

Abstract
The giant sarcomere protein titin is a major determinant of cardiomyocyte stiffness and contributor to cardiac strain sensing. Titin-based forces are highly regulated in health and disease, which aids in the regulation of myocardial function, including cardiac filling and output. Due to the enormous size, complexity, and malleability of the titin molecule, titin properties are also vulnerable to dysregulation, as observed in various cardiac disorders. This review provides an overview of how cardiac titin properties can be changed at a molecular level, including the role isoform diversity and post-translational modifications (acetylation, oxidation, and phosphorylation) play in regulating myocardial stiffness and contractility. We then consider how this regulation becomes unbalanced in heart disease, with an emphasis on changes in titin stiffness and protein quality control. In this context, new insights into the key pathomechanisms of human cardiomyopathy due to a truncation in the titin gene (TTN) are discussed. Along the way, we touch on the potential for titin to be therapeutically targeted to treat acquired or inherited cardiac conditions, such as HFpEF or TTN-truncation cardiomyopathy.
Keywords: Heart failure, Cardiomyopathy, Sarcomere, Mechanical function, Signalling
Graphical Abstract
Graphical Abstract.

1. Introduction
The heart needs to continually pump throughout a lifetime and meet the ever-changing requirements of the body. Some changes take place relatively slowly throughout development, ageing, and disease, while others occur almost on a beat-to-beat basis. How the heart adapts to these changes continues to be the subject of an entire research field, as many details remain incompletely understood. For example, increased ventricular filling and pressure induce stretch and stress signalling pathways within the heart, which are complex, intertwined in many ways, and challenging to study—notably in vivo.1 At the level of the cardiac cells, a myriad of proteins has been identified which participate in mechanical and chemical signalling in various ways. The focus of this review is the mechanosensitive protein titin2 in the sarcomeres, the contractile units of the cardiomyocytes (CMs). We zoom in on titin’s properties as a molecular spring whose elasticity can be extensively modulated under physiological and pathophysiological conditions, resulting in larger modulations in cardiac function, such as diastolic filling or length-dependent activation (LDA), the basis of the Frank-Starling law. Titin has several mechanisms it can exploit to physiologically modulate myocardial stiffness and distensibility, including post-translational modifications (PTMs, e.g. acetylation, oxidation, and phosphorylation) and isoform switch (Figure 1). Other possibilities to fine-tune the mechanical properties of the cardiac sarcomere/CMs/heart involve the binding of specific titin regions to Ca2+, chaperones, or protein ligands, such as actin (not reviewed here, but see Ref.2). However, titin’s giant size and complexity leaves it vulnerable to dysregulation, which can result in the development of cardiac disease. To date, the dysregulation of titin, its binding partners, and associated signalling pathways have been implicated in various forms of heart disease, such as heart failure (HF) with preserved ejection fraction (HFpEF), HF with reduced ejection fraction (HFrEF), aortic stenosis, and ischaemic injury (Figure 1). Moreover, next-generation sequencing has revealed pathogenic variants in the titin gene (TTN) as a major cause of inherited cardiomyopathies,3 the pathomechanisms of which have only recently become clearer,4 as discussed further below in this review. The overall goal of our review is to explore how cardiac titin properties can be changed at a molecular level, with an emphasis on titin stiffness and protein quality control (PQC), and how this regulation becomes unbalanced in heart disease. We also highlight new insight suggesting that titin can be therapeutically targeted to help treat acquired or inherited cardiac conditions, such as HFpEF or TTN-truncation cardiomyopathy.
Figure 1.

Known changes in cardiac titin properties under physiological stress and as a cause or consequence of heart disease.
2. Structure, expression, and isoform diversity of titin
Spanning the half-sarcomere from the Z-disc to the M-band as the ‘third’ sarcomeric filament system (along with myosin and actin filaments), titin is well placed to assist in the regulation of both passive and, to some extent, active force development of the heart (Figure 2A).5,6 The intrinsic properties of titin vary along its length, depending on the location within the sarcomere: A-band titin is bound to myosin filaments and is functionally inextensible, I-band titin lays slightly oblique between the Z-disc and the I/A-band junction and is elastic, and the ends of the titin filament are anchored at the Z-disc (via α-actinin, actin, and telethonin) and M-band (via myomesin). The specific amino acid sequences and domain structures in each region determine the structural and functional roles of these titin segments.2 A-band titin is largely comprised of super-repeats of relatively stable fibronectin-type 3 and immunoglobulin-like (Ig) domains7 providing a template for thick-filament assembly and A-band length,8 while the M-band region engages in protein–protein interactions that serve structural and regulatory functions.9,10 I-band titin is variable and contains several extensible elements, such as the PEVK-repeats (motifs rich in proline, glutamic acid, valine, and lysine), Ig-domain regions, and unique sequences, notably the N2B-unique sequence (N2Bus), enabling titin’s spring-like properties.11,12 Unique sequence elements are also present in Z-disc and M-band titin.
Figure 2.

Titin isoforms in cardiac muscle, isoform switch, and consequences for titin-based stiffness. (A) Architecture of cardiac titin N2B and N2BA isoforms in the half-sarcomere. Note the abbreviated A-band region (double-line break). (B) Titin isoforms (‘Full length’ is a theoretical variant); only main I-band segments are shown. Dotted lines indicate alternative splicing. (C) Titin species separated on a typical Coomassie-stained titin gel (1.8% SDS–PAGE) of adult human heart tissue. (D) The proportion of N2BA (green) vs. N2B (red) titin isoforms can change under numerous conditions. This isoform switch determines a change in titin spring stiffness. AngII, angiotensin II; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; Ig, immunoglobulin-like domain; PEVK, segment rich in proline (P), glutamic acid (E), valine (V), and lysine (K) residues; RBM20, RNA-binding motif protein 20; T3, thyroid hormone.
Part of the variability within I-band titin comes from the high degree of differential splicing that occurs in this region.13 Of note, this continues to cause inconsistencies with the nomenclature when assigning specific domain numbers to I-band titin. Further adding to the confusion is that commonly referred-to databases for human titin, such as UniProtKB and NCBI (canonical sequence accession numbers, Q8WZ42-1 and NP_001243779.1, respectively), falsely recognize some PEVK-repeats or globular Ig domains as insertion sequences. Human TTN has 364 exons and the complete meta-transcript 363 exons, from which several common transcripts (isoforms) are generated (Figure 2B). A good overview is provided at http://cardiodb.org/titin. Because the frequency at which an exon is incorporated into the titin molecule varies greatly in the elastic I-band region, the different isoforms have different spring length and variable compliance.2,13,14
The principal cardiac isoforms are the Z-disc-anchored N2B, N2BA, and Novex-3 variants,15 as well as the recently identified C-terminal Cronos isoform driven by an internal promoter (Figure 2B).16 On a typical Coomassie-stained titin-protein gel of human heart tissue (Figure 2C), these isoforms appear together with a proteolytic fragment, T2 (2.4 MDa). The two most common cardiac isoforms, N2B (3 MDa) and N2BA (3.2–3.8 MDa), determine titin extensibility and myofibrillar passive stiffness.12,17 The N2BA isoforms exist in many splice variants that have different I-band length. The N2B isoform (named after the N2B element coded by TTN exon 49) is shorter and less compliant than the N2BA isoforms, as its spring segment has only 6 PEVK-repeats, no ‘middle’-Ig domains, and no N2A element (Figure 2B).12–14 The short isoform Novex-3 (∼650 kDa), which splices-in exon 48 (thus introducing a stop), does not appear to be relevant for titin stiffness; however, the exon 48-encoded Novex-3 region may have a role as a structural and regulatory element, e.g. through its interaction with obscurin.15 Cronos (2.3 MDa; close in size to the T2 fragment) is more highly expressed in developing CMs than in adult hearts,18 where it makes up only ∼10% of the total titin-protein pool (human adult hearts).4 In human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), Cronos has been shown to enable partial sarcomere formation in the absence of full-length titin.18 However, Cronos failed to rescue cardiac sarcomerogenesis in adult mice lacking Z-disc-anchored titin.19 Moreover, Cronos transcript and protein content were found to be unaltered in end-stage failing hearts from dilated cardiomyopathy (DCM) patients vs. non-failing human donor hearts.4
2.1 Titin splicing regulation and mechanical consequences
The titin N2B vs. N2BA isoform expression pattern is mediated, at least in part, by the splicing factor RNA-binding motif protein-20 (RBM20).20 RBM20 suppresses the splicing-in of I-band titin exons,21,22 thereby promoting expression of the shorter, stiffer N2B isoform (Figure 2D, inset).20,23 RBM20 is also regulated by titin splicing itself: spliced-out regions of TTN can form functional motifs of circular RNA, such as cTTN1, which regulate the activity of RBM20 and that of another splicing factor, SRSF10.24 RMB20-deficient rats or mice have been found to produce aberrantly large N2BA-isoforms and no N2B, resulting in reduced myocardial passive stiffness and dilated ventricles.20,21,23 In humans, RBM20 pathogenic variants are associated with the expression of oversized N2BA isoforms,4,20,25 greater titin compliance,26 and irregular calcium homeostasis27 leading to the development of DCM.20,26 Interestingly, both insulin and thyroid hormone, T3, modulate RBM20 expression and activity pathways.28,29 This could explain why T330,31 and insulin32 (and also Angiotensin II) promote the expression of the N2B titin isoform, e.g. during heart development.30 Long and compliant, foetal N2BA titin isoforms render titin-based stiffness very low in the embryonic heart, but a transition to shorter and less stretchable (adult) N2BA isoforms and N2B in the pre-/perinatal period greatly increases this stiffness (Figure 2D). Thus, long-term cardiac stiffness regulation can be achieved by changing the relative abundance of the N2BA and N2B isoforms, known as isoform switching.
2.2 Titin–isoform switch in heart disease
Titin–isoform transitions also regulate titin-based stiffness in heart disease. A healthy adult human heart (left ventricle) has an N2BA: N2B ratio of 30:70 to 40:60,33 and this ratio remains relatively constant during normal ageing.34 Isoform switching occurs in many forms of HF, including HFrEF (e.g. DCM or chronic ischaemic heart disease), HFpEF, and aortic stenosis (Figure 2D). Various studies have found that relatively more N2BA and less N2B isoform is present in failing vs. non-failing human hearts, such as in HFrEF,4,33,35,36 HFpEF,37 and aortic stenosis.37,38 An increased proportion of N2BA is correlated with an increased end-diastolic volume35 and lower titin-based passive stiffness (Figure 2D).33,35,36 However, earlier studies reported a reduced N2BA:N2B ratio in aortic stenosis39 and HFpEF patients,40 whereas no change in titin–isoform composition was found in human hypertrophic cardiomyopathy and sometimes even in human DCM or aortic stenosis.41–44 Conversely, the N2BA:N2B ratio has sometimes (e.g. Refs45,46) but not consistently been found to be reduced in animal models of HF. These discrepancies may be related to the stage of the heart disease during which the cardiac muscle samples were studied, the type of HF syndrome, species characteristics, heart chamber-specific differences,47 and/or methodological limitations. It is still incompletely understood whether titin–isoform switch causes or compensates for cardiac stiffness changes that occur during disease progression.
Our knowledge of the influence titin–isoform switch can have on cardiac stiffness has made it a potential therapeutic target for the treatment of HF. Successful attempts have been made to modulate the titin–isoform composition in animal models by reducing RBM20 expression, increasing the N2BA to N2B ratio and lowering cardiac stiffness.23,48,49 However, RBM20 is responsible for the splicing of many other cardiac proteins and the reduction of RBM20 will have consequences for the function of at least some of them, including crucial Ca2+-handling proteins.27,50 The lack of titin specificity therefore limits the therapeutic scope of RBM20 inhibition. Regardless, targeting titin–isoform switching might improve prognosis and symptoms of some HF patients, potentially making it a therapeutic approach in the future.
3. Cardiac titin stiffness as a regulator of active contraction
The mechanical properties of titin not only determine a significant portion of CM passive stiffness but also regulate active contraction.6 A prime example is LDA, which is characterized by an immediate increase in the Ca2+ sensitivity of the myofilaments with a CM stretch. A consistent observation is the correlation between I-band titin length or titin-based stiffness and LDA: CMs with a short/stiff titin spring show a larger increase in myofilament Ca2+ sensitivity upon cell stretching than those with a long/soft titin spring.51,52 Similarly, RBM20-deficient rat CMs expressing long and very compliant N2BA titin have a blunted LDA,53–55 whereas mouse CMs lacking extensible I-band titin regions show increased titin-based stiffness and improved LDA compared to wildtype (wt) CMs.56 Therefore, the titin–isoform switch towards more compliant N2BA present in failing hearts33 is expected to reduce LDA and (via the Frank–Starling mechanism) cardiac output. These and other observations have suggested that stretch effects mediated by titin cause alterations to both thick- and thin-filament properties; however, the underlying molecular mechanisms remain a matter of debate.6,57–64 Titin also has the potential to more directly support the work output of the contractile system by storing and releasing elastic energy via Ig-domain unfolding–refolding transitions, which occur under physiological (low) stretch forces.6,65,66 While the magnitude of this contribution remains to be tested in vivo, the mechanism may be relevant to synchronize the mechanical activities of the actomyosin and titin systems.6,66,67 Collectively, available data leave no doubt that the mechanical design of titin aids active contraction of cardiac muscle in multiple ways. It will be worth studying how the connectivity between titin spring stiffness and cardiac contractility may change in failing hearts.
4. Titin PTMs
Cardiac filling and output are optimized according to the actual demands of the body, and these adjustments also employ changes to titin stiffness. The question is how do you fine-tune stiffness in such a large protein potentially on almost a beat-to-beat basis? One of the quickest ways to adjust protein activity is through PTMs. For titin, phosphorylation, oxidation, and more recently acetylation have been shown to modulate its spring stiffness. Differences in these modifications have also been detected under pathophysiological conditions, giving rise to novel treatment strategies aiming at reversing pathological passive stiffness of patient hearts.
4.1 Acetylation–deacetylation
Lysine acetylation/deacetylation has traditionally been studied as a modification of histones and a regulator of transcription but it is increasingly becoming recognized as a modulator of cytoplasmic proteins, particularly relating to cellular metabolism.68 Acetylation regulation has become of interest to the cardiac community, not least because of its association with obesity,69 a worsening global burden of disease and a major comorbidity in HF. Acetylation has been abundantly detected along the length of titin (Figure 3A);70 however, both the native acetylation state of titin and how it changes in CMs require additional studies, especially on human hearts. Proteomic analysis has revealed an increase in titin acetylation in obese mice69 but no significant difference in acetylation levels in HFpEF models compared with the experimental controls.70,71
Figure 3.
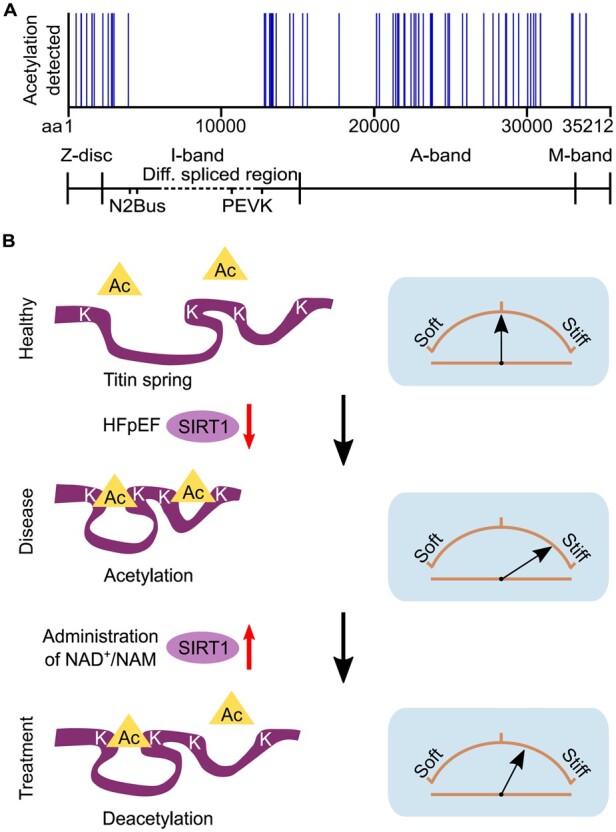
Locations and proposed mechanism of titin acetylation. (A) Titin acetylation sites detected in rat heart tissue by mass spectrometry (data obtained from Ref.70). (B) Locally increased acetylation of elastic titin in HFpEF may be due to reduced deacetylase activity of SIRT1; this increases titin-based stiffness. Treatment with NAD+/NAM restores SIRT1 activity to reverse this process. Brown arches depict titin spring stiffness. aa, amino acid.
Proposed treatments targeting acetylation pathways have also uncovered changes to titin acetylation, which may play a role in the modulation of cardiac stiffness. The caloric restriction mimetic, nicotinamide (NAM), is a precursor of nicotinamide adenine dinucleotide (NAD+), which is required for the activation of deacetylase enzymes, such as sirtuin-1 (SIRT1). NAD+ has been suggested as a potential therapeutic for HFpEF.70 Direct application of SIRT1 on skinned rat CMs was found to reduce their stiffness.70 This was suggested to be a contributing factor of improved diastolic function in HFpEF after treatment with NAM.70 However, conflicting results in similar animal models have shown that the use of histone deacetylase inhibitors can also ameliorate diastolic dysfunction and increase myofibril relaxation.71,72 although changes in the acetylation state of titin after treatment were not assessed.
Another open question is how deacetylation causes a decrease in titin stiffness. The additional negative charge added to positively charged lysines during acetylation has been shown to stabilize enzymes and decrease the rate of unfolding.68,73 Based on our findings,70 we propose that the increased acetylation of the titin spring, e.g. in HFpEF due to reduced SIRT1 activity, promotes intramolecular interactions within elastic titin via addition of negative charges in a positively charged environment; as a consequence, titin becomes stiffer (Figure 3B). Conversely, the increased deacetylation of titin, e.g. by elevated SIRT1 activity following NAD+/NAM treatment,70 partially reverses this stiffening (Figure 3B). Additionally, titin acetylation may play a role in competitive PTM crosstalk with titin ubiquitination as both occur on lysine residues.68 The exact role acetylation plays in titin regulation and its association with cardio-metabolic disorders provides some exciting prospects for future research and therapeutics.
4.2 Oxidation–reduction
Oxidative stress is a generalized term for an imbalance between reactive oxygen species (ROS) and antioxidants. While low levels of ROS are required for normal cellular function and regulation, higher levels are associated with the pathogenesis of many diseases including cardiovascular disorders.74 Excess ROS leads to the modification of various cellular processes including modifications of amino acids containing thiol groups (commonly cysteines) in either a reversible or irreversible manner. Titin is a prime target of oxidation and I-band titin oxidation–reduction modulates titin-based stiffness.75
Mass spectrometric analyses of cardiac titin have detected oxidation sites along the entire molecule (Figure 4A).76 In mouse hearts exposed to oxidative stress, the highest relative increase in titin oxidation was found within I-band ‘hotspots’, specifically, the N2Bus and Ig domains of the distal I-band region.76 However, both the location of oxidation site and the type of oxidation are critical in determining the consequence on titin stiffness: titin domains can undergo either S-glutathionylation, which leads to an increase in titin compliance, or disulphide bonding, which causes increased titin stiffness (Figure 4B). These modifications are reversible, e.g. under the influence of reductants like dithiothreitol or glutathione.76–78
Figure 4.

Titin oxidation sites and mechanisms of how S-glutathionylation and disulphide bonding alter titin stiffness in health and disease. (A) Titin oxidation sites detected in mouse heart tissue by mass spectrometry (data obtained from Ref.76). (B) Changes in titin stiffness are dependent on whether more S-glutathionylation or more disulphide bonding occurs in unfolded I-band titin domains. Increased compliance is mediated via the S-glutathionylation mechanism involving Ig domains and increased stiffness via the disulphide bonding mechanism involving Ig domains or the N2Bus. Brown arches depict titin spring stiffness. (C) Observed changes in I-band titin oxidation under cardiac stress (increased preload/afterload) or in diseased states (ischaemia, HFpEF) modulate titin stiffness in a complex manner. S-glutathionylation under cardiac stress can cause controlled in-register aggregation of distal I-band titin, whereas disease states can be associated with a general increase in titin oxidation. aa, amino acid; HFpEF, heart failure with preserved ejection fraction; Ig, immunoglobulin-like; UnDOx, unfolded domain oxidation.
Mechanistically relevant is that titin oxidation within Ig domains may rarely occur spontaneously but may rather require cysteines buried within the Ig domains (cryptic cysteines) to be exposed through domain unfolding77,78—a process we have coined UnDOx (unfolded domain oxidation).76 If UnDOx occurs, these domains cannot refold back to their native state, which affects titin-based stiffness. Importantly, the type of oxidation that occurs at the cryptic cysteines determines the effect on stiffness.76 UnDOx, through S-glutathionylation, prevents the Ig domain from refolding, thereby maintaining a longer contour length and lowering titin stiffness (Figure 4B, left).76–78 Additionally, this unfolded oxidized state allows for the controlled homotypic interactions and in-register aggregation of the distal I-band region (Figure 4C), which may aid stiffness regulation and force propagation.76 Alternatively, UnDOx can involve disulphide bond formation (Figure 4B, top right). A disulphide bridge can arise between any two (of a diad, triad, or quadripartite group of) cysteines within an unfolded Ig domain and prevent the extension of the domain to its full contour length,78 therefore increasing the passive stiffness of titin.76 It has been suggested that the formation of disulphide bonds in titin may fine-tune muscle work production by providing additional power during Ig-domain refolding under a (low) stretch force.65 The formation of disulphide bonds in Ig domains may also be aided by intermediatory S‐sulfenylation,79 but due to the highly volatile nature of S‐sulphenylation, there is yet to be any evidence showing that this occurs in vivo. On the other hand, disulphide bond formation also occurs within the N2Bus region of I-band titin (Figure 4B, bottom right) and contributes to increased titin stiffness by producing additional scaffolding to this normally disordered I-band region and preventing it from full extension.80,81
Both S-glutathionylation and disulphide bonding are a natural consequence of exertion and increased metabolism. Therefore, low levels are detected in cardiac titin under healthy conditions, but they rise with an increase in cardiac preload or afterload (Figure 4C).76 However, in the case of cardiac ischaemia or chronic HF due to metabolic syndrome (e.g. HFpEF), there is a loss in the balance of these oxidative modifications and what was once a normal short-term regulatory process becomes an extensive modification and marker of disease (Figure 4C).76 Targeting titin oxidation therapeutically could be useful in the treatment for these cardiac conditions to modulate myocardial stiffness.
4.3 Phosphorylation–dephosphorylation
Phosphorylation is a well-established PTM in many cellular processes. In the heart, it regulates cardiac output on multiple levels. Phosphorylation is also the best-studied titin PTM, modulating the molecule’s stiffness.82 Over 300 titin phosphorylation sites have been detected using proteomic (mass spectrometric) techniques, with some phosphosites consistently being identified in multiple studies (Figure 5A).83 At this stage, it appears that the titin phosphorylation-dependent changes in CM stiffness are related to where the titin spring becomes phosphorylated and the net protein charge present in that titin region (Figure 5B). Caution needs to be taken with this assumption, however, due to the bias in where along the titin molecule phosphorylation has been assessed historically.
Figure 5.

Where and how titin becomes phosphorylated, how this affects titin stiffness, and the role titin phosphorylation changes play in heart disease. (A) The number of times a titin phosphorylation site has been referenced (ref.) in the literature from either high-throughput (mass spectrometry; blue) or low-throughput, site-specific (antibody detection, mutagenesis; red) detection in human heart tissue. Data obtained from www.phosphosite.org.83 Note that phosphosite S4062 is currently not detected in this database but has been added based on the publication by Ref.95 (B) Specific phosphorylation sites detected in different regions of titin, known PKs/phosphatases involved, and differential effects of I-band titin phosphorylation on titin stiffness. (C) Hypo-phosphorylation of the N2Bus and hyper-phosphorylation of constitutively expressed PEVK titin are thought to increase titin stiffness in HF. (D) Proposed treatment strategies in pre-clinical tests aimed at correcting titin phosphorylation in animal models of heart disease, to reduce titin-based cardiac stiffness. ‘Trial’ highlights results of large clinical trials on human HF patients using the respective drugs. Brown arches depict titin spring stiffness. aa, amino acid; BNP, B-type natriuretic peptide; CaM, calmodulin; CaMKIIδ, Ca2+/calmodulin-dependent protein kinase IIδ; cGMP, cyclic guanosine monophosphate; ERK, extracellular signal-regulated kinase; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; Ig, immunoglobulin-like; LV, left ventricular; NRG1, neuregulin 1; PDE-5, phosphodiesterase type 5; PKA, protein kinase A; PKCα, protein kinase Cα; PKD, protein kinase D; PKG, protein kinase G; PP5, serine/threonine protein phosphatase 5; sGC, soluble guanylyl cyclase; TK, titin kinase domain; UnDOx, unfolded domain oxidation.
The gigantic size of titin currently makes it impossible to recombinantly express the whole protein. Therefore, small regions of titin in specific locations, such as the Z-disc or M-band, or with unique mechanical properties, such as the PEVK and N2Bus regions, have been the focus of most low-throughput approaches (e.g. antibody-specific targeting or mutagenesis of phosphosites). Initial studies determined that phosphorylation within the Z-disc region could be attained by cyclin-dependent kinase (cdc2) and extracellular signal‐regulated kinase (ERK)284 and similarly, cdc2 could also phosphorylate the M-band titin region.85 Additionally, early studies showed that the titin kinase domain (TK) also required phosphorylation of tyrosine residue 32341, as well as the binding of Ca2+/calmodulin, to enable activation (Figure 5B, right);86 however, TK currently is considered an inactive pseudo-kinase.87 Since then, two main regions, the N2Bus and PEVK segments, have been the focus of most titin phosphorylation studies. There is much compelling evidence to suggest that phosphorylation at these regions plays a crucial role in titin-stiffness regulation.
Interestingly, the phosphorylation of the N2Bus and PEVK regions has opposing effects (Figure 5B). Increased phosphorylation of the N2Bus causes increased titin compliance through an increase in persistence length, as determined by single molecule atomic force microscopy stretch experiments.88,89 Conversely, phosphorylation of the PEVK region leads to increased titin stiffness.90,91 The mechanisms behind this change in stiffness are still not completely clear; however, we have previously suggested that the difference may be due to the N2Bus region and the PEVK having different localized net charges.82 The N2Bus has a net negative charge and therefore, the introduction of additional negative phosphate groups could cause electrostatic repulsion within the N2Bus region, leading to improved extensibility and a decrease in titin-based force. Conversely, the positive net charge of the PEVK region (notably: only the constitutively expressed and not the differentially spliced segment, which has a net negative charge!) would see increased intramolecular interactions, reduced extensibility, and increased force (Figure 5B). Firm experimental proof for this ‘charge theory’ is still lacking.
To complicate matters further, there is an array of protein kinases (PKs) that can phosphorylate multiple sites in titin. At least four main phosphoserines have been identified in the human N2Bus region (Figure 5B): S4010, S4062, S4099, and S4185 (according to human titin consensus sequence, UniProKB #Q8WZ42-1). The first three are evolutionary conserved (mouse equivalents S3991, S4043, and S4080, respectively). These phosphosites have been identified using site-specific methods, such as mutagenesis of phosphorylated residues or antibody-specific targeting. The N2Bus sites have been determined to be phosphorylated by various PKs, such as PKA and PKG,44,46,88,92 PKD,93 ERK2,89,94 and/or CaMKIIδ.95,96 These phosphosites have also been shown to be dephosphorylated by serine/threonine protein phosphatase 5,97 a highly regulated protein phosphatase with low basal activity.98 Similarly, within titin’s PEVK region, several sites have been detected, which are phosphorylated by PKCα, CaMKIIδ, and/or PKD, in particular conserved phosphoserines S11878 and S12022 (mouse equivalents S12742 and S12884).44,46,90,91,93,95 In part, this would imply conflicting signalling as both PKD and CaMKIIδ phosphorylate N2Bus and PEVK titin—which in theory should have opposite effects on titin stiffness (Figure 5B). However, both kinases cause a decrease in titin-based force of isolated cardiac preparations,89,93,95 suggesting the effect on N2Bus dominates. However, caution is required when interpreting these results, considering the number of phosphorylation sites that have been detected but not yet thoroughly investigated in titin (Figure 5A). Indeed, at least one of the above PKs, CaMKIIδ, can also phosphorylate Ig domains in I-band titin, but only after domain unfolding, and the process is further promoted by UnDOx through S-glutathionylation (Figure 5B).76 This additional possibility for titin regulation by phosphorylation may aid in stabilizing the unfolded state of the domain and support mechano-chemical signalling events. In summary, titin-stiffness regulation by phosphorylation is complex and involves several signalling hotspots within I-band titin and various PKs/phosphatases; currently, it is the best-understood mode of titin regulation by PTMs backed by a large body of evidence.
4.3.1 Titin phosphorylation in heart disease
Irregular phosphorylation of titin is also highly implicated in HF. Frequently, HF is associated with pathologically increased myocardial and CM passive stiffness, at least in HFpEF patients.40 Some of this stiffness increase can be explained by hypo-phosphorylation of phosphoserines within the N2Bus region of titin and hyper-phosphorylation of select sites within the PEVK region, e.g. S11878 (Figure 5C).44,46,97 However, exceptions exist depending on the form of human HF and specific heart chamber investigated, or the animal model used.44,82,95,99 A detailed review of this topic has recently been compiled.82 Generally, it is more informative to determine site-specific titin phosphorylation rather than global titin phosphorylation, considering the huge number of potential phosphosites in the full-length titin protein.82 However, given where phosphorylation occurs affects how titin stiffness is modulated, more site-specific information on titin phosphorylation is needed to better understand the diseased state. Altogether, reduced phosphorylation of titin’s N2Bus region appears to be a frequent alteration in various HF types, and this alteration may be an important contributor to increased CM stiffness in disease.
4.3.2 Titin phosphorylation as a therapeutic target
Disruption in phosphorylation regulation is a common feature in cardiac diseases not only affecting titin but also many other cardiac proteins. This makes phosphorylation regulation an attractive target for therapeutic treatment. Pathologically increased myocardial passive stiffness is a sign of diastolic dysfunction often seen in HF patients when associated with comorbidities, including diabetes mellitus.43,100 Interestingly, conventional therapeutics for diabetes, including metformin and insulin, have been found to improve diastolic function in these patients. Specifically, metformin and insulin increased the activity of titin-targeting PKs (ERK1/2; PKCα; PKA), improving phosphorylation of the N2Bus (while marginally increasing PEVK phosphorylation) and causing a reduction in titin-based stiffness (Figure 5D).100,101 Similarly, chronic admission of the cardiac growth factor neuregulin-1 was shown to activate ERK1/2 and PKG while suppressing PKCα, resulting in hyper-phosphorylation of N2Bus and hypo-phosphorylation of PEVK titin, both of which are conducive in reducing titin stiffness (Figure 5D).100
In this context, a main focus has been on the cyclic guanosine monophosphate (cGMP)-PKG pathway, because dysregulation of this pathway is strongly associated with cardiac remodelling and HF. There are now multiple substances available which stimulate this pathway (see Ref.102 for a recent, comprehensive review on the role cGMP plays in the heart). Phosphodiesterase-5A inhibitors (such as sildenafil) and B-type natriuretic peptide (BNP), which boost cGMP levels, showed promise as a potential treatment for diastolic dysfunction in pre-clinical tests (Figure 5D). In regards to titin, both sildenafil and BNP increased (total) titin phosphorylation, reduced CM stiffness and increased left ventricular (LV) distensibility in a dog model of diastolic dysfunction.46,103 However, the use of PDE5 inhibitors in the RELAX trial failed to show significant improvement of diastolic function in HFpEF patients that received the treatment (Figure 5D).104 Similar trials are underway with PDE9A inhibitors, which also aim to increase the cGMP concentration in the heart and benefit cardiac function.105 In mouse models of diastolic function, PDE9A inhibition reduced LV diastolic stiffness through reduction of CM stiffness; however, titin phosphorylation was not measured.106 Furthermore, soluble guanylyl cyclase (sGC) activators/stimulators are considered a promising treatment for HFpEF, having been found to raise cGMP levels leading to increased PKG (and also PKA and ERK2) activity, while reducing PKCα and CaMKIIδ activity.107,108 This again resulted in increased N2Bus and total titin phosphorylation, reduced CM stiffness and reduced LV stiffness (Figure 5D). However, these results were not replicated in either the VITALITY or the SOCRATES clinical trials where no improvement in diastolic function was seen in HFpEF patients treated with sGC-stimulator.109,110
Although these treatments showed promise in the laboratory setting, discrepancies between dosages and metabolic differences in animals vs. humans may in part be the cause of clinical trial failures.102 This highlights one of the many barriers in finding effective new treatments for HF. Additionally, the complexity of pathways, such as cGMP-PKG, means that the regulation of intermediate steps in the pathway can also be unintentionally changed and counteract the desired effect of the treatment.102 Therefore, increasing local cGMP levels within titin microdomains by exploiting compartmentalization might prove more useful, but such microdomains are yet to be determined. Regardless, these treatments may still be successful for a subset of patients with only modestly impaired cGMP-PKG signalling.
5. Titin-PQC
Maintaining a protein as large as titin over a lifetime requires a sophisticated PQC system. Titin is thought to be turned over at least every ∼3 days in cell culture,111 but the protein’s half-life in adult mice is 2–3 weeks.19,112 This enables sarcomere maintenance and CM remodelling during development and repair of stress-related protein damage in adulthood.113 However, with advancing age, the cellular PQC machinery, including the titin-directed PQC systems, may slow down.34 Consequently, defective titin protein could accumulate intracellularly and form cytoplasmic aggregates that are cytotoxic. As shown for other proteins, this may be one of the contributing factors to the development of cardiac disease in the elderly.113
An initial step in PQC is to stabilize or repair (refold) damaged (unfolded) proteins under stress with the aid of chaperones.114 This appears to be particularly important for the elastic I-band titin as it contains many regions that unfold during a stretch. Small heat shock proteins (sHSPs) are chaperones that protect unfolded proteins from permanent damage but do not necessarily mediate the refolding (which requires an ATP-dependent chaperone).115 The sHSPs alpha-B-crystallin (HSPB5) and HSP27 (HSPB1) are abundant in the cytosol of CMs and are also present at the Z-disc under physiological conditions.115,116 Upon physiological stretch, as well as in failing hearts (e.g. ischaemia; cardiomyopathy), these chaperones are up-regulated and play a protective role by translocating to the N2Bus, N2A, and proximal/middle I-band Ig-domain regions of titin (Figure 6A and B).117–119 There, they stabilize unfolded segments and prevent aggregation.117,118,120 Moreover, the ATP-dependent HSP90 associates with titin’s N2B region97 and also with the N2A element if first methylated by the co-chaperone and methyltransferase Smyd2, a direct N2A ligand (Figure 6A).121,122 This interaction protects I-band integrity.121–124 The protective roles of HSPs on titin may include the prevention of pathological stiffening;117 however, their stabilizing function could also impede the elastic properties of titin and contribute to increased stiffness.123
Figure 6.

Titin-PQC pathways. (A) Interaction of different regions in cardiac titin (N2BA isoform) with components of the PQC machinery: UPS, ubiquitin-proteasome system (purple), proteases (blue), chaperones (green), and autophagy-lysosomal pathways (yellow). (B) Heart disease-related changes in PQC pathways, effects on titin, and suggestions for therapeutic approaches that could be taken to improve titin properties but are still speculative. CRYAB, alpha-B-crystallin; HSP27, heat shock protein 27; HSP90, heat shock protein 90; Ig, immunoglobulin-like domain; Mdm2, mouse-double-minute 2 homolog; MMP2, matrix metalloproteinase-2; MuRF1/2, muscle RING-finger protein-1/2; Nbr1, neighbor-of-BRCA1-gene-1; PEVK, segment rich in proline (P), glutamic acid (E), valine (V), and lysine (K) residues; Smyd2, SET and MYND domain containing protein 2; SQSTM1/p62, sequestosome 1; T-CAP, telethonin; Ub, ubiquitin.
If chaperones fail to protect the protein, it then becomes marked for degradation and turnover.125 For titin turnover to occur, this large protein likely needs to be (partially) released from its binding partners within the sarcomere.34 Proteases, such as calpain-1 and matrix-metalloproteinase (MMP)-2 can bind to the proximal I-band and Z-disc or M-band regions of titin (Figure 6A) and aid in the pre-digestion of titin.126–128 Under ischaemic conditions or in the presence of anthracyclines used in cancer treatment (e.g. doxorubicin), there is an increase in the expression and activity of these proteases leading to increased titin breakdown and cardiac remodelling (Figure 6B).129–131 Protection from titin degradation by these proteases would be possible by using protease inhibitors, as is known from in vitro work.
Protease pre-digestion of titin is thought to further expose binding sites for targeted degradation. Two interlinked pathways, the ubiquitin-proteasome (UPS) and autophagy-lysosomal systems, regulate the subsequent degradation steps.132 Through the UPS, damaged or aged titin molecules can become ubiquitinated by ubiquitin E3 ligase(s) and thus marked for proteasomal degradation. The E3 ligase mouse-double-minute 2 homolog interacts with the titin-capping protein telethonin,133 but it is unknown whether it ubiquitinates titin. Other E3 ligases, the muscle ring-finger proteins (MuRF)-1 and -2, bind in the titin A-/M-band transition zone and to the TK (Figure 6A).87,134–138 They preferentially ubiquitinate A-band proteins, including the TK.137,138 In diseased states, protective effects on sarcomere proteins (including titin?) and contractile improvements have been observed with MuRF1-interfering small molecules as a potential therapeutic approach.139 It is likely that one or more other, not yet identified, E3 ligase ubiquitinate titin. The TK is a well-established hub for protein–protein interactions as it binds the Nbr1/SQSTM1(p62) complex (Figure 6A),140 which acts as an autophagy receptor for ubiquitinated proteins.141 Autophagosome activity reduces with age; however, the role of autophagy in heart disease is incompletely understood:142 both increased and reduced activities have been reported (Figure 6B). A more common finding is that the activity of the UPS is reduced in HF and cardiomyopathy, resulting in the accumulation of ubiquitinated but not degraded proteins.143 These alterations also include titin (Figure 6B).4,144,145 In end-stage failing human DCM hearts due to a TTN-truncating variant (TTNtv), increased ubiquitination of wt-titin was found, whereas truncated (tr-)titin proteins were barely ubiquitinated and stably expressed (Figure 6B).4 Truncated titin accumulated in cytoplasmic aggregates.4 Thus, titin-degradation pathways appeared to be deregulated in TTNtv hearts. The changes observed in TTNtv-DCM patient hearts also included reduced MuRF1 expression, whereas autophagy was not impaired or even activated.4 Taken together, despite recent advances, many details of the titin turnover and degradation processes are still poorly understood.
5.1 PQC as a therapeutic point of influence
Several approaches have been made to target the PQC machinery therapeutically in cardiac diseases, which may also work for disorders related to titin dysfunction. Chaperone induction in mice has been shown to improve cardiomyopathy associated with muscular dystrophy146 and perhaps can also be employed to enhance titin protection against ischaemia or DCM (Figure 6B).117 Moreover, autophagy activators, such as spermidine, have been shown to be protective against age-related cardiovascular disease, including animal models of diastolic dysfunction.147 Further, the inhibition of MMP-2 or calpain proteases might be advantageous as a prophylactic therapy during cancer treatment, e.g. to reduce doxorubicin-induced cardiotoxicity (Figure 6B). Given the dysfunction of the UPS in several forms of heart disease, modulators of the UPS may show promise as a potential treatment, as demonstrated for UPS inhibitors.148 However, the toxicity of proteasome inhibitors may limit their therapeutic value.143 Interestingly, in hiPSC-CMs with a TTNtv, UPS-inhibition raised wt-titin-protein expression (as well as tr-titin-protein content) and boosted contractility.4 Increased wt-titin levels may be beneficial as they promote the formation of sarcomeres.4,19 At this stage, caution needs to be taken with any speculations on the therapeutic benefit of protease or UPS inhibitors, due to their unspecific nature and our limited understanding of their complex cellular interactions.
5.2 PQC and pathomechanisms of TTN-truncation cardiomyopathy
Importantly, deregulated PQC of titin is central to the pathomechanisms of TTN-truncation cardiomyopathy.4 Earlier, groundbreaking work established a titin truncation as the most frequent genetic cause of human DCM: 15–25% of most DCM patient cohorts studied carry a heterozygous TTNtv—by far the largest share among all cardiomyopathy gene variants known.3,149,150 Similarly, a TTNtv is the most common genetic predisposition in other types of inherited cardiac disorders, including restrictive, non-compaction, and peripartum cardiomyopathy, but also in acquired cardiomyopathies, such as those induced by alcohol abuse or cancer treatment.151 Heterozygous TTNtv can occur anywhere along titin; however, the prevalence of having a TTN truncation and the odds of getting DCM from a TTNtv vary depending on the location of the pathogenic variant (Figure 7A and B). The odds ratio is highest if the truncation is in the A-band segment (prevalence in DCM 10.74% vs. control 0.24%, odds ratio 49.8), followed by truncations in constitutive exons of I-band TTN (Figure 7B).3,149,150 In contrast, the odds ratio is lowest for central I-band titin (prevalence in DCM 0.24% vs. control 0.17%, odds ratio 1.5).150 This is a consequence of the extensive alternative splicing of I-band TTN, where exon usage is low for all regions not expressed in the N2B titin isoform (Figure 7A). Thus, while TTNtv are found in 0.5–3% of the healthy (control) population, many of these variants occur in I-band titin exons with low percentage spliced-in (PSI) and do not cause DCM (Figure 7B).3,149,150
Figure 7.

Pathomechanisms observed in human cardiomyopathy due to a TTNtv. (A) Titin domain architecture (according to human titin meta-transcript) and TTN exon usage in adult human hearts.150 PSI above 90% indicates (nearly) constitutive expression, lower PSI values are due to differential splicing. (B) Location of heterozygous TTNtv detected by next-generation sequencing in the general population and in DCM patients, and odds ratio indicating the chance to get DCM if the TTNtv is in a specific titin region (marked by red boxes).150 (C) Recently identified pathomechanisms of human TTNtv-DCM.4 (D) Specific treatment strategies suggested.4
Previous suggestions about the possible pathomechanisms of TTNtv-DCM included sarcomere insufficiency,152 modest nonsense-mediated decay of TTNtv-mRNA,150 or translational deregulation,153 whereas a poison-peptide (dominant-negative) mechanism was considered unlikely because truncated titin proteins were detected only in hiPSC-CMs152 but not in adult heart tissue.149–151 However, recent studies have unequivocally shown that tr-titin proteins are stably expressed in human end-stage failing, adult TTNtv-DCM hearts.4,154 Their concentration reaches up to 50% of the total titin-protein pool but is highly variable. Strikingly, the higher the tr-titin-protein content of a TTNtv-heart, the younger the (adult) patients at the time of transplantation, suggesting that these proteins are disease-relevant.4 The tr-titin proteins (unlike the wt-titin proteins) are not built into the sarcomeres at meaningful amounts but are sequestered in aggregates.4 Thus, a poison-peptide mechanism is likely part of the pathomechanisms of TTNtv-DCM (Figure 7C).
Apart from a dominant-negative mechanism, TTNtv-DCM patient hearts contain less wt-titin protein than DCM hearts without TTNtv or non-failing (donor) hearts, which demonstrates titin haploinsufficiency.4,154 However, nonsense-mediated decay of TTNtv-mRNA is not a prominent feature of TTNtv-DCM patient hearts (Figure 7C).4,150,153 With wt-titin being lost, the CMs of TTNtv-DCM hearts also have fewer sarcomeres per unit area than non-TTNtv-DCM hearts,4 confirming sarcomere insufficiency152 and explaining chronic contractile deficiency.4,152
Recent findings4,154 underscored that TTNtv-DCM hearts have a problem with intracellular PQC (Figure 7C; cf. section above). With increasing patient age, the UPS may be overwhelmed by the large amounts of tr-titin protein produced and partially shut down, whereas E3-ubiquitin ligases, such as MuRF1 become down-regulated.4 Conversely, the intracellular aggregate formation may promote autophagy. Interestingly, disease modelling in hiPSC-CMs with a TTNtv demonstrated that UPS-inhibition, but not autophagy-modulation, increased both tr- and wt-titin-protein content, with larger effects on tr-titin.4 Reversal of the titin haploinsufficiency by UPS-inhibition improved TTNtv-hiPSC-CMs contractility, despite the raised tr-titin-protein content. If the TTNtv was repaired by CRISPR/Cas9 gene-editing, the titin haploinsufficiency was corrected, tr-titin proteins were absent, and contractility was fully recovered.4 These findings can be exploited for new therapies of TTNtv-related cardiomyopathies (Figure 7D).
In summary, several key pathomechanisms come together in TTNtv-patient hearts, providing a rationale for phenotypic diversity. Disease mechanisms include titin haploinsufficiency as a life-long condition and truncated titin-protein enrichment with aggregate formation, as well as aberrant PQC, presumably both as additional late-onset pathomechanisms. Having a TTNtv may represent a major risk factor to get DCM later in life, especially when other stressors hit.
6. Conclusion and outlook
New technologies emerging over the last decade (e.g. next-generation sequencing, 2D and 3D culture of hiPSC-CMs, gene-editing, or ‘omics’) have allowed amazing progress to made also in the titin field. This has greatly improved our understanding of the role titin plays in HF and the disease mechanisms of TTN-truncation cardiomyopathy. However, much work still lies ahead of us. For example, TTN pathogenic variants include not only truncations but also missense variants whose pathophysiological relevance is only slowly evolving.155 Missense and truncation variants can occur together in the same patient, typically amplifying the pathophenotype.155 Moreover, the role of titin–isoform switch and titin PTMs in HF (notably HFpEF) remains fuzzy, as it is not yet clear whether these changes are a cause or consequence of the disease, and whether targeting these titin properties specifically (if possible) could improve the syndrome. The ageing population is also highlighting the importance of proteostasis regulated by PQC, and we are only just starting to appreciate how deregulated PQC in ageing and disease may affect titin. Similarly, the discovery that circular RNA of titin can also play a regulatory role in cellular function potentially opens the door to a whole new relevance of titin. Finally, this giant protein may still carry more molecular mysteries awaiting discovery. Perhaps we will know soon, how titin processes a stretch signal to boost active myocardial contraction and support increased cardiac output.
Acknowledgements
We thank the past and present members of the Linke lab for their research contributions to the titin topic.
Contributor Information
Christine M Loescher, Department of Cardiology I, Coronary, Peripheral Vascular Disease and Heart Failure, University Hospital Münster, Albert-Schweitzer-Campus 1, Building A1, 48149 Münster, Germany.
Anastasia J Hobbach, Department of Cardiology I, Coronary, Peripheral Vascular Disease and Heart Failure, University Hospital Münster, Münster, Germany.
Wolfgang A Linke, Department of Cardiology I, Coronary, Peripheral Vascular Disease and Heart Failure, University Hospital Münster, Albert-Schweitzer-Campus 1, Building A1, 48149 Münster, Germany.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB1002 TPA08) and Interdisziplinären Zentrum für Klinische Forschung Münster (Li1/029/20) to W.A.L.
Data availability
This review article does not contain new original data.
References
- 1. Münch J, Abdelilah-Seyfried S. Sensing and responding of cardiomyocytes to changes of tissue stiffness in the diseased heart. Front Cell Dev Biol 2021;9:642840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res 2008;77:637–648. [DOI] [PubMed] [Google Scholar]
- 3. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fomin A, Gärtner A, Cyganek L, Tiburcy M, Tuleta I, Wellers L, Folsche L, Hobbach AJ, von Frieling-Salewsky M, Unger A, Hucke A, Koser F, Kassner A, Sielemann K, Streckfuß-Bömeke K, Hasenfuss G, Goedel A, Laugwitz KL, Moretti A, Gummert JF, dos Remedios CG, Reinecke H, Knöll R, van Heesch S, Hubner N, Zimmermann WH, Milting H, Linke WA. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci Transl Med 2021, in press. [DOI] [PubMed] [Google Scholar]
- 5. Granzier HL, Hutchinson KR, Tonino P, Methawasin M, Li FW, Slater RE, Bull MM, Saripalli C, Pappas CT, Gregorio CC, Smith JE. Deleting titin's I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci USA 2014;111:14589–14594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Linke WA. Titin gene and protein functions in passive and active muscle. Annu Rev Physiol 2018;10:389–411. [DOI] [PubMed] [Google Scholar]
- 7. Labeit S, Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 1995;270:293–296. [DOI] [PubMed] [Google Scholar]
- 8. Tonino P, Kiss B, Strom J, Methawasin M, Smith JE III, Kolb J, Labeit S, Granzier H. The giant protein titin regulates the length of the striated muscle thick filament. Nat Commun 2017;8:1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gautel M, Djinović-Carugo K. The sarcomeric cytoskeleton: from molecules to motion. J Exp Biol 2016;219:135–145. [DOI] [PubMed] [Google Scholar]
- 10. Lange S, Pinotsis N, Agarkova I, Ehler E. The M-band: the underestimated part of the sarcomere. Biochim Biophys Acta Mol Cell Res 2020;1867:118440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Linke WA, Ivemeyer M, Olivieri N, Kolmerer B, Rüegg JC, Labeit S. Towards a molecular understanding of the elasticity of titin. J Mol Biol 1996;261:62–71. [DOI] [PubMed] [Google Scholar]
- 12. Linke WA, Rudy DE, Centner T, Gautel M, Witt C, Labeit S, Gregorio CC. I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J Cell Biol 1999;146:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC, Granzier H, Labeit S. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res 2000;86:1114–1121. [DOI] [PubMed] [Google Scholar]
- 14. Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res 2004;94:284–295. [DOI] [PubMed] [Google Scholar]
- 15. Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 2001;89:1065–1072. [DOI] [PubMed] [Google Scholar]
- 16. Zou J, Tran D, Baalbaki M, Tang LF, Poon A, Pelonero A, Titus EW, Yuan C, Shi C, Patchava S, Halper E, Garg J, Movsesyan I, Yin C, Wu R, Wilsbacher L, Liu J, Hager RL, Coughlin SR, Jinek M, Pullinger CR, Kane JP, Hart DO, Kwok P-Y, Deo RC. An internal promoter underlies the difference in disease severity between N- and C-terminal truncation mutations of Titin in zebrafish. Elife 2015;4:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H, Linke WA, Oberhauser AF, Carrion-Vazquez M, Kerkvliet JG, Lu H, Marszalek PE, Fernandez JM. Reverse engineering of the giant muscle protein titin. Nature 2002;418:998–1002. [DOI] [PubMed] [Google Scholar]
- 18. Zaunbrecher RJ, Abel AN, Beussman K, Leonard A, von Frieling-Salewsky M, Fields PA, Pabon L, Reinecke H, Yang X, Macadangdang J, Kim D-H, Linke WA, Sniadecki NJ, Regnier M, Murry CE. Cronos titin is expressed in human cardiomyocytesand necessary for normal sarcomere function. Circulation 2019;140:1647–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swist S, Unger A, Li Y, Vöge A, von Frieling-Salewsky M, Asa S, Cacciani N, Braun T, Larsson L, Linke WA. Maintenance of sarcomeric integrity in adult muscle cells crucially depends on Z-disc anchored titin. Nat Commun 2020;11:4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, Dauksaite V, Vakeel P, Klaassen S, Gerull B, Thierfelder L, Regitz-Zagrosek V, Hacker TA, Saupe KW, Dec GW, Ellinor PT, MacRae CA, Spallek B, Fischer R, Perrot A, Özcelik C, Saar K, Hubner N, Gotthardt M. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 2012;18:766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li S, Guo W, Dewey CN, Greaser ML. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res 2013;41:2659–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, Adami E, Rintisch C, Dauksaite V, Radke MH, Selbach M, Barton PJR, Cook SA, Rajewsky N, Gotthardt M, Landthaler M, Hubner N. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest 2014;124:3419–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Methawasin M, Strom JG, Slater RE, Fernandez V, Saripalli C, Granzier H. Experimentally increasing the compliance of titin through RNA binding Motif-20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fraction. Circulation 2016;134:1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tijsen AJ, Cócera Ortega L, Reckman YJ, Zhang X, van der Made I, Aufiero S, Li J, Kamps SC, van den Bout A, Devalla HD, van Spaendonck-Zwarts KY, Engelhardt S, Gepstein L, Ware JS, Pinto YM. Titin circular RNAs create a back-splice motif essential for SRSF10 splicing. Circulation 2021;143:1502–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Streckfuss-Bömeke K, Tiburcy M, Fomin A, Luo X, Li W, Fischer C, Özcelik C, Perrot A, Sossalla S, Haas J, Vidal RO, Rebs S, Khadjeh S, Meder B, Bonn S, Linke WA, Zimmermann WH, Hasenfuss G, Guan K. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 2017;113:9–21. [DOI] [PubMed] [Google Scholar]
- 26. Beqqali A, Bollen IA, Rasmussen TB, van den Hoogenhof MM, van Deutekom HW, Schafer S, Haas J, Meder B, Sørensen KE, van Oort RJ, Mogensen J, Hubner N, Creemers EE, van der Velden J, Pinto YM. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc Res 2016;112:452–463. [DOI] [PubMed] [Google Scholar]
- 27. van den Hoogenhof MMG, Beqqali A, Amin AS, van der Made I, Aufiero S, Khan MAF, Schumacher CA, Jansweijer JA, van Spaendonck-Zwarts KY, Remme CA, Backs J, Verkerk AO, Baartscheer A, Pinto YM, Creemers EE. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 2018;138:1330–1342. [DOI] [PubMed] [Google Scholar]
- 28. Zhu C, Yin Z, Ren J, McCormick RJ, Ford SP, Guo W. RBM20 is an essential factor for thyroid hormone-regulated titin isoform transition. J Mol Cell Biol 2015;7:88–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu C, Yin Z, Tan B, Guo W. Insulin regulates titin pre-mRNA splicing through the PI3K-Akt-mTOR kinase axis in a RBM20-dependent manner. Biochim Biophys Acta Mol Basis Dis 2017;1863:2363–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krüger M, Sachse C, Zimmermann WH, Eschenhagen T, Klede S, Linke WA. Thyroid hormone regulates developmental titin isoform transitions via the phosphatidylinositol-3-Kinase/ AKT pathway. Circ Res 2008;102:439–447. [DOI] [PubMed] [Google Scholar]
- 31. Wu Y, Peng J, Campbell KB, Labeit S, Granzier H. Hypothyroidism leads to increased collagen-based stiffness and re-expression of large cardiac titin isoforms with high compliance. J Mol Cell Cardiol 2007;42:186–195. [DOI] [PubMed] [Google Scholar]
- 32. Krüger M, Babicz K, von Frieling-Salewsky M, Linke WA. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J Mol Cell Cardiol 2010;48:910–916. [DOI] [PubMed] [Google Scholar]
- 33. Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA. Titin isoform switch in ischemic human heart disease. Circulation 2002;106:1333–1341. [DOI] [PubMed] [Google Scholar]
- 34. Salcan S, Bongardt S, Barbosa DM, Efimov IR, Rassaf T, Krüger M, Kötter S. Elastic titin properties and protein quality control in the aging heart. Biochim Biophys Acta Mol Cell Res 2020;1867:118532. [DOI] [PubMed] [Google Scholar]
- 35. Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004;110:155–162. [DOI] [PubMed] [Google Scholar]
- 36. Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ, Linke WA. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res 2004;95:708–716. [DOI] [PubMed] [Google Scholar]
- 37. Borbély A, Falcao-Pires I, van Heerebeek L, Hamdani N, Édes I, Gavina C, Leite-Moreira AF, Bronzwaer JGF, Papp Z, van der Velden J, Stienen GJM, Paulus WJ, Hypophosphorylation of the stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res 2009;104:780–786. [DOI] [PubMed] [Google Scholar]
- 38. Gotzmann M, Grabbe S, Schöne D, von Frieling-Salewsky M, Dos Remedios CG, Strauch J, Bechtel M, Dietrich JW, Tannapfel A, Mügge A, Linke WA. Alterations in titin properties and myocardial fibrosis correlate with clinical phenotypes in hemodynamic subgroups of severe aortic stenosis. JACC Basic Transl Sci 2018;3:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Williams L, Howell N, Pagano D, Andreka P, Vertesaljai M, Pecor T, Frenneaux M, Granzier H. Titin isoform expression in aortic stenosis. Clin Sci 2009;117:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Heerebeek L, Borbély A, Niessen HWM, Bronzwaer JGF, van der Velden J, Stienen GJM, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 2006;113:1966–1973. [DOI] [PubMed] [Google Scholar]
- 41. Chaturvedi RR, Herron T, Simmons R, Shore D, Kumar P, Sethia B, Chua F, Vassiliadis E, Kentish JC. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation 2010;121:979–988. [DOI] [PubMed] [Google Scholar]
- 42. Hoskins AC, Jacques A, Bardswell SC, McKenna WJ, Tsang V, dos Remedios CG, Ehler E, Adams K, Jalilzadeh S, Avkiran M, Watkins H, Redwood C, Marston SB, Kentish JC. Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy. J Mol Cell Cardiol 2010;49:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Falcão-Pires I, Hamdani N, Borbély A, Gavina C, Schalkwijk CG, van der Velden J, van Heerebeek L, Stienen GJM, Niessen HWM, Leite-Moreira AF, Paulus WJ. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation 2011;124:1151–1159. [DOI] [PubMed] [Google Scholar]
- 44. Kötter S, Gout L, von Frieling-Salewsky M, Müller AE, Helling S, Marcus K, dos Remedios C, Linke WA, Krüger M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res 2013;99:648–656. [DOI] [PubMed] [Google Scholar]
- 45. Shapiro BP, Lam CS, Patel JB, Mohammed SF, Kruger M, Meyer DM, Linke WA, Redfield MM. Acute and chronic ventricular-arterial coupling in systole and diastole: insights from an elderly hypertensive model. Hypertension 2007;50:503–511. [DOI] [PubMed] [Google Scholar]
- 46. Hamdani N, Bishu KG, von Frieling-Salewsky M, Redfield MM, Linke WA. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res 2013;97:464–471. [DOI] [PubMed] [Google Scholar]
- 47. Zakeri R, Moulay G, Chai Q, Ogut O, Hussain S, Takahama H, Lu T, Wang XL, Linke WA, Lee HC, Redfield MM. Left atrial remodeling and atrioventricular coupling in a canine model of early heart failure with preserved ejection fraction. Circ Heart Fail 2016;9:e003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bull M, Methawasin M, Strom J, Nair P, Hutchinson K, Granzier H. Alternative splicing of titin restores diastolic function in an HFpEF-like genetic murine model (TtnΔIAjxn). Circ Res 2016;119:764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liss M, Radke MH, Eckhard J, Neuenschwander M, Dauksaite V, von Kries J-P, Gotthardt M. Drug discovery with an RBM20 dependent titin splice reporter identifies cardenolides as lead structures to improve cardiac filling. PLoS One 2018;13:e0198492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lennermann D, Backs J, van den Hoogenhof MMG. New insights in RBM20 cardiomyopathy. Curr Heart Fail Rep 2020;17:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cazorla O, Wu Y, Irving TC, Granzier H. Titin-based modulation of calcium sensitivity of active tension in mouse skinned cardiac myocytes. Circ Res 2001;88:1028–1035. [DOI] [PubMed] [Google Scholar]
- 52. Fukuda N, Sasaki D, Ishiwata S, Kurihara S. Length dependence of tension generation in rat skinned cardiac muscle: role of titin in the Frank-Starling mechanism of the heart. Circulation 2001;104:1639–1645. [DOI] [PubMed] [Google Scholar]
- 53. Patel JR, Pleitner JM, Moss RL, Greaser ML. Magnitude of length-dependent changes in contractile properties varies with titin isoform in rat ventricles. Am J Physiol Heart Circ Physiol 2012;302:H697–H708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Methawasin M, Hutchinson KR, Lee EJ, Smith JE III, Saripalli C, Hidalgo CG, Ottenheijm CA, Granzier H. Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 2014;129:1924–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ait-Mou Y, Hsu K, Farman GP, Kumar M, Greaser ML, Irving TC, de Tombe PP. Titin strain contributes to the Frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc Natl Acad Sci USA 2016;113:2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee EJ, Peng J, Radke M, Gotthardt M, Granzier HL. Calcium sensitivity and the Frank-Starling mechanism of the heart are increased in titin N2B region-deficient mice. J Mol Cell Cardiol 2010;49:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Campbell KS. Impact of myocyte strain on cardiac myofilament activation. Pflugers Arch 2011;462:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hanft LM, Greaser ML, McDonald KS. Titin-mediated control of cardiac myofibrillar function. Arch Biochem Biophys 2014;552-553:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sequeira V, van der Velden J. The Frank-Starling Law: a jigsaw of titin proportions. Biophys Rev 2017;9:259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marcucci L, Washio T, Yanagida T. Titin-mediated thick filament activation, through a mechanosensing mechanism, introduces sarcomere-length dependencies in mathematical models of rat trabecula and whole ventricle. Sci Rep 2017;7:5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bullard B, Pastore A. Through thick and thin: dual regulation of insect flight muscle and cardiac muscle compared. J Muscle Res Cell Motil 2019;40:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boldt K, Han SW, Joumaa V, Herzog W. Residual and passive force enhancement in skinned cardiac fibre bundles. J Biomech 2020;109:109953. [DOI] [PubMed] [Google Scholar]
- 63. Kawai M, Jin JP. Mechanisms of Frank-Starling law of the heart and stretch activation in striated muscles may have a common molecular origin. J Muscle Res Cell Motil 2021;42:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Park-Holohan SJ, Brunello E, Kampourakis T, Rees M, Irving M, Fusi L. Stress-dependent activation of myosin in the heart requires thin filament activation and thick filament mechanosensing. Proc Natl Acad Sci USA 2021;118:e2023706118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Eckels EC, Haldar S, Tapia-Rojo R, Rivas-Pardo JA, Fernández JM. The mechanical power of titin folding. Cell Rep 2019;27:1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rivas-Pardo JA, Eckels EC, Popa I, Kosuri P, Linke WA, Fernández JM. Work done by titin protein folding assists muscle contraction. Cell Rep 2016;14:1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eckels EC, Tapia-Rojo R, Rivas-Pardo JA, Fernández JM. The work of titin protein folding as a major driver in muscle contraction. Annu Rev Physiol 2018;80:327–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 2019;20:156–174. [DOI] [PubMed] [Google Scholar]
- 69. Romanick SS, Ulrich C, Schlauch K, Hostler A, Payne J, Woolsey R, Quilici D, Feng Y, Ferguson BS. Obesity-mediated regulation of cardiac protein acetylation: parallel analysis of total and acetylated proteins via TMT-tagged mass spectrometry. Biosci Rep 2018;38:BSR20180721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Abdellatif M, Trummer-Herbst V, Koser F, Durand S, Adão R, Vasques-Nóvoa F, Freundt JK, Voglhuber J, Pricolo MR, Kasa M, Türk C, Aprahamian F, Herrero-Galán E, Hofer SJ, Pendl T, Rech L, Kargl J, Anto-Michel N, Ljubojevic-Holzer S, Schipke J, Brandenberger C, Auer M, Schreiber R, Koyani CN, Heinemann A, Zirlik A, Schmidt A, von Lewinski D, Scherr D, Rainer PP, von Maltzahn J, Mühlfeld C, Krüger M, Frank S, Madeo F, Eisenberg T, Prokesch A, Leite-Moreira AF, Lourenço AP, Alegre-Cebollada J, Kiechl S, Linke WA, Kroemer G, Sedej S. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci Transl Med 2021;13:eabd7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, Wu J, Jeong MY, Lin YH, Borghetti G, Baker ST, Zhao H, Pfleger J, Blass S, Rainer PP, von Lewinski D, Bugger H, Mohsin S, Graier WF, Zirlik A, McKinsey TA, Birner-Gruenberger R, Wolfson MR, Houser SR. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci Transl Med 2020;12:eaay7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, Monzani V, Saripalli C, Mascagni P, Reece TB, Ambardekar AV, Granzier HL, Dinarello CA, McKinsey TA. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med 2018;10:eaao0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shaw BF, Schneider GF, Bilgiçer B, Kaufman GK, Neveu JM, Lane WS, Whitelegge JP, Whitesides GM. Lysine acetylation can generate highly charged enzymes with increased resistance toward irreversible inactivation. Protein Sci 2008;17:1446–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sharifi-Rad M, Anil Kumar NV, Zucca P, Varoni EM, Dini L, Panzarini E, Rajkovic J, Tsouh Fokou PV, Azzini E, Peluso I, Prakash Mishra A, Nigam M, El Rayess Y, Beyrouthy ME, Polito L, Iriti M, Martins N, Martorell M, Docea AO, Setzer WN, Calina D, Cho WC, Sharifi-Rad J. Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Front Physiol 2020;11:694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Beckendorf L, Linke WA. Emerging importance of oxidative stress in regulating striated muscle elasticity. J Muscle Res Cell Motil 2015;36:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Loescher CM, Breitkreuz M, Li Y, Nickel A, Unger A, Dietl A, Schmidt A, Mohamed BA, Kötter S, Schmitt JP, Krüger M, Krüger M, Toischer K, Maack C, Leichert LI, Hamdani N, Linke WA. Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx). Proc Natl Acad Sci USA 2020;117:24545–24556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Warren CM, Solaro RJ, Linke WA, Fernández JM. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 2014;156:1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Giganti D, Yan K, Badilla CL, Fernandez JM, Alegre‐Cebollada J. Disulfide isomerization reactions in titin immunoglobulin domains enable a mode of protein elasticity. Nat Commun 2018;9:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Beedle AE, Lynham S, Garcia‐Manyes S. Protein S‐sulfenylation is a fleeting molecular switch that regulates non‐enzymatic oxidative folding. Nat Commun 2016;7:12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Grutzner A, Garcia‐Manyes S, Kotter S, Badilla CL, Fernandez JM, Linke WA. Modulation of titin‐based stiffness by disulfide bonding in the cardiac titin N2‐B unique sequence. Biophys J 2009;97:825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Leake MC, Grutzner A, Krüger M, Linke WA. Mechanical properties of cardiac titin's N2B‐region by single‐molecule atomic force spectroscopy. J Struct Biol 2006;155:263–272. [DOI] [PubMed] [Google Scholar]
- 82. Koser F, Loescher C, Linke WA. Posttranslational modifications of titin from cardiac muscle: how, where, and what for? Febs J 2019;286:2240–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 2015;43:D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gautel M, Goulding D, Bullard B, Weber K, Fürst DO. The central Z-disk region of titin is assembled from a novel repeat in variable copy numbers. J Cell Sci 1996;109:2747–2754. [DOI] [PubMed] [Google Scholar]
- 85. Gautel M, Leonard K, Labeit S. Phosphorylation of KSP motifs in the C-terminal region of titin in differentiating myoblasts. Embo J 1993;12:3827–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mayans O, van der Ven PF, Wilm M, Mues A, Young P, Fürst DO, Wilmanns M, Gautel M. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature 1998;395:863–869. [DOI] [PubMed] [Google Scholar]
- 87. Bogomolovas J, Gasch A, Simkovic F, Rigden DJ, Labeit S, Mayans O. Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open Biol 2014;4:140041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Krüger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, Dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res 2009;104:87–94. [DOI] [PubMed] [Google Scholar]
- 89. Perkin J, Slater R, Del FG, Lanzicher T, Hidalgo C, Anderson B, Smith JE III, Sbaizero O, Labeit S, Granzier H. Phosphorylating titin's cardiac N2B element by ERK2 or CaMKIIdelta lowers the single molecule and cardiac muscle force. Biophys J 2015;109:2592–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res 2009;105:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Anderson BR, Bogomolovas J, Labeit S, Granzier H. The effects of PKCα phosphorylation on the extensibility of titin’s PEVK element. J Struct Biol 2010;170:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Michel K, Herwig M, Werner F, Špiranec Spes K, Abeßer M, Schuh K, Dabral S, Mügge A, Baba HA, Skryabin BV, Hamdani N, Kuhn M. C-type natriuretic peptide moderates titin-based cardiomyocyte stiffness. JCI Insight 2020;5:e139910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Herwig M, Kolijn D, Lódi M, Hölper S, Kovács Á, Papp Z, Jaquet K, Haldenwang P, Dos Remedios C, Reusch PH, Mügge A, Krüger M, Fielitz J, Linke WA, Hamdani N. Modulation of titin-based stiffness in hypertrophic cardiomyopathy via protein kinase D. Front Physiol 2020;11:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Raskin A, Lange S, Banares K, Lyon RC, Zieseniss A, Lee LK, Yamazaki KG, Granzier HL, Gregorio CC, McCulloch AD, Omens JH, Sheikh F. A novel mechanism involving four‐and‐a‐half LIM domain protein‐1 and extracellular signal‐regulated kinase‐2 regulates titin phosphorylation and mechanics. J Biol Chem 2012;287:29273–29284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Krüger M, Backs J, Linke WA. Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res 2013;112:664–674. [DOI] [PubMed] [Google Scholar]
- 96. Hidalgo CG, Chung CS, Saripalli C, Methawasin M, Hutchinson KR, Tsaprailis G, Labeit S, Mattiazzi A, Granzier HL. The multifunctional Ca(2+)/calmodulin-dependent protein kinase II delta (CaMKIIdelta) phosphorylates cardiac titin's spring elements. J Mol Cell Cardiol 2013;54:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Krysiak J, Unger A, Beckendorf L, Hamdani N, von Frieling-Salewsky M, Redfield MM, dos Remedios CG, Sheikh F, Gergs U, Boknik P, Linke WA. Protein phosphatase 5 regulates titin phosphorylation and function at a sarcomere-associated mechanosensor complex in cardiomyocytes. Nat Commun 2018;9:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yang J, Roe SM, Cliff MJ, Williams MA, Ladbury JE, Cohen PT, Barford D. Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. Embo J 2005;24:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rain S, Bos DS, Handoko ML, Westerhof N, Stienen G, Ottenheijm C, Goebel M, Dorfmuller P, Guignabert C, Humbert M, Bogaard H, Dos Remedios C, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, van der Velden J, de Man FS. Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J Am Heart Assoc 2014;3:e000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hopf A-E, Andresen C, Kötter S, Isić M, Ulrich K, Sahin S, Bongardt S, Röll W, Drove F, Scheerer N, Vandekerckhove L, De Keulenaer GW, Hamdani N, Linke WA, Krüger M. Diabetes-induced cardiomyocyte passive stiffening is caused by impaired insulin-dependent titin modification and can be modulated by neuregulin-1. Circ Res 2018;123:342–355. [DOI] [PubMed] [Google Scholar]
- 101. Slater RE, Strom JG, Methawasin M, Liss M, Gotthardt M, Sweitzer N, Granzier HL. Metformin improves diastolic function in an HFpEF-like mouse model by increasing titin compliance. J Gen Physiol 2019;151:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Blanton RM. cGMP Signaling and modulation in heart failure. J Cardiovasc Pharmacol 2020;75:385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bishu K, Hamdani N, Mohammed SF, Kruger M, Ohtani T, Ogut O, Brozovich FV, Burnett JC, Linke WA, Redfield MM. Sildenafil and B-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation 2011;124:2882–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, RELAX Trial . Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013;309:1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015;519:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Methawasin M, Strom J, Borkowski T, Hourani Z, Runyan R, Smith JE III, Granzier H. Phosphodiesterase 9a inhibition in mouse models of diastolic dysfunction. Circ Heart Fail 2020;13:e006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kolijn D, Kovács Á, Herwig M, Lódi M, Sieme M, Alhaj A, Sandner P, Papp Z, Reusch PH, Haldenwang P, Falcao-Pires I, Linke WA, Jaquet K, van Linthout S, Mügge A, Tschöpe C, Hamdani N. Enhanced cardiomyocyte function in hypertensive rats with diastolic dysfunction and human heart failure patients after acute treatment with soluble guanylyl cyclase (sGC) activator. Front Physiol 2020;11:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Alogna A, Schwarzl M, Manninger M, Hamdani N, Zirngast B, Kloth B, Steendijk P, Verderber J, Zweiker D, Westermann D, Blankenberg S, Maechler H, Tschöpe C, Linke WA, Marsche G, Pieske BM, Post H. Acute stimulation of the soluble guanylate cyclase does not impact on left ventricular capacitance in normal and hypertrophied porcine hearts in vivo. Am J Physiol Heart Circ Physiol 2018;315:H669–H680. [DOI] [PubMed] [Google Scholar]
- 109. Armstrong PW, Lam CSP, Anstrom KJ, Ezekowitz J, Hernandez AF, O'Connor CM, Pieske B, Ponikowski P, Shah SJ, Solomon SD, Voors AA, She L, Vlajnic V, Carvalho F, Bamber L, Blaustein RO, Roessig L, Butler J, VITALITY-HFpEF Study Group . Effect of vericiguat vs placebo on quality of life in patients with heart failure and preserved ejection fraction: the VITALITY-HFpEF randomized clinical trial. JAMA 2020;324:1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Pieske B, Maggioni AP, Lam CSP, Pieske-Kraigher E, Filippatos G, Butler J, Ponikowski P, Shah SJ, Solomon SD, Scalise A-V, Mueller K, Roessig L, Gheorghiade M. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur Heart J 2017;38:1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Isaacs WB, Kim IS, Struve A, Fulton AB. Biosynthesis of titin in cultured skeletal muscle cells. J Cell Biol 1989;109:2189–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fornasiero EF, Mandad S, Wildhagen H, Alevra M, Rammner B, Keihani S, Opazo F, Urban I, Ischebeck T, Sakib MS, Fard MK, Kirli K, Centeno TP, Vidal RO, Rahman RU, Benito E, Fischer A, Dennerlein S, Rehling P, Feussner I, Bonn S, Simons M, Urlaub H, Rizzoli SO. Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat Commun 2018;9:4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hnia K, Clausen T, Moog-Lutz C. Shaping striated muscles with ubiquitin proteasome system in health and disease. Trends Mol Med 2019;25:760–774. [DOI] [PubMed] [Google Scholar]
- 114. Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 2008;47:7001–7011. [DOI] [PubMed] [Google Scholar]
- 115. Dimauro I, Antonioni A, Mercatelli N, Caporossi D. The role of αB-crystallin in skeletal and cardiac muscle tissues. Cell Stress Chaperones 2018;23:491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mymrikov EV, Seit-Nebi AS, Gusev NB. Large potentials of small heat shock proteins. Physiol Rev 2011;91:1123–1159. [DOI] [PubMed] [Google Scholar]
- 117. Kötter S, Unger A, Hamdani N, Lang P, Vorgerd M, Nagel-Steger L, Linke WA. Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J Cell Biol 2014;204:187–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, Ding L, Labeit S, Horwitz J, Leonard KR, Linke WA. Association of the chaperone αB-crystallin with titin in heart muscle. J Biol Chem 2004;279:7917–7924. [DOI] [PubMed] [Google Scholar]
- 119. Golenhofen N, Arbeiter A, Koob R, Drenckhahn D. Ischemia-induced association of the stress protein alpha B-crystallin with I-band portion of cardiac titin. J Mol Cell Cardiol 2002;34:309–319. [DOI] [PubMed] [Google Scholar]
- 120. Zhu Y, Bogomolovas J, Labeit S, Granzier H. Single molecule force spectroscopy of the cardiac titin N2B element: effects of the molecular chaperone αB-crystallin with disease-causing mutations. J Biol Chem 2009;284:13914–13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Donlin LT, Andresen C, Just S, Rudensky E, Pappas CT, Kruger M, Jacobs EY, Unger A, Zieseniss A, Dobenecker M-W, Voelkel T, Chait BT, Gregorio CC, Rottbauer W, Tarakhovsky A, Linke WA. Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev 2012;26:114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Voelkel T, Andresen C, Unger A, Just S, Rottbauer W, Linke WA. Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim Biophys Acta Mol Cell Res 2013;1833:812–822. [DOI] [PubMed] [Google Scholar]
- 123. Unger A, Beckendorf L, Böhme P, Kley R, von Frieling-Salewsky M, Lochmüller H, Schröder R, Fürst DO, Vorgerd M, Linke WA. Translocation of molecular chaperones to the titin springs is common in skeletal myopathy patients and affects sarcomere function. Acta Neuropathol Commun 2017;5:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Munkanatta Godage DNP, VanHecke GC, Samarasinghe KTG, Feng HZ, Hiske M, Holcomb J, Yang Z, Jin JP, Chung CS, Ahn YH. SMYD2 glutathionylation contributes to degradation of sarcomeric proteins. Nat Commun 2018;9:4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bozaykut P, Ozer NK, Karademir B. Regulation of protein turnover by heat shock proteins. Free Radic Biol Med 2014;77:195–209. [DOI] [PubMed] [Google Scholar]
- 126. Raynaud F, Fernandez E, Coulis G, Aubry L, Vignon X, Bleimling N, Gautel M, Benyamin Y, Ouali A. Calpain 1-titin interactions concentrate calpain 1 in the Z-band edges and in the N2-line region within the skeletal myofibril. Febs J 2005;272:2578–2590. [DOI] [PubMed] [Google Scholar]
- 127. Coulis G, Becila S, Herrera-Mendez CH, Sentandreu MA, Raynaud F, Richard I, Benyamin Y, Ouali A. Calpain 1 binding capacities of the N1-line region of titin are significantly enhanced by physiological concentrations of calcium. Biochemistry 2008;47:9174–9183. [DOI] [PubMed] [Google Scholar]
- 128. Ali MA, Cho WJ, Hudson B, Kassiri Z, Granzier H, Schulz R. Titin is a target of matrix metalloproteinase-2: implications in myocardial ischemia/reperfusion injury. Circulation 2010;122:2039–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Neuhof C, Neuhof H. Calpain system and its involvement in myocardial ischemia and reperfusion injury. World J Cardiol 2014;6:638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Chan BYH, Roczkowsky A, Cho WJ, Poirier M, Sergi C, Keschrumrus V, Churko JM, Granzier H, Schulz R. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovasc Res 2021;117:188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R, Sawyer DB. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem 2004;279:8290–8299. [DOI] [PubMed] [Google Scholar]
- 132. Kocaturk NM, Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front Cell Dev Biol 2018;6:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Tian L-F, Li H-Y, Jin B-F, Pan X, Man J-H, Zhang P-J, Li W-H, Liang B, Liu H, Zhao J, Gong W-L, Zhou T, Zhang X-M. MDM2 interacts with and downregulates a sarcomeric protein. TCAP. Biochem Biophys Res Commun 2006;345:355–361. [DOI] [PubMed] [Google Scholar]
- 134. Pizon V, Iakovenko A, van der Ven PF, Kelly R, Fatu C, Fürst DO, Karsenti E, Gautel M. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J Cell Sci 2002;115:4469–4482. [DOI] [PubMed] [Google Scholar]
- 135. Gregorio CC, Perry CN, McElhinny AS. Functional properties of the titin/connectin-associated proteins, the muscle-specific RING-finger proteins (MURFs), in striated muscle. J Muscle Res Cell Motil 2005;26:389–400. [DOI] [PubMed] [Google Scholar]
- 136. Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 2001;306:717–726. [DOI] [PubMed] [Google Scholar]
- 137. Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J Mol Biol 2005;350:713–722. [DOI] [PubMed] [Google Scholar]
- 138. Bogomolovas J, Fleming JR, Franke B, Manso B, Simon B, Gasch A, Markovic M, Brunner T, Knöll R, Chen J, Labeit S, Scheffner M, Peter C, Mayans O. Titin kinase ubiquitination aligns autophagy receptors with mechanical signals in the sarcomere. EMBO Rep 2021;22:e48018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Adams V, Bowen TS, Werner S, Barthel P, Amberger C, Konzer A, Graumann J, Sehr P, Lewis J, Provaznik J, Benes V, Büttner P, Gasch A, Mangner N, Witt CC, Labeit D, Linke A, Labeit S. Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J Cachexia Sarcopenia Muscle 2019;10:1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, Gunnarsson LG, Hughes SM, Marchand S, Sejersen T, Richard I, Edström L, Ehler E, Udd B, Gautel M. The kinase domain of titin controls muscle gene expression and protein turnover. Science 2005;308:1599–1603. [DOI] [PubMed] [Google Scholar]
- 141. Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. Febs J 2015;282:4672–4678. [DOI] [PubMed] [Google Scholar]
- 142. Ghosh R, Vinod V, Symons JD, Boudina S. Protein and mitochondria quality control mechanisms and cardiac aging. Cells 2020;9:933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gilda JE, Gomes AV. Proteasome dysfunction in cardiomyopathies. J Physiol 2017;595:4051–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Higashikuse Y, Mittal N, Arimura T, Yoon SH, Oda M, Enomoto H, Kaneda R, Hattori F, Suzuki T, Kawakami A, Gasch A. Perturbation of the titin/MURF1 signaling complex is associated with hypertrophic cardiomyopathy in a fish model and in human patients. Dis Model Mech 2019;12:dmm041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kötter S, Kazmierowska M, Andresen C, Bottermann K, Grandoch M, Gorressen S, Heinen A, Moll JM, Scheller J, Gödecke A, Fischer JW, Schmitt JP, Krüger M. Titin-based cardiac myocyte stiffening contributes to early adaptive ventricular remodeling after myocardial infarction. Circ Res 2016;119:1017–1102. [DOI] [PubMed] [Google Scholar]
- 146. Kennedy TL, Swiderski K, Murphy KT, Gehrig SM, Curl CL, Chandramouli C, Febbraio MA, Delbridge LM, Koopman R, Lynch GS. BGP-15 improves aspects of the dystrophic pathology in mdx and dko mice with differing efficacies in heart and skeletal muscle. Am J Pathol 2016;186:3246–3260. [DOI] [PubMed] [Google Scholar]
- 147. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, Tong M, Ruckenstuhl C, Dammbrueck C, Gross AS, Herbst V, Magnes C, Trausinger G, Narath S, Meinitzer A, Hu Z, Kirsch A, Eller K, Camona-Gutierrez D, Büttner S, Pietrocola F, Knittelfelder O, Schrepfer E, Rockenfeller P, Simonini C, Rahn A, Horsch M, Moreth K, Beckers J, Fuchs H, Gailus-Durner V, Neff F, Janik D, Rathkolb B, Rozman J, de Angelis MH, Moustafa T, Haemmerle G, Mayr M, Willeit P, von Frieling-Salewsky M, Pieske B, Scorrano L, Pieber T, Pechlaner R, Willeit J, Sigrist SJ, Linke WA, Mühlfeld C, Sadoshima J, Dengjel J, Kiechl S, Kroemer G, Sedej S, Madeo F. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 2016;22:1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Schlossarek S, Singh SR, Geertz B, Schulz H, Reischmann S, Hübner N, Carrier L. Proteasome inhibition slightly improves cardiac function in mice with hypertrophic cardiomyopathy. Front Physiol 2014;5:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O'Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O'Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 2015;7:270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, Rackham OJ, van Heesch S, Pua CJ, Kui M, Walsh R, Tayal U, Prasad SK, Dawes TJ, Ko NS, Sim D, Chan LL, Chin CW, Mazzarotto F, Barton PJ, Kreuchwig F, de Kleijn DP, Totman T, Biffi C, Tee N, Rueckert D, Schneider V, Faber A, Regitz-Zagrosek V, Seidman JG, Seidman CE, Linke WA, Kovalik JP, O'Regan D, Ware JS, Hubner N, Cook SA. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 2017;49:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ware JS, Cook SA. Role of titin in cardiomyopathy: from DNA variants to patient stratification. Nat Rev Cardiol 2018;15:241–252. [DOI] [PubMed] [Google Scholar]
- 152. Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, Haghighi A, Homsy J, Hubner N, Church G, Cook SA, Linke WA, Chen CS, Seidman JG, Seidman CE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015;349:982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. van Heesch S, Witte F, Schneider-Lunitz V, Schulz JF, Adami E, Faber AB, Kirchner M, Maatz H, Blachut S, Sandmann CL, Kanda M, Worth CL, Schafer S, Calviello L, Merriott R, Patone G, Hummel O, Wyler E, Obermayer B, Mücke MB, Lindberg EL, Trnka F, Memczak S, Schilling M, Felkin LE, Barton PJR, Quaife NM, Vanezis K, Diecke S, Mukai M, Mah N, Oh SJ, Kurtz A, Schramm C, Schwinge D, Sebode M, Harakalova M, Asselbergs FW, Vink A, de Weger RA, Viswanathan S, Widjaja AA, Gärtner-Rommel A, Milting H, Dos Remedios C, Knosalla C, Mertins P, Landthaler M, Vingron M, Linke WA, Seidman JG, Seidman CE, Rajewsky N, Ohler U, Cook SA, Hubner N. The Translational Landscape of the Human Heart. Cell 2019;178:242–260. [DOI] [PubMed] [Google Scholar]
- 154. McAfee Q, Yingxian Chen C, Yang Y, Caporizzo M, Morley M, Babu A, Jeong S, Brandimarto J, Bedi KC Jr, Flam E, Cesare J, Cappola TP, Margulies K, Prosser B, Arany Z. Truncated titin proteins in dilated cardiomyopathy. Sci Transl Med 2021. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Rees M, Nikoopour R, Fukuzawa A, Kho AL, Fernandez-Garcia MA, Wraige E, Bodi I, Deshpande C, Özdemir Ö, Daimagüler HS, Pfuhl M, Holt M, Brandmeier B, Grover S, Fluss J, Longman C, Farrugia ME, Matthews E, Hanna M, Muntoni F, Sarkozy A, Phadke R, Quinlivan R, Oates EC, Schröder R, Thiel C, Reimann J, Voermans N, Erasmus C, Kamsteeg EJ, Konersman C, Grosmann C, McKee S, Tirupathi S, Moore SA, Wilichowski E, Hobbiebrunken E, Dekomien G, Richard I, Van den Bergh P, Domínguez-González C, Cirak S, Ferreiro A, Jungbluth H, Gautel M. Making sense of missense variants in TTN-related congenital myopathies. Acta Neuropathol 2021;141:431–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This review article does not contain new original data.