

Abbreviations
- Ac‐H3
acetylation‐histone 3
- Ac‐H4
acetylation‐histone 4
- AIFM1
apoptosis inducing factor, mitochondrion‐associated, 1
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BUN
blood urea nitrogen
- CypA
Cyclophilin A
- DAPI
4′, 6‐diamidino‐2‐phenylindole
- DMSO
dimethyl sulfoxide
- EMT
epithelial–mesenchymal transition
- FDA
Food and Drug Administration
- HDAC
histone deacetylase
- HDACI
histone deacetylase inhibitor
- HMGB1
high mobility group protein B1
- LDH
lactate dehydrogenase
- PCNA
proliferation cell nuclear antigen
- PI
propidine iodide
- RT‐PCR
reverse transcription‐polymerase chain reaction
- SAHA
suberoylanilide hydroxamic acid
- siRNA
short interfering RNA
- TEM
transmission electron microscope
- ZBG
Zn binding group
Dear Editor,
Breast cancer is one of the most lethal cancers in women, with many patients still succumbing to this disease [1]. Accumulating evidence demonstrates that histone deacetylase inhibitors (HDACIs) are a promising therapeutic intervention for breast cancer [2], and many of them have shown favorable anti‐cancer activities in both preclinical and clinical settings [3]. However, most current HDACIs only exhibit limited efficacy against solid tumors with toxic side effects and readily produce drug resistance [4]. Therefore, it is necessary to develop new HDACIs with improved anti‐tumor activities and decreased toxicities for breast cancer therapeutics and investigate their mechanism of action.
To improve the physicochemical properties of new generation HDACIs, the coumarin unit, as a promising pharmacophore in anti‐cancer drug discovery, was incorporated into hydroxamate HDACIs, and a series of new coumarin‐based derivatives were synthesized. After our initial screening, a compound coded YF349 with excellent HDAC inhibitory activity was identified (Supplementary Figure S1A, B). A hallmark index of HDAC inhibition is the increased acetylation of histones H3 and H4 [5]. As shown in Supplementary Figure S1C, D, YF349 significantly increased the acetylation of hH3 and Ac‐H4 compared with Suberoylanilide Hydroxamic Acid (SAHA). SAHA was used as a positive control in our experiments, as it is the first HDACI approved by the US Food and Drug Administration (FDA) for the clinical treatment of breast cancer [6]. Collectively, these results suggested that YF349 was a bona fide HDACI and had potential anti‐breast cancer activity.
The chemical structure of YF349 is shown in Figure 1A. We next examined the anti‐breast cancer activity of YF349 in vitro. As shown in Figure 1B, YF349 significantly inhibited the growth of breast cancer cells compared with SAHA. In addition, YF349 significantly inhibited colony formation and the invasion ability of breast cancer cells (Figure 1C, D and Supplementary Figure S2A). Additionally, YF349 induced obvious cell death in breast cancer cells compared with SAHA treatment at the same dosage (Figure 1E). Finally, YF349 significantly altered the expression of proliferation cell nuclear antigen (PCNA), apoptosis‐related proteins (cleaved‐poly ADP‐ribose polymerase [PARP]), and epithelial–mesenchymal transition (EMT)‐related proteins (Supplementary Figure S2B). We then investigated the anti‐breast cancer effect of YF349 in vivo. As shown in Figure 1F, G and Supplementary Figure S2C‐E, YF349 significantly inhibited the tumor growth and metastasis compared with the control group and SAHA treatment at the same concentration. Meanwhile, YF349 significantly increased the levels of Ac‐H3 and Ac‐H3, which confirmed the HDAC inhibitory effect of YF349 on breast cancer in vivo (Supplementary Figure S2F). We then sought to investigate the potential toxicity of YF349. As shown in Supplementary Figure S3A, B, no significant changes in body weight and the major organs of mice treated with YF349 were observed. Histological analyses revealed no obvious damage to major organs (Supplementary Figure S3C). In addition, levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are representative indicators of liver function, and blood urea nitrogen (BUN) is an indicator of kidney and liver conditions. As shown in Supplementary Figure S3D, YF349 treatment did not significantly affect the ALT, AST and BUN levels. Taken together, these results indicate that YF349 significantly inhibited breast cancer cell growth and metastasis both in vitro and in vivo and showed few adverse effects on the experimental mice at a therapeutic concentration.
FIGURE 1.
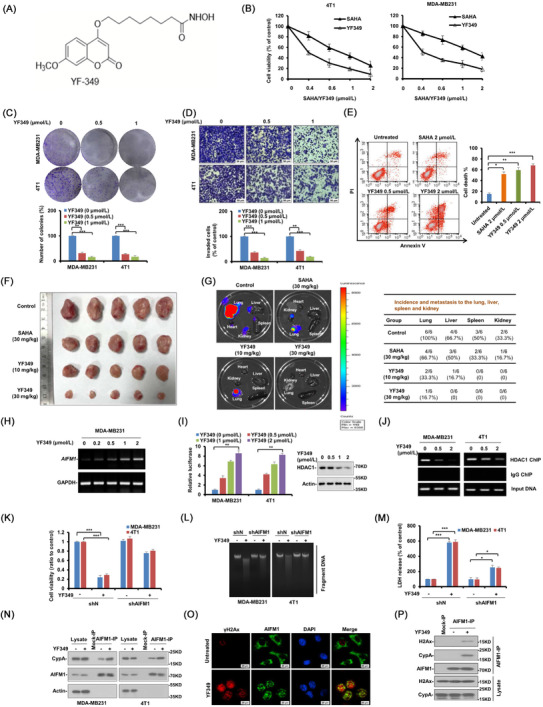
YF349 exerts promising anti‐breast cancer activity by triggering AIFM1‐dependent programmed necrosis. (A) Chemical structure of the new HDACI, YF349. (B) Breast cancer cells (MDA‐MB231 and 4T1) were treated with YF349 or SAHA. After 48 h, MTS assay was performed. The bars indicate mean ± SD. (C) MDA‐MB231 and 4T1 cells were seeded on 6‐well plates. After 12 h, cells were treated with indicated concentrations of YF349. On day 10, the number of colonies was counted in experiments repeated three times. Results represent the average of three replications. (D) MDA‐MB231 and 4T1 cells were treated with different concentrations of YF349 and allowed to invade through matrigel. Images were obtained after 12 h of incubation (upper). The invaded cell number was counted and expressed as % untreated control (lower). Data were shown as mean ± SD from three independent experiments. Scale: 50 μm. (E) 4T1 cells were treated with different dosages of YF349 or SAHA for 48 h. Cell death was assessed by Annexin V/PI staining and flow cytometry. (F) MDA‐MB231‐luc cells (1×105) were injected into the mammary fat pad of nude mice. Mice were divided into 5 groups (n = 5 per group) on day 7. Mice were administrated with YF349 or SAHA every day. After 35 days, all mice were sacrificed. (G) Ex vivo bioluminescence images were obtained in each group to determine the effects of YF349 against distant metastasis. Metastasis in distant organs was quantified. (H) MDA‐MB231 cells were treated with YF349 with the indicated concentrations for 24 h, RNA samples were prepared using Trizol and total RNA was converted to cDNA using oligodT primer. The relative expression of AIFM1 was analysed by RT‐PCR with GAPDH as an internal control. PCR products were separated on 1.2% agarose gel and stained with ethidium bromide. (I) Human AIFM1 promoter (1343 to 141 bp upstream of ATG) luciferase reporter was transfected in MDA‐MB231 and 4T1 cells, and 12 h later, the cells were treated with increased concentrations of YF349 for 24 h, and then the relative luciferase activity was analyzed. Data were presented as mean ± SD, ** P < 0.01. Representative western blotting shows the expression of HDAC1 and Actin. (J) YF349 blocks HDAC1 binding to the AIFM1 promoter. MDA‐MB231 and 4T1 cells were treated with YF349 or not, and cells were processed for chromatin immunoprecipitation using HDAC1 antibody. Co‐precipitated chromatin DNA was analyzed by PCR using a pair of primers that amplify the 463 to 318 bp region of the AIFM1 promoter. (K) Cell viability was analyzed using MTS assay in AIFM1‐knockdown cells under YF349 treatment or not (*** P < 0.001). (L) The large‐scale DNA fragmentations in AIFM1‐knockdown cells treated with YF349 or not were detected on agarose gel electrophoresis. (M) The LDH release assay was performed in AIFM1‐knockdown cells under YF349 treatment or not (*** P < 0.001). (N) MDA‐MB231 and 4T1 cells were treated with YF349 for 24 h, and the co‐localization of AIFM1 and CypA was examined by immunoprecipitation assay. (O) 4T1 cells were treated with 2 μmol/L YF349 for 12 h and then washed with PBS three times. AIFM1 (green), γH2AX (red) and DAPI (blue) were detected by immunofluorescence staining (scale bar 20 μm). (P) MDA‐MB231 cells treated with or without YF349 were lysed and immunoprecipitated using AIFM1 antibody followed by anti‐H2AX and anti‐CypA western blot.
We then identified the modality of breast cancer cell death caused by YF349. As shown in Supplementary Figure S4A, a significant portion of cells appeared necrotic, and the cell death modality induced by YF349 was similar to that caused by the necrosis‐inducer shikonin. Moreover, pretreatment with the pan‐caspase inhibitor z‐VAD‐fmk did not prevent YF349‐induced cell death, while this treatment inhibited the proteasome inhibitor MG132‐induced cell death (Supplementary Figure S4B). In addition, cells treated with YF349 presented smeared DNA bands on an agarose gel in the DNA large fragment assay (Supplementary Figure S4C). The morphological characteristics of necrosis were also confirmed by transmission electron microscope (TEM) under YF349 treatment (Supplementary Figure S4D). In addition, YF349 promoted the cellular release of high mobility group protein B1 (HMGB1) and lactate dehydrogenase (LDH), which were shown to be necrosis markers [7] (Supplementary Figure S4E, F). Moreover, increased concentration of YF349 significantly upregulated the proportion of necrotic cells (Supplementary Figure S4G). Overall, these results indicated that YF349 induced necrosis in breast cancer cells.
To further explore the detailed mechanisms of the anti‐breast cancer effect of YF349. RNA‐sequencing analysis was performed to identify differentially expressed genes between MDA‐MB231 cells treated with or without YF349. Supplementary Table S1 shows the differentially expressed genes after YF349 treatment. We then analyzed the top 10 upregulated genes (cut‐off, fold change > 4.8 and P < 0.05) listed in Supplementary Table S1. As shown in Supplementary Figure S5A, among the top 10 upregulated genes, the mRNA level of AIFM1 (apoptosis‐inducing factor, mitochondrion‐associated, 1) was significantly increased upon YF349 treatment compared with other genes. Supplementary Figure S4 showed that YF349 induced obvious necrosis of breast cancer cells, and AIFM1 has been known to be a key regulator of necrosis [8]. Therefore, we speculated that AIFM1 may play an essential role in the anti‐tumor effect of YF349 on breast cancer.
A previous study reported that the increased total AIFM1 expression in cells led to increased sensitivity to cell death [9]. As shown in Figure 1H, YF349 significantly increased the mRNA level of AIFM1. An earlier study has demonstrated that HDAC1 could bind to the promoter region of AIFM1 and thus repress AIFM1 expression [10]. The molecular docking model showed that YF349 could interact with the active site of HDAC1 (Supplementary Figure S5B). These findings provide evidence that the increased mRNA level of AIFM1 induced by YF349 may be through the competitive interference of HDAC1 binding to the promoter of AIFM1. As shown in Figure 1I, J and Supplementary Figure S5C‐E, the promoter activity of AIFM1 was significantly upregulated by YF349, and YF349 significantly disrupted the binding of HDAC1 to the AIFM1 promoter. Taken together, these results indicated that YF349 upregulated the AIFM1 expression by disrupting HDAC1 binding to the AIFM1 promoter.
We then clarified the role of AIFM1 in the YF349‐induced necrosis in breast cancer cells. As shown in Figure 1K‐M and Supplementary Figure S6A, B, AIFM1 knockdown cells were less sensitive to YF349‐induced necrosis. These results indicated that YF349 induced‐necrosis of breast cancer cells is in an AIFM1‐dependent manner. Furthermore, we found that YF349 promoted the formation of the AIFM1‐Cyclophilin A (CypA)‐γH2Ax complex in breast cancer cells (Figure 1N‐P and Supplementary Figure S6C, D). Collectively, these results demonstrated that YF349 remarkably induced the nuclear translocation of AIFM1 and significantly promoted the formation of the AIFM1‐CypA‐γH2Ax complex.
In conclusion, we identified a novel HDACI, YF349, which displayed promising anti‐breast cancer activity both in vitro and in vivo. Further mechanistic studies revealed that YF349 increased the AIFM1 expression via inducing the disassociation of HDAC1 from the AIFM1 promoter, subsequently accelerating the nuclear translocation of AIFM1, promoting the formation of the AIFM1‐CypA‐γH2Ax complex, and finally inducing AIFM1‐mediated necrosis of breast cancer cells. Collectively, our work highlighted the anti‐breast cancer therapeutic potential of a new HDACI, YF349, via triggering AIFM1‐dependent necrosis. Our results suggest that the HDAC1‐AIFM1‐CypA‐γH2Ax signal axis can be a novel therapeutic target of breast cancer, and YF349 could be a promising preclinical drug candidate for breast cancer treatment.
AUTHOR CONTRIBUTIONS
Author contributions: PPS contributed to experimental design, data acquisition, analysis and interpretation and drafted the manuscript. FFY contributed to data acquisition, analysis and interpretation, and drafted the manuscript. PPS made sequencing and statistical analysis. JY and LRW collected clinical data and collected samples. YHQ and HRQ participated in the synthesis and purification of target compounds. PPS and LRW performed the animal experiments, model improvement and data analysis. HZ and SJZ contributed to the conception, experimental design, data interpretation, and critical revision of the manuscript. All authors reviewed and edited the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This work was supported by the National Natural Science Foundation of China (No. 91849209, 81803016, 81703360, 81903539); the Natural Science Foundation of Shandong Province (No. ZR2019HB012, ZR2021MC189); the China Postdoctoral Science Foundation (No. 2019M650157).
AVAILABILITY OF DATA AND MATERIALS
All data needed to evaluate the conclusions are presented in the paper or the Supplementary Materials. The raw data of the RNA‐seq results were submitted to the Gene Expression Omnibus database, with the approval number of GSE203025.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal experiments were performed according to the guidelines approved by the Institutional Animal Care and performed following the guidelines for Animal Experimentation of Qingdao University and approved by the Ethics Committee of Medical College of Qingdao University (QDU‐AEC‐2022093).
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank Cuiyun Liu from The Affiliated Hospital of Qingdao University for supporting this study and for guidance and insightful discussions; We thank Yanyan Gao from the Institute of Translational Medicine, Qingdao University for technical support; We thank the participants and staff of Shanghai Key Laboratory of Regulatory Biology, Institute of Biomedical Sciences and School of Life Sciences, East China Normal University.
Peipei Shan and Feifei Yang contributed equally to this work.
Contributor Information
Peipei Shan, Email: shanpeipei@qdu.edu.cn.
Hua Zhang, Email: bio_zhangh@ujn.edu.cn.
Sujie Zhu, Email: zhusujie@qdu.edu.cn.
REFERENCES
- 1. Cesca MG, Vian L, Cristovao‐Ferreira S, Ponde N, de Azambuja E. HER2‐positive advanced breast cancer treatment in 2020. Cancer Treat Rev. 2020;88:102033. [DOI] [PubMed] [Google Scholar]
- 2. Huang M, Zhang J, Yan C, Li X, Zhang J, Ling R. Small molecule HDAC inhibitors: Promising agents for breast cancer treatment. Bioorg Chem. 2019;91:103184. [DOI] [PubMed] [Google Scholar]
- 3. Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90(1):85–94. [DOI] [PubMed] [Google Scholar]
- 4. Qiu T, Zhou L, Zhu W, Wang T, Wang J, Shu Y, et al. Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol. 2013;9(2):255–69. [DOI] [PubMed] [Google Scholar]
- 5. Somech R, Izraeli S, A JS. Histone deacetylase inhibitors–a new tool to treat cancer. Cancer Treat Rev. 2004;30(5):461–72. [DOI] [PubMed] [Google Scholar]
- 6. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T‐cell lymphoma. Oncologist. 2007;12(10):1247–52. [DOI] [PubMed] [Google Scholar]
- 7. Minsart C, Liefferinckx C, Lemmers A, Dressen C, Quertinmont E, Leclercq I, et al. New insights in acetaminophen toxicity: HMGB1 contributes by itself to amplify hepatocyte necrosis in vitro through the TLR4‐TRIF‐RIPK3 axis. Sci Rep. 2020;10(1):5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Artus C, Boujrad H, Bouharrour A, Brunelle MN, Hoos S, Yuste VJ, et al. AIF promotes chromatinolysis and caspase‐independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;29(9):1585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, et al. Essential role of the mitochondrial apoptosis‐inducing factor in programmed cell death. Nature. 2001;410(6828):549–54. [DOI] [PubMed] [Google Scholar]
- 10. Burton TR, Eisenstat DD, Gibson SB. BNIP3 (Bcl‐2 19 kDa interacting protein) acts as transcriptional repressor of apoptosis‐inducing factor expression preventing cell death in human malignant gliomas. J Neurosci. 2009;29(13):4189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
All data needed to evaluate the conclusions are presented in the paper or the Supplementary Materials. The raw data of the RNA‐seq results were submitted to the Gene Expression Omnibus database, with the approval number of GSE203025.