

SUMMARY
The effects of fasting-mimicking diet (FMD) cycles in reducing many aging and disease risk factors indicate it could affect Alzheimer’s disease (AD). Here, we show that FMD cycles reduce cognitive decline and AD pathology in E4FAD and 3xTg AD mouse models, with effects superior to those caused by protein restriction cycles. In 3xTg mice, long-term FMD cycles reduce hippocampal Aβ load and hyperphosphorylated tau, enhance genesis of neural stem cells, decrease microglia number, and reduce expression of neuroinflammatory genes, including superoxide-generating NADPH oxidase (Nox2). 3xTg mice lacking Nox2 or mice treated with the NADPH oxidase inhibitor apocynin also display improved cognition and reduced microglia activation compared with controls. Clinical data indicate that FMD cycles are feasible and generally safe in a small group of AD patients. These results indicate that FMD cycles delay cognitive decline in AD models in part by reducing neuroinflammation and/or superoxide production in the brain.
Graphical abstract
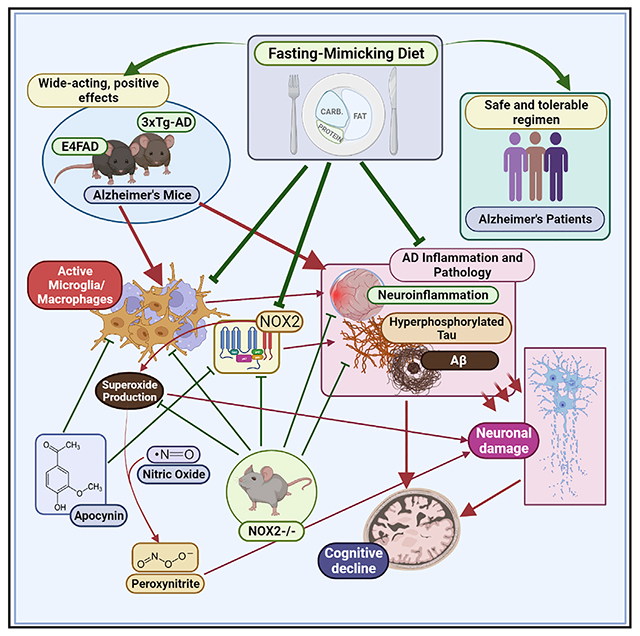
In brief
Rangan et al. show that cycles of a fasting-mimicking diet (FMD) attenuate cognitive decline and reduce Alzheimer’s disease pathology, reduce neuroinflammation, and enhance neurogenesis in AD mouse models. They also show that a similar FMD is safe and feasible in a small group of AD patients.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by the accumulation of amyloid-beta (Aβ) via Aβ oligomers (oAβ) that can be toxic in their fibrillar form (Gong et al., 2003) or aggregate to form amyloid plaques and promote the generation of hyperphosphorylated tau protein (Bloom, 2014). This distinct neuropathology can lead to inflammation and oxidative damage, synaptic degeneration, and neuronal death, ultimately affecting the learning and memory functions of the cerebral cortex and hippocampus (Cline et al., 2018). The 3xTg-AD mouse model (3xTg) exhibits both Aβ and tau pathology, characteristic of the human disease (Oddo et al., 2003; Sterniczuk et al., 2010). In contrast, the EFAD-Tg mouse model (Youmans et al., 2012) can have different human APOE alleles (APOE2, APOE3, APOE4) knocked into the 5xFAD-Tg mice (Oakley et al., 2006), allowing investigation of the role of different APOEs on AD pathology (Lewandowski et al., 2020).
The efficacy of drugs thus far approved for AD treatment is limited (Connelly et al., 2019; Schneider et al., 2011). The more recent effort to remove Aβ by an antibody-based intervention appears to be promising and has led to cognitive improvements in AD patients, but the high incidence of amyloid-related imaging Abnormalities (ARIAa), particularly in APOE4 carriers treated with the higher and effective doses, may limit its efficacy and safety (Plotkin and Cashman, 2020). Thus, there is a need for broader-acting but also safer interventions, particularly considering the advanced ages of subjects diagnosed with dementias, possibly to be combined with more targeted therapies.
Increasing evidence suggests that different forms of dietary interventions may be effective in limiting AD progression in mouse models. Caloric restriction (CR) generally refers to a 20%–40% reduction in total calorie intake, without lowering micronutrient intake, whereas dietary restriction refers to the restriction of a particular macronutrient (proteins, carbohydrates, or fats) with or without a reduction in calorie intake (Mirzaei et al., 2016). CR studies previously conducted in PS1 mutant knockin mice concluded that an alternate-day fasting (intermittent fasting [IF]) regimen of 3 months reduced excitotoxic damage to hippocampal CA1 and CA3 neurons compared with mice that were fed ad libitum (Zhu et al., 1999), while caloric restriction for 14 weeks in amyloid precursor protein (APP) and PS1 transgenic mice led to a reduction in the accumulation of Aβ plaques and decreased Aβ plaque-associated astrocyte activation (Patel et al., 2005). CR regimens in other AD mouse models slowed the progression of Aβ deposition in the hippocampus and in cerebral cortex (Mouton et al., 2009; Halagappa et al., 2007). A long-term study of 3xTg mice undergoing either CR or IF for either 7 or 14 months concluded that both CR and IF dietary regimens ameliorate age-related behavioral deficits by mechanisms that may or may not be related to Aβ and tau pathologies (Halagappa et al., 2007). However, chronic dietary restrictions are associated with both safety and compliance concerns, particularly in the elderly population, which represents the great majority of AD subjects. For that purpose, our group has investigated the role of alternative and less restrictive interventions in murine AD or cognitive decline models, which may be feasible for human testing including intermittent essential amino acid/protein restriction (Parrella et al., 2013) and periodic fasting and fasting-mimicking diets (Brandhorst et al., 2015). Notably, in human clinical trials, FMD cycles either caused no loss or an increase in lean body mass and function (Wei et al., 2017; Caffa et al., 2020). We previously showed that, after 4 months of protein restriction cycles, alternated with normal feeding, male 3xTg mice exhibited improved behavior performance and reduced phosphorylated tau compared with ad libitum-fed animals, and these improvements were accompanied by reduced IGF-1 signaling during the restricted period (Parrella et al., 2013).
Inflammation and oxidative stress play a role in AD pathology, by damaging neurons and contributing to the accumulation of Aβ (Block, 2008). In turn, this leads to the activation of microglia cells, which respond by changing morphology, by upregulating or by synthetizing de novo surface receptors, and by secreting pro-inflammatory cytokines and reactive species of oxygen (ROS), such as nitric oxide (NO) and superoxide (Schlachetzki and Hull, 2009). The NADPH oxidase enzymatic complex (NOX2), the major producer of superoxide in microglia cells, was proposed as an attractive therapeutic target for the development of interventions against AD (Block, 2008). In fact, we have previously shown that Aβ can stimulate both an increase in superoxide production in neurons (Longo et al., 2000) and the generation of highly neurotoxic microglial/NADPH oxidase-derived peroxynitrite (Xie et al., 2002).
The fasting-mimicking diet (FMD) is a low-calorie/low-protein but high-unsaturated-fat diet that has an effect on stress resistance/longevity-related markers similar to those caused by water-only fasting, but that minimizes the burden of prolonged fasting as it provides both macro- and micronutrients. Here we investigated the effect of bi-monthly cycles of an FMD, administered at an early age in male and female 3xTg and in female E4FAD mice. We show that FMD cycles improve cognitive performance, reduce AD-associated neuropathology, increase markers of neural stem cell regeneration, and reduce microglial activation and neuroinflammation, including Nox2 (NADPH oxidase) expression levels. Nox2 deficiency or treatment with the NADPH oxidase inhibitor apocynin was sufficient to reduce microglia density/activation and delay cognitive decline, indicating that microglial-dependent toxicity is important for age-dependent cognitive impairment.
RESULTS
FMD cycles improve cognitive behavior in female E4FAD mice
We first evaluated the effects of FMD cycles specifically on female E4FAD mice expressing the human ApoE4 isoform, since, in these mice, pathology rapidly develops in the cortex and hippocampus, reaching significant levels by 6 months of age (Youmans et al., 2012; Cacciottolo et al., 2016). Using a variety of AD-relevant readouts (Maldonado Weng, 2019), the hierarchy of pathology in the EFAD mice is male E3FAD < female E3FAD ~ male E4FAD < female E4FAD.
Female E4FAD mice were enrolled in the study at approximately 2.5 months of age, during which baseline behavior tests (spontaneous alternation behavior [SAB] and elevated plus maze [EPM]) were conducted. Mice were then assigned to either a standard rodent chow diet group (control) or to a 5-day FMD twice a month starting at 3 months of age, with the standard chow diet fed to the mice between FMD cycles. The dietary regimens were halted at ~7–7.5 months of age once the end point behavioral tests were completed (Figure 1A).
Figure 1. FMD cycles improve cognitive behavior in female E4FAD mice.
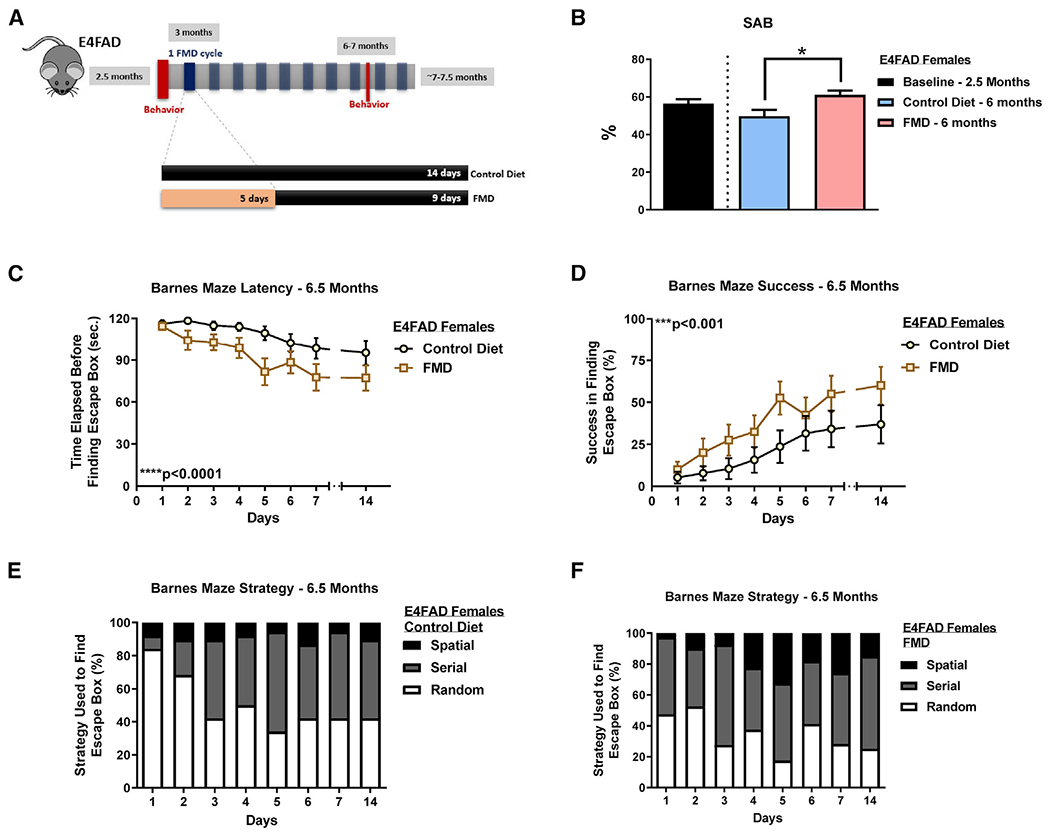
(A) Experimental diet and behavior schedule for female E4FAD mice starting at 2.5 months of age through 7–7.5 months of age.
(B) SAB percentage for E4FAD females at baseline (2.5 months) and at 6 or 6.5 months after ~3 months of diet (baseline, n = 19; control, 6 months, n = 9; FMD, 6 months, n = 11).
(C) Latency (seconds lapsed before finding escape box) between E4FAD FMD females (n = 20) and E4FAD females on control diet (n = 19) in the Barnes maze at approximately 6.5–7 months.
(D) Success rate in finding the escape box between E4FAD FMD females (n = 20) and E4FAD females on control diet (n = 19) in the Barnes maze at approximately 6.5–7 months.
(E) Strategies (random, serial, and spatial) used by female E4FAD control group (n = 19) to locate escape box.
(F) Strategies (random, serial, and spatial) used by female E4FAD FMD group (n = 20) to locate escape box. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ****p < 0.0001, unpaired two-tailed student’s t test (SAB). Two-way ANOVA (Barnes maze).
At baseline, female E4FAD mice obtained an SAB score of 56.4% ± 10.5% (mean ± SD; Figure 1B). After ~3 months of FMD cycles (at 6 months of age), the FMD group displayed significantly higher SAB scores compared with mice fed a control diet (p < 0.05; Figure 1B). We also analyzed changes in anxiety using the EPM, in which more time spent in the open arms of the maze reflects lower levels of anxiety and increased exploratory behavior (Young, 1996). E4FAD mice that underwent FMD treatment spent more time in the open arms compared with age-matched control (animals from both groups spent comparable times in the open arms at baseline; Figure S1A).
We also assessed spatial memory using the Barnes maze in 6.5- to 7-month-old E4FAD females. After the 7-day training period, during the retention test at day 14, FMD-treated mice performed better than those from the control group both in terms of latency (p < 0.0001; Figure 1C) and success (p < 0.001; Figure 1D), with mice from the FMD group taking less time to find the escape box and achieving a higher success in finding the escape box in the time allotted. There was no significant difference between control versus the FMD group in errors made or in deviation from the escape box (Data S1B and S1C). A larger percentage of mice from the FMD group also progressively increased utilization of a spatial strategy versus the random strategy taken by control diet mice (Figures 1E and 1F). Altogether, the behavioral tests suggest that bi-monthly FMD cycles improve visual attention, working memory, and spatial memory; ameliorate anxiety-associated behaviors; and increase exploratory activity in E4FAD female mice.
FMD cycles reduce hippocampal and cortex Aβ load, Aβ peptides, and neuroinflammatory cytokines, while increasing hippocampal neurogenesis markers in E4FAD mice
In the E4FAD model, we assessed hippocampal and cortical Aβ immunoreactivity in 7- to 7.5-month-old female mice after ~4 months of bi-monthly FMD cycles (Figure 2A and S5A). Compared with E4FAD females that were fed an ad libitum diet, those receiving the FMD showed reduced Aβ load in the subiculum (p < 0.01; Figure 2B) and cortex (p < 0.001; Figure 2D), but not in the CA1 region of the hippocampus (Figure 2C). We also measured the levels of Aβ38, Aβ40, and of Aβ42 in Tris-buffered saline (TBS)-soluble cortex extracts, but did not detect changes between the E4FAD control and FMD groups (Figures S1B–S1D). In the triton-soluble (TBS-X) cortex extracts, which were examined to assess the amount of Aβ in a membrane-associated state (Kostylev et al., 2015), we observed a significant reduction in all three peptides in the E4FAD mice treated with periodic FMD (Aβ38, p < 0.05, Figure 2E; Aβ40, p < 0.01, Figure 2F; Aβ42, p < 0.05, Figure 2G). These results indicate that FMD cycles either reduce the generation or contribute to the clearance of different Aβ peptides.
Figure 2. FMD cycles reduce hippocampal and cortex Aβ load, Aβ peptides, and neuroinflammatory cytokines, while increasing hippocampal neurogenesis markers in E4FAD mice.
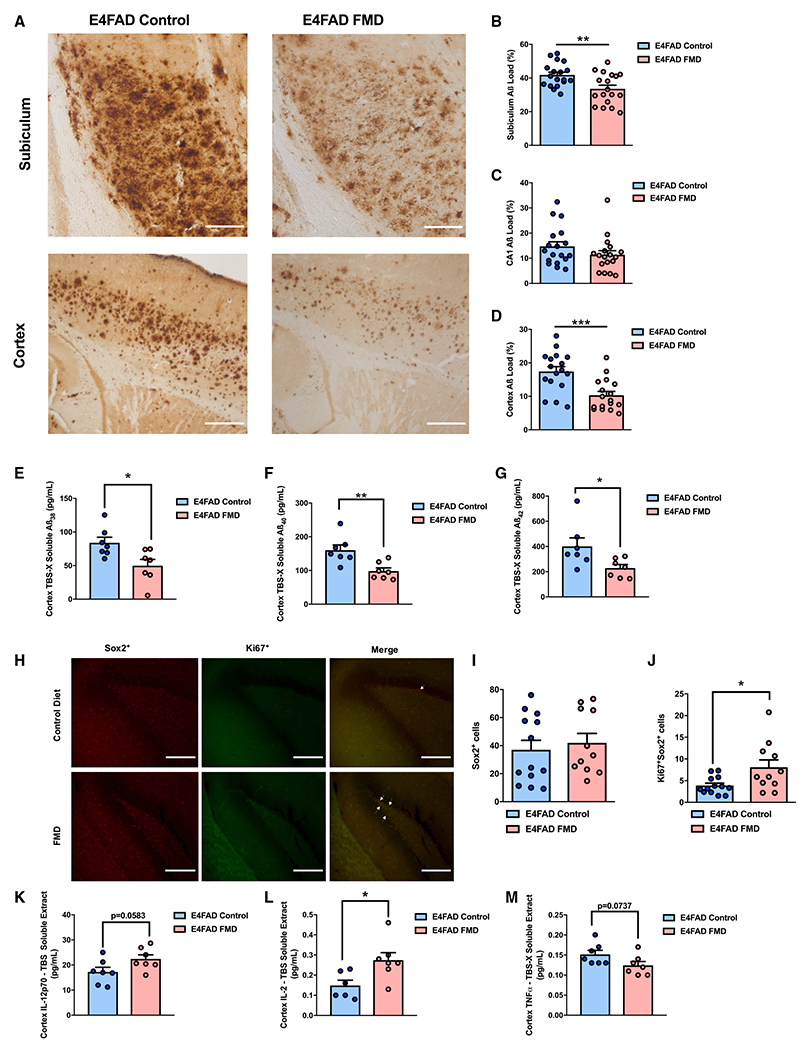
(A) Representative images showing Aβ immunoreactivity in subiculum and cortical regions of female E4FAD control and FMD groups.
(B) Quantification of subiculum Aβ load (%) for female E4FAD control (n = 18) and FMD (n = 18) groups.
(C) Quantification of CA1 Aβ load (%) for female E4FAD control (n = 18) and FMD (n = 18) groups.
(D) Quantification of cortex Aβ load (%) for female E4FAD control (n = 17) and FMD (n = 17) groups.
(E) Quantification of triton-soluble Aβ38 for female E4FAD control (n = 7) and FMD (n = 7) groups.
(F) Quantification of triton-soluble Aβ40 for female E4FAD control (n = 7) and FMD (n = 7) groups.
(G) Quantification of triton-soluble Aβ42 for female E4FAD control (n = 7) and FMD (n = 7) groups.
(H) Representative images showing Sox2+, Ki67+, and co-stain hippocampal immunohistochemistry for ~7–7.5-month-old female E4FAD control and FMD groups. White arrows indicate the Ki67+/Sox2+ foci.
(I) Quantification of ~7–7.5-month-old E4FAD female Sox2+ (control [n = 13] and FMD [n = 11]) cells in the dentate gyrus (DG) after ~4 months of FMD cycles.
(J) Quantification of ~7–7.5-month-old E4FAD female Ki67+Sox2+ (control [n = 13] and FMD [n = 11]) cells in the DG after ~4 months of FMD cycles.
(K) Quantification of IL-2p70 in TBS-soluble cortex extract in control (n = 7) and FMD (n = 7) ~7–7.5-month-old female E4FAD mice.
(L) Quantification of IL-2 in TBS-soluble cortex extract in control (n = 6) and FMD (n = 7) ~7–7.5-month-old female E4FAD mice.
(M) Quantification of TNFα in detergent-soluble/triton-soluble cortex extract in control (n = 7) and FMD (n = 7) ~7–7.5-month-old female E4FAD mice. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001, unpaired two-tailed Student’s t test. Images were taken at 20× magnification. Scale bar, 100 μm.
Next, we measured neurogenesis markers in female E4FAD mice after 4 months of FMD cycles. Specifically, we measured cells that stained positive for Ki67+ (an endogenous marker of proliferation) in the dentate gyrus (DG) (Khuu et al., 2018; Ma et al., 2014; Kee et al., 2002), or that expressed both Ki67 and Sox2, a neuronal stem cell marker, in the hippocampus of control and of FMD-treated female E4FAD mice (Figure 2H). Sox2+ levels remained unchanged between the two groups (Figure 2I), while there was a significant increase in Ki67+Sox2+ cells in mice receiving the FMD compared with the control animals (p < 0.05; Figure 2J), consistent with adult neurogenesis in FMD-treated mice.
To exclude the possibility that the Sox2+ cells may be proliferating astrocytes, immunofluorescence staining was carried out on sections from E4FAD control and FMD mice using Sox2+ and GFAP+ antibodies. There was no significant difference in the number of Sox2+ cells, GFAP+ Sox2+ cells, and the percentage of GFAP+Sox2+ cells/total Sox2+ cells in the DG between the E4FAD control (n = 3) and FMD (n = 4) groups (Figures S1F, S1G, S1I, and S1J). However, there was a non-significant trend for an increase in the number of GFAP+ cells in the DG in the E4FAD FMD mice compared with the E4FAD control mice (p = 0.0715; Figures S1F and S1H).
To investigate neuroinflammation in the E4FAD model, we performed a multi-plex analysis on several cytokines in TBS-soluble and triton-soluble cortex extracts from female E4FAD mice treated or untreated for 4 months with FMD cycles (ending at ~7–7.5 months of age). E4FAD females that underwent four FMD cycles displayed a non-significant trend toward an increase in interleukin (IL)-12p70 (a cytokine) (Méndez-Samperio, 2010), and an increase in IL-2 levels compared with age-matched controls (p = 0.0583, Figure 2K; p < 0.05, Figure 2L). IL-2 was previously shown to reduce the levels of AD pathology and to improve cognitive behavior in AD transgenic mouse models (Gao et al., 2017; Alves et al., 2017), while IL-2 knockout mice have an impairment in spatial learning and memory (Petitto et al., 1999). Tumor necrosis factor alpha (TNFα) levels in human AD brains are normally elevated (Dong et al., 2015). In our triton-soluble cortex extracts, we observed a non-significant trend toward TNFα reduction in E4FAD FMD females compared with control mice (p = 0.0737; Figure 2M). Because FMD cycles caused a significant reduction in Aβ peptides in E4FAD FMD females (Figures 2E–2G), we asked whether there could be an interaction between CD11b and Aβ, since CD11b was previously shown to form a complex with Aβ in AD subjects and to facilitate its clearance (Zabel et al., 2013). Our co-immunoprecipitation (coIP analysis) of CD11b and Aβ42 binding did not reveal a significant difference in Aβ42 fold change between the E4FAD control and FMD groups, at least in the triton-soluble cortex extracts, which harbor membrane-associated Aβ (Figure S1E).
Taken together, these data suggest that 4 months of bi-monthly FMD cycles in female E4FAD mice mitigate Aβ hippocampal and cortical load, reduce Aβ38/40/42 and increase IL-2 expression in cortex extracts. There is also evidence for a modest increase in neurogenesis after FMD treatment.
FMD cycles improve cognitive behavior in 3xTg mice
We also studied the effects of FMD cycles on 3xTg male and female mice starting at 3.5 months of age. In addition to a group on a standard rodent chow diet (control) and a group receiving FMD treatment for 4–5 days, we studied a group that consumed a 7-day protein-restricted diet (approximately 4% of calories obtained from proteins [4% PR]), based on our previous studies (Parrella et al., 2013; Levine et al., 2014) (Figure 3A). FMD males underwent a 4-day cycle versus a 5-day cycle, since 3xTg males were found to lose weight more rapidly and to a higher extent compared with female mice. 3xTg male and female control mice weighed an average of 40.3 g and 34.7 g respectively at 18.5 months. 3xTg male and female mice in the FMD and 4% PR groups regained most of their weight lost during the diet cycles upon refeeding (Figures S4A–S4F). Midpoint behavioral assays SAB in the Y-Maze and Novel Object Recognition [NOR]) were conducted when mice reached 10.5 months of age (after 7 months of dietary treatment), and approximately half-way through the study, with a final behavioral assay (Barnes maze) conducted toward the end of the study, prior to sacrifice, when mice were approximately 18 months of age (Data S1, 2A to 7D). Survival was monitored until 18 months of age. No significant difference in percent survival was detected between control, FMD, and 4% PR groups at 18 months (Figures 3B and 3C).
Figure 3. FMD cycles improve cognitive behavior in 3xTg mice.
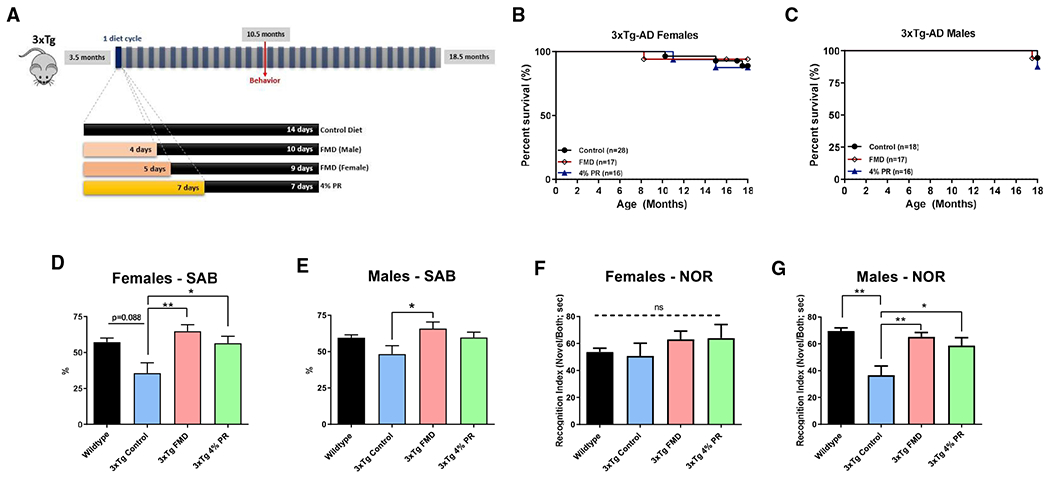
(A) Experimental diet and behavior schedule for 3xTg males and females starting at 3.5 months of age through 18.5 months of age.
(B) Kaplan-Meier survival curves for 3xTg females in control (n = 25/28), FMD (n = 16/17), and 4% PR (n = 14/16).
(C) Kaplan-Meier survival curves for 3xTg males in control (n = 17/18), FMD (n = 16/17), and 4% PR (n = 14/16) groups.
(D) SAB score (%) for 8.5-month-old C57B/6 WT females (n = 8), 10.5-month-old 3xTg female control (n = 14), FMD (n = 14), and 4% PR (n = 16) groups.
(E) SAB score (%) for 8.5-month-old C57B/6 WT males (n = 8), 10.5-month-old 3xTg male control (n = 13), FMD (n = 16), and 4% PR (n = 15) groups.
(F) Recognition index (RI) scored as a percentage for trial 2 (time in seconds spent exploring novel object versus old object) of NOR task for 8.5-month-old C57B/6 WT females (n = 7), and 10.5-month-old 3xTg female control (n = 14), FMD (n = 13), and 4% PR (n = 13) groups.
(G) RI for trial 2 of NOR task for 8.5-month-old C57B/6 WT males (n = 8), and 10.5-month-old 3xTg male control (n = 14), FMD (n = 14), and 4% PR (n = 16) groups.
Figures 1B and 1C: n = mice that survived to 18 months of age/total mice enrolled in study for group.
Figures 1D–1G: 8.5-month-old C57B/6 WT male and female data shown here are the same data as from Figure 5 and S3 and are shown as a comparison with aging 3xTg mice with or without previous treatment cycles. Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01, one-way ANOVA and Bonferroni post test.
3xTg mice receiving FMD or 4% PR diets had higher SAB scores compared with the 3xTg that received the control diet or wild-type (WT) mice on the control diet (3xTg FMD versus 3xTg control, p < 0.01; 3xTg 4% PR versus 3xTg control, p < 0.05; Figure 3D). For the male mice, there was a significant increase in SAB in the 3xTg FMD group (p < 0.05; Figure 3E), but not in the 4% PR group compared with the 3xTg control. No significant difference in the total number of arm entries (a measure of activity) was seen among the groups in both sexes, except when the 3xTg mice groups were compared with a sex-matched WT (**p < 0.01, ***p < 0.001; Figures S2A and S2B), indicating that all groups were similarly active.
In the NOR assay, there were no significant differences among the groups, male or female, in the first trial of the test, indicating no significant biases were present in mice activity among the groups (Figures S2C and S2D). In trial 2, during which one of the identical objects was replaced with a novel object, 3xTg mice displayed a major reduction in recognition index (RI), which did not occur in 3xTg mice treated with either FMD or 4% PR cycles, among the male 3xTg groups (3xTg FMD versus 3xTg control, p < 0.01; 3xTg 4% PR versus 3xTg control, p < 0.05; Figure 3G). There were no differences in RI among the female 3xTg groups (Figure 3F). These results suggests that both FMD and protein restriction cycles can improve cognitive performance in 3xTg mice, although FMD cycles appear to have a more consistent effect, which is in most cases not dependent on sex.
We also assessed male and female 18-month-old 3xTg mice after ~14.5 months of FMD and 4% PR dietary regimens as well as age-matched controls (Data S1- 2A and 7D). Of the various combinations tested, a non-significant trend for a reduction in latency (time spent to locate escape box) was observed in female 3xTg FMD compared with female 3xTg controls (Data S1- 2D). The female 3xTg FMD group also displayed a non-significant trend for an increased success rate in finding the escape box compared with the 3xTg female controls (Data S1–2E).
Together, these behavioral tests suggest that FMD cycles improve visual attention and working and spatial memory in aged 3xTg mice. However, in old mice, the accumulation of pathology appears to minimize these effects of the FMD in 3xTg mice.
FMD cycles slow the progression of AD-associated pathology, increase levels of hippocampal neurogenesis markers, and regulate microglia levels and activation in aged 3xTg mice
Since Aβ accumulation and hyperphosphorylated tau are well-established markers for AD (Lo et al., 2013), we assessed hippocampal Aβ load and the number of hippocampal neurons that are AT8+, a marker for abnormally phosphorylated tau (Parrella et al., 2013), in female and male 3xTg mice at the end of the study. We stained for Aβ in the subiculum and CA1 hippocampal regions of aged female 3xTg mice after ~15 months of dietary regimens in approximately 18.5-month-old mice (Figure 4A, top and middle; Figure S5B, top). We observed that female 3xTg mice treated with the periodic FMD had a reduced Aβ load in the subiculum (p < 0.01; Figure 4B) and in CA1 regions (p < 0.05; Figure 4C) compared with control 3xTg mice. A similar reduction in Aβ load was observed in the subiculum of 4% PR female 3xTg mice compared with the control 3xTg females (p < 0.01; Figure 4B). The number of AT8+ neurons in the combined subiculum and CA1 regions from female 3xTg groups (Figure 4A, bottom; Figure S5B, bottom) was reduced in FMD-treated 3xTg females compared with the control 3xTg mice (p < 0.01; Figure 4D), while there was no significant difference in AT8+ neuron number between the 4% PR 3xTg and the 3xTg control animals (Figure 4D). In the subiculum and CA1 hippocampal regions of aged male 3xTg mice after ~15 months of dietary regimens (18.5 months of age) (Figure 4E, top and middle; Figure S5C, top), there were no significant reductions in subiculum (Figure 4F) or CA1 (Figure 4G) Aβ load between the FMD and control 3xTg groups. A non-significant trend toward a reduction in Aβ load in the CA1 was observed in the 4% PR male 3xTg group with the control 3xTg males (p = 0.0525; Figure 4G). Concerning hyperphosphorylated tau counts, the number of AT8+ neurons in the combined subiculum and CA1 regions for the male 3xTg groups (Figure 4E, bottom; Figure S5C, bottom) was reduced in the FMD 3xTg mice compared with the 3xTg controls (p < 0.05; Figure 4H). A similar reduction in the number of AT8+ neurons was observed in the 4% PR animals and FMD group with the 3xTg controls (Figure 4E, bottom) (p < 0.05; Figure 4H). These results indicate that FMD cycles reduce both Aβ load and/or hyperphosphorylated tau in female and male mice, in part through the temporary reduction of protein intake.
Figure 4. FMD cycles slow the progression of AD-associated pathology, increase levels of hippocampal neurogenesis markers, and regulate microglia levels and activation in aged 3xTg mice.
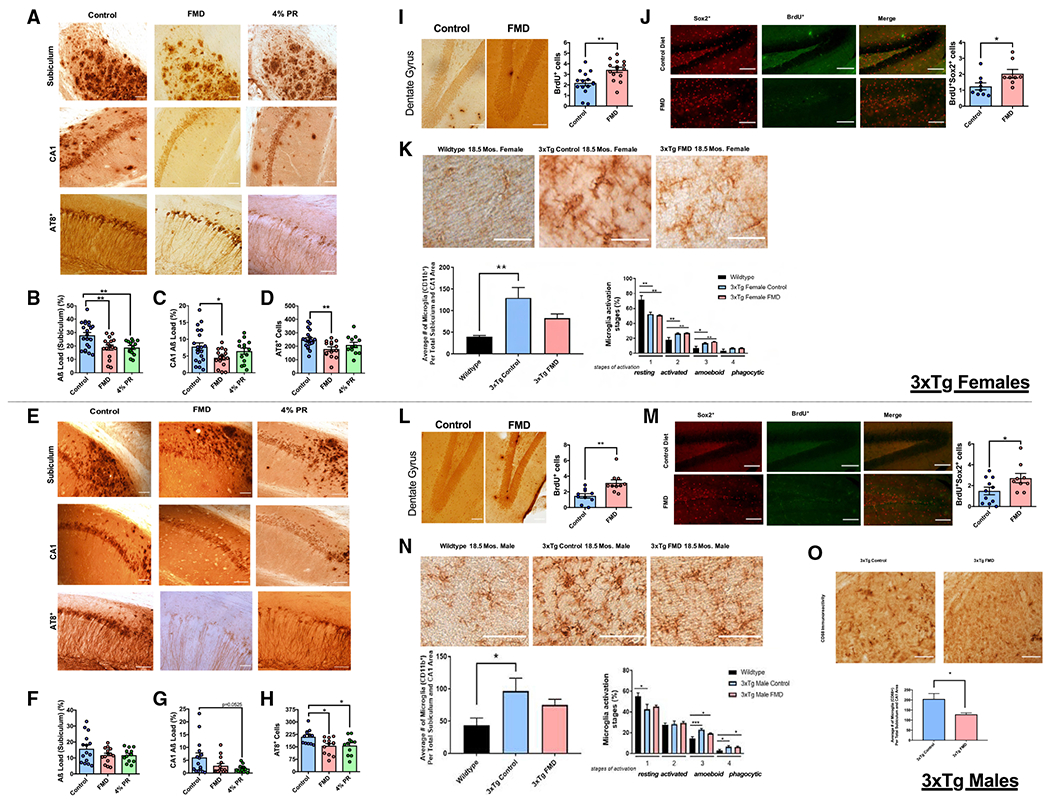
(A) Representative images showing Aβ immunoreactivity and AT8-positive neurons (recognizes abnormally phosphorylated tau) in subiculum or CA1 hippocampus regions of female control, FMD, and 4% PR 3xTg mice.
(B) Quantification of subiculum Aβ load (%) for female 3xTg control (n = 21), FMD (n = 16), and 4% PR (n = 14) mice.
(C) Quantification of CA1 Aβ load (%) for female 3xTg control (n = 20), FMD (n = 16), and 4% PR (n = 14) mice.
(D) Quantification of AT8+ neurons in the subiculum and CA1 for female 3xTg control (n = 20), FMD (n = 14), and PR (n = 12) mice.
(E) Representative images showing Aβ immunoreactivity and AT8+ neurons in subiculum or CA1 hippocampus regions of male 3xTg control, FMD, and 4% PR mice.
(F) Quantification of subiculum Aβ load (%) for male 3xTg control (n = 15), FMD (n = 14), and 4%PR (n = 12) mice.
(G) Quantification of CA1 Aβ load (%) for male 3xTg control (n = 15), FMD (n = 14), and 4%PR (n = 12) mice.
(H) Quantification of AT8+ neurons in the subiculum and CA1 for male 3xTg control (n = 12), FMD (n = 13), and 4%PR (n = 11) mice.
(I) Representative images showing BrdU+ DAB-immunohistochemistry in DG of hippocampus for 18.5-month-old female 3xTg control and FMD groups (left). Quantification of BrdU+ cells within the SGZ and inner third of the granule cell layer of the DG for 18.5-month-old female 3xTg control (n = 15) and FMD (n = 15) groups (right).
(J) Representative images showing Sox2+, BrdU+, and co-stain hippocampal immunohistochemistry for 18.5-month-old female 3xTg control and FMD groups (left). Quantification of 18.5-month-old 3xTg female BrdU+Sox2+ (control [n = 9] and FMD [n = 8]) cells within the SGZ and inner third of the granule cell layer of the DG after ~15 months of FMD cycles (right).
(K) Representative images showing CD11b-ir microglia in hippocampus sections of 18.5-month-old female C57B/6 WT, 3xTg control, and 3xTg FMD mice (top). Quantification of density of CD11b-ir cells in hippocampus CA1 and subiculum combined brain regions of C57B/6 WT, 3xTg control, and 3xTg FMD groups (bottom left; n = 5–7 animals per group). Percentage of different microglia activation stages (from 1 to 4) of C57B/6 WT, 3xTg control, and 3xTg FMD mice (bottom right; n = 5–7 animals per group).
(L) Representative images showing BrdU+ DAB-immunohistochemistry in DG of hippocampus for 18.5-month-old male 3xTg control and FMD groups (left). Quantification of BrdU+ cells within the SGZ and inner third of the granule cell layer of the DG for 18.5-month-old male 3xTg control (n = 10) and FMD (n = 10) groups (right).
(M) Representative images showing Sox2+, BrdU+, and co-stain hippocampal immunohistochemistry for 18.5-month-old male 3xTg control and FMD groups (left). Quantification of 18.5-month-old 3xTg male BrdU+Sox2+ (control [n = 11] and FMD [n = 9]) cells within the SGZ and inner third of the granule cell layer of the DG after ~15 months of FMD cycles (right).
(N) Representative images showing CD11b-ir microglia in hippocampus sections of 18.5-month-old male C57B/6 WT, 3xTg control, and 3xTg FMD mice (top). Quantification of density of CD11b-ir cells in hippocampus CA1 and subiculum combined brain regions of C57B/6 WT, 3xTg control, and 3xTg FMD (bottom left; n = 5–8 animals per group). Percentage of different microglia activation stages (from 1 to 4) of C57B/6 WT, 3xTg control, and 3xTg FMD mice (bottom right; n = 5–8 animals per group).
(O) Representative images showing CD68+ microglia in hippocampus sections of 18.5-month-old male 3xTg control and 3xTg FMD mice (top; n = 2–3 animals per group). Quantification of density of CD68+ cells in hippocampus CA1 and subiculum combined brain regions of 3xTg control and 3xTg FMD (bottom; n = 2–3 animals per group. Data are presented as mean ± SEM. (B–D and F–H) *p < 0.05 and **p < 0.01, one-way ANOVA. (I–O) *p < 0.05 and **p < 0.01, unpaired two-tailed Student’s t test. Images were taken at 20× magnification. Scale bar, 100 μm.
Previous work from our group indicates that FMD cycles in aged WT mice can promote neurogenesis (Brandhorst et al., 2015). A greater decline in neurogenesis was previously observed in aging 3xTg mice versus age-matched WT controls (Rodríguez et al., 2008). We measured bromodeoxyuridine (BrdU) incorporation within the subgranular zone (SGZ) and inner third of the granule cell layer of the DG (DG) in 18.5-month-old male and female 3xTg mice to determine whether FMD cycles had any effect on neurogenesis (FMD and 4% PR; Figures 4I and 4J and 4L and 4M and S2E–S2J). Using DAB immunohistochemistry, we observed that, among the female 3xTg groups, the FMD group but not the 4% PR group displayed an increase in the number of BrdU+ cells in the DG compared with that in 3xTg female controls (p < 0.01; Figure 4I) (Figure S2F). Among the 3xTg males, both the FMD and 4% PR groups showed an increase in the number of BrdU+ cells in the DG as well, compared with the male controls (p < 0.01, Figure 4L; p < 0.05, Figure S2H).
Because FMD cycles increased BrdU+ DG cells, we evaluated whether BrdU+ cells in these groups were also Sox2+, as evidence for type I and type II neural stem cells (NSCs) (Ming and Song, 2005) (Figures 4J and 4M). In both the female and male 3xTg control and FMD groups, we did not observe changes in the number of Sox2+ cells in the DG (Figures S2I and S2J), although we did see a modest but significant increase in BrdU+Sox2+ levels in the FMD groups compared with the controls (p < 0.05; Figures 4J and 4M). These results indicate that FMD cycles can cause modest increases in the generation of NSCs in agreement with the results for the E4FAD mice.
To assess neuroinflammation, we stained for CD11b, a marker for activated microglia and macrophages in the hippocampi of 18.5-month-old WT male and female mice and compared them with the control and FMD 18.5-month-old 3xTg female and male groups (Figure 4K, top; Figure 4N, top). Among females, there was a major increase in CD11b+ cells in the combined subiculum and CA1 regions of the 3xTg mice on a standard diet compared with microglia density in WT controls (p < 0.01; Figure 4K, bottom left). FMD cycles reduced CD11b cells to a level that was no longer significantly higher than that of the WT mice (Figure 4K, bottom left). Female 3xTg mice that were subjected either to the control diet or to the periodic FMD showed a lower proportion of resting state microglia and a higher proportion of activated and amoeboid microglia compared with the age-matched WT mice (stage 3: WT versus control, *p < 0.05; WT versus FMD, **p < 0.01; Figure 4K, bottom right). Similarly, among male mice, there was a major increase in microglia density in the combined subiculum and CA1 regions of the 3xTg mice on a standard diet compared with that in WT controls (p < 0.05; Figure 4N, bottom left). FMD cycles reversed this increase in microglia (Figure 4N, bottom left). The 3xTg mice on the control or FMD diets displayed either a lower or a trend toward a lower proportion of resting state microglia and a higher proportion of amoeboid microglia compared with the age-matched WT mice on the standard diet (stage 1, WT versus control,*p < 0.05; stage 3, WT versus control, ***p < 0.0001; WT versus FMD,*p < 0.05; stage 4, WT versus control and WT versus FMD, *p < 0.05; Figure 4N, bottom right). FMD cycles also reduced the average number of cells expressing CD68+, another microglia marker in the hippocampus of male 3xTg mice (p < 0.05; Figure 4O, bottom).
Overall, these data indicate that a long-term regimen of FMD cycles can reduce AD-associated pathology in aged male and female 3xTg mice, possibly by reducing microglia density and by modulating microglia activation state. Notably, others have shown that perivascular macrophages and not microglia may be responsible for oxidative damage and pathology in AD mouse models (Park et al., 2017), raising the possibility that macrophages in addition to or instead of microglia may be responsible for these effects. Stem cell-dependent generation of neurons could also contribute to the positive effects of the FMD on cognition in 3xTg mice.
Short-term treatment with FMD cycles improves memory, mitigates pathology progression, reduces microglia activation, and reduces the expression of neuroinflammation genes, microglial activation, and tau phosphorylation in 3xTg mice
We also administered FMD cycles over a short-term period to mice ~6.5 months of age, when AD pathology is increasing rapidly and begins to influence cognitive behavior and neuroinflammation (Oddo et al., 2003). Male and female 3xTg mice from the FMD group were administered a 4-day FMD, for five cycles (4 days of diet, 10 days of refeeding), and were sacrificed after the fifth cycle, and before refeeding (Figure 5A).
Figure 5. Short-term treatment with FMD cycles improves memory, mitigates pathology progression, reduces microglia activation, and reduces the expression of neuroinflammation genes, microglial activation, and reduced tau phosphorylation in 3xTg mice.
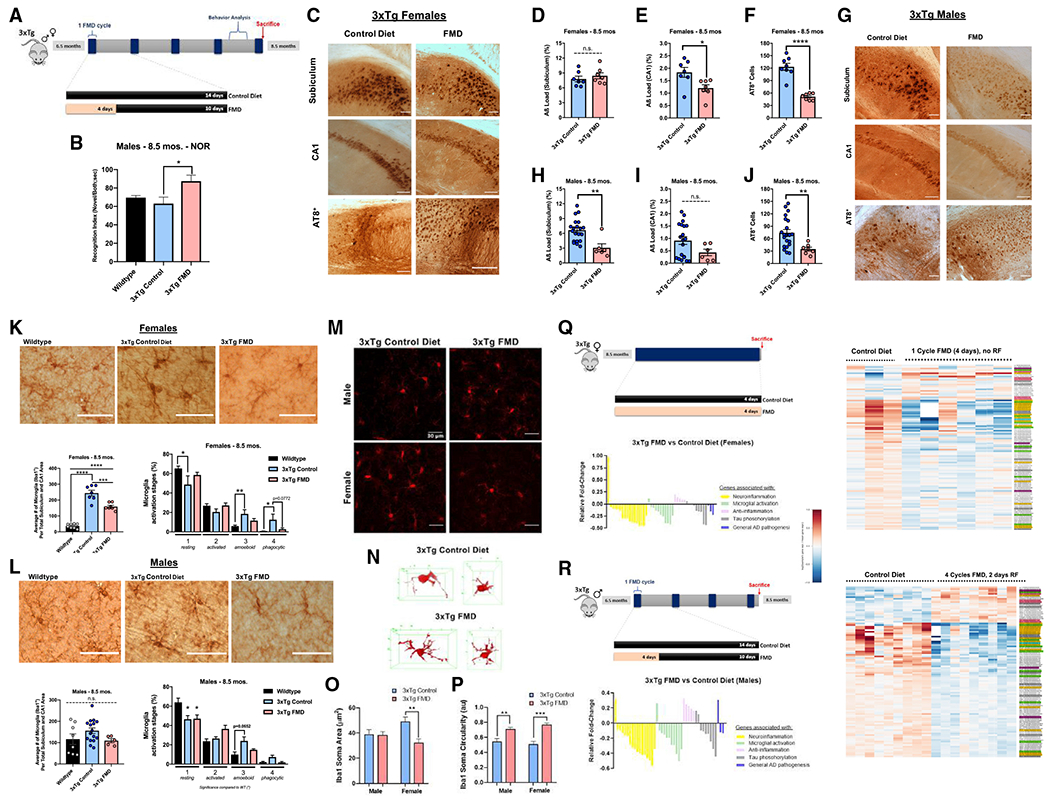
(A) Experimental diet and behavior schedule for 3xTg males and females starting at 6.5 months of age through approximately 8.5 months of age for five FMD cycles.
(B) RI for trial 2 of NOR task for 8.5-month-old C57B/6 WT males (n = 8) and 8.5-month-old 3xTg male control (n = 10) and FMD after four cycles of FMD and 7 days of refeeding (n = 5).
(C) Representative images showing subiculum and CA1 Aβ immunoreactivity and AT8+ neurons in hippocampus for 8.5-month-old 3xTg female control and FMD groups.
(D) Quantification of subiculum Aβ load (%) for 3xTg female control (n = 7) and FMD (n = 7) groups.
(E) Quantification of CA1 Aβ load (%) for 3xTg female control (n = 7) and FMD (n = 7) groups.
(F) Quantification of AT8+ neurons in the subiculum and CA1 for 3xTg female control (n = 8) and FMD (n = 8) groups.
(G) Representative images showing subiculum and CA1 Aβ immunoreactivity and AT8+ neurons in hippocampus for 8.5-month-old 3xTg male control and FMD groups.
(H) Quantification of subiculum Aβ load (%) for 3xTg male control (n = 20) and FMD (n = 6) groups.
(I) Quantification of CA1 Aβ load (%) for 3xTg male control (n = 20) and FMD (n = 6) groups.
(J) Quantification of AT8+ neurons in the subiculum and CA1 for 3xTg male control (n = 21) and FMD (n = 7) groups.
(K) Representative images showing Iba1-stained microglia in hippocampus sections of 8.5-month-old female C57B/6 WT and 3xTg control and FMD groups (top). Quantification of density of Iba1+ microglia in the CA1 and subiculum hippocampus regions of C57B/6 WT females (n = 8) and 3xTg female control (n = 7) and FMD (n = 7) groups (bottom left). Percentage of different microglia activation stages (from 1, resting, to 4, most activated) of C57B/6 WT females (n = 8) and 3xTg female control (n = 7) and FMD (n = 7) groups (bottom right).
(L) Representative images showing Iba1-stained microglia in hippocampus sections of 8.5-month-old male C57B/6 WT and 3xTg control and FMD groups (top). Quantification of density of Iba1+ microglia in the CA1 and subiculum hippocampus regions of C57B/6 WT males (n = 8) and 3xTg male control (n = 16) and FMD (n = 5) groups (bottom left). Percentage of different microglia activation stages (from 1, resting, to 4, most activated) of C57B/6 WT males (n = 8) and 3xTg male control (n = 16) and FMD (n = 5) groups (bottom right).
(M) Representative images of confocal stack immune reactive for Iba1 microglia (red) in the prefrontal cortex for male and female 3xTg control diet and FMD cohorts. Scale bars, 100 μm.
(N) Example 3D skeletonized microglial projections for 3xTg control diet and 3xTg FMD cohorts.
(O and P) Quantification of Iba1 immuno-reactive soma area (O) and circularity (P) in male (M) and female (F) 3×Tg mice with control diet (open bars) and FMD (filled bars) from high-magnification images (n = 2/group).
(Q) Experimental timeline and gene expression (top left and right) in cortex samples of 8.5-month-old female 3xTg controls (n = 3) and 3xTg females after one 4-day cycle of FMD, with no refeeding (n = 6). Yellow-, green-, blue-, rose-, gray-, and lavender-highlighted gene names in heatmap correspond with genes associated with neuroinflammation, microglial activation, neuroinflammation and microglial activation, anti-neuroinflammation, tau phosphorylation, and general association with AD pathogenesis, respectively. Relative fold change (Log2-transformed fold-change values, centered at 0) of gene expression in FMD group versus control diet group (bottom left) for genes associated with neuroinflammation, microglial activation, anti-neuroinflammation, tau phosphorylation, and general association with AD pathogenesis. Values in histogram were calculated from fold change in Table S1. “0” value is equivalent to no change between FMD group and control diet group.
(R) Experimental timeline and gene expression (top left and right) in cortex samples of 8.5-month-old male 3xTg controls (n = 9) and 3xTg males after four cycles of FMD and 2 days of refeeding (n = 9). Yellow-, green-, blue-, rose-, gray-, and lavender-highlighted gene names in heatmap correspond with genes associated with neuroinflammation, microglial activation, neuroinflammation and microglial activation, anti-neuroinflammation, tau phosphorylation, and general association with AD pathogenesis, respectively. Relative fold change (Log2-transformed fold-change values, centered at 0) of gene expression in FMD group versus control diet group (bottom left) for genes associated with neuroinflammation, microglial activation, anti-neuroinflammation, tau phosphorylation, and general association with AD pathogenesis. Values in histogram were calculated from fold change in Table S1. “0” value is equivalent to no change between FMD group and control diet group. Data are presented as mean ± SEM. (B) *p < 0.05, compared with WT; one-way ANOVA followed by Tukey’s multiple comparisons test. (D-F and H-J) *p < 0.05, **p < 0.01, ****p < 0.0001; unpaired two-tailed student’sttest. (K and L) *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; one-way ANOVA followed by Tukey’s multiple comparisons test. (M-P) Two mice were analyzed per group, n = 20–35 cells per group. **p < 0.01, ***p < 0.001; two-way ANOVA followed by Tukey’s multiple comparisons test. Images were taken at 20× magnification unless otherwise noted. Scale bar, (C, G, K, L) 100 μm and (G) 30 μm. RNA-seq libraries were sequenced 1 × 50 bp on an Illumina HiSeq3000 system
Cognitive behavior was assessed after four cycles, when mice were approximately 8.5 months old and before the fifth cycle. SAB results showed no significant differences among the male or female cohorts (Figures S3A and S3B). However, 3xTg mice did not display a reduction in SAB and NOR performance compared with WT controls, suggesting that the effect of the FMD may represent an improvement even compared with WT control mice, in agreement with our previous studies (Brandhorst et al., 2015). NOR results instead suggest the 3xTg FMD males had a significantly higher average RI score compared with that in 3xTg control male mice (p < 0.05; Figure 5B). No significant differences were observed among the female groups (Figure S3C). There were no significant differences among the groups, female or male, in the total number of arm entries (a measure of activity) in the Y-maze (Figures S3D and S3E) as well as in the first trial of the NOR test, indicating no significant biases were present in mice activity among the groups (Figures S3F and S3G).
Among the female groups, no significant changes in Aβ load were observed in the subiculum (Figures 5D and S5D, top), but, in the CA1 region, there was a significant reduction in Aβ in the 3xTg FMD females compared with controls (p < 0.05; Figure 5E). AT8+ hyperphosphorylated tau was significantly reduced in the hippocampus of 3xTg FMD females (p < 0.0001; Figure 5F and S5D, bottom). Aβ load was significantly reduced in the subiculum of 3xTg FMD males compared with that in 3xTg control diet males (p < 0.01; Figures 5H and S5E, top), whereas no significant changes were observed in CA1 Aβ load (Figure 5I). A significant reduction in hyperphosphorylated tau was observed in the 3xTg FMD males compared with that in controls (p < 0.01; Figure 5J and S5E, bottom).
We also stained hippocampal tissue for ionized calcium-binding adaptor protein-1 (Iba1), a 17-kDa actin-binding protein that is specifically and constitutively expressed in all microglia (Hovens et al., 2014), counted Iba1 density, and categorized the stages of microglial activation based on criteria established in previous studies (Kreutzberg, 1996; Crews and Vetreno, 2016) (Figures 5K and 5L, top). 3xTg female mice displayed a major increase in microglial number compared with WT controls (p < 0.0001; Figure 5K, bottom left), which was reduced in 3xTg FMD females (p < 0.001; Figure 5K, bottom left). There was a significant reduction in microglia at stage 1 of activation in the 3xTg female controls compared with WT (p < 0.05; Figure 5K, bottom right). Microglia at stage 3 and 4 of activation were significantly increased in the 3xTg control group compared with WT (stage 3, p < 0.01; stage 4, p < 0.05; Figure 5K, bottom right). 3xTg FMD females displayed a non-significant trend for the reduction in microglia at stage 4 compared with the 3xTg control diet group (p = 0.0772; Figure 5K, bottom right). Among the groups in the male cohort, there was no significant increase in microglia number among any of the male groups (Figure 5L; bottom left). Both 3xTg male groups having significantly fewer microglia at stage 1 compared with WT males (p < 0.05; Figure 5L, bottom right). In summary, resting-state microglia were reduced and different forms of activated microglia were increased in 3xTg compared with WT mice. In this set of short-term treatment experiments, the FMD appears to allow a general state of microglial activation while reducing the highly active phagocytic microglia, possibly in part because of the reduction in Aβ and AT8+ hyperphosphorylated tau in the 3xTg FMD-treated mice.
In order to investigate the effect of FMD cycles on amyloid pathology, we isolated microglia from primary mixed glia cultures from the whole brains (except the cerebellum) of 8.5-month-old male and female 3xTg mice five cycles of FMD or control diet, and added oligomeric Aβ42 to the microglia to assess whether FMD can enhance the uptake of oligomeric Aβ42 by IBA-1 positive microglia compared with the control diet. We found that the oligomeric Aβ42 localizes to the cytosol of the microglial cells (Figures S7A and S7C). IBA-1 positive microglia isolated from the brain of 8.5-month-old 3xTg male mice after FMD cycles starting at 6.5 months of age internalized significantly more Aβ42 compared with the IBA-1 positive microglia from the control group (p < 0.05; Figure S7B). A possible link between FMD and reduced Aβ accumulation could be via the increase in oligomeric Aβ42 internalization by IBA-1 positive microglia.
We next investigated the presence of any changes in the characteristics of microglia after short-term FMD cycles in male and female 3xTg mice. Using quantitative confocal microscopy methodology previously established (Stephen et al., 2015, 2019), representative images of confocal stack immune-reactive for Iba1 microglia in the prefrontal cortex (Figure 5M) and 3D skeletonized microglial projections (Figure 5N) were used to quantify the Iba1 immuno-reactive soma area (Figure 5O) and circularity (Figure 5P). Resting microglia cells tend to be smaller, rounder cells with elaborate ramifications, whereas activated microglia tend to be bigger and more amoeboid-like in shape with retracted processes (Davis et al., 2017). Iba1 soma area was significantly reduced in the 3xTg FMD females compared with 3xTg control diet females (p < 0.01; Figure 5O), while soma circularity was significantly increased in the dietary intervention groups of both sexes compared with sex-matched controls (p < 0.01, 3xTg FMD males versus 3xTg control males; p < 0.001, 3xTg FMD females versus 3xTg control females; Figure 5P). These results are consistent with an effect of FMD cycles in modulating the microglial activation state observed in 3xTg mice.
To further test the hypothesis that reduced or modified neuroinflammation and/or microgliosis mediate part of the protective effects of FMD cycles, we performed mRNA sequencing from homogenized cortex samples from hemi-brains of female 3xTg mice ~8.5 months old after only one cycle of a 4-day FMD and before refeeding (Figure 5Q, top left) as well as in male 3xTg ~8.5-month-old mice after four cycles of a 4-day FMD and after 2 days of refeeding (Figure 5R, top left). For 104 genes, expression was altered significantly by the FMD in the male cohort (Figure 5R, right). When the expression of these genes was assessed in the female cohort, a similar pattern emerged (Figure 5Q, right). Relative fold change indicated an effect of FMD in consistently reducing the expression of genes associated with neuroinflammation, microglial activation, tau phosphorylation, and AD pathogenesis, and increasing in the expression of genes associated with anti-inflammatory functions, based on characteristics from the Ensembl genome database (Figures 5Q and 5R, bottom left; Table S1). Of the genes that were downregulated in the FMD groups, Fosb, Gdi1, Hspa8, Smdt1, Nr4a1, Nr4a3, Fosl2, Trib1, Egr1/2/3/4, Tiparp, Fbxo33, Hspa1b, and Prmt1 were all previously found to be linked to increased oxidative stress, pro-inflammatory cytokine secretion, and/or upregulation of gene expression after ischemia (Nomaru et al., 2015; Lopes et al., 2016; Wu et al. (2019b); Bonam et al., 2019; Azevedo et al., 2018; Lyons and West, 2011; Close et al., 2019; Vilkeviciute et al., 2019; Giri et al., 2005; Mengozzia et al., 2012; Marballi and Gallitano, 2018; Wu et al., 2019a; Flood et al., 2004; Clarimón et al., 2003; Liu et al., 2019) (Table S1; Data S1 pages 3–17). Of these genes, Egr1 was also suggested to have a role in stimulating microglial activation (Raj et al., 2015), along with Sertad1, Gadd45b, Ndel1, Hmgcr, Spry2, Arc, Prdx5, Siah2, and Hspa1a (Kuhn et al., 2006; Tamboli et al., 2010; Abels et al., 2019; Rosi, 2011; Sun et al., 2010; Park et al., 2016) (Table S1; Data S1, pages 20–28). On the other hand, the genes that were upregulated in the FMD group, including Cd33, Inpp5d, Stab1, Tia1, and Rsrp1, are associated with anti-inflammatory roles (Malik et al., 2015; Park et al., 2009; Chen and Liu, 2017; Stephens et al., 2019) (Table S1; Data S1, pages 33–37). Mertk, a gene associated with activated microglia that phagocytose dying, stressed, or excess neurons (Nomura et al., 2017), was also upregulated in the FMD groups (Table S1; Data S1, page 29). Thus, these results, taken together, are consistent with an effect of the FMD in decreasing the activation and oxidant production in microglia but possibly also macrophages or other brains cells, including smooth muscle cells and endothelial cells, which have been linked to inflammation and the expression of many genes identified here (Chow et al., 2007; Park et al., 2017).
The diet groups also showed reductions in Nedd8, Cacybp, Fos, Phf13, and Junb, with the male cohort showing an additional reduction in Ppme1. (Table S1; Data S1, pages 39–44), which were all previously linked to the localization and/or increased phosphorylation of tau (Mori et al., 2004; Wasik et al., 2013; Anderson et al., 1994; Vázquez-Higuera et al., 2011; Park et al., 2018; Chu et al., 2013; Fang et al., 2016). The expression of Hook1, which encodes the protein that localizes to tau aggregates, was slightly increased in the dieting mice groups (Table S1; Data S1, page 45), although it has been previously demonstrated that the expression of Hook1 proteins is reduced in AD (Herrmann et al., 2015). Rheb, a direct activator of mammalian target of rapamycin (mTOR) (Lafourcade et al., 2013), was significantly downregulated in FMD-treated male 3xTg mice, with a similar trend seen with dieting 3xTg females (Table S1; Data S1, page 46) in agreement with the demonstrated effect of mTOR inhibition in neuroprotection in various in vivo models of neurodegenerative disease, and of the mTOR inhibitor rapamycin in ameliorating tau pathology (Caccamo et al., 2013). Yod1, which is specifically associated with macro-autophagy, and codes for the cofactor YOD1, which binds with p97 to promote lysosomal clearance (Papadopoulos et al., 2017), was significantly increased in the FMD 3xTg male cohort compared with male 3xTg controls, although this effect was not observed in female 3xTg mice (Table S1; Data S1, page 47). Notably, full-length tau is preferentially degraded via macro-autophagy, which involves the activation of AMP-activated protein kinase (AMPK), and in turn reduces mTOR signaling (Zare-shahabadi et al., 2015). Hspa8, Atpif1, and Fez1, genes associated with autophagy, had reduced expression in both males after four FMD cycles and 2 days of refeeding and in females after 4 days of FMD and no refeeding, indicating that chronic autophagy activation is unlikely to be responsible for the clearance of tau or Aβ.
Other genes generally associated with AD pathogenesis were also affected by diet. Plcg2 was increased in FMD group males, with no effect in FMD group females (Table S1; Data S1, page 49). It has been suggested that the activation of Plcg2, rather than inhibition, could be therapeutically beneficial in treating AD (Magno et al., 2018). Otud1 encodes a deubiquitinase (DUB) enzyme, a class of enzymes that have been suggested as potential targets for treating AD (Baillie et al., 2017; Yuan et al., 2018). In FMD-treated mice, Otud1 was significantly reduced in males, with a similar trend seen in females (Table S1; Data S1, page 50). Erf, which encodes a transcription factor related to the E26 transformation-specific (ETS) family of proteins, was found to be reduced in FMD-treated mice (Table S1, Data S1, page 51). ETS-domain proteins have been linked to regulating neuronal functions, especially by activating the transcription of early-onset AD genes such as PSEN1 (Pandey et al., 2019).
Taken together, these results suggest that short-term FMD cycles reduce inflammation, improve short-term memory, and ameliorate AD-associated pathology in 3xTg mice. These effects may be mediated by wide-acting effects involving the modulation of microglia and possibly other pro-inflammatory cell types, including perivascular macrophages (Park et al., 2017), resulting in reduced inflammation, Aβ, and hyperphosphorylated tau, while allowing microglia to contribute to the scavenging of damaged cells, organelles, and macromolecules.
Short-term cycles of FMD regulate Nox2 cortex levels in 3xTg and E4FAD mice, while Nox2 deletion or inhibition improves cognitive behavior, mitigates pathology progression, and reduces microglia activation
We have previously shown that O2− and iron contribute to neurotoxicity in Aβ-treated neuronal cell lines (Longo et al., 2000), and our studies in vitro demonstrated that peroxynitrite (ONOO−), formed by the reaction between NO and O2−, is a major mediator of the neurotoxicity promoted by microglia activated by Aβ or lipopolysaccharide (LPS), suggesting that it can mediate the toxicity caused by chronically activated microglia in brains affected by AD (Xie et al., 2002). In fact, targeting Nox2 reduced neurovascular and cognitive dysfunctions in mice overexpressing the Swedish mutation of the APP (Park et al., 2008). Altogether, this suggests that, while microglia cells play a key role in neuronal protection and repair, at the same time, their production of O2− leading to the subsequent production of ONOO− may play a central role in neurotoxicity (Figure 6A).
Figure 6. Short-term cycles of FMD regulate Nox2 cortex levels in 3xTg and E4FAD mice, while NOX2 deletion or inhibition improves cognitive behavior, mitigates pathology progression, and reduces microglia activation.
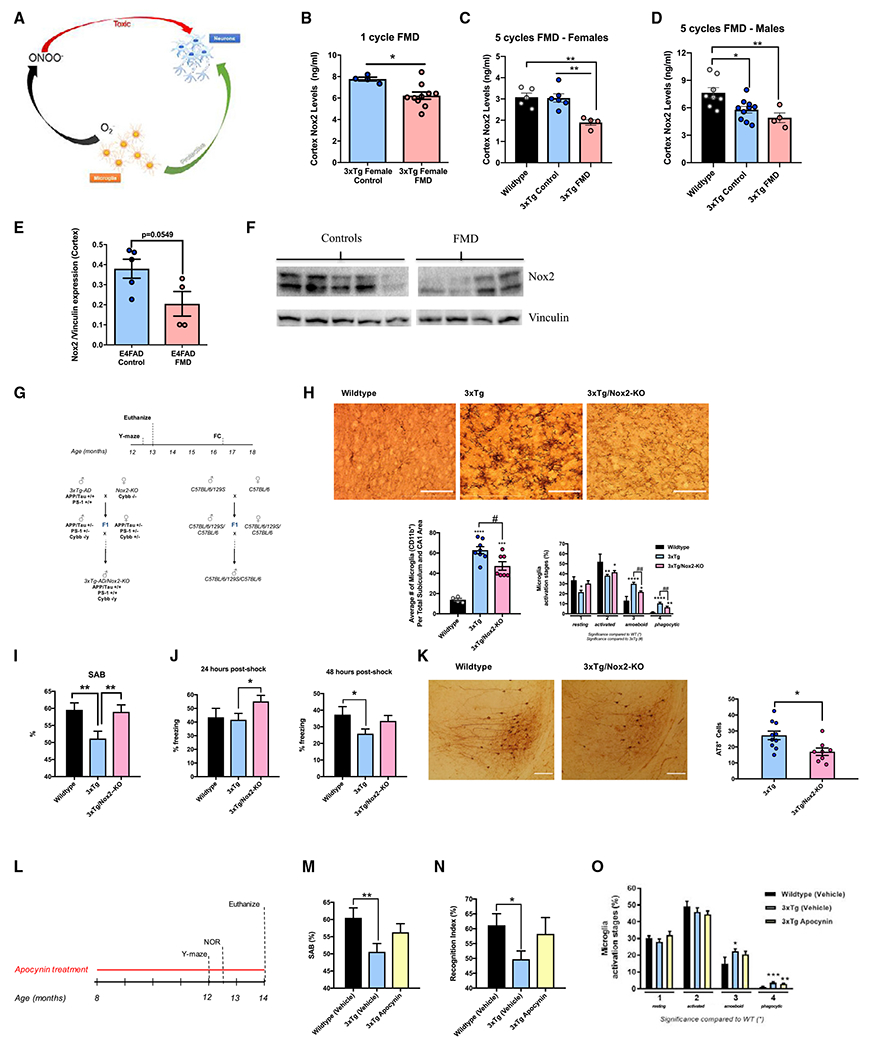
(A) Diagram describing the dual positive and negative function of microglia. Microglia are involved in neuronal development and repair, but their production of O2− into ONOO− can promote toxicity and lead to neurodegeneration.
(B) Quantification of Nox2 (ng/mL) in cortex extract of 8.5-month-old female 3xTg controls (n = 4) and 3xTg females after one 4-day cycle of FMD with no refeeding (n = 10).
(C) Quantification of Nox2 (ng/mL) in cortex extract of 8.5-month-old female C57B/6 WT (n = 5) and 8.5-month-old 3xTg female control (n = 6) and FMD after five cycles of FMD and no refeeding after last cycle (n = 4) groups.
(D) Quantification of Nox2 (ng/mL) in cortex extract of 8.5-month-old male C57B/6 WT (n = 8) and 8.5-month-old 3xTg male control (n = 10) and FMD after five cycles of FMD and no refeeding after last cycle (n = 4) groups.
(E) Quantification of Nox2 levels in cortex extract of control (n = 5) and FMD (n = 4) ~7-7.5-month-old female E4FAD mice after 4 months of biweekly FMD cycles (measured as Nox2/Vinculin protein expression levels).
(F) Western blot image used for quantifying Nox2 levels in whole-cortex extract of control (n = 5) and FMD (n = 4) ~7- to 7.5-month-old female E4FAD mice. Vinculin was loading control (bottom).
(G) 3xTg/Nox2-KO mice generation and experimental design. The experimental design of the tests conducted on 3xTg/Nox2-KO and control mice is depicted, as well as a schematic representation of the breeding strategy used to develop 3xTg/Nox2-KO mice and corresponding WT (mixed background 129/B6/B6). Some mice were euthanized for pathology at 13–14 months, while those used for fear-conditioning (FC) tests were aged until 15–18 months.
(H) Representative images showing CD11b-ir microglia in hippocampus sections of 13- to 14-month-old male WT (129/B6 background), 3xTg, and 3xTg/Nox2-KO mice (top). Quantification of density of CD11b-ir cells in hippocampus CA1 and subiculum combined brain regions of WT (129/B6 background), 3xTg, and 3xTg/Nox2-KO (bottom left; n = 4–8 animals per group). Percentage of different microglia activation stages (from 1 to 4) of WT (129/B6 background), 3xTg, and 3xTg/Nox2-KO mice (bottom right; n = 4–8 animals per group).
(I) WT (129/B6 background), 3xTg, and 3xTg/Nox2-KO male mice were tested with the Y-maze apparatus (12.5 months of age). SAB scores obtained through Y-maze task are shown (n = 7–23 per group).
(J) WT (129/B6 background), 3xTg, and 3xTg/Nox2-KO male mice (15–18 months of age) FC tests. Freezing times (%) 24-h post shock for FC are shown (n = 6–22 per group) (left). WT (129/B6 background), 3xTg, and 3xTg/Nox2-KO male mice (15–18 months of age) underwent FC tests. Freezing times (%) 48-h post shock for FC are shown (n = 6–22 per group) (right).
(K) Representative images showing AT8 antibody (recognizes abnormally phosphorylated tau) immunoreactivity in hippocampus of 13- to 14-month-old male 3xTg and 3xTg/Nox2-KO mice (left). Quantification of total AT8-immuno-reactive cells in hippocampus of 13- to 14-month-old male 3xTg and 3xTg/Nox2-KO mice (n = 10–13/group) (right).
(L) Experimental design of apocynin treatment. Eight-month-old WT (129/B6 background) and 3xTg mice were treated with apocynin-dissolved drinkable water or apocynin-free water for 6 months. During the final 4 weeks of treatment, the animals were tested using the Y-maze apparatus, and NOR and Rotarod assays. After completion of the behavioral tasks, the mice were euthanized and their brains analyzed.
(M) Comparison of SAB scores between WT (129/B6 background) vehicle, 3xTg vehicle, and 3xTg apocynin-treated mice using the Y-maze apparatus (n = 10–13 animals per group).
(N) Comparison of RI values between WT (129/B6 background) vehicle, 3xTg vehicle, and 3xTg apocynin- treated mice during NOR test (n = 10–13 animals per group).
(O) Percentage of different microglia activation stages (from 1 to 4) of WT (129/B6 background) vehicle, 3xTg vehicle, and 3xTg apocynin-treated mice (n = 5–10 animals/group). Data are presented as mean ± SEM. (H and O) *p < 0.05, **p < 0.01, ***p < 0.001 compared with WT; #p < 0.05, ##p < 0.01 for 3xTg/Nox2-KO versus 3xTg, one-way ANOVA followed by Tukey’s multiple comparisons test (H, bottom left) and Fisher’s least significant difference test (H, bottom right, and O). (B, E, and K) *p < 0.05, unpaired two-tailed student’s t test. (C and D) *p < 0.05, **p < 0.01, ***p < 0.001 compared with WT; one-way ANOVA followed by Tukey’s multiple comparisons test. (I, J, M, and N) *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA followed by Fisher’s least significant difference test. Images were taken at 20× magnification. Scale bar, 100 μm.
Thus, we assessed whether the expression of Nox2 was affected at different time points of FMD administration. After one cycle of a 4-day FMD to young female 3xTg mice, aged ~8.5 months (Figure 5Q, top left), we observed a significant reduction in Nox2 levels compared with an age- and sex-matched control (p < 0.05; Figure 6B). In female mice, Nox2 levels were significantly reduced in the 3xTg group that received five cycles of the FMD compared with both female WT mice and with the 3xTg control diet group (p < 0.01; Figure 6C). In the male cohort, there was a significant reduction in Nox2 levels in both 3xTg males on the control diet or that received five FMD cycles compared with WT male mice on a standard diet (p < 0.01, WT versus 3xTg FMD; p < 0.05, WT versus 3xTg control; Figure 6D). We did not observe a significant difference in Nox2 between the FMD and control groups after four cycles of FMD and 2 days of refeeding in male 3xTg mice (p = 0.1497; Figure S3H), although we observed a non-significant trend toward a reduction of Nox2 levels after refeeding in FMD-treated female E4FAD mice (p = 0.0549; Figures 6E and 6F). Based on the results of the effects of FMD in mediating Nox2 levels and the potential role of Nox2 as a mediator of AD pathology, we hypothesized that the knockout of NADPH oxidase could protect against cognitive decay and neuropathology in the 3xTg mouse model.
Thus, we generated 3xTg/Nox2-KO mice by crossing Nox2-KO (Cybb−/−) mice with 3xTg mice (Figure 6G). Similar to 18.5-month-old male 3xTg mice treated with FMD cycles (Figure 4N), 13.5- to 14-month-old 3xTg/Nox2-KO male mice displayed a partial reversal of the increase in microglia density in the combined subiculum and CA1 regions compared with 3xTg mice. Activated microglia cells as detected by CD11b (Lynch, 2009) were reduced in 3xTg/Nox2-KO mice, compared with 3xTg mice (#p < 0.05; Figure 6H, bottom left), while both groups had significantly elevated levels of microglia in the combined subiculum and CA1 regions compared with the WT controls (****p < 0.0001, 3xTg and ***p < 0.001, 3xTg/Nox2-KO; Figure 6H, bottom left). When microglia activation stages were examined, based on a four-stage classification (Zhang et al., 2011; Parrella et al., 2013), 3xTg mice showed a higher proportion of microglia in the highly activated stages (3 and 4) compared with the WT group (stage 1, *p < 0.05; stage 2, **p < 0.01; stage 3, ****p < 0.0001; stage 4, ****p < 0.0001; Figure 6H, bottom right). In contrast, 3xTg/Nox2-KO mice showed a similar resting and stage 2 activation state, but a lower high activation state compared with 3xTg (stage 3, ##p < 0.01; stage 4, ##p < 0.01; Figure 6H, bottom right).
With our progeny of male 3xTg/Nox2-KO mice (Figure 6G), we assessed short-term working memory as well as contextual and tone learning with the Y-maze apparatus and fear-conditioning (FC) behavioral assay, respectively. Mice of strains C57B/6/Nox2-KO, 3xTg, 3xTg/Nox2-KO, and corresponding WT mice (C57B/6 and 129/B6) were assessed in these cognitive tasks. There was a significant reduction in SAB scores with 3xTg male mice compared with 12.5-month-old, age-matched WT mice and the 3xTg/Nox2-KO mice (p < 0.01; Figure 6I), and no significant changes in the number of arm entries were apparent among the groups (Figure S3J). FC tests revealed that memory in the 3xTg/Nox2-KO group improved compared with the 3xTg group (p < 0.05; Figure 6J, left) at both 24- and 48-h post shock (p < 0.05; Figure 6J, right) with no changes in the 3xTg/Nox2-KO group compared with WT. Following Y-maze testing, the mice were tested with FC, based on previously established protocols (Liu et al., 2004). When tested with the FC test on day 1 for baseline measurements, no significant differences in freezing time among the groups was apparent (Figure S3K). On day 2, 24 h post shock, memory in the 3xTg/Nox2-KO improved compared with the 3xTg group (p < 0.05; Figure 6J, left). On day 3, in the novel environment before re-exposure to the tone, no significant changes in freezing time were seen among the WT, 3xTg, and 3xTg/Nox2-KO mice (Figure S3I). Upon re-exposure to the tone, 3xTg mice displayed decreased freezing behavior compared with the WT group (p < 0.05; Figure 6J, right). These results indicate that Nox2 inactivation was able to delay the decline in working memory and associative learning in 3xTg/Nox2-KO mice.
Although Aβ accumulation was not modified by the inactivation of NADPH oxidase (Figure S3I), we observed a significant reduction in AT8+ hyperphosphorylated tau in the 3xTg/Nox2-KO mice compared with 3xTg male controls (p < 0.05; Figure 6K), indicating that reduced NADPH oxidase activity and O2−/ONOO− generation represents only part of the effects of FMD/refeeding cycles.
Apocynin is extensively used as an inhibitor of NADPH oxidase activity and of the concomitant production of ROS, including peroxynitrite, both in vitro and in vivo. In vivo treatment for periods of 6 months were reported (Figure 6L; Stefanska and Pawliczak, 2008; Simonyi et al. 2012; ‘t Hart et al., 2014). The effect of apocynin treatment on 3xTg cognitive dysfunction was assessed using the Y-maze apparatus and the NOR assay. When we tested these mice at ~12 months of age on the Y maze, 3xTg mice exhibited a significant working memory deficit in comparison with WT mice, whereas SAB performance in 3xTg mice treated with apocynin was similar to that of control mice, in agreement with our results with 3xTg/Nox2-KO mice (p < 0.01, 3xTg vehicle versus WT vehicle; Figure 6M). We did not find significant differences in the number of arm entries among WT and 3xTg groups, suggesting that the drug treatment does not interfere with the activity levels of the rodents (Figure S3M). Similarly, for NOR, 3xTg mice had a significantly lower RI score compared with the WT group, whereas 3xTg apocynin-treated mice did not (p < 0.05, 3xTg vehicle versus WT vehicle; Figure 6N), which is also in agreement with the results obtained with 3xTg/Nox2-KO mice. There were no significant differences among the groups in the first trial of the test, indicating no biases were present in mice activity among the groups (Figure S3N). Although apocynin-treated WT mice were assessed in all of the above parameters, there were no significant differences between apocynin-treated WT and the WT vehicle groups (Figures S6A–S6D).
Notably, mouse treatment with apocynin did not modulate Aβ accumulation and did not affect tau hyperphosphorylation in the hippocampus of 3xTg mice (Figures S6E–S6H). Apocynin treatment had minor effects in dampening the increase in amoeboid and phagocytic states observed in the hippocampus of 3xTg mice (stage 4, ***p < 0.001 3xTg vehicle versus WT vehicle, **p < 0.01 3xTg Apocynin versus WT vehicle; Figure 6O).
These results indicate that NADPH oxidase activity and probably O2−/ONOO− generation contribute to cognitive decline, but not to Aβ accumulation in Alzheimer’s mouse models.
Safety and feasibility of FMD cycles in patients diagnosed with amnestic mild cognitive impairment or early-stage AD
To begin to assess the feasibility and safety of FMD cycles in patients diagnosed with amnestic mild cognitive impairment (aMCI) or mild AD, we started a phase I/II randomized and placebo-controlled (single-blind) clinical study to test the effects of monthly FMD cycles in 40 patients with aMCI or mild AD and adequate nutritional status (see STAR Methods for the inclusion criteria of this trial). Twenty-eight patients have been enrolled to date (13 males, 15 females; average age 71 years, range 55–80 years; Figure 7A). Patients were diagnosed with aMCI or with early-stage AD (Mini-Mental Status Examination [MMSE] score 18–23) according to the international diagnostic criteria and regularly followed up at the Geriatric Unit of the San Martino Hospital (Genoa, Italy) or of the Santa Maria della Misericordia Hospital (Perugia, Italy). After screening and a baseline assessment addressing cognitive performance, functional status and caregiver burden (same assessments are repeated at 6- and 12-month points of study), 12 patients were randomly included in the FMD (active) group, while the other 16 patients were assigned to the placebo group (Figure 7A). The placebo diet assigned to patients in the control arm consists of replacing lunch or dinner with a meal based on pasta or rice with vegetables for 5 days a month, without supplements, whereas patients in the FMD arm complete FMD cycles that last 5 days, with supplements noted for fasting-mimicking, neuroprotective, anti-inflammatory, and antioxidant properties, including olive oil, coconut oil, algal oil, nuts, caffeine, and cocoa, given to patient in between FMD cycles for 25 days and while on a normal diet (Figure 7A).
Figure 7. Safety and feasibility of FMD cycles as a treatment for patients with aMCI or mild AD.
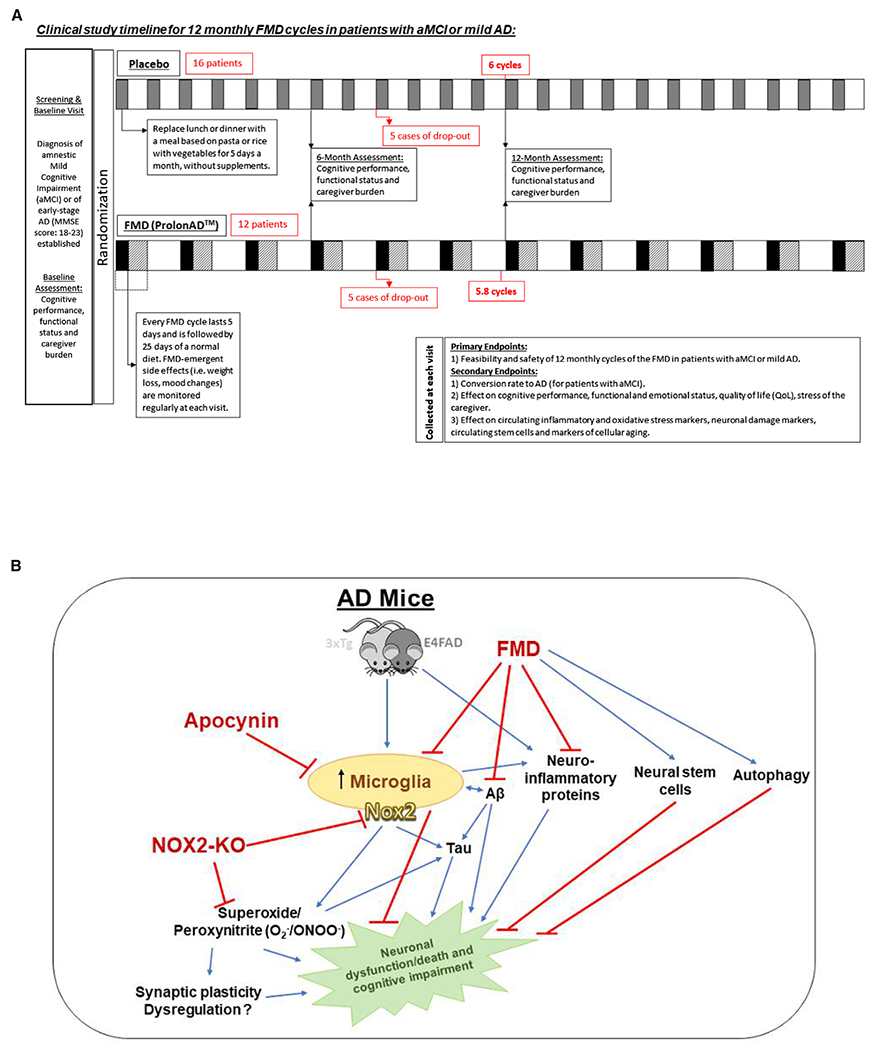
(A) A phase I/II randomized and placebo-controlled (single-blind) clinical study for 40 patients with aMCI or mild AD was designed for 12 monthly FMD cycles; 28/40 enrolled patients were randomly assigned to a placebo (control) diet arm (n = 16) or an FMD (ProLonAD) arm (n = 12). In the placebo arm, a gray square indicates that, for 5 days a month, patients are assigned a diet in which lunch or dinner is replaced with a meal based on pasta or rice with vegetables, without added supplements. In the FMD arm, a black square indicates a 5-day FMD cycle, followed by a striped box that indicates 25 days of a normal diet. White box indicates an in-between period between FMD cycles. Primary and secondary endpoints collected at each visit (per FMD cycles completed) is described in lower right-hand corner.
(B) Graphical representation of the effects of FMD on the E4FAD and the 3xTg mouse models of AD, through which it mediates AD-associated pathology, neuroinflammation, NSCs, and Nox2 levels.
Patients from the FMD arm and from the placebo control arm have received an average of 5.8 (range 1–12) and of 6 (range 1–12) diet cycles, respectively. Five cases of drop-out were recorded in the FMD arm (41.6%) after an average of four FMD cycles. These were due to poor acceptance of the FMD components (n = 2), worsening of the nutritional status (n = 2), or to personal reasons (n = 1). On the other hand, in the placebo diet arm, five cases of drop-out (31.3%) were also recorded (also after an average of four placebo diet cycles), as a result of worsening of the nutritional status (n = 3), poor acceptance of the prescribed diet (n = 1), or personal reasons (n = 1). The FMD-emergent adverse events (graded according to Common Terminology Criteria for Adverse Events 5.0) that have been observed so far are all mild or moderate. They include fatigue (grade 1 [G1]; n = 5, 41.6%), headache (G1 or G2; n = 4, 33.3%), hypotension (G2; n = 1), irritability (G2; n = 1), autoimmune reaction (G2; n = 1; transient worsening of a pre-existing pemphigus), abdominal pain (G1; n = 2), depression (G1; n = 1), and paresthesia (G1; n = 1). The number of patients enrolled in the clinical trial, the number of FMD cycles, and the adverse events for each patient have been included in Table S2.
Overall, diet compliance has also been satisfactory during the periods between FMD cycles, when patients take several supplements during the day. Thus, these initial data suggest that 5-day FMD cycles administered once a month have been feasible and overall safe in a small group of patients with aMCI/early AD. However, further monitoring of the enrolled patients and completing the foreseen patient accrual for this clinical study are both necessary to confirm these conclusions. As more patients are enrolled, and cognitive assessments or secondary endpoints are assessed after multiple FMD cycles, more information will be available to determine whether FMD cycles can slow down cognitive decline, delay the conversion rate to AD (for patients with aMCI), and affect biomarkers of inflammation, oxidative stress, neuronal damage, circulating stem cells, and markers of cellular aging.
DISCUSSION
Results from two AD transgenic mouse models indicate that FMD cycles reduce the levels of key pathological markers, including Aβ and hyperphosphorylated tau, as well as microglia density and markers for neuroinflammation to improve cognition. The results obtained with the 3xTg/Nox2-KO mice and with apocynin treatment support our hypothesis that FMD cycles induce positive effects on the 3xTg and E4FAD models in part by modulating the activity of microglia and possibly brain macrophages allowing it to perform protective functions, including the scavenging of Aβ, while at the same time reducing production of toxic O2−/ONOO−.
We show that, in males, there is a significant improvement in oligomeric Aβ42 uptake by microglia isolated from FMD-treated mice. In females, we instead only observed a trend for this effect (Figure S7A–S7D). A recent study showed that microglia activation is higher in AppNL –G-F female mice compared with male mice in response to amyloidosis (Biechele et al., 2020). There is also a higher proportion of activated response microglia (ARMs) cells in 6-month-old and older AppNL–G-F female mice compared with male mice, indicating that the female microglia in an amyloid model of AD are activated earlier than those in males (Sala Frigerio et al., 2019). This could explain the differences in the effects of FMD on the uptake of oligomeric Aβ42 in male versus female microglia. Based on current studies but also on our past studies with periodic essential amino acid restriction, we believe that FMD cycles can affect Aβ levels, but that they protect AD mice by altering both Aβ-dependent and Ab-independent effects, including those on hyperphosphorylated tau and neuroinflammation.
Increasing evidence suggests that the NADPH oxidase complex has a crucial and specific role in modulating microglia activation status, and that ROS production by NADPH oxidase and other sources is involved in neurotoxicity and Aβ-dependent microglia proliferation through the release of pro-inflammatory cytokines (Jekabsone et al., 2006). Together with our previous results showing that both O2− and ONOO− promote Aβ- and microglial-dependent neurotoxicity (Longo et al., 2000; Xie et al., 2002), these results suggest that part of the protective effects of the FMD cycles are associated with reduced microglial and Nox2 activation/levels and therefore reduced O2− and ONOO− generation and toxicity, which may contribute to cognitive decline by damaging neurons, increasing tau phosphorylation but potentially also by interfering with synaptic plasticity (Figure 7B). In fact, O2− and other ROS are known to promote synaptic plasticity raising the possibility that a high and continuous stimulation of long-term potentiation (LTP) could eventually interfere with learning and memory (Massaad and Klann, 2011). Notably, because microglia and perivascular macrophages share key markers, and considering that both can have central roles in superoxide production and neuroinflammation related to AD (Park et al., 2017), further studies are needed to determine the relative role of brain microglia and macrophages but also of endothelial cells, astrocytes, and neurons in the effects described here, and specifically in the contribution of NO and O2−.
The fact that the great majority of significantly downregulated genes in the FMD 3xTg male and female groups compared with 3xTg on the standard diet were in the neuroinflammation, microglial activation, tau phosphorylation, and AD pathogenesis groups, and that the major set of upregulated genes was in the anti-inflammatory group, is consistent with our model (Figure 7B). One notable finding was that the family of Egr(1–4) genes were all downregulated in FMD-treated 3xTg mouse cortex compared with 3xTg controls (Table S1; Data S1, pages 11–13, 31).
In mammals, Egr1 expression is upregulated by the master energy sensor AMPK (Benboubker et al., 2014; Berasi et al., 2006; Andrade et al., 2013), which itself is activated in low-calorie conditions, as well as by drugs such as metformin (Hardie, 2011; Draznin et al., 2012; Blagosklonny, 2009). Interestingly, we previously investigated the role of mammalian EGR1 in cellular protection and its link to glucose and PKA/AMPK, finding that glucose restriction protects cardiomyocytes through AMPK/EGR1 activation (Di Biase et al., 2017). In the context of neurological disorders and injury, it has been suggested that Egr1 is expressed during microglial activation (Raj et al., 2015) and that its inhibition through either siRNA or pharmacological agents may reduce microglial activation and possibly ameliorate the neuroinflammation imposed by AD (Giri et al., 2005). Thus, starvation conditions appear to modulate EGR1 expression differentially in different cell types and tissues, but, in the nervous system, its downregulation is associated with reduced inflammation and pathology. The downregulation of Egr genes that is related to neuroinflammation could also be affected by Egr expression changes in neurons not necessarily related to neuroinflammation. Notably, early growth response 1 (Egr1) gene expression was upregulated in the brain endothelial cells of aged mice (Zhao et al., 2020), and the brains of mice lacking the transcription factor Egr-1 had significantly lower levels of Aβ and β-secretase 1 (BACE-1) compared with WT mice. It was found that Egr-1 promotes Aβ synthesis via transcriptional activation of BACE-1 (Qin et al., 2016).
In previous studies, we showed that FMD cycles alternated with normal diet refeeding can promote stem cell-dependent regeneration in the nervous and other systems (Brandhorst et al., 2015; Choi et al., 2016; Rangan et al., 2019; Cheng et al., 2017). Here, we also show that FMD cycles increase the expression and generation of NSCs (as indicated by BrdU+Sox2+ expression), but the contribution of NSCs to the cognitive improvements or connection with Nox2 remains to be investigated.
In summary, we propose a role for periodic FMD cycles in reducing the expression and activity of neuroinflammatory and toxic microglial/macrophage genes/proteins, including Nox2, and consequently in reducing O2−/ONOO− while preserving the ability of microglia and possibly other cell types to remove Aβ and other accumulated or damaged proteins or cellular components, consistent with studies in a different AD mouse model (Bruce-Keller, et al., 2011). Our ongoing, placebo-controlled, randomized clinical trial in patients with MCI or early-stage AD provides initial evidence indicating that FMD cycles were feasible and safe overall in the 12 patients assigned to the FMD arm thus far. The enrollment of all patients in this trial will provide more conclusive data on the portion of the patient population for which FMD cycles are feasible and safe, but may also provide initial evidence of their effect against cognitive decline and AD progression.
Limitations of the study
It is important to note that a limitation of the RNA sequencing data (Table S1; RNA sequencing [RNA-seq] supplemental file) is that it was obtained from the analysis of RNA from hemi-cortex and not from specific cell types such as neurons or microglia. Thus, in future studies, it will be important to identify the source of high Egr gene(s) expression in AD to determine whether their expression is upstream or downstream of neuroinflammation, pathology, and/or cognitive impairment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the lead contact, Valter D. Longo (vlongo@usc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. RNA-seq data have been deposited at SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Barnes maze data have been deposited at Mendeley data and are publicly available as of the date of publication. The DOI is listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request (vlongo@usc.edu).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-beta amyloid (1:300; IHC) | Thermo Fisher Scientific | Cat#71-5800; RRID: AB_2533989 |
| Mouse anti-Phospho-Tau (Ser202, Thr205) (AT8) (1:1000; IHC) | Thermo Fisher Scientific | Cat#MN1020; RRID: AB_223647 |
| Rat anti-Bromodeoxyuridine/BrdU (BU1/75 (ICR1)) (1:500; IHC) | Novus Biologicals | Cat#NB500-169; RRID: AB_10002608 |
| Rat anti-mouse CD11b (1:500; IHC) | Bio-Rad | Cat#MCA711GT; RRID: AB_1100616 |
| Mouse anti-Iba1 (1:500; IHC) | Wako | Cat#016-26721; RRID: AB_2811160 |
| Rat anti-Mouse CD68 (1:500; IHC) | Bio-Rad | Cat#MCA1957GA; RRID: AB_324217 |
| Biotinylated goat anti-rabbit IgG (for beta amyloid, 1:500; IHC) | Vector Laboratories | Cat#BA-1000; RRID: AB_2313606 |
| Biotinylated horse anti-mouse IgG, rat adsorbed (for AT8 and Iba1, 1:500; IHC) | Vector Laboratories | Cat#BA-2001; RRID: AB_2336180 |
| Biotin-SP-AffiniPure donkey anti-rat IgG (H+L) (for BrdU, 1:250; IHC) | Jackson ImmunoResearch | Cat#712-065-150; RRID: AB_2340646 |
| Donkey anti rat IgG (H+L), biotin (for CD11b, 1:250; IHC) | Thermo Fisher Scientific | Cat#A18743; RRID: AB_2535520 |
| Rat anti-Ki-67 Monoclonal Antibody (SolA15) (1:200; IHC) | Thermo Fisher Scientific | Cat#14-5698-82; RRID: AB_10854564 |
| Rabbit anti-SOX2 (1:200; IHC) | Abcam | Cat#ab97959; RRID: AB_2341193 |
| Mouse anti-GFAP (1:200; IHC) | Cell Signaling | Cat#3670; RRID: AB_561049 |
| Donkey anti-rat-488 (for BrdU, Ki67 and GFAP, 1:400; IHC) | Thermo Fisher Scientific | Cat#A-21208; RRID: AB_2535794 |
| Donkey anti-rabbit-594 (for Sox2, 1:400; IHC) | Thermo Fisher Scientific | Cat#A-21207; RRID: AB_141637 |
| Rat anti-mouse CD11b antibody (1 μg/μL; IP) | Bio-Rad | Cat#MCA711GT; RRID:AB_321292 |
| Rabbit anti-NOX2 (1:5000; WB) | Abcam | Cat#ab129068; RRID: AB_11144496 |
| Rabbit anti-Aβ42 (1:500; WB) | Abcam | Cat#ab201060; RRID: AB_2818982 |
| Mouse anti-Vinculin (1:1000; WB) | Millipore | Cat#MAB3574; RRID: AB_2304338 |
| Chemicals, peptides, and recombinant proteins | ||
| Paraformaldehyde | Sigma-Aldrich | Cat#158127; CAS:30525-89-4 |
| 5’-Bromo-2’-deoxyuridine (BrdU) | Sigma-Aldrich | Cat#B5002; CAS:59-14-3 |
| Normal Donkey Serum | Jackson ImmunoResearch | Cat#017-000-121 |
| Polyvinyl alcohol mounting medium with DABCO, antifading | Sigma-Aldrich | Cat#10981 |
| Hoechst 33342 Solution | Thermo Fisher Scientific | Cat#62249 |
| VECTASHIELD® HardSet™ Antifade Mounting Medium with DAPI | Vector Laboratories | Cat#H-1500 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat#89900 |
| DAB (3,3’-diaminobenzidine) Peroxidase (HRP) Substrate Kit, With Nickel | Vector Laboratories | Cat#SK-4100 |
| Apocynin | Sigma-Aldrich | Cat#178385; CAS:498-02-2 |
| VECTASTAIN® Elite Avidin-Biotin Complex (ABC)-HRP Detection Kit, Peroxidase (Standard) | Vector Laboratories | Cat#PK-6100 |
| NuPAGE™ LDS Sample Buffer (4X) | Invitrogen | Cat#NP0007 |
| HyGLO™ Chemiluminescent HRP Detection Reagent | Denville Scientific | Cat#E2500 |
| RNase-Free DNase Set | Qiagen | Cat#79254 |
| Papain, Lyophilized | Worthington Biochemical | Cat#LS003118; CAS: 9001-73-4 |
| HiLyte™ Fluor 555-labeled Amyloidβ 42 (Aβ42) | AnaSpec | Cat#AS-60480-01 |
| Poly-D Lysine | Sigma-Aldrich | Cat#P7280 |
| Percoll PLUS | Cytiva | Cat#17-5445-02 |
| Critical commercial assays | ||
| Dynabeads™ Protein G Immunoprecipitation Kit | Thermo Fisher Scientific | Cat#10007D |
| Rat CYBB / NOX2 / gp91phox ELISA Kit | LSBio | Cat#LS-F39030 |
| V-PLEX Proinflammatory Panel 1 (mouse) Kit | Meso Scale Diagnostics | Cat#K15048D-1 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat#74804 |
| V-PLEX Plus Aβ Peptide Panel 1 (4G8) | Meso Scale Diagnostics | Cat#K15199G-1 |
| D1000 ScreenTape Assay | Agilent Technologies | N/A |
| KAPA mRNA HyperPrep kit | Roche-Sequencing | N/A |
| Deposited data | ||
| RNA-Seq of hemi-cortex of the 8.5 month old 3xTg young mice study | This paper | SRA: PRJNA864838 |
| Barnes Maze data of E4FAD mice | This paper | Mendeley Data: https://doi.org/10.17632/p2c5z4vjsw.1 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: 3xTg | Frank LaFerla, University of California, Irvine | N/A |
| Mouse: E4FAD | Mary Jo LaDu, University of Illinois | N/A |
| Mouse: Nox2-KO | The Jackson Laboratory | JAX: 002365 |
| Mouse: 3xTg/Nox2-KO | Generated from 3xTg and Nox2KO | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism v.8 | Graphpad | https://www.graphpad.com/ |
| HTSeq | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| EdgeR | McCarthy et al., 2012; Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| FastQC v0.11.8 | Andrews, 2010 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic v0.38 | Bolger et al., 2014 | http://www.usadellab.org/cms/index.php?page=trimmomatic |
| STAR v2.6.1d | Dobin et al., 2013 | http://code.google.com/p/rna-star/ |
| gProfileR v0.6.8 | Reimand et al., 2007 | http://biit.cs.ut.ee/gprofiler/ |
| Imagelab 6.0 software | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Other | ||
| Mouse diet: Fasting mimicking diet (FMD) | Cheng et al., 2017 | N/A |
| Mouse diet: 4% protein-restricted diet (4% PR) | Parrella et al., 2013 | N/A |
| Human diet: ProlonAD TM | Propriety formulation belonging to L-Nutra | https://www.prolon.it |
| Control Diet: PicoLab Rodent Diet 20 | LabDiet | Cat#5053 |
| HydroGel | ClearH2O | N/A |
| Barnes Maze | San Diego Instruments | N/A |
| All-in-One Fluorescence Microscope | Keyence | BZ-X710 |
| Olympus BX50 Microscope | Olympus | N/A |
| Olympus DP73 Camera | Olympus | N/A |
| Zeiss Laser Scanning Microscope-780 upright microscope | Zeiss | N/A |
| 2200 TapeStation | Agilent Technologies | N/A |
| HiSeq3000 | Illumina | N/A |
| Vibratome | Leica | VT1000S |
| Janus G3 liquid handler | Perkin-Elmer | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Southern California. Animals were maintained in a pathogen-free environment, housed in clear shoebox cages in groups of up to five animals per cage with constant temperature, humidity and 12 h/12 h light/dark cycles, and unlimited access to water. Body weight and food intake of individual animals was measured routinely: every day during diet weeks and at least once in the middle of a refeeding week. Mice that appeared weak and/or showed signs of illness were not included in any experiment.
3xTg mice (long-term study)
3xTg breeding pairs were obtained from Frank LaFerla, PhD (UC Irvine) under an MTA agreement. For the long-term study using 3xTg mice, male and female 3xTg mice were divided into the ad libitum-fed control (Control) group, the FMD group, and a 7-day protein-restricted diet (approximately 4% protein in composition relative to carbohydrates and fat) group (4% PR) starting at 3 months of age, until approximately 18.5 months of age. Wildtype mice (C57BL/6) were purchased from Jackson Laboratory (Bar Harbor, ME) at 6 months of age and aged to 18 months at the University of Southern California.
3xTg mice (short-term study)
For the short-term study using 3xTg mice, male 3xTg mice were divided into the ad libitum-fed control (Control) group and the FMD group starting at ~6.5 months of age, until approximately ~8.5 months of age. FMD cycles were 4 days long followed by 10 days of refeeding. Male 3xTg mice were sacrificed 2 days after refeeding, after the 4th FMD cycle. Female 3xTg mice were divided into the ad libitum-fed control (Control) group and the FMD group starting at 8.5 months of age to assess 1 cycle of FMD and sacrificed after 4 days of FMD prior to refeeding. In another short-term study, male and female 3xTg mice were given FMD starting at 6.5 to ~8.5 months for a total of 5 cycles and the mice were euthanized after the 5th cycle before refeeding. Wildtype mice (C57BL/6) were purchased from Jackson Laboratory (Bar Harbor, ME) at 8.5 months of age.
E4FAD mice
E4FAD breeding trios were obtained from Mary Jo LaDu, PhD (University of Illinois) under an MTA agreement. Female E4FAD were divided into the ad libitum-fed control (Control) group or the FMD group starting at 3 months of age, until approximately 7–7.5 months of age.
3xTg/Nox2-KO mice
The following male mice were used in this study: 3xTg, Nox2–KO, 3xTg/Nox2-KO and corresponding wildtypes [WT: C57BL/6 (B6), C57BL/6/129S (129/B6) and C57BL/6/129S /C57BL/6 (129/B6/B6), respectively]. For simplicity, in reference to this study, only the following groups are represented in the data of this manuscript: 129/B6 (referred to as Wildtype), 3xTg, and 3xTg/Nox2-KO.
Nox2-KO mice present a null allele of the X-linked Cybb gene which encodes the Nox2 subunit of the NADPH oxidase complex and lack phagocyte superoxide production (Pollock et al., 1995). Nox2-KO mice and corresponding WT (C57BL/6) were purchased from Jackson Laboratory (Bar Harbor, ME). Colonies of 3xTg mice and related WT (C57BL/6/129S) were established and bred at the University of Southern California. 3xTg/Nox2-KO mice have been generated in our laboratory as described below:
3xTg males were crossed with females KO for Cybb, the gene coding for the Nox2 subunit of NADPH oxidase. Nox2-KO mice and corresponding wildtype (C57BL/6) were purchased from Jackson Laboratory. Since the Cybb gene is X-linked, all the male progenies were hemizygous for the genes related to AD and lacked Nox2. The F1 male offspring were back-crossed to homozygosity for APP/Tau and PS-1 genes. The correct genotype was determined by PCR (Guoet al., 1999; Pollock et al., 1995) or qPCR by comparing ΔCt values of each unknown sample against known homozygous and hemizygous controls. Similarly, in order to obtain the correct 3xTg/Nox2-KO background, C57BL/6/129S males (129/B6, 3xTg background) were crossed with C57BL/6 (B6, Nox2-KO background). To equalize the genetic background the F1 offspring were backcrossed for four generations. To detect possible side effects caused by Nox2 deletion, 3xTg/Nox2-KO mice were constantly monitored for abnormal changes. Although we did not observe apparent signs of distress in 3xTg/Nox2-KO mice, animals showing first signs of skin lesion were immediately excluded from the study.
FMD and aMCI or mild AD clinical trial
A phase I/II randomized and placebo controlled (single-blind) clinical study of a Fasting Mimicking Diet (FMD) in 40 patients diagnosed with amnestic Mild Cognitive Impairment (aMCI) or mild Alzheimer Disease (AD) was designed as follows:
The inclusion criteria are:
-
1)
Written informed consent;
-
2)
Age 55–80;
-
3)
Diagnosis of aMCI or mild AD according to the international definition criteria (Petersen, 2004; McKhann et al., 2011);
-
4)
Adequate nutritional status according to the Mini Nutritional Assessment - MNA® Elderly (12–14 points)
-
5)
Body Mass Index (BMI) ≥ 20 kg/m2.
-
5)
Phase Angle (PhA) > 4.7° at the bioimpedance analysis;
-
6)
Normal organ function.
The exclusion criteria are:
-
1)
Age >80.
-
2)
Diabetes Mellitus.
-
3)
Liver or renal failure.
-
4)
Food allergies to the components of FMD;
-
5)
Therapy with vitamin K antagonists;
-
6)
Patients who live alone or are not adequately supported by the family context;
-
7)
Other experimental therapies.
The primary endpoints of the study are the feasibility and safety of 12 monthly cycles of the FMD in patients with aMCI or mild AD. Feasibility is defined as the consumption of at least one FMD cycle every two months with the possibility of admitting the consumption of only 50% of the planned diet and/or a maximum consumption of 10 Kcal/kg body weight of food not provided in only one of the 5 days of each cycle. FMD-emergent side effects are monitored regularly at each visit. Secondary endpoints are: 1) conversion rate to AD (for patients with aMCI); 2) effect on cognitive performance, functional and emotional status, quality of life (QoL), stress of the caregiver; 3) effect on circulating inflammatory and oxidative stress markers, neuronal damage markers (Neurofilament Light, NfL), circulating stem cells and markers of cellular aging (e.g. evaluation of the telomerase activity of lymphocytes).
The FMD tested in this study (ProlonAD TM) is a plant-based dietary regimen that has fasting-mimicking properties, while ensuring an adequate supply of macro- and micronutrients. Every FMD cycle lasts 5 days and is followed by 25 days of a normal diet. The composition of ProlonAD TM is equal to that of the ProlonTM diet (L-Nutra, Los Angeles, CA, https://www.prolon.it) and provides ~43–47% carbohydrates, ~ 44–46% fats and ~ 9–11% proteins per day in the form of vegetable soups, broths, bars, olives, crackers, herbal teas, with the addition of specific supplements (like olive oil, coconut oil, algal oil, nuts, caffeine and cocoa) that substantially increase the daily calories (300–500 kcal) compared to ProlonTM and seem to have neuroprotective, anti-inflammatory and antioxidant properties. The placebo diet assigned to patients in the control arm will simply consist in taking a meal based on pasta or rice with vegetables to replace lunch or dinner for 5 days a month, without supplements.
Patients, which are all at low nutritional risk, receive rigorous monitoring of their nutritional status every cycle (monthly) through the collection of anthropometric data, evaluation of body composition by bioimpedance analysis and assessment of muscle strength with the handgrip strength test; moreover, they are invited to perform a light/moderate daily physical exercise to promote protein anabolism and maintenance of muscle mass. In each visit FMD-related side effects are recorded, and a venous sampling is made for the evaluation of markers of inflammation, neuronal injury, cellular aging and oxidative stress.
The cognitive performance, functional status and caregiver burden is evaluated at the beginning of the study, at 6 months and at 12 months with a complete neuropsychological and geriatric assessment [Free and Cued Selective Reminding Test (FCRST), Addenbrook’s Cognitive Examination- Revised (ACE-R), Stroop Test, Babcock Test, Digit Span, Trail Making Test A and B, Raven Test, Token Test, Barthel Index, Rockwood’s Frailty Index, CDR-Sum of the boxes, Activities of Daily Living (ADL), Instrumental Activities of Daily Living (IADL), Center for Epidemiologic Studies Depression Scale (CESD-R), Caregiver Burden Inventory, Neuropsychiatric Inventory (NPI)].
METHOD DETAILS
Rodent Diets
Mice were fed ad libitum with PicoLab Rodent Diet 20 (LabDiet). The FMD is based on a nutritional screen that identified ingredients that allow nourishment during periods of low-calorie consumption (Brandhorst et al., 2015). The FMD formulation fed to the AD mouse models consists of two different components designated as day 1 diet and day 2–4/5 diet (in the long-term study, male 3xTg mice consumed 4 days of diet and female 3xTg mice consumed 5 days of diet): The day 1 diet consists of a mix of various low-calorie broth powders, a vegetable medley powder, extra virgin olive oil, and essential fatty acids; day 2–4/5 diet consists of the same ingredients as in day 1, with the addition of glycerol. The diet formulations also contain hydrogel (ClearH2O) to achieve binding and to allow the supply of the food in the cage feeders. Day 1 is composed of approximately 55% fat, 36% carbohydrate and 9% protein, with mice consuming anywhere between 12.36 kJ-17.31 kJ. Day 2–4/5 is composed of approximately 43.3% fat, 47.4% carbohydrate, and 9.3% protein, with mice consuming anywhere between 8.17kJ – 11.44 kJ a day.
The 4% protein-restricted diet is based on previous amino acid restriction studies with 3xTg mice (Parrella et al., 2013). A 7-day diet composed of approximately 75.67% carbohydrate, 3.90% casein protein, and 20.43% fat was fed to 3xTg male and female mice in the 4% PR group, with mice consuming anywhere between 19.36 kJ – 27.10 kJ per day.
Mice consumed all the supplied food on each day of the FMD regimen and showed no signs of food aversion. At the end of either diet, we supplied PicoLab Rodent Diet 20 chow ad libitum for 10, 9, or 7 days before starting another FMD or 4% PR cycle. Prior to the FMD, animals were transferred into fresh cages to avoid feeding on residual chow and coprophagy.
BrdU injections
5′-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich) was administered to male and female 3xTg mice in the long-term study at a 50 mg/kg dose for 7 consecutive days before mice were sacrificed. BrdU powder was dissolved in boiling saline and prepared fresh each day of injection.
Apocynin treatment
Male 3xTg and corresponding wildtype (129/B6 background) mice were housed 3–5 per cage and, starting at 8 months of age, they were treated with 1 mg/mL apocynin (Sigma-Aldrich) dissolved in drinking water or apocynin-free water for 6 months. Dose and route of administration of the drug were chosen according to previous reports (Harraz et al., 2008). The mice were assigned to the experimental groups by simple randomization. The time point of drug treatment was chosen based on previous studies showing that at 8 months of age 3xTg rodents display detectable memory deficits and their brains show Aβ and tau neuropathology (Mastrangelo and Bowers 2008; Oddo et al., 2003). Apocynin powder was dissolved into hot (~60°C) sterile water and then allowed to cool to room temperature before being given to the mice. The drug was replaced every week since between 5-7 days any decay of apocynin is detectable (Harraz et al., 2008). Considering the average daily water intake of approximately 4 mL/mouse ((Bachmanov et al., 2002) and Figure S6I) the mg/kg dose was estimated approximately 150 mg/kg body weight/day.
In order to detect possible adverse effects related to the apocynin treatment, the animals were weighed weekly and monitored for abnormal changes during the treatment period. During the final three weeks of treatment, while they were still under treatment, the mice were tested with the indicated behavioral tests. At the end of the treatment the animals were euthanized as described previously. Since we did not find significant differences between wildtype mice treated with apocynin or vehicle (apocynin-free water) in any of the parameters analyzed, the results obtained with apocynin-treated wildtype mice are not shown.
Behavior tests
SAB using the Y-maze
Short-term working memory was examined by a spatial novelty preference task in the Y-maze (Parrella et al., 2013). The Y-maze was made of black plexi-glass and was designed with three identical arms (50 × 9 × 10 cm), radiating from a central triangle (8 cm on each side) and spaced 120° apart from each other. Mice were placed at the bottom of one of the arms of the maze at the beginning of the test and allowed to freely explore the environment for 8 min. The total number of arm entries and arm choices were recorded. Arm choices are defined as forepaws fully entering the arm. SAB was calculated as the proportion of alternations, defined as an arm choice different from the previous two choices, to the total number of alternations.
Novel object recognition – 3xTg study
The novel object recognition task was used to assess the ability of rodents to recognize a novel object in a familiar environment (Parrella et al., 2013). The test includes a habituation phase (5 min on day one) and trial phases (5 min each on the second day) for each mouse. Briefly, in the habituation phase, the mouse was placed into a rectangular cage (50 x 50 x 40 cm) made of black plexi-glass for 5 min on day one without any objects. The following day, mice underwent two test trials, the duration of each trial being 5 min. Mice were always placed in the apparatus facing the wall at the middle of the front segment. Exploration of the objects is defined as any physical contact with an object (whisking, sniffing, rearing on or touching the object) as well as positioning its nose toward the object at a distance of less than 2 cm. Sitting or standing on top of the object is not counted toward the exploration time. In trial 1, mice explored the arena with two identical objects in presence. After the first exploration period, mice were placed back in their home cage. To control for odor cues, the open field arena and the objects were thoroughly cleaned with 70% ethanol, dried, and ventilated for a few minutes between mice. After a 5-min* delay interval (related to Figure 5 et al., 2003), mice were placed back in the apparatus for the second trial, but with one of the objects from the first trial placed with a new one. Recognition index (RI) was calculated as time the animals spent exploring the novel object to the total time spent exploring both the objects (Parrella et al., 2013).
Novel object recognition – Apocynin study
The mice treated with apocynin or apocynin-free water were tested for short-term spatial memory using a maze consisting of an opaque plastic box measuring 61 cm (length) x 36 cm (width) x 30 cm (height). Briefly, on the first day of test (habituation day) the mice were placed in the box and allowed to explore the field for 5 min. Twenty-four hours later (test day) habituated mice were placed again into the box in the presence of two identical, non-toxic objects and were allowed to freely explore them for 5 min (trial 1). The time spent exploring the objects was recorded considering exploration as any physical contact with an object and/or approach with obvious orientation to it within 5 cm. At the end of the trial 1 the animals were returned to their home cage. After 3 min the mice were returned to the testing field where one of the familiar objects was replaced by a novel object. The mice could explore the arena for 5 min and the time exploring the objects monitored again. Recognition index (RI) was calculated as time the animals spent exploring the novel object to the total time spent exploring both the objects (Parrella et al., 2013).
Barnes Maze
The Barnes maze protocol used here was based on previous protocols (Barnes, 1988; Michán et al., 2010). The maze consists of a platform with 20 holes (San Diego Instruments) and 20 boxes underneath each hole. A nestlet was placed in one box (escape box, “EB”), which was big enough for the mouse to enter/hide. All three walls around Barnes maze have different reference cues to learn the position of the escape hole. A unique position for the EB based on the position of the reference cues was randomly assigned to each mouse and this position was always located underneath the same hole for a specific animal. All mice were trained once daily on days 0 to 7. During training sessions, mice could freely explore the maze until either entering the EB or after 2 min time elapsed. On Day 0, the mice were either identified as active or inactive based on whether the mice move around the center of the maze or explored the holes at the periphery of the maze. If the mouse did not enter the EB by itself, it was gently guided to and allowed to stay in the EB for 30 s. After the training session, mice were tested twice daily for 7 days. Testing was like training, but if after 2 min the mouse did not find the EB, it was directly returned to its cage. Mice were acclimated to the behavior room an hour before the test began each day. The buzzer with a noise level of 80 dB was always switched on when the mice are in the behavior room except when the mouse finds the escape box.
Success rate (100%, finding the escape box [EB] within 2 min; 0%, not finding the EB within 2 min), latency (time to enter the EB), number of errors (nose pokes and head deflections over false holes), deviation (how many holes away from the EB was the first error), and strategies used to locate the EB were recorded and averaged from two tests to obtain daily values. Search strategies were classified as random (crossings through the maze center), serial (searches in clockwise or counterclockwise direction), or spatial (navigating directly to the EB with both error and deviation scores of no more than 3). Retention was assessed by testing once on day 14. Mice that did not move in any of the trials on any day were excluded from the study.
Elevated Plus Maze
The Elevated Plus Maze is used to assess anxiety behaviors, being based on rodent exploratory behavior and by a rodent’s natural aversion against open space. The avoidance of elevated open arms is an indication of the intensity of anxiety (Carroll et al., 2010). The maze has the shape of a cross formed by two alternate open and two alternate closed arms extending from a central platform, each arm measuring 30 cm in length, 5 cm in width, and 15 cm in height. During the test the mouse was placed onto the center field and allowed to freely explore the maze for 5 min, and the time spent in the open arms, corresponding to lower anxiety levels, was measured (Parrella et al., 2013).
Fear conditioning (FC) test
The FC test was used to measure contextual and tone learning in the 3xTg/Nox2-KO mice. The apparatus and the protocol used for FC test have been described previously (Liu et al., 2004). Both the initial training and the contextual testing were conducted in the same conditioning chamber, while tone testing was performed in a novel chamber. The conditioning chamber (27 x 28 x 30.5 cm) was built of plexi-glass (front and back) and aluminum (top and sides), whereas the floor consisted of stainless-steel rods connected to a shock generator (Precision Controlled Animal Shocker; Coulbourn Instruments, Allentown, PA). The chamber was equipped with a speaker and a house light. For tone testing in a novel environment, the new chamber consisted of an aluminum box (27 x 21.5 x 17 cm). A house light and the speaker were placed on top of the box. Briefly, on day 1 of training (training day), the animals were placed in the conditioning chamber. After a 3 min baseline period, the mice were exposed to 3 mild tone-footshock pairings (tone, 20 s, 80 dB, 2 kHz; footshock, 1 s, 0.5 Ma at the termination of the tone; intertrial interval, 1 min). The delivery of the footshock was controlled by software (LabLinc Operant Control Software; Coulbourn Instruments). One minute after the third shock, mice were returned to their home cages. Twenty-four hours later (day 2), contextual learning of the mice was assessed by returning the mice to the conditioning chamber for 8 min (context test). 48 h later, mice were evaluated for cued fear conditioning to the tone in a novel context. Mice were placed in a new chamber and, following 3 min of exploration (day 3, first 3 min), the same tone delivered on day 1 was played for 8 min (day 3, final 8 min). The chamber was cleaned with 70% ethanol between individual sessions on day 1 and day 2 and with 5% ammonium hydroxide on day 3. Mice behavior was monitored using a video camera mounted in front of the test chambers. Freezing behavior, defined as the lack of all body movement except for that associated with respiration, was scored during the three days of tests by two observers blind to mouse identity.
Immunohistochemistry
Adult mice were anesthetized with isoflurane, punctured in the heart for serum, followed by intracardial perfusion with 4% paraformaldehyde (PFA). The tissues were removed immediately and post-fixed in 4% PFA for 24 h (long-term 3xTg mice study) and stored in 0.05% sodium azide. For E4FAD and short-term 3xTg studies, mice were perfused with saline. Brains were removed immediately and cut in half. One half was post-fixed in 4% PFA for 48 h and stored in 0.05% sodium azide afterwards, while the other half was dissected for the cortex and hippocampus, both of which were flash-frozen at −80C until further processing. Brains from 3xTg/Nox2 mice were collected, immersion-fixed in fresh 4% paraformaldehyde/0.1 M PBS for 48 h and then stored at 4°C in 0.1 M PBS/0.2% sodium azide until further processing. Hemi-brains were cut in a sagittal direction (40 μm) using a vibratome VT1000S (Leica) and stored in 0.05% sodium azide solution until ready for immunohistochemistry (IHC). Every 6th section was immunostained in the subsequent protocols.
3,3′-diaminobenzidine (DAB) Staining.
For 3,3′-diaminobenzidine (DAB)-based protocols, sections were washed in TBS, pre-treated for antigen retrieval when necessary, incubated in methanol and H2O2 and blocked in 2% BSA for 1 h before incubating with primary antibodies overnight. The following day, sections would be washed in TBS, incubate with secondary antibodies, ABC Vector Elite (Vector Laboratories), and DAB kits (Vector Laboratories) before finally being mounted on slides and air-dried. Primary antibodies used for this protocol were beta amyloid (1:300; Thermo Fisher Scientific), Phospho-Tau (Ser202, Thr205) Antibody (AT8) (1:1000; Thermo Fisher Scientific), BrdU (1:500; Novus Biologicals), CD11b (1:500; Bio-Rad),Iba1 (1:500; Wako) and CD68 (1:500; Bio-Rad) . Secondary antibodies used for this protocol were biotinylated goat anti-rabbit IgG (for beta amyloid, 1:500; Vector Laboratories), biotinylated horse anti-mouse IgG Antibody, rat adsorbed (for AT8 and Iba1, 1:500; Vector Laboratories), biotin-SP-AffiniPure donkey anti-rat IgG (H + L) (for BrdU, 1:250; Jackson ImmunoResearch); donkey anti rat IgG (H + L), biotin (for CD11b, 1:250; Thermo Fisher Scientific) and biotin-SP-AffiniPure donkey anti rat IgG (H + L) (for CD68, 1:500; Jackson ImmunoResearch).
Immunofluorescent staining
For fluorescent IHC, sections were rinsed 3 times in phosphate buffered saline (PBS) for 5 min and denatured in 2N HCl at 37°C for 20 min. Sections were neutralized with 0.1 M boric acid for 10 min and blocked with 2% Normal Donkey Serum (NDS; Jackson ImmunoResearch) for 1 h at room temperature. Sections were incubated with primary antibodies overnight in a solution containing 2% NDS and 0.3% triton at 4°C. The following day, sections were rinsed 3 times in PBS for 10 min, followed by incubation with secondary antibodies. After subsequent washes in PBS, nuclei were stained with Hoechst 33342 (Thermo Fisher), washed in PBS, and mounted on slides (except for GFAP + Sox2+ staining as the mounting medium contained DAPI). Slides were cover-slipped with antifading polyvinyl alcohol mounting medium with DABCO (Sigma-Aldrich) or VECTASHIELD® HardSet™ Antifade Mounting Medium with DAPI (Vector Laboratories) immediately after. Primary antibodies used for this protocol were BrdU (1:200; Novus Biologicals), Ki67 (1:200; Thermo Fisher Scientific), Sox2 (1:200; Abcam) and GFAP (1:200; Cell Signaling). Secondary antibodies used for this protocol were donkey anti-rat-488 (for BrdU, Ki67 and GFAP, 1:400; Thermo Fisher Scientific) and donkey anti-rabbit-594 (for Sox2, 1:400; Thermo Fisher Scientific). Co-expression was confirmed by fluorescent microscopy. For quantification, stereological counting methods were used.
Aβ
To enhance Aβ immunoreactivity (IR), sections were rinsed for 5 min in 99% formic acid. Aβ IR was calculated as load values. Briefly, selected fields of nonoverlapping immunolabeled sections of hippocampus (one-two fields for subiculum and two-three for CA1–Cornu Ammonis area 1) were captured at 20× and digitized using a video capture system coupled to a microscope (Olympus BX50 microscope and Olympus DP73 camera). Using ImageJ software (NIH), software, images were converted into binary/negative data and the positive pixels (equivalent to IR area) were quantified (Carroll et al., 2010).
Tau
AT8-immunoreactive neurons were defined as cells showing strong AT8 immunolabeling over most of the cell surface. The positive cells were counted within the hippocampal CA1 and subiculum regions. Images were captured with a video capture system coupled to a microscope (Olympus BX50 microscope and Olympus DP73 camera) at 20×.
CD11b, Iba1 and CD68 (microglia staining)
CD11b-immunoreactive (ir), Iba1 positive and CD68 positive microglia cells were defined as cells covered by CD11b, Iba1 or CD68 immunostaining over the cell body and processes. Sections were either treated with 10 mM EDTA (pH 6) or Citric Acid Buffer (pH 6) at 95°C for 10 min before proceeding with staining. CD11b-ir or Iba1 cells were counted in the subiculum and CA1 hippocampal regions. There was no pre-treatment with 10 mM EDTA (pH 6) or Citric Acid Buffer (pH 6) for required the CD68 Antibody.CD68+ cells were counted in the subiculum and CA1 hippocampal regions.
The stage of cells activation was identified by their morphology. Briefly, we defined four stages of microglia activation based on criteria established in previous studies (Zhang et al., 2011; Kreutzberg, 1996; Crews and Vetreno, 2016):
Stage 1: Resting microglia. Rod-shaped soma with many long thin ramified processes.
Stage 2: Activated ramified microglia. Elongated cell body, the processes are thicker.
Stage 3: Amoeboid microglia showing a marked cellular hypertrophy and short and thick processes.
Stage 4: Phagocytic cells. Round cells, processes are not detectable.
Cells in the different activation stages were counted and plotted as percentage of the total CD11b-ir or Iba1 cell number.
Images were captured with a video capture system coupled to a microscope (Olympus BX50 microscope and Olympus DP73 camera) at 20×.
Neurogenesis markers
The levels of BrdU (DAB and fluorescent), Sox2, and Ki67 were quantified in the dentate gyrus. Images were captured at 20× and captured with a video capture system coupled to a microscope (Olympus BX50 microscope and Olympus DP73 camera) or with the BZ-X710 All-in-One Fluorescence Microscope (Keyence) and analyzed with ImageJ (NIH). Individual BrdU+, Sox2+, Ki67+ and GFAP+ cells, as well as BrdU+Sox2+, Ki67+Sox2+ and GFAP+Sox2+ co-stained cells, were counted if they were located within the subgranular zone (SGZ) and up to the inner third of the granule cell layer. Every sixth section of the mouse hemi-brain containing the hippocampus was stained. The numbers quantified are the averages taken from the total number of sections stained for each hemi-brain.
Aβ protein extraction
Soluble Aβ peptides (Aβ38/40/42) were extracted from the cortex of hemi-brains of female E4FAD 7–7.5 months old, after undergoing 4 months of FMD treatment. A serial extraction process previously described was used to produce TBS-soluble and TBS plus triton-soluble fractions (Youmans et al., 2011). The amount of soluble Aβ per isoform per sample in the resulting extracts were evaluated using the V-PLEX Plus Aβ Peptide Panel 1 (4G8) (Meso Scale Diagnostics). Plate results were read using the MSD Technology Platform and associated software.
Immunoprecipitation
Magnetic Dynabeads (Invitrogen) were re-suspended and 50 μL was transferred to 8 tubes. Beads were separated from supernatant on a magnet and the supernatant was removed. 2.5 μL of rat anti-mouse CD11b antibody (1 μg/μL, Bio-Rad, MCA711GT) was diluted in 200 μL of Ab Binding and Washing Buffer and the immunoprecipitation procedure was carried out using the Dynabeads™ Protein G Immunoprecipitation Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions. 500 ug of target antigen (Aβ42) was added to the magnetic bead antibody complex. After elution with elution buffer, samples were mixed with 10 μL of pre-mixed NuPAGE™ LDS Sample Buffer and NuPAGE Sample Reducing Agent (Invitrogen) to ensure the resuspension of the magnetic bead-Ab-Ag complex. Samples were heated for 10 min at 70°C. The tubes were placed on a magnet, the supernatant was transferred to a clean tube and prepared for western blot as described in the Western Blot section.
Western Blot
The cortex from E4FAD mouse brains was homogenized in Eppendorf tubes (kept on ice), using 300 ul of RIPA buffer (Thermo Fischer Scientific) with freshly added protease and phosphatase inhibitor cocktails. The samples were placed on an orbital shaker at 4°C overnight and spun the next day at 12000 rpm for 20 min at 4°C. The supernatant from each sample was collected and the protein concentration was assayed using BSA as working standard. Equal amounts of protein (40 μg) were heat-denaturized in NuPAGE LDS sample- loading buffer (Invitrogen) for 5 min at 95°C, resolved by SDS-PAGE and transferred to PVDF membranes (Sigma-Aldrich, MO, USA). The membranes were blocked with Tris-buffered saline (TBS) containing 0.05% Tween and 5% non-fat dry milk (NOX2, Aβ42) and Tris-buffered saline (TBS) containing 0.05% Tween and 2% BSA (Vinculin) and then incubated overnight with antibodies directed against NOX2 (Rabbit monoclonal, 1:5000, Abcam, ab129068), Aβ42 (Rabbit monoclonal, 1:500, Abcam, ab201060) or Vinculin (Mouse monoclonal, 1:1000, Millipore, MAB3574). Peroxidase conjugated IgG was used as secondary antibody. Membrane-bound immune complexes were detected by HyGLO™ Chemiluminescent HRP Detection Reagent (Denville Scientific). Protein loading was normalized according to Vinculin expression. Quantification was performed by densitometric analysis using Imagelab 6.0 software (Bio-Rad).
Nox2 ELISA
Nox2 activity in cortex lysates was measured using a Rat CYBB / NOX2 / gp91phox ELISA Kit (LSBio, Seattle, WA, USA, LS-F39030) and performed according to manufacturer’s instructions.
Brain cytokine analysis
Inflammatory cytokines in the brain were evaluated using aliquots of the female E4FAD hemi-brain TBS-soluble and TBS plus triton-soluble extracts used in the Aβ protein extraction experiment. The amount of IL-10, IL-12p70, IL-1β, IL-6, TNFα, IL-2, KC/GRO, IL-4, and IL-5 cytokines per sample in the resulting extracts were evaluated using V-PLEX Proinflammatory Panel 1 (mouse) Kit (Meso Scale Diagnostics). Plate results were read using the MSD Technology Platform and associated software.
Microglia confocal microscopy
Images were captured using a confocal microscope (Zeiss Laser Scanning Microscope-780 upright microscope) with Zeiss ZEN imaging software. Laser and detector settings were unchanged across acquisition sessions. 3D images of sections labeled with anti-Iba1 were collected with optimal resolution z-stack depths. Stack section depth for low magnification images was 2.2 μm and 0.4 μm for high magnification images. A 63x oil immersion objective (1.4 NA) was used to acquire region of interest (ROI) stacks (192.8 μm x 192.8 μm, 512 x 512 pixels, 16 bit). Analysis was conducted in a researcher-blinded manner with a custom ImageJ plugin (Stephen et al., 2015) and de-noised using background subtraction rolling ball radius of 50 pixels. Average intensity stack projections were used for analysis (unless otherwise stated). Microglial circularity, a measure of roundness/activation, was calculated using the formula 4π x area / (perimeter)2. Data represents average of all cells per group. Microglial soma size was measured by manually identifying, thresholding, outlining, and measuring Iba1-immuno-reactive cell bodies (~ 30 cells/group) using ImageJ.
RNAseq library preparation and analyses
Murine-derived hemi-cortex samples were prepared as lysates using the RNeasy Lipid Tissue Mini Kit (Qiagen) and RNase-Free DNase Set (Qiagen), according to manufacturer’s protocols. RNAseq libraries were prepared using KAPA mRNA HyperPrep kit (Roche-Sequencing) on a custom-built Janus G3 liquid handler (Perkin-Elmer). The quality of the final libraries was assessed with the D1000 assay on a 2200 TapeStation (Agilent Technologies) and the pooled samples were sequenced 1 × 50 bp on an Illumina HiSeq3000 system, yielding 24–52 M reads per sample for 31 samples. Reads were quality checked with FastQC v0.11.8 (Andrews, 2010), filtered with Trimmomatic v0.38 (Bolger et al., 2014), and aligned to the Mus musculus GRCm38 reference genome with GENCODE M22 annotations using STAR v2.6.1d (Dobin et al., 2013) with a single-pass approach, retaining uniquely mapped reads with less than 5 mismatches. Gene expression was quantified using HTSeq (Anders et al., 2015) over exons in union mode, and analyzed using the R package EdgeR (McCarthy et al., 2012; Robinson et al., 2010). Four samples that appeared to be technical outliers in the FastQC and gene expression analyses were discarded. Gene expressions for the remaining 18 male and 9 female samples were separately TMM-normalized and between-diet group differences were assessed using EdgeR’s GLM approach. Gene ontology analyses were performed using the R packages gProfileR v0.6.8 (Reimand et al., 2007) and GO.db (Carlson, 2019). GO terms for the significantly expressed genes are listed in Table S1.
In vitro microglia-Aβ42 assay
Mixed glia were cultured from the brains of 8.5 month old 3xTg male and female mice administered either a control diet or 5 cycles of FMD with 4 days of refeeding (n = 1–2 mice/group). The mice started the diet at 6.5 months till 8.5 months of age. The brains were perfused using DPBS and the whole brain (except the cerebellum) from each of the 5 mice (3 female mice-2 FMD and 1 Control and 2 male mice- 1 FMD and 1 Control) were placed in 5 mL of DMEM/F12 (Gibco) and stored on ice. Whole brains were mechanically and enzymatically dissociated by first mincing with a sterile razor blade and then incubated with 2 mL of serum free DMEM/12 media containing 10 units/mL papain for 25 min followed by the addition of 40 ul of Dnase (10 mg/mL stock, final 0.2 ug/mL) for 5 min at 37°C. Enzymatic activity was quenched by addition of 500 μL of fetal bovine serum followed by addition of 2 mL of serum free media at room temperature. Once the serum free media was added, large clumps of tissue were broken up by pipetting using a 1 mL pipette. After the cell pellets settle for two minutes, the supernatant was transferred to a fresh falcon tube. This step was repeated twice more. Isotonic Percoll was added to the cell suspension (final concentration: 20%). Isotonic Percoll (ISP) was prepared by mixing nine parts of Percoll (Percoll PLUS, Cytiva) with one-part 10× HBSS.
To collect the glial-enriched fraction, the cell suspension was centrifuged for 20 min at 700×g in a 20% ISP/cell suspension. The supernatant was aspirated using a 5 mL stereological pipette, and the cell pellets were re suspended in 8 mL of DMEM/F12 supplemented with 10% FBS and a penicillin G and streptomycin cocktail. The resultant suspension was transferred through a 100 μm cell strainer using a 5 mL stereological pipette on petri plates coated with 10 μg/mL of Poly-D Lysine. Cultures were maintained at 37°C in a humidified incubator for 10 days with room air supplemented with 5% CO2. The media was replaced 3 times per week.
After the primary mixed glial cultures were incubated for 10 days, 2500 microglial cells per well were seeded on 96 well plates coated with 10 ug/mL of Poly-D Lysine and incubated for two days at 37°C in a humidified incubator with room air supplemented with 5% CO2.
HiLyte™ Fluor 555-labeled Amyloidβ 42 (Aβ42) (AnaSpec, Liège, Belgium) was dissolved in 100% 1,1,1,3,3,3-Hexafluoro-2-propanol and subsequently dried followed by evaporation. The peptide film was dissolved in 10 mM NaOH and neutralized by adding pH 7.4 PBS (final concentration of Aβ42: 20 μM). After the sample was vortexed for 30s, Aβ42 was incubated at 4°C for 24 h for oligomerization and was used for assay immediately where it was added to the seeded cells for an hour to quantify the levels of Amyloid beta uptake in FMD compared to the control group. The microglia were fixed with 4% PFA and stained with IBA-1 (1:200; Wako) to confirm the cell type. The Secondary antibody used for this protocol was donkey anti-mouse-488 (for IBA-1, 1:500; Thermo Fisher Scientific). Images are taken with the BZ-X710 All-in-One Fluorescence Microscope (Keyence) and analyzed with ImageJ (NIH). The area of Amyloid beta 42 to IBA-1 area was quantified to identify the percentage of Aβ 42 uptake in each group.
QUANTIFICATION AND STATISTICAL ANALYSIS
The software used for statistical analysis was GraphPad Prism v.8. The figure legends describe the statistical tests used, value of n for each experimental group, and what n represents for each experiment. All statistical analyses were two-sided and p values <0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Differences between the means of two groups were tested with unpaired 2-tailed student’s t-test comparison, and one-way or two-way ANOVA followed by Bonferroni, Tukey’s, or Fisher’s post-test among multiple groups. Unless otherwise specified in figure legends, all data are expressed as the mean ± SEM. All samples represent biological replicates. No samples were excluded from analysis, and animals were only excluded if they fell ill based on protocol. Sample size estimates were not used. Animals were randomly assigned to experimental groups.
ADDITIONAL RESOURCES
Clinical trial registry (NCT05480358) for the human FMD trial data described in the manuscript can be found here. (description:https://clinicaltrials.gov/ct2/show/NCT05480358?term=NCT05480358&cntry=IT&draw=2&rank=1).
Supplementary Material
Highlights.
FMD cycles reduce cognitive decline and AD pathology in preclinical mouse models
FMD cycles decrease hippocampal microglia number and neuroinflammation
3xTg mice lacking Nox2 show improved cognition and microglia activation
Clinical data suggest FMD cycles are safe and feasible in a small group of AD patients
ACKNOWLEDGMENTS
We thank Amy Christensen-Pacheco, Emilly R. Rosario, and Jenna C. Carroll for providing 3xTg breeding pairs, assistance with protocols, and for loaning laboratory equipment to process tissue samples, as well as the late Professor Richard F. Thompson for thorough advice on fear conditioning tests. We thank the lab of Dr. Caleb Finch for providing our initial set of E4FAD breeding trios. We thank Gerardo Navarrete for assistance with tissue collection and preparation of FMD and 4% PR diet. We thank Steffi Bolton, Melissa Carpenter, Sebastian Densley, Andrew Ingalls, Naveet Sandhu, Daral Maesincee, Claire Ngyuen, Vanessa Ocon, Gab Pasia, and Chae Sutherland for their technical assistance; Steven Nomura, Chanpreet Singh, Min Wei, Zheng-Yi Zhou, Tom Maxim, Inyoung Choi, Sebastian Brandhorst, Caleb E. Finch, and Todd E. Morgan for their technical assistance and/or advice on Nox2-KO experiments; and Tomomitsu Iida for technical assistance and advice on the in vitro microglia experiments. The graphical abstract was created with BioRender.com. This study was funded in part by NIH/NIA grants AG20642, AG025135, and P01 AG034906 to V.D.L.; AG058068 to C.J.P.; the NIA T32 training grant AG052374 to P.R.; and the PE-2016-02362694 and PE-2016-02363073 grants by the Italian Ministry of Health to P.O., P.M., F.M., and to V.D.L. The LaDu lab is funded by NIH (NIA) R01 AG056472, R01 AG057008, UH2/3 NS10012, R56 AG058655, 1R44 AG060826, institutional funds from the College of Medicine at the University of Illinois, Chicago, and generous philanthropic contributions. V.D.L. has equity interest in L-Nutra, a company that develops medical food.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111417.
DECLARATION OF INTERESTS
V.D.L. has equity interest in L-Nutra, which develops and sells medical food for the prevention and treatment of diseases. A patent on this work has been filed by the University of Southern California, which has equity interest in L-Nutra (Fasting-mimicking Diet (FMD) as an Intervention for Alzheimer’s Disease (AD), US 62/840,762, Filed Apr 15, 2019).
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects.
REFERENCES
- ‘t Hart BA, Copray S, and Philippens I (2014). Apocynin, a low molecular oral treatment for neurodegenerative disease. BioMed Res. Int, 298020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abels ER, Maas SLN, Nieland L, Wei Z, Cheah PS, Tai E, Kolsteeg CJ, Dusoswa SA, Ting DT, Hickman S, et al. (2019). Glioblastoma-associated microglia reprogramming is mediated by functional transfer of extracellular miR-21. Cell Rep. 28, 3105–3119.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves S, Churlaud G, Audrain M, Michaelsen-Preusse K, Fol R, Souchet B, Braudeau J, Korte M, Klatzmann D, and Cartier N (2017). Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer’s disease mice. Brain 140, 826–842. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AJ, Cummings BJ, and Cotman CW (1994). Increased immunoreactivity for Jun- and Fos-related proteins in Alzheimer’s disease: association with pathology. Exp. Neurol 125, 286–295. [DOI] [PubMed] [Google Scholar]
- Andrade J, Quinn J, Becker RZ, and Shupnik MA (2013). AMP-activated protein kinase is a key intermediary in GnRH-stimulated LHbeta gene transcription. Mol. Endocrinol 27, 828–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Azevedo H, Khaled NA, Santos P, Bertonha FB, and Moreira-Filho CA (2018). Temporal analysis of hippocampal CA3 gene co-expression networks in a rat model of febrile seizures. Disease models & mechanisms 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmanov AA, Reed DR, Beauchamp GK, and Tordoff MG (2002). Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet 32, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie JK, Arner E, Daub C, De Hoon M, Itoh M, Kawaji H, Lassmann T, Carninci P, Forrest ARR, Hayashizaki Y, et al. (2017). Analysis of the human monocyte-derived macrophage transcriptome and response to lipopolysaccharide provides new insights into genetic aetiology of inflammatory bowel disease. PLoS Genet. 13, e1006641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA (1988). Spatial learning and memory processes: the search for their neurobiological mechanisms in the rat. Trends Neurosci. 11, 163–169. [DOI] [PubMed] [Google Scholar]
- Benboubker L, Dimopoulos MA, Dispenzieri A, Catalano J, Belch AR, Cavo M, Pinto A, Weisel K, Ludwig H, Bahlis N, et al. (2014). Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N. Engl. J. Med 371, 906–917. [DOI] [PubMed] [Google Scholar]
- Berasi SP, Huard C, Li D, Shih HH, Sun Y, Zhong W, Paulsen JE, Brown EL, Gimeno RE, and Martinez RV (2006). Inhibition of gluconeogenesis through transcriptional activation of EGR1 and DUSP4 by AMP-activated kinase. J. Biol. Chem 281, 27167–27177. [DOI] [PubMed] [Google Scholar]
- Biechele G, Franzmeier N, Blume T, Ewers M, Luque JM, Eckenweber F, Sacher C, Beyer L, Ruch-Rubinstein F, Lindner S, et al. (2020). Glial activation is moderated by sex in response to amyloidosis but not to tau pathology in mouse models of neurodegenerative diseases. J. Neuroinflammation 17, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV (2009). Validation of anti-aging drugs by treating age-related diseases. Aging 1, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML (2008). NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 9 (Suppl 2), S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS (2014). Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonam SR, Ruff M, and Muller S (2019). HSPA8/HSC70 in immune disorders: a molecular rheostat that adjusts chaperone-mediated autophagy substrates. Cells 8, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, et al. (2015). A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 22, 86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Gupta S, Knight AG, Beckett TL, McMullen JM, Davis PR, Murphy MP, Van Eldik LJ, St Clair D, and Keller JN (2011). Cognitive impairment in humanized APP×PS1 mice is linked to Aβ (1-42) and NOX activation. Neurobiol. Dis 44, 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Magrì A, Medina DX, Wisely EV, López-Aranda MF, Silva AJ, and Oddo S (2013). mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell 12, 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacciottolo M, Christensen A, Moser A, Liu J, Pike CJ, Smith C, LaDu MJ, Sullivan PM, Morgan TE, Dolzhenko E, et al. (2016). The APOE4 allele shows opposite sex bias in microbleeds and Alzheimer’s disease of humans and mice. Neurobiol. Aging 37, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffa I, Spagnolo V, Vernieri C, Valdemarin F, Becherini P, Wei M, Brandhorst S, Zucal C, Driehuis E, Ferrando L, et al. (2020). Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 583, 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M (2019). GO.db: a Set of Annotation Maps Describing the Entire Gene Ontology. R package version 3.8.2
- Carroll JC, Rosario ER, Villamagna A, and Pike CJ (2010). Continuous and cyclic progesterone differentially interact with estradiol in the regulation of Alzheimer like pathology in female 3x Transgenic-Alzheimer’s disease mice. Endocrinology 151, 2713–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Liu B (2017). Relationships between stress granules, oxidative stress, and neurodegenerative diseases. Oxid. Med. Cell. Longev, 1809592–1809610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CW, Villani V, Buono R, Wei M, Kumar S, Yilmaz OH, Cohen P, Sneddon JB, Perin L, and Longo VD (2017). Fasting-mimicking diet Promotes Ngn3-Driven β-Cell regeneration to reverse diabetes. Cell 168, 775–788.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IY, Piccio L, Childress P, Bollman B, Ghosh A, Brandhorst S, Suarez J, Michalsen A, Cross AH, Morgan TE, et al. (2016). A diet mimicking fasting promotes regeneration and reduces autoimmunity and multiple sclerosis symptoms. Cell Rep. 15, 2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, Van Nostrand W, Miano JM, and Zlokovic BV (2007). Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc. Natl. Acad. Sci. USA 104, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Li JG, Ceballos-Diaz C, Golde T, and Praticò D (2013). The influence of 5-lipoxygenase on Alzheimer’s disease-related tau pathology: in vivo and in vitro evidence. Biol. Psychiatry 74, 321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarimón J, Bertranpetit J, Boada M, Tàrraga L, and Comas D (2003). HSP70-2 (HSPA1B) is associated with noncognitive symptoms in late-onset Alzheimer’s disease. J. Geriatr. Psychiatry Neurol 16, 146–150. [DOI] [PubMed] [Google Scholar]
- Cline EN, Bicca MA, Viola KL, and Klein WL (2018). The amyloid-beta oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis 64, S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close AF, Dadheech N, Villela BS, Rouillard C, and Buteau J (2019). The orphan nuclear receptor Nor1/Nr4a3 is a negative regulator of β-cell mass. J. Biol. Chem 294, 4889–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly PJ, Adams F, Tayar ZI, and Khan F (2019). Peripheral vascular responses to acetylcholine as a predictive tool for response to cholinesterase inhibitors in Alzheimer’s disease. BMC Neurol. 19, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, and Vetreno RP (2016). Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 233, 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, Salinas-Navarro M, Cordeiro MF, Moons L, and De Groef L (2017). Characterizing microglia activation: a spatial statistics approach to maximize information extraction. Sci. Rep 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Biase S, Shim HS, Kim KH, Vinciguerra M, Rappa F, Wei M, Brandhorst S, Cappello F, Mirzaei H, Lee C, and Longo VD (2017). Fasting regulates EGR1 and protects from glucose- and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol. 15, e2001951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). Star : ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Dekens D, De Deyn P, Naudé P, and Eisel U (2015). Targeting of tumor necrosis factor alpha receptors as a therapeutic strategy for neurodegenerative disorders. Antibodies 4, 369–408. [Google Scholar]
- Draznin B, Wang C, Adochio R, Leitner JW, and Cornier MA (2012). Effect of dietary macronutrient composition on AMPK and SIRT1 expression and activity in human skeletal muscle. Horm. Metab. Res 44, 650–655. [DOI] [PubMed] [Google Scholar]
- Fang Y, Yao L, Li C, Wang J, Wang J, Chen S, Zhou XF, and Liao H (2016). The blockage of the Nogo/NgR signal pathway in microglia alleviates the formation of Aβ plaques and tau phosphorylation in APP/PS1 transgenic mice. J. Neuroinflammation 13, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood WD, Moyer RW, Tsykin A, Sutherland GR, and Koblar SA (2004). Nxf and Fbxo33: novel seizure-responsive genes in mice. Eur. J. Neurosci 20, 1819–1826. [DOI] [PubMed] [Google Scholar]
- Gao W, Li F, Zhou Z, Xu X, Wu Y, Zhou S, Yin D, Sun D, Xiong J, Jiang R, and Zhang J (2017). IL-2/Anti-IL-2 complex attenuates inflammation and BBB disruption in mice subjected to traumatic brain injury. Front. Neurol 8, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri RK, Rajagopal V, Shahi S, Zlokovic BV, and Kalra VK (2005). Mechanism of amyloid peptide induced CCR5 expression in monocytes and its inhibition by siRNA for Egr-1. Am. J. Physiol. Cell Physiol 289, 264–276. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, and Klein WL (2003). Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 100, 10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, and Mattson MP (1999). Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat. Med 5, 101–106. [DOI] [PubMed] [Google Scholar]
- Halagappa VKM, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, and Mattson MP (2007). Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis 26, 212–220. [DOI] [PubMed] [Google Scholar]
- Hardie DG (2011). AMP-activated protein kinase - an energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schöneich C, and Engelhardt JF (2008). SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Invest 118, 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann L, Wiegmann C, Arsalan-Werner A, Hilbrich I, Jäger C, Flach K, Suttkus A, Lachmann I, Arendt T, and Holzer M (2015). Hook proteins: association with Alzheimer pathology and regulatory role of hook3 in amyloid beta generation. PLoS One 10, e0119423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovens I, Nyakas C, and Schoemaker R (2014). A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: cell body to cell size ratio. Neuroimmunol. Neuroinflamm 1, 82. https://bioconductor.org/packages/release/data/annotation/html/GO.db.html. [Google Scholar]
- Jekabsone A, Mander PK, Tickler A, Sharpe M, and Brown GC (2006). Fibrillar beta-amyloid peptide Abeta1-40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J. Neuroinflammation 3, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, and Wojtowicz JM (2002). The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J. Neurosci. Methods 115, 97–105. [DOI] [PubMed] [Google Scholar]
- Khuu MA, Pagan CM, Nallamothu T, Hevner RF, Hodge RD, Ramirez JM, and Garcia AJ (2018). Intermittent hypoxia disrupts adult neurogenesis and synaptic plasticity in the dentate gyrus. J. Neurosci 39, 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostylev MA, Kaufman AC, Nygaard HB, Patel P, Haas LT, Gunther EC, Vortmeyer A, and Strittmatter SM (2015). Prion-Protein-interacting amyloid-β oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J. Biol. Chem 290, 17415–17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW (1996). Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318. [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Francescutti-Verbeem DM, and Thomas DM (2006). Dopamine quinones activate microglia and induce a neurotoxic gene expression profile. Ann N Y Acad Sci 1074, 31–41. [DOI] [PubMed] [Google Scholar]
- Lafourcade CA, Lin TV, Feliciano DM, Zhang L, Hsieh LS, and Bordey A (2013). Rheb activation in subventricular zone progenitors leads to heterotopia, ectopic neuronal differentiation, and rapamycin-sensitive olfactory micronodules and dendrite hypertrophy of newborn neurons. J. Neurosci 33, 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, Suarez JA, Brandhorst S, Balasubramanian P, Cheng CW, Madia F, Fontana L, Mirisola MG, Guevara-Aguirre J, Wan J, et al. (2014). Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 19, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski CT, Maldonado Weng J, and LaDu MJ (2020). Alzheimer’s disease pathology in ApoE transgenic mouse models: the who, what, when, where, why, and how. Neurobiol. Dis 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Jiang J, and Zhao L (2019). Protein arginine methyltransferase-1 deficiency restrains depression-like behavior of mice by inhibiting inflammation and oxidative stress via Nrf-2. Biochem. Biophys. Res. Commun 518, 430–437. [DOI] [PubMed] [Google Scholar]
- Liu IYC, Lyons WE, Mamounas LA, and Thompson RF (2004). Brain-derived neurotrophic factor plays a critical role in contextual fear conditioning. J. Neurosci 24, 7958–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo AC, Iscru E, Blum D, Tesseur I, Callaerts-Vegh Z, Buée L, De Strooper B, Balschun D, and Dhooge R (2013). Amyloid and tau neuropathology differentially affect prefrontal synaptic plasticity and cognitive performance in mouse models of alzheimers disease. J. Alzheimers Dis 37, 109–125. [DOI] [PubMed] [Google Scholar]
- Longo VD, Viola KL, Klein WL, and Finch CE (2000). Reversible inactivation of superoxide-sensitive aconitase in Abeta1-42-treated neuronal cell lines. J. Neurochem 75, 1977–1985. [DOI] [PubMed] [Google Scholar]
- Lopes S, Teplytska L, Vaz-Silva J, Dioli C, Trindade R, Morais M, Webhofer C, Maccarrone G, Almeida OFX, Turck CW, et al. (2016). Tau deletion prevents stress-induced dendritic atrophy in prefrontal cortex: role of synaptic mitochondria. Cereb. Cortex 27, 2580–2591. [DOI] [PubMed] [Google Scholar]
- Lynch MA (2009). The multifaceted profile of activated microglia. Mol. Neurobiol 40, 139–156. [DOI] [PubMed] [Google Scholar]
- Lyons MR, and West AE (2011). Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog. Neurobiol 94, 259–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CY, Yao M.j., Zhai Q.w., Jiao J.w., Yuan X.b., and Poo M.m. (2014). SIRT1 suppresses self-renewal of adult hippocampal neural stem cells. Development 141, 4697–4709. [DOI] [PubMed] [Google Scholar]
- Magno L, Lessard CB, Martins M, Lang V, Cruz P, Asi Y, Katan M, Bilsland J, Lashley T, Chakrabarty P, et al. (2018). Alzheimer’s disease Phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimer’s Res. Ther 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado Weng J, Parikh I, Naqib A, York J, Green SJ, Estus S, and LaDu MJ (2019). Synergistic effects of APOE and sex on the gut microbiome of young EFAD transgenic mice. Mol. Neurodegener 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, LaDu MJ, Fardo DW, Rebeck GW, and Estus S (2015). Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener 10, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marballi KK, and Gallitano AL (2018). Immediate early genes anchor a biological pathway of proteins required for memory formation, long-term depression and risk for schizophrenia. Front. Behav. Neurosci 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad CA, and Klann E (2011). Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal 14, 2013–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrangelo MA, and Bowers WJ (2008). Detailed immunohistochemical characterization of temporal and spatial progression of Alzheimer’s disease-related pathologies in male triple-transgenic mice. BMC Neurosci. 9, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-Samperio P (2010). Role of interleukin-12 family cytokines in the cellular response to mycobacterial disease. Int. J. Infect. Dis 14, e366–e371. [DOI] [PubMed] [Google Scholar]
- Mengozzi M, Cervellini I, Villa P, Erbayraktar Z, Gökmen N, Yilmaz O, Erbayraktar S, Manohasandra M, Van Hummelen P, Vandenabeele P, et al. (2012). Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc. Natl. Acad. Sci. USA 109, 9617–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michán S, Li Y, Chou MMH, Parrella E, Ge H, Long JM, Allard JS, Lewis K, Miller M, Xu W, et al. (2010). SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci 30, 9695–9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G.l., and Song H (2005). Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci 28, 223–250. [DOI] [PubMed] [Google Scholar]
- Mirzaei H, Di Biase S, and Longo VD (2016). Dietary interventions, cardiovascular aging, and disease: animal models and human studies. Circ. Res 118, 1612–1625. [DOI] [PubMed] [Google Scholar]
- Mori F, Nishie M, Piao YS, Kito K, Kamitani T, Takahashi H, and Wakabayashi K (2004). Accumulation of NEDD8 in neuronal and glial inclusions of neurodegenerative disorders. Neuropathol. Appl. Neurobiol 31, 53–61. [DOI] [PubMed] [Google Scholar]
- Mouton PR, Chachich ME, Quigley C, Spangler E, and Ingram DK (2009). Caloric restriction attenuates amyloid deposition in middle-aged dtg APP/PS1 mice. Neurosci. Lett 464, 184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomaru H, Sakumi K, Katogi A, Ohnishi YN, Kajitani K, Tsuchimoto D, Nestler EJ, and Nakabeppu Y (2015). Fosb gene products contribute to excitotoxic microglial activation by regulating the expression of complement C5a receptors in microglia. Glia 62, 1284–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura K, Vilalta A, Allendorf DH, Hornik TC, and Brown GC (2017). Activated microglia desialylate and phagocytose cells via neuraminidase, galectin-3, and mer tyrosine kinase. J. Immunol 198, 4792–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, and LaFerla FM (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. [DOI] [PubMed] [Google Scholar]
- Pandey RS, Graham L, Uyar A, Preuss C, Howell GR, and Carter GW (2019). Genetic perturbations of disease risk genes in mice capture transcriptomic signatures of late-onset Alzheimer’s disease. Mol. Neurodegener 14, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, et al. (2017). VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 36, 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Lee KW, Oh S, Yan R, Zhang J, Beach TG, Adler CH, Voronkov M, Braithwaite SP, Stock JB, and Mouradian MM (2018). Protein phosphatase 2A and its methylation modulating enzymes LCMT-1 and PME-1 are dysregulated in tauopathies of progressive supranuclear palsy and Alzheimer’s disease. J. Neuropathol. Exp. Neurol 77, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Choi H, Kim B, Chae U, Lee DG, Lee SR, Lee S, Lee HS, and Lee DS (2016). Peroxiredoxin 5 (Prx5) decreases LPS-induced microglial activation through regulation of Ca2+/calcineurin-Drp1-dependent mitochondrial fission. Free Radic. Biol. Med 99, 392–404. [DOI] [PubMed] [Google Scholar]
- Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pistik R, Younkin L, Younkin S, Zhou P, Carlson G, et al. (2017). Brain perivascular macrophages initiate the neurovascular dysfunction of alzheimer Aβ peptides. Circ. Res 121, 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, and Iadecola C (2008). Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A 105, 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Jung MY, Lee SJ, Kang KB, Gratchev A, Riabov V, Kzhyshkowska J, Kim IS, and Kim I (2009). Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J. Cell Sci 122, 3365–3373. [DOI] [PubMed] [Google Scholar]
- Parrella E, Maxim T, Maialetti F, Zhang L, Wan J, Wei M, Cohen P, Fontana L, and Longo VD (2013). Protein restriction cycles reduce IGF-1 and phosphorylated tau, and improve behavioral performance in an alzheimer’s disease mouse model. Aging Cell 12, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J, Morgan DG, Morgan TE, and Finch CE (2005). Caloric restriction attenuates Aβ-deposition in alzheimer transgenic models. Neurobiol. Aging 26, 995–1000. [DOI] [PubMed] [Google Scholar]
- Petersen RC (2004). Mild cognitive impairment as a diagnostic entry. J. Intern. Med 256, 183–194. [DOI] [PubMed] [Google Scholar]
- Petitto JM, Jackson AJ, McNamara RK, Gendreau PL, and Huang Z (1999). Impaired learning and memory and altered hippocampal neurodevelopment resulting from interleukin-2 gene deletion. J. Neurosci. Res 56, 441–446. [DOI] [PubMed] [Google Scholar]
- Plotkin SS, and Cashman NR (2020). Passive immunotherapies targeting Aβ and tau in Alzheimer’s disease. Neurobiol. Dis 144, 105010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, and Dinauer MC (1995). Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat. Genet 9, 202–209. [DOI] [PubMed] [Google Scholar]
- Qin X, Wang Y, and Paudel HK (2016). Early growth response 1 (Egr-1) is a transcriptional activator of β-secretase 1 (BACE-1) in the brain. J. Biol. Chem 291, 22276–22287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj DDA, Moser J, Pol SMA, Os RP, Holtman IR, Brouwer N, Oeseburg H, Schaafsma W, Wesseling EM, Dunnen W, et al. (2015). Enhanced microglial pro-inflammatory response to lipopolysaccharide correlates with brain infiltration and blood–brain barrier dysregulation in a mouse model of telomere shortening. Aging Cell 14, 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan P, Choi I, Wei M, Navarrete G, Guen E, Brandhorst S, Enyati N, Pasia G, Maesincee D, Ocon V, et al. (2019). Fasting-mimicking diet modulates microbiota and promotes intestinal regeneration to reduce inflammatory bowel disease pathology. Cell Rep. 26, 2704–2719.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Kull M, Peterson H, Hansen J, and Vilo J (2007). g: Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35, W193–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, Laferla FM, Oddo S, and Verkhratsky A (2008). Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of alzheimer’s disease. PLoS One 3, e2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosi S (2011). Neuroinflammation and the plasticity-related immediate-early gene Arc. Brain Behav. Immun 25, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen WT, Woodbury ME, Srivastava G, et al. (2019). The major risk factors for alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 27, 1293–1306.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlachetzki JCM, and Hüll M (2009). Microglial activation in alzheimer’s disease. Curr. Alzheimer Res 6, 554–563. [DOI] [PubMed] [Google Scholar]
- Schneider LS, Dagerman KS, Higgins JPT, and McShane R (2011). Lack of evidence for the efficacy of memantine in mild alzheimer disease. Arch. Neurol 68, 991–998. [DOI] [PubMed] [Google Scholar]
- Simonyi A, Serfozo P, Lehmidi TM, Cui J, Gu Z, Lubahn DB, Sun AY, and Sun GY (2012). The neuroprotective effects of apocynin. Front. Biosci 4, 2183–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanska J, and Pawliczak R (2008). Apocynin: Molecular Aptitudes (Mediators of inflammation), p. 106507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen T-L, Cacciottolo M, Balu D, Morgan TE, LaDu MJ, Finch CE, and Pike CJ (2019). APOE genotype and sex affect microglial interactions with plaques in Alzheimer’s disease mice. Acta neuropathologica communications 7, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen T-L, Higgs NF, Sheehan DF, Al Awabdh S, López-Doménech G, Arancibia-Carcamo IL, and Kittler JT (2015). Miro1 regulates activity-driven positioning of mitochondria within astrocytic processes apposed to synapses to regulate intracellular calcium signaling. J. Neurosci 35, 15996–16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens KE, Zhou W, Ji Z, Chen Z, He S, Ji H, Guan Y, and Taverna SD (2019). Sex differences in gene regulation in the dorsal root ganglion after nerve injury. BMC Genom. 20, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterniczuk R, Antle MC, Laferla FM, and Dyck RH (2010). Characterization of the 3xTg mouse model of alzheimers disease: part 2. behavioral and cognitive changes. Brain Res. 1348, 149–155. [DOI] [PubMed] [Google Scholar]
- Sun HN, Kim SU, Huang SM, Kim JM, Park YH, Kim SH, Yang HY, Chung KJ, Lee TH, Choi HS, et al. (2010). Microglial peroxiredoxin V acts as an inducible anti-inflammatory antioxidant through cooperation with redox signaling cascades. J. Neurochem 114, 39–50. [DOI] [PubMed] [Google Scholar]
- Tamboli IY, Barth E, Christian L, Siepmann M, Kumar S, Singh S, Tolksdorf K, Heneka MT, Lütjohann D, Wunderlich P, and Walter J (2010). Statins promote the degradation of extracellular amyloid β-peptide by microglia via stimulation of exosome-associated IDE secretion. J. Biol. Chem 285, 37405–37414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Higuera JL, Mateo I, Sánchez-Juan P, Rodríguez-Rodríguez E, Pozueta A, Calero M, Dobato JL, Frank-García A, Valdivieso F, Berciano J, et al. (2011). Genetic variation in the tau protein phosphatase-2A pathway is not associated with Alzheimer’s disease risk. BMC Res. Notes 4, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilkeviciute A, Kriauciuniene L, Chaleckis R, Deltuva VP, and Liutkeviciene R (2019). COL10A1 (rs1064583) gene variants with predisposition to age-related macular degeneration. Dis. Markers 2019, 5631083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasik U, Schneider G, Mietelska-Porowska A, Mazurkiewicz M, Fabczak H, Weis S, Zabke C, Harrington CR, Filipek A, and Niewiadomska G (2013). Calcyclin binding protein and Siah-1 interacting protein in Alzheimer’s disease pathology: neuronal localization and possible function. Neurobiol. Aging 34, 1380–1388. [DOI] [PubMed] [Google Scholar]
- Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, Groshen S, Mack WJ, Guen E, Di Biase S, et al. (2017). Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med 9, eaai8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Han B, Wu S, Yang L, Leng S, Li M, Liao J, Wang G, Ye Q, Yuan Z, et al. (2019a). Circular RNA TLK1 aggravates neuronal injury and neurological deficits after ischemic stroke via miR-335-3p/TIPARP. J. Neurosci 39, 7369–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Fang K, Wang W, Lin W, Guo L, and Wang J (2019b). Identification of key genes and pathways for alzheimer’s disease via combined analysis of genome-wide expression profiling in the hippocampus. Biophys. Rep 5, 98–109. [Google Scholar]
- Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, and Longo VD (2002). Peroxynitrite mediates neurotoxicity of amyloid beta-peptide1-42- and lipopolysaccharide-activated microglia. J. Neurosci 22, 3484–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Leung S, Zhang J, Maus E, Baysac K, Bu G, Vassar R, Yu C, and LaDu MJ (2011). Amyloid-β42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J. Neurosci. Methods 196, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck G, et al. (2012). APOE4-specific changes in Aß accumulation in a new transgenic mouse model of alzheimer disease. J. Biol. Chem 287, 41774–41786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SN (1996). Behavioral effects of dietary neurotransmitter precursors: basic and clinical aspects. Neurosci. Biobehav. Rev 20, 313–323. [DOI] [PubMed] [Google Scholar]
- Yuan T, Yan F, Ying M, Cao J, He Q, Zhu H, and Yang B (2018). Inhibition of ubiquitin-specific proteases as a novel anticancer therapeutic strategy. Front. Pharmacol 9, 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel M, Schrag M, Crofton A, Tung S, Beaufond P, Van Ornam J, Di-Ninni A, Vinters HV, Coppola G, and Kirsch WM (2013). A shift in microglial β-amyloid binding in Alzheimer’s disease is associated with cerebral amyloid angiopathy. Brain Pathol 23, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zare-shahabadi A, Masliah E, Johnson GVW, and Rezaei N (2015). Autophagy in Alzheimer’s disease. Rev. Neurosci 26, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang XJ, Tian LP, Pan J, Lu GQ, Zhang YJ, Ding JQ, and Chen SD (2011). CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. J. Neuroinflammation 8, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Li Z, Vong JSL, Chen X, Lai HM, Yan LYC, Huang J, Sy SKH, Tian X, Huang Y, et al. (2020). Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain. Nat. Commun 11, 4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Guo Q, and Mattson MP (1999). Dietary restriction protects hippocampal neurons against the death-promoting action of a presenilin-1 mutation. Brain Res. 842, 224–229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. RNA-seq data have been deposited at SRA and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Barnes maze data have been deposited at Mendeley data and are publicly available as of the date of publication. The DOI is listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request (vlongo@usc.edu).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-beta amyloid (1:300; IHC) | Thermo Fisher Scientific | Cat#71-5800; RRID: AB_2533989 |
| Mouse anti-Phospho-Tau (Ser202, Thr205) (AT8) (1:1000; IHC) | Thermo Fisher Scientific | Cat#MN1020; RRID: AB_223647 |
| Rat anti-Bromodeoxyuridine/BrdU (BU1/75 (ICR1)) (1:500; IHC) | Novus Biologicals | Cat#NB500-169; RRID: AB_10002608 |
| Rat anti-mouse CD11b (1:500; IHC) | Bio-Rad | Cat#MCA711GT; RRID: AB_1100616 |
| Mouse anti-Iba1 (1:500; IHC) | Wako | Cat#016-26721; RRID: AB_2811160 |
| Rat anti-Mouse CD68 (1:500; IHC) | Bio-Rad | Cat#MCA1957GA; RRID: AB_324217 |
| Biotinylated goat anti-rabbit IgG (for beta amyloid, 1:500; IHC) | Vector Laboratories | Cat#BA-1000; RRID: AB_2313606 |
| Biotinylated horse anti-mouse IgG, rat adsorbed (for AT8 and Iba1, 1:500; IHC) | Vector Laboratories | Cat#BA-2001; RRID: AB_2336180 |
| Biotin-SP-AffiniPure donkey anti-rat IgG (H+L) (for BrdU, 1:250; IHC) | Jackson ImmunoResearch | Cat#712-065-150; RRID: AB_2340646 |
| Donkey anti rat IgG (H+L), biotin (for CD11b, 1:250; IHC) | Thermo Fisher Scientific | Cat#A18743; RRID: AB_2535520 |
| Rat anti-Ki-67 Monoclonal Antibody (SolA15) (1:200; IHC) | Thermo Fisher Scientific | Cat#14-5698-82; RRID: AB_10854564 |
| Rabbit anti-SOX2 (1:200; IHC) | Abcam | Cat#ab97959; RRID: AB_2341193 |
| Mouse anti-GFAP (1:200; IHC) | Cell Signaling | Cat#3670; RRID: AB_561049 |
| Donkey anti-rat-488 (for BrdU, Ki67 and GFAP, 1:400; IHC) | Thermo Fisher Scientific | Cat#A-21208; RRID: AB_2535794 |
| Donkey anti-rabbit-594 (for Sox2, 1:400; IHC) | Thermo Fisher Scientific | Cat#A-21207; RRID: AB_141637 |
| Rat anti-mouse CD11b antibody (1 μg/μL; IP) | Bio-Rad | Cat#MCA711GT; RRID:AB_321292 |
| Rabbit anti-NOX2 (1:5000; WB) | Abcam | Cat#ab129068; RRID: AB_11144496 |
| Rabbit anti-Aβ42 (1:500; WB) | Abcam | Cat#ab201060; RRID: AB_2818982 |
| Mouse anti-Vinculin (1:1000; WB) | Millipore | Cat#MAB3574; RRID: AB_2304338 |
| Chemicals, peptides, and recombinant proteins | ||
| Paraformaldehyde | Sigma-Aldrich | Cat#158127; CAS:30525-89-4 |
| 5’-Bromo-2’-deoxyuridine (BrdU) | Sigma-Aldrich | Cat#B5002; CAS:59-14-3 |
| Normal Donkey Serum | Jackson ImmunoResearch | Cat#017-000-121 |
| Polyvinyl alcohol mounting medium with DABCO, antifading | Sigma-Aldrich | Cat#10981 |
| Hoechst 33342 Solution | Thermo Fisher Scientific | Cat#62249 |
| VECTASHIELD® HardSet™ Antifade Mounting Medium with DAPI | Vector Laboratories | Cat#H-1500 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat#89900 |
| DAB (3,3’-diaminobenzidine) Peroxidase (HRP) Substrate Kit, With Nickel | Vector Laboratories | Cat#SK-4100 |
| Apocynin | Sigma-Aldrich | Cat#178385; CAS:498-02-2 |
| VECTASTAIN® Elite Avidin-Biotin Complex (ABC)-HRP Detection Kit, Peroxidase (Standard) | Vector Laboratories | Cat#PK-6100 |
| NuPAGE™ LDS Sample Buffer (4X) | Invitrogen | Cat#NP0007 |
| HyGLO™ Chemiluminescent HRP Detection Reagent | Denville Scientific | Cat#E2500 |
| RNase-Free DNase Set | Qiagen | Cat#79254 |
| Papain, Lyophilized | Worthington Biochemical | Cat#LS003118; CAS: 9001-73-4 |
| HiLyte™ Fluor 555-labeled Amyloidβ 42 (Aβ42) | AnaSpec | Cat#AS-60480-01 |
| Poly-D Lysine | Sigma-Aldrich | Cat#P7280 |
| Percoll PLUS | Cytiva | Cat#17-5445-02 |
| Critical commercial assays | ||
| Dynabeads™ Protein G Immunoprecipitation Kit | Thermo Fisher Scientific | Cat#10007D |
| Rat CYBB / NOX2 / gp91phox ELISA Kit | LSBio | Cat#LS-F39030 |
| V-PLEX Proinflammatory Panel 1 (mouse) Kit | Meso Scale Diagnostics | Cat#K15048D-1 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat#74804 |
| V-PLEX Plus Aβ Peptide Panel 1 (4G8) | Meso Scale Diagnostics | Cat#K15199G-1 |
| D1000 ScreenTape Assay | Agilent Technologies | N/A |
| KAPA mRNA HyperPrep kit | Roche-Sequencing | N/A |
| Deposited data | ||
| RNA-Seq of hemi-cortex of the 8.5 month old 3xTg young mice study | This paper | SRA: PRJNA864838 |
| Barnes Maze data of E4FAD mice | This paper | Mendeley Data: https://doi.org/10.17632/p2c5z4vjsw.1 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: 3xTg | Frank LaFerla, University of California, Irvine | N/A |
| Mouse: E4FAD | Mary Jo LaDu, University of Illinois | N/A |
| Mouse: Nox2-KO | The Jackson Laboratory | JAX: 002365 |
| Mouse: 3xTg/Nox2-KO | Generated from 3xTg and Nox2KO | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism v.8 | Graphpad | https://www.graphpad.com/ |
| HTSeq | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| EdgeR | McCarthy et al., 2012; Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| FastQC v0.11.8 | Andrews, 2010 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic v0.38 | Bolger et al., 2014 | http://www.usadellab.org/cms/index.php?page=trimmomatic |
| STAR v2.6.1d | Dobin et al., 2013 | http://code.google.com/p/rna-star/ |
| gProfileR v0.6.8 | Reimand et al., 2007 | http://biit.cs.ut.ee/gprofiler/ |
| Imagelab 6.0 software | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Other | ||
| Mouse diet: Fasting mimicking diet (FMD) | Cheng et al., 2017 | N/A |
| Mouse diet: 4% protein-restricted diet (4% PR) | Parrella et al., 2013 | N/A |
| Human diet: ProlonAD TM | Propriety formulation belonging to L-Nutra | https://www.prolon.it |
| Control Diet: PicoLab Rodent Diet 20 | LabDiet | Cat#5053 |
| HydroGel | ClearH2O | N/A |
| Barnes Maze | San Diego Instruments | N/A |
| All-in-One Fluorescence Microscope | Keyence | BZ-X710 |
| Olympus BX50 Microscope | Olympus | N/A |
| Olympus DP73 Camera | Olympus | N/A |
| Zeiss Laser Scanning Microscope-780 upright microscope | Zeiss | N/A |
| 2200 TapeStation | Agilent Technologies | N/A |
| HiSeq3000 | Illumina | N/A |
| Vibratome | Leica | VT1000S |
| Janus G3 liquid handler | Perkin-Elmer | N/A |