomics data. BMC Bioinf. 21, 119. 10.1186/s12859020-3427-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer JG (2021). Deep learning neural network tools for proteomics. Cell Rep. Methods 1, 100003. 10.1016/j.crmeth.2021.100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desaire H (2022). How (not) to generate a predictive biomarker panel using machine learning. J. Proteome Res. 21, 2071–2074. 10.1021/acs.jproteome.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simon R, Radmacher MD, Dobbin K, and McShane LM (2003). Pittfalls in the use of DNA microarray data for diagnostic and prognostic classification. J. Natl. Cancer Inst. 95, 14–18. 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]
- 23.Desaire H, Patabandige MW, and Hua D (2021). The local-balanced model for improved machine learning outcomes on mass spectrometry data sets and other instrumental data. Anal. Bioanal. Chem. 413, 1583–1593. 10.1007/s00216-020-03117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392. 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 25.Käll L, Canterbury JD, Weston J, Noble WS, and MacCoss MJ (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925. 10.1038/NMETH1113. [DOI] [PubMed] [Google Scholar]
- 26.Anderson DC, Li W, Payan DG, and Noble WS (2003). A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and SEQUEST scores. J. Proteome Res. 2, 137–146. 10.1021/pr0255654. [DOI] [PubMed] [Google Scholar]
- 27.Demichev V, Messner CB, Vernardis SI, Lilley KS, and Ralser M (2020). DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44. 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou XX, Zeng WF, Chi H, Luo C, Liu C, Zhan J, He SM, and Zhang Z (2017). pDeep: predicting MS/MS spectra of peptides with deep learning. Anal. Chem. 89, 12690–12697. 10.1021/acs.analchem.7b02566. [DOI] [PubMed] [Google Scholar]
- 29.Gessulat S, Schmidt T, Zolg DP, Samaras P, Schnatbaum K, Zerweck J, Knaute T, Rechenberger J, Delanghe B, Huhmer A, et al. (2019). Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 16, 509–518. 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- 30.Chen X, Qiu JD, Shi SP, Suo SB, Huang SY, and Liang RP (2013). Incorporating key position and amino acid residue features to identify general and species-specific Ubiquitin conjugation sites. Bioinformatics 29, 1614–1622. 10.1093/bioinformatics/btt196. [DOI] [PubMed] [Google Scholar]
- 31.Dorl S, Winkler S, Mechtler K, and Dorfer V (2018). PhoStar: identifying tandem mass spectra of phosphorylated peptides before database search. J. Proteome Res. 17, 290–295. 10.1021/acs.jproteome.7b00563. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Wang S, Zhang YH, Wei L, Xu X, Huang T, and Cai YD (2018). Prediction of nitrated tyrosine residues in protein sequences by extreme learning machine and feature selection methods. Comb. Chem. High Throughput Screen. 21, 393–402. 10.2174/1386207321666180531091619. [DOI] [PubMed] [Google Scholar]
- 33.AL-barakati HJ, Saigo H, Newman RH, and Kc DB (2019). RF-GlutarySite: a random forest based predictor for glutarylation sites. Mol. Omics 15, 189–204. 10.1039/c9mo00028c. [DOI] [PubMed] [Google Scholar]
- 34.AL-barakati H, Thapa N, Hiroto S, Roy K, Newman RH, and Kc D (2020). RF-MaloSite and DL-Malosite: methods based on random forest and deep learning to identify malonylation sites. Comput. Struct. Biotechnol. J. 18, 852–860. 10.1016/j.csbj.2020.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solovyeva EM, Kopysov VN, Pereverzev AY, Lobas AA, Moshkovskii SA, Gorshkov MV, and Boyarkin OV (2019). Method for identification of threonine isoforms in peptides by ultraviolet photofragmentation of cold ions. Anal. Chem. 91, 6709–6715. 10.1021/acs.analchem.9b00770. [DOI] [PubMed] [Google Scholar]
- 36.Kao HJ, Huang CH, Bretaña NA, Lu CT, Huang KY, Weng SL, and Lee TY (2015). A two-layered machine learning method to identify protein O-GlcNAcylation sites with O-GlcNAc transferase substrate motifs. BMC Bioinf. 16, S10. 10.1186/1471-2105-16-S18-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu SW, Liang SY, Pu TH, Chang FY, and Khoo KH (2013). Sweet-Heart - an integrated suite of enabling computational tools for automated MS2/MS3 sequencing and identification of glycopeptides. J. Proteomics 84, 1–16. 10.1016/j.jprot.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 38.Go EP, Zhang S, Ding H, Kappes JC, Sodroski J, and Desaire H (2021). The opportunity cost of automated glycopeptide analysis: case study profiling the SARS-CoV-2 S glycoprotein. Anal. Bioanal. Chem. 413, 7215–7227. 10.1007/s00216-021-03621-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun RX, Luo L, Wu L, Wang RM, Zeng WF, Chi H, Liu C, and He SM (2016). pTop 1.0: a high-accuracy and high-efficiency search engine for intact protein identification. Anal. Chem. 88, 3082–3090. 10.1021/acs.analchem.5b03963. [DOI] [PubMed] [Google Scholar]
- 40.McIlwain SJ, Wu Z, Wetzel M, Belongia D, Jin Y, Wenger K, Ong IM, and Ge Y (2020). Enhancing top-down proteomics data analysis by combining deconvolution results through a machine learning strategy. J. Am. Soc. Mass Spectrom. 31, 1104–1113. 10.1021/jasms.0c00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diamandis EP (2004). Analysis of serum proteomic patterns for early cancer diagnosis: drawing attention to potential problems. J. Natl. Cancer Inst. 96, 353–356. 10.1093/jnci/djh056. [DOI] [PubMed] [Google Scholar]
- 42.Geyer PE, Holdt LM, Teupser D, and Mann M (2017). Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 13, 942. 10.15252/msb.20156297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kearney P, Boniface JJ, Price ND, and Hood L (2018). The building blocks of successful translation of proteomics to the clinic. Curr. Opin. Biotechnol. 51, 123–129. 10.1016/j.copbio.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diamandis EP (2012). The failure of protein cancer biomarkers to reach the clinic: why, and what can be done to address the problem? BMC Med. 10, 87. 10.1186/1741-7015-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinn TP (2021). Stool studies don’t pass the sniff test: a systematic review of human gut microbiome research suggests widespread misuse of machine learning. Preprint at arXiv. 10.48550/arXiv.2107.03611. [DOI] [Google Scholar]
- 46.Klein O, Kanter F, Kulbe H, Jank P, Denkert C, Nebrich G, Schmitt WD, Wu Z, Kunze CA, Sehouli J, et al. (2019). MALDI-imaging for classification of epithelial ovarian cancer histotypes from a tissue microarray using machine learning methods. Proteomics Clin. Appl. 13, 1700181. 10.1002/prca.201700181. [DOI] [PubMed] [Google Scholar]
- 47.Tarazona S, Balzano-Nogueira L, Gómez-Cabrero D, Schmidt A, Imhof A, Hankemeier T, Tegnér J, Westerhuis JA, and Conesa A (2020). Harmonization of quality metrics and power calculation in multi-omic studies. Nat. Commun. 11, 3092. 10.1038/s41467-020-16937-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ransohoff DF (2004). Opinion - rules of evidence for cancer molecular-marker discovery and validation. Nat. Rev. Cancer 4, 309–314. 10.1038/nrc1322. [DOI] [PubMed] [Google Scholar]
- 49.Ntoutsi E, Fafalios P, Gadiraju U, Iosifidis V, Nejdl W, Vidal M, Ruggieri S, Turini F, Papadopoulos S, Krasanakis E, et al. (2020). Bias in data-driven artificial intelligence systems—an introductory survey. WIREs Data Mining Knowl. WIREs Data Mining Knowl. Discov. 10, e1356. 10.1002/widm.1356. [DOI] [Google Scholar]
- 50.Khan MJ, Desaire H, Lopez OL, Kamboh MI, and Robinson RAS (2021). Why inclusion matters for Alzheimer’s disease biomarker discovery in plasma. J. Alzheimer’s Dis.79,1327–1344. 10.3233/JAD-201318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rohart F, Gautier B, Singh A, and Lê Cao KA (2017). mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 13, e1005752. 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonçalves JPL, Bollwein C, Schlitter AM, Martin B, Märkl B, Utpatel K, Weichert W, and Schwamborn K (2021). The impact of histological annotations for accurate tissue classification using mass spectrometry imaging. Metabolites 11, 752. 10.3390/metabo11110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karpievitch YV, Nikolic SB, Wilson R, Sharman JE, and Edwards LM (2014). Metabolomics data normalization with EigenMS. PLoS One 9, e116221. 10.1371/journal.pone.0116221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Välikangas T, Suomi T, and Elo LL (2018). A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 19, 1–11. 10.1093/bib/bbw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ioannidis JPA, and Khoury MJ (2011). Improving validation practices in “omics” research. Science 334, 1230–1232. 10.1126/science.1211811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muraoka S, DeLeo AM, Yang Z, Tatebe H, Yukawa-Takamatsu K, Ikezu S, Tokuda T, Issadore D, Stern RA, and Ikezu T (2021). Proteomic profiling of extracellular vesicles separated from plasma of former National Football League players at risk for chronic traumatic encephalopathy. Aging Dis. 12, 1363–1375. 10.14336/AD.2020.0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bostanci N, Selevsek N, Wolski W, Grossmann J, Bao K, Wahlander A, Trachsel C, Schlapbach R, Öztürk VÖ, Afacan B, et al. (2018). Targeted proteomics guided by label-free quantitative proteome analysis in saliva reveal transition signatures from health to periodontal disease. Mol. Cell. Proteomics 17, 1392–1409. 10.1074/mcp.RA118.000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bahado-Singh RO, Sonek J, McKenna D, Cool D, Aydas B, Turkoglu O, Bjorndahl T, Mandal R, Wishart D, Friedman P, et al. (2019). Artificial intelligence and amniotic fluid multiomics: prediction of perinatal outcome in asymptomatic women with short cervix. Ultrasound Obstet. Gynecol. 54, 110–118. 10.1002/uog.20168. [DOI] [PubMed] [Google Scholar]
- 59.Virreira Winter S, Karayel O, Strauss MT, Padmanabhan S, Surface M, Merchant K, Alcalay RN, and Mann M (2021). Urinary proteome profiling for stratifying patients with familial Parkinson’s disease. EMBO Mol. Med. 13, e13257. 10.15252/emmm.202013257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kriegsmann M, Casadonte R, Maurer N, Stoehr C, Erlmeier F, Moch H, Junker K, Zgorzelski C, Weichert W, Schwamborn K, et al. (2020). Mass spectrometry imaging differentiates chromophobe renal cell carcinoma and renal oncocytoma with high accuracy. J. Cancer 11, 6081–6089. 10.7150/jca.47698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin B, Gonçalves JPL, Bollwein C, Sommer F, Schenkirsch G, Jacob A, Seibert A, Weichert W, Märkl B, and Schwamborn K (2021). A mass spectrometry imaging based approach for prognosis prediction in UICC stage I/II colon cancer. Cancers 13, 5371. 10.3390/cancers13215371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim Y, Jeon J, Mejia S, Yao CQ, Ignatchenko V, Nyalwidhe JO, Gramolini AO, Lance RS, Troyer DA, Drake RR, et al. (2016). Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer. Nat. Commun. 7, 11906. 10.1038/ncomms11906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahn HS, Kim JH, Jeong H, Yu J, Yeom J, Song SH, Kim SS, Kim IJ, and Kim K (2020). Differential urinary proteome analysis for predicting prognosis in type 2 diabetes patients with and without renal dysfunction. Int. J. Mol. Sci. 21, 4236. 10.3390/ijms21124236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Captur G, Heywood WE, Coats C, Rosmini S, Patel V, Lopes LR, Collis R, Patel N, Syrris P, Bassett P, et al. (2020). Identification of a multiplex biomarker panel for hypertrophic cardiomyopathy using quantitative proteomics and machine learning. Mol. Cell. Proteomics 19, 114–127. 10.1074/mcp.RA119.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]