

Abstract
Background
CT-P16 is a candidate bevacizumab biosimilar.
Objective
This double-blind, multicenter, parallel-group, phase III study aimed to establish equivalent efficacy between CT-P16 and European Union-approved reference bevacizumab (EU-bevacizumab) in patients with metastatic or recurrent non-squamous non-small cell lung cancer (nsNSCLC).
Patients and Methods
Patients with stage IV or recurrent nsNSCLC were randomized (1:1) to receive CT-P16 or EU-bevacizumab (15 mg/kg every 3 weeks; ≤ 6 cycles) with paclitaxel (200 mg/m2) and carboplatin (area under the curve 6.0; both for 4–6 cycles), as induction therapy. Patients with controlled disease after induction therapy continued with CT-P16 or EU-bevacizumab maintenance therapy. The primary endpoint was objective response rate (ORR) during the induction period. Time-to-event analyses, pharmacokinetics, safety, and immunogenicity were also evaluated. Results obtained after 1 year of follow-up are presented.
Results
Overall, 689 patients were randomized (CT-P16, N = 342; EU-bevacizumab, N = 347). ORR was 42.40% (95% confidence interval [CI] 37.16–47.64) and 42.07% (95% CI 36.88–47.27) for CT-P16 and EU-bevacizumab, respectively. The risk difference (0.40 [95% CI − 7.02 to 7.83]) and risk ratio (1.0136 [90% CI 0.8767–1.1719]) for ORR fell within predefined equivalence margins (− 12.5 to + 12.5%, and 0.7368 to 1.3572, respectively), demonstrating equivalence between CT-P16 and EU-bevacizumab. Median response duration, time to progression, progression-free survival, and overall survival were comparable between treatment groups. Safety profiles were similar: 96.2% (CT-P16) and 93.0% (EU-bevacizumab) of patients experienced treatment-emergent adverse events. Pharmacokinetics and immunogenicity were comparable between groups.
Conclusions
Equivalent efficacy and similar pharmacokinetics, safety, and immunogenicity support bioequivalence of CT-P16 and EU-bevacizumab in patients with nsNSCLC.
Trial registration number
Supplementary Information
The online version contains supplementary material available at 10.1007/s40259-022-00552-8.
Key Points
| This phase III study compared candidate bevacizumab biosimilar CT-P16 and European Union-approved reference bevacizumab (EU-bevacizumab) in patients with metastatic or recurrent non-squamous non-small cell lung cancer. |
| Equivalent efficacy between CT-P16 and EU-bevacizumab was demonstrated for the primary endpoint: objective response rate following induction therapy. |
| After 1 year of follow-up in this ongoing study, findings also suggest comparable survival outcomes, pharmacokinetics, safety, and immunogenicity profiles between CT-P16 and EU-bevacizumab. |
Introduction
Bevacizumab is a recombinant, humanized immunoglobulin G1 monoclonal antibody (mAb) that targets vascular endothelial growth factor (VEGF), a potent angiogenic growth factor important for tumor growth and survival [1, 2]. Bevacizumab inhibits the binding of VEGF to cell surface receptors, reducing tumor vascularization and limiting tumor growth [3]. In the US and Europe, reference bevacizumab (Avastin®; Roche, Basel, Switzerland/Genentech, Inc., South San Francisco, CA, USA) is approved for use in treating a number of advanced solid tumors, including metastatic non-small cell lung cancer (NSCLC), where it is administered in combination with chemotherapy in the first-line setting [1, 3–5].
Bevacizumab provided an important breakthrough in treatment options for advanced non-squamous NSCLC (nsNSCLC), beyond traditional chemotherapy regimens, and continues to offer an effective treatment option for many patients [5–9]. Regulatory approval for reference bevacizumab followed demonstration of significant overall survival (OS) and progression-free survival (PFS) benefits with bevacizumab plus platinum-based chemotherapy, compared with chemotherapy alone, in patients with nsNSCLC [10, 11]. However, a major limitation of mAb treatments is their high cost, and analyses have failed to demonstrate the cost-effectiveness or cost utility of adding bevacizumab to chemotherapy for the treatment of NSCLC [12, 13]. Limited access to bevacizumab through public healthcare services is a global problem, particularly in emerging markets, leading to geographical inequalities and rationing of treatments, potentially leading to poorer clinical outcomes [14–16].
Biosimilars are biological medicines that are highly similar to an already approved product, and are typically associated with cost savings of 20–35% compared with their reference products [15]. Thus, bevacizumab biosimilars have the potential to alleviate the financial burden of biological therapy on healthcare systems and improve access to effective treatments for patients with advanced cancers, including nsNSCLC [16]. Regulatory approval of biosimilars requires evidence of biosimilarity, i.e., that there is no clinically meaningful difference in quality, activity, safety, and efficacy between a biosimilar and the approved reference product [17, 18]. Biosimilarity is assessed based on the totality of evidence generated via a stepwise program of non-clinical and clinical studies evaluating quality, pharmacokinetics, pharmacodynamics, efficacy, safety, and immunogenicity [17, 18]. Several biosimilars of bevacizumab have been approved for the treatment of advanced or recurrent NSCLC in Europe and the US [19–28]. CT-P16 (Celltrion, Inc., Incheon, Republic of Korea) is a candidate bevacizumab biosimilar that has previously demonstrated pharmacokinetic equivalence to European Union-approved reference bevacizumab (EU-bevacizumab) and US-licensed reference bevacizumab in a phase I clinical trial (NCT03247673) [29].
This phase III study aims to determine whether the efficacy of CT-P16 is equivalent to that of EU-bevacizumab in patients with metastatic or recurrent nsNSCLC. The study is also evaluating whether pharmacokinetic parameters, safety, immunogenicity, and quality of life are comparable between treatment groups. The first part of the study has been completed, including outcomes for the primary efficacy endpoint, and these data are presented in this article, along with available data for secondary endpoints, as part of a planned interim analysis upon completion of 1 year of follow-up for the last patient enrolled. Full data for the entire study period will become available upon study completion.
Methods
Study Design
This ongoing, randomized, active-controlled, double-blind, parallel-group, phase III study (NCT03676192) was conducted at 164 hospitals or clinics in 21 countries (electronic supplementary material [ESM] Table S1). Screening of eligible participants occurred within 28 days prior to randomization, or up to 8 weeks for patients with central nervous system (CNS) metastases to provide sufficient time for CNS treatment. During the induction period, patients were randomized (1:1) to receive either CT-P16 15 mg/kg or EU-bevacizumab 15 mg/kg; both treatments were administered intravenously every 3 weeks for ≤ 6 cycles. Patients in both treatment groups received concurrent intravenous paclitaxel 200 mg/m2 and intravenous carboplatin area under the curve 6.0 every 3 weeks for 4–6 cycles. Patients with controlled disease (i.e., complete response [CR], partial response [PR], or stable disease) at the end of the induction period entered the maintenance period and received monotherapy with their assigned treatment until disease progression or intolerable toxicity. The end of treatment (EOT) visit occurred 3 weeks after the final dose of study treatment in the induction or maintenance period; subsequently, patients entered the follow-up period, in which they were followed up every 9 weeks until death or end of the study.
An interactive web response system (IWRS) was used for randomization. A computer-generated randomization schedule was prepared for the IWRS by an unblinded statistician. Patients were randomized in blocks stratified by country, sex (female vs. male), disease status (recurrence vs. metastatic), and Eastern Cooperative Oncology Group (ECOG) performance status (0 vs. 1). Patients and investigators (including local and central outcome assessors) were blinded to treatment group. Blinding and concealment of permuted block size will be maintained until completion of this ongoing study, to avoid bias.
Key protocol amendments made after the study start date are detailed below or listed in the ESM Methods. The study was monitored by an independent data safety monitoring board.
Participants
Full eligibility criteria are listed in the ESM Methods. Briefly, eligible individuals were male or female, aged ≥ 18 years, with histologically or cytologically confirmed stage IV or recurrent NSCLC. Patients were required to have one or more measurable lesions (per Response Evaluation Criteria In Solid Tumours [RECIST] criteria version 1.1); an ECOG performance status of 0 or 1; adequate hematologic, hepatic, and renal functions; and negative epidermal growth factor receptor (EGFR) mutation and anaplastic lymphoma kinase (ALK) rearrangement test results. Key exclusion criteria included NSCLC with predominantly squamous histology; previous systemic therapy, surgery, or radiotherapy for NSCLC; therapeutic use of parenteral anticoagulants or thrombolytic agents; untreated CNS metastases or CNS metastasis with bleeding risk; hemoptysis; uncontrolled hypertension, diabetes, or cardiac disease; a history of vascular disease; or pregnancy or lactation.
Endpoints
The primary efficacy endpoint was objective response rate (ORR) based on best overall response (BOR), per RECIST criteria version 1.1, achieved during the 6-cycle induction period and confirmed, if necessary, by subsequent assessment up to Cycle 3 of the maintenance period. Central review results were used for the primary analysis and local review results were used for a sensitivity analysis.
Secondary efficacy endpoints included response duration, time to progression (TTP), and PFS, each based on central review results, and OS. Response duration was defined as the time from initial response to progressive disease (PD), recurrence, or death from any cause in patients achieving a CR or PR; during the study, the planned analysis was updated to add death to the description and to add a requirement for CR or PR to be confirmed through subsequent assessment.
Safety evaluations included the incidence and severity of treatment-emergent adverse events (TEAEs) and serious TEAEs. TEAEs of special interest (TEAESIs) were captured using standardized Medical Dictionary for Regulatory Activities queries, thus evaluated across system organ classes, and included hypersensitivity/infusion-related reactions, gastrointestinal perforations and fistulae, wound healing complications, hypertension, posterior reversible encephalopathy syndrome, proteinuria, arterial or venous thromboembolism, hemorrhages, congestive heart failure, or ovarian failure/fertility. The full list of prespecified safety endpoints is available in the ESM Methods. Pharmacokinetic evaluation comprised assessment of trough serum concentrations of CT-P16 (Ctrough). Immunogenicity was assessed in terms of the incidence of both antidrug antibodies (ADAs), antibodies that bind to the biologic agent (in this study, bevacizumab) in human serum, and neutralizing antibodies (NAbs), ADAs that bind to the biologic agent and neutralize its biologic activity [30].
Assessments
RECIST criteria version 1.1 were used for tumor evaluation. Computed tomography (CT) scans were conducted at screening, at the end of Cycles 2, 4, and 6 during the induction period, at the end of every third cycle during the maintenance period, and at EOT. Brain CT or magnetic resonance imaging, and bone scans, were conducted at screening and at later timepoints if brain or bone metastases were present at screening or if new lesions were suspected, respectively. Images for tumor response were assessed via both central review (by a central independent reviewer) and local review (by investigators).
Blood samples were collected predose for pharmacokinetic analyses on Day 1 of each cycle and at the end of Cycle 6 during the induction period, at the end of every third cycle during the maintenance period, and at EOT. Serum CT-P16 and EU-bevacizumab concentrations were measured using a validated electrochemiluminescent assay with a lower limit of quantification of 50 ng/mL.
Adverse events (AEs) were monitored throughout and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0. Clinical laboratory tests and physical examinations were performed at screening, on Day 1 of each cycle, and at EOT.
Blood samples were collected for immunogenicity assessments on Day 1 of Cycle 1 and at the end of Cycles 2, 4, and 6 during the induction period, at the end of every third cycle during the maintenance period, and at EOT. The presence of ADAs and NAbs was detected using validated electrochemiluminescent assays. Blood samples were first screened for ADAs with a positive control concentration range of 50–2000 ng/mL, and if found to be positive, underwent further testing in a confirmatory assay. Samples with confirmed positive ADA results were further screened for NAbs using three positive controls (low: 125 ng/mL; titer: 500 ng/mL; high: 10,000 ng/mL).
Statistical Analysis
A total of 305 patients per treatment group was estimated to provide 80% power to demonstrate similarity in efficacy between CT-P16 and EU-bevacizumab based on an expected ORR of 38%. This calculation was based on two sets of statistical assumptions, to meet the requirements of both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA): an equivalence margin of −12.5 to +12.5% using a 95% confidence interval (CI; two one-sided alpha of 0.025) for the risk difference (RD) in ORR (EMA assumption; predefined in the original protocol), and an equivalence margin of 0.7368–1.3572 using a 90% CI (two one-sided alpha of 0.05) for the risk ratio (RR) in ORR (FDA assumption; included as a protocol amendment). Therefore, 678 patients (n = 339 per group) were required to allow for an anticipated dropout rate of 10%.
The primary analysis of the primary efficacy endpoint (ORR based on BOR during the induction period) was performed using a logistic regression model for RD and a log-binomial regression model for RR. Covariates comprised region (Europe, Middle East, and Africa vs. America vs. Asia), sex (male vs. female), disease status at baseline (recurrent vs. metastatic), and ECOG performance status at baseline (0 vs. 1). Treatment groups (CT-P16 vs. EU-bevacizumab) were considered as a fixed effect. Equivalence in terms of efficacy would be achieved if the 95% CI for the RD for ORR was bounded by the interval −12.5 to +12.5% (EMA assumption) and if the 90% CI for the RR for ORR was entirely within the predefined equivalence margin of 0.7368–1.3572 (FDA assumption). Patients with missing values for ORR were considered non-responders. For secondary efficacy variables, continuous data were summarized using descriptive statistics, and categorical data were summarized using numbers and percentages. Analysis sets are defined in the ESM Methods. All statistical analyses were conducted using Statistical Analysis System (SAS®) software version 9.4 or higher (SAS Institute Inc., Cary, NC, USA).
Results
Patient Characteristics
This ongoing study started on 1 February 2019 (first patient randomized) and has a planned follow-up period of approximately 3 years from enrollment of the last patient. The data cut-off was 22 April 2021 for the primary endpoint (completion of the study induction period) and 21 September 2021 for all other analyses reported herein.
A total of 1530 patients were screened, of whom 689 met the eligibility criteria and were randomized to receive either CT-P16 (n = 342) or EU-bevacizumab (n = 347) and initiated induction treatment (Fig. 1). Overall, 499 (72.4%) patients completed the induction period (CT-P16: n = 258 [75.4%]; EU-bevacizumab: n = 241 [69.5%]). Subsequently, a total of 466 (67.6%) patients entered the maintenance period (CT-P16: n = 239 [69.9%]; EU-bevacizumab: n = 227 [65.4%]). The proportions of patients who discontinued study treatment during each study period were generally similar between treatment groups; the most common reason for discontinuation during the induction and maintenance periods was PD.
Fig. 1.

Patient disposition. aTwo patients who were randomized to the EU-bevacizumab group had EGFR mutations associated with lack of responsiveness to EGFR TKI therapy (EGFR exon 20 insertions). The patients were permitted to be enrolled by the investigator and sponsor since TKI therapy was not an option for their treatment. bOne patient in the CT-P16 group accidentally missed a CT-P16 dose in Cycle 4 but was considered as having completed the induction period and entered the maintenance period. cTwo patients in each treatment group discontinued as treatment could not be resumed within 6 weeks of the last dose; one patient in the CT-P16 group discontinued due to a change in the patient’s financial status. AE adverse event, EGFR epidermal growth factor receptor, EU-bevacizumab European Union-approved reference bevacizumab, FU follow-up, PD progressive disease, TKI tyrosine kinase inhibitor
The two treatment groups were well balanced with respect to baseline characteristics, medical history, and previous treatments for NSCLC (Table 1). Overall, patients had a median age of 62 years (range 26–82), 244 (35.4%) were female, and most were White (n = 528 [76.6%]) or Asian (n = 114 [16.5%]). Most patients had metastatic disease (stage IVA: n = 311 [45.1%]; stage IVB: n = 319 [46.3%]), and the most common final pathological diagnosis was adenocarcinoma (n = 676 [98.1%]). Overall, 64 (9.3%), 83 (12.0%), and 16 (2.3%) patients had received at least one prior surgical procedure, radiotherapy, or anticancer therapy for NSCLC, respectively.
Table 1.
Baseline characteristics (intent-to-treat population)
| Parameter | CT-P16 [N = 342] | EU-bevacizumab [N = 347] | Overall [N = 689] |
|---|---|---|---|
| Median age, years (range) | 62 (32–82) | 62 (26–82) | 62 (26–82) |
| Female | 119 (34.8) | 125 (36.0) | 244 (35.4) |
| Race | |||
| American Indian or Alaska Native | 9 (2.6) | 9 (2.6) | 18 (2.6) |
| Asian | 59 (17.3) | 55 (15.9) | 114 (16.5) |
| Black or African American | 2 (0.6) | 1 (0.3) | 3 (0.4) |
| White | 264 (77.2) | 264 (76.1) | 528 (76.6) |
| Other | 8 (2.3) | 18 (5.2) | 26 (3.8) |
| Median weight, kg (range) | 67 (36–131) | 69 (35–126) | 68 (35–131) |
| ECOG PS | |||
| Grade 0 | 105 (30.7) | 110 (31.7) | 215 (31.2) |
| Grade 1 | 237 (69.3) | 237 (68.3) | 474 (68.8) |
| Disease status | |||
| Recurrent | 25 (7.3) | 33 (9.5) | 58 (8.4) |
| Metastatic | 317 (92.7) | 314 (90.5) | 631 (91.6) |
| Final pathological diagnosis | |||
| Adenocarcinoma | 336 (98.2) | 340 (98.0) | 676 (98.1) |
| Large cell carcinoma | 3 (0.9) | 2 (0.6) | 5 (0.7) |
| NSCC NOS | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Adenosquamous carcinoma | 2 (0.6) | 2 (0.6) | 4 (0.6) |
| Othera | 0 | 2 (0.6) | 2 (0.3) |
| Clinical stage | |||
| IIIBb | 0 | 1 (0.3) | 1 (0.1) |
| IVA | 147 (43.0) | 164 (47.3) | 311 (45.1) |
| IVB | 170 (49.7) | 149 (42.9) | 319 (46.3) |
| Recurrent | 25 (7.3) | 33 (9.5) | 58 (8.4) |
| Prior treatment | |||
| Surgery | 27 (7.9) | 37 (10.7) | 64 (9.3) |
| Radiotherapy | 41 (12.0) | 42 (12.1) | 83 (12.0) |
| Anticancer systemic therapyc | 6 (1.8) | 10 (2.9) | 16 (2.3) |
Data are expressed as n (%) unless otherwise specified
ECOG PS Eastern Cooperative Oncology Group performance status, EU-bevacizumab European Union-approved reference bevacizumab, NSCC NOS non-small cell carcinoma not otherwise specified
aTwo patients in the EU-bevacizumab group had signet ring cell carcinoma
bOne patient in the EU-bevacizumab group was enrolled as stage IIIB adenocarcinoma and was excluded from the per-protocol population due to violation of inclusion criterion
cCytotoxic chemotherapy was the only authorized prior anticancer systemic therapy, of which cisplatin was the most frequently reported (CT-P16: n = 4 [1.2%]; EU-bevacizumab: n = 6 [1.7%])
Efficacy
The primary endpoint, ORR based on BOR during the induction period, was 42.40% (95% CI 37.16–47.64) in the CT-P16 group compared with 42.07% (95% CI 36.88–47.27%) in the EU-bevacizumab group (intent-to-treat [ITT] population) [Table 2]. Equivalence was demonstrated between CT-P16 and EU-bevacizumab as the 95% CI for the RD (0.40 [95% CI − 7.02 to 7.83]) and the 90% CI for the RR (1.0136 [90% CI 0.8767–1.1719]) were contained within the predefined equivalence margins of −12.5 to +12.5% and 0.7368–1.3572, respectively. A similar proportion of patients in each treatment group achieved a CR (CT-P16: n = 2 [0.6%]; EU-bevacizumab: n = 3 [0.9%]) or a PR (CT-P16: n = 143 [41.8%]; EU-bevacizumab: n = 143 [41.2%]) as their BOR. In the per-protocol (PP) population, results for BOR and ORR were similar to those in the ITT population; the RD and RR were both contained within the predefined equivalence margins, supporting the primary analysis (Table 2). In a sensitivity analysis of the primary endpoint using local review, ORR during the induction period was 43.86% (95% CI 38.60–49.12) in the CT-P16 group compared with 39.19% (95% CI 34.06–44.33%) in the EU-bevacizumab group (ITT population) [ESM Fig. S1]. The RD (4.87 [95% CI − 2.53 to 12.26]) and RR (1.1234 [90% CI 0.9683–1.3032]) supported the demonstration of equivalence in the primary analysis.
Table 2.
Objective response rate during the induction period (primary endpoint; central review)
| Parameter | CT-P16 | EU-bevacizumab |
|---|---|---|
|
Intent-to-treat population (primary analysis) |
N = 342 | N = 347 |
| Best overall response [n (%)] | ||
| Complete response | 2 (0.6) | 3 (0.9) |
| Partial response | 143 (41.8) | 143 (41.2) |
| Stable disease | 156 (45.6) | 140 (40.3) |
| Progressive disease | 17 (5.0) | 19 (5.5) |
| Non-evaluable | 3 (0.9) | 3 (0.9) |
| Missing | 21 (6.1) | 39 (11.2) |
| Objective response rate, % (95% CI) | 42.40 (37.16–47.64) | 42.07 (36.88–47.27) |
| Risk difference for CT-P16:EU-bevacizumab (95% CI) | 0.40 (−7.02 to 7.83) | |
| Risk ratio for CT-P16:EU-bevacizumab (90% CI) | 1.0136 (0.8767–1.1719) | |
|
Per-protocol population (supportive analysis) |
N = 318 | N = 303 |
| Best overall response [n (%)] | ||
| Complete response | 2 (0.6) | 3 (1.0) |
| Partial response | 142 (44.7) | 140 (46.2) |
| Stable disease | 154 (48.4) | 138 (45.5) |
| Progressive disease | 17 (5.3) | 19 (6.3) |
| Non-evaluable | 3 (0.9) | 3 (1.0) |
| Objective response rate, % (95% CI) | 45.28 (39.81–50.75) | 47.19 (41.57–52.82) |
| Risk difference for CT-P16:EU-bevacizumab (95% CI) | −1.90 (−9.80 to 6.00) | |
| Risk ratio for CT-P16:EU-bevacizumab (90% CI) | 0.9962 (0.8387–1.1132) | |
CI confidence interval, EU-bevacizumab European Union-approved reference bevacizumab
By the time of data cut-off, median (range) follow-up duration was 11.91 (0.03–29.64) months (CT-P16: 12.15 [0.03–29.64] months; EU-bevacizumab: 11.35 [0.03–28.52] months). Median response duration, TTP, PFS, and OS had been reached in both groups. In patients achieving either a CR or PR, which was confirmed by subsequent assessment, median response duration was similar between treatment groups (CT-P16: 7.2 months [95% CI 6.3–8.2]; EU-bevacizumab: 6.3 months [95% CI 5.8–7.5]). Median TTP was similar in both groups (CT-P16: 8.5 months [95% CI 8.3–10.0]; EU-bevacizumab: 8.3 months [95% CI 7.4–9.1]). Median PFS was 7.9 months (95% CI 6.9–8.3) in the CT-P16 group versus 7.2 months (95% CI 6.5–8.3) in the EU-bevacizumab group (hazard ratio [HR] 0.92, 95% CI 0.77–1.10) [Fig. 2a]. Correspondingly, median OS was 17.1 months (95% CI 14.6–18.7) in the CT-P16 group compared with 15.6 months (95% CI 13.4–18.0) in the EU-bevacizumab group (HR 0.95, 95% CI 0.77–1.19) [Fig. 2b].
Fig. 2.
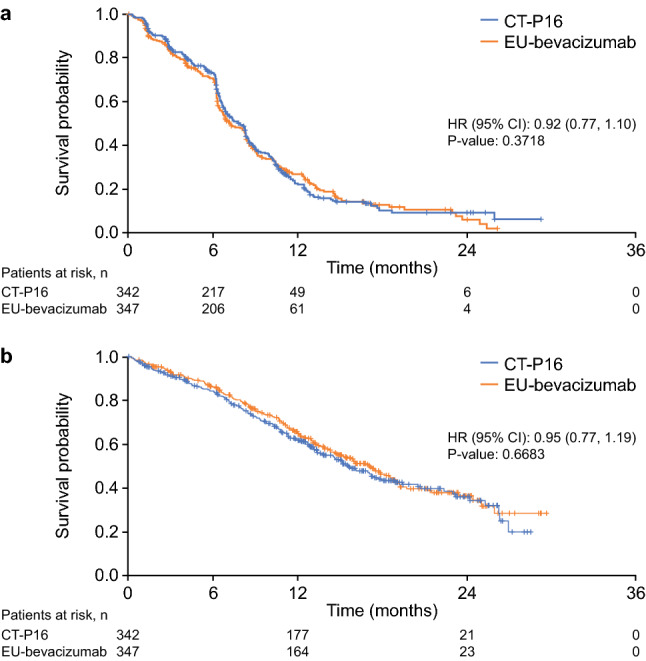
Kaplan–Meier plots depicting a progression-free survival as assessed by central review, and b overall survival (intent-to-treat population). CI confidence interval, EU-bevacizumab European Union-approved reference bevacizumab, HR hazard ratio
Pharmacokinetics
Mean bevacizumab Ctrough was generally similar for CT-P16 and EU-bevacizumab on Day 1 of each cycle up to data cut-off (data for the induction period are shown in ESM Fig. S2).
Safety
Safety data are presented up to the data cut-off date of 21 September 2021; the study is ongoing and final safety data will be available after study completion. Over this period, a similar number of patients in the CT-P16 and EU-bevacizumab groups experienced one or more TEAEs (CT-P16: n = 332 [96.2%]; EU-bevacizumab: n = 320 [93.0%]) [Table 3]. The majority of TEAEs were grade 1 or 2 in severity. TEAEs considered to be related to the study drug by the investigator were reported in 178 (51.6%) and 174 (50.6%) patients in the CT-P16 and EU-bevacizumab groups, respectively. Overall, the most frequently reported TEAEs in either treatment group were alopecia (CT-P16: n = 220 [63.8%]; EU-bevacizumab: n = 218 [63.4%]) and anemia (CT-P16: n = 109 [31.6%]; EU-bevacizumab: n = 93 [27.0%]) (ESM Table S2).
Table 3.
Summary of TEAEs (safety population)
| CT-P16 [N = 345a] |
EU-bevacizumab [N = 344] |
|
|---|---|---|
| Patients with one or more TEAE of any cause | ||
| Any | 332 (96.2) | 320 (93.0) |
| Grade 3 or higher | 151 (43.8) | 144 (41.9) |
| Serious | 69 (20.0) | 73 (21.2) |
| Leading to discontinuation | 55 (15.9) | 55 (16.0) |
| Leading to death | 23 (6.7) | 24 (7.0) |
| Patients with one or more TEAESI | ||
| Hypersensitivity/IRRs | 11 (3.2) | 16 (4.7) |
| GI perforations/fistulae | 3 (0.9) | 5 (1.5) |
| Wound healing complications | 1 (0.3) | 0 |
| Hypertension | 44 (12.8) | 39 (11.3) |
| PRES | 1 (0.3) | 0 |
| Proteinuria | 42 (12.2) | 38 (11.0) |
| Arterial thromboembolism | 2 (0.6) | 4 (1.2) |
| Venous thromboembolism | 10 (2.9) | 5 (1.5) |
| Hemorrhages | 40 (11.6) | 37 (10.8) |
| Congestive heart failure | 3 (0.9) | 2 (0.6) |
| Ovarian failure/fertility | 0 | 0 |
Data are expressed as n (%)
EU-bevacizumab European Union-approved reference bevacizumab, GI gastrointestinal, IRR infusion-related reaction, PRES posterior reversible encephalopathy syndrome, TEAE treatment-emergent adverse event, TEAESI TEAE of special interest
aThree patients who were randomized to the EU-bevacizumab treatment group were included in the CT-P16 safety population as the patients incorrectly received CT-P16 during the treatment period
The proportions of patients with serious TEAEs, TEAEs leading to discontinuation, or TEAEs leading to death were similar between treatment groups (Table 3). Three deaths in the CT-P16 group (n = 1 each due to pulmonary hemorrhage, sepsis, and subarachnoid hemorrhage) and seven deaths in the EU-bevacizumab group (n = 2 due to pulmonary hemorrhage and n = 1 each due to cardiac arrest, cerebral infarction, lung abscess, septic shock, and sudden death) were considered by the investigator to be related to the study drug. The proportions of patients experiencing at least one TEAESI were low and well balanced between treatment groups (Table 3). In both groups, the most frequent TEAESI was hypertension (CT-P16: n = 44 [12.8%]; EU-bevacizumab: n = 39 [11.3%]) (Table 3). Most TEAESIs of hypertension were grade 2–3 in severity.
Clinical laboratory findings demonstrated that, at the time of analysis, there was no evidence of any intergroup differences in clinical chemistry, hematology, coagulation, and urinalysis parameters. In general, there were no notable differences between treatment groups in the proportions of patients with clinically notable vital sign results or clinically significant electrocardiogram results.
Immunogenicity
Five (1.4%) patients in the CT-P16 group and seven (2.0%) patients in the EU-bevacizumab group tested positive for ADAs at baseline. One (0.3%) patient in the CT-P16 group tested positive for NAbs at baseline. At the time of analysis, the proportion of patients with a positive ADA result at any time during the study was similar between treatment groups (CT-P16: n = 78 [22.6%]; EU-bevacizumab: n = 83 [24.1%]). NAb tests were only performed for patients with positive ADA results; the proportion of patients with a positive NAb result at any time during the study was low and similar between treatment groups (CT-P16: n = 8 [2.3%]; EU-bevacizumab: n = 8 [2.3%]).
Discussion
This phase III clinical trial was the first to compare the candidate biosimilar CT-P16 with reference bevacizumab. Equivalent efficacy was demonstrated between CT-P16 and EU-bevacizumab in terms of the primary endpoint—ORR based on BOR during the induction period—in patients with metastatic or recurrent nsNSCLC. Comparable efficacy between treatment groups was also demonstrated for secondary efficacy endpoints evaluable at the time of analysis, including PFS and OS, further supporting bioequivalence between CT-P16 and reference bevacizumab. In addition, Ctrough levels were comparable between groups at all timepoints assessed. The proportion of patients with ADAs at baseline was low in both treatment groups, and a similar proportion of patients in each group had positive ADA results during the study. Overall, the safety profile of CT-P16 was comparable with that of EU-bevacizumab and no new safety concerns have been identified to date.
In the present study, the ORR based on BOR during the induction period was approximately 42% in both treatment groups, which was comparable with historical clinical trial data for reference bevacizumab administered in combination with paclitaxel and carboplatin in patients with stage IIIB/IV NSCLC (35–54%) [10, 31–35]. In contrast, median OS and PFS in the present study (17.1 months and 7.9 months, respectively, with CT-P16, and 15.6 months and 7.2 months, respectively, with EU-bevacizumab) were longer than reported with bevacizumab plus paclitaxel and carboplatin in the pivotal phase III study (12.3 months and 6.2 months, respectively) [10]. Similar patient populations were enrolled in the two studies (although a slightly lower proportion of females was enrolled in the present study), and identical dosing regimens were employed. Thus, the apparently longer durations of PFS and OS observed in the present study may be accounted for by advances in medical practices (such as supportive therapies, and for OS, the availability of new subsequent lines of therapy) since the pivotal phase III study for the reference product was conducted. More recent studies evaluating biosimilars of bevacizumab may better reflect the current standard of care for patients with nsNSCLC. Accordingly, median PFS and OS reported with reference bevacizumab in biosimilar studies are more comparable with findings from the current study, ranging from 7.6 to 8.6 months and from 15.8 months to not reached after 18 months of follow-up, respectively [31–35].
The strengths of the current study include the randomized, double-blind design, and that it was conducted in multiple countries. The study used well-established efficacy measures and endpoints, and utilized central review by blinded independent reviewers for efficacy assessment, alongside local review to corroborate the central review findings. Findings from the sensitivity analysis, which analyzed ORR according to local review of tumor images, were in line with those from the primary analysis by central review in the CT-P16 group (42.40% and 43.86%, respectively) and EU-bevacizumab group (42.07% and 39.19%, respectively). A potential limitation of this study is that participants were mostly White or Asian; Black or African American patients only accounted for 0.4% of the enrolled ITT population. Therefore, the findings may lack generalizability in populations that are not White or Asian.
Another potential limitation of the current study is that patients received EU-bevacizumab or CT-P16 in combination with chemotherapy only, without the addition of another targeted therapy or immune checkpoint inhibitor, which might be indicated for some patients [36, 37]. However, ALK- and EGFR-targeted therapies would not have been suitable for participants in the current study, as patients with ALK rearrangements or EGFR mutations were ineligible. In addition, the comparison of CT-P16 and EU-bevacizumab, both in combination with chemotherapy, is aligned with the approaches taken for the evaluation of other bevacizumab biosimilars licensed by the FDA and/or EMA to date [31–33, 35, 38]. Despite the advent of immunotherapies, bevacizumab remains an important element of the nsNSCLC treatment landscape [9], including for patients with contraindications for immune checkpoint inhibitor therapy (such as autoimmune disease) [36]. Chemotherapy with or without bevacizumab plus an immune checkpoint inhibitor is widely used as a standard treatment for patients with nsNSCLC in developed countries. However, there are some limitations to the use of these regimens globally, such as financial barriers and patient access issues, particularly in developing countries [16, 39, 40]. As a bevacizumab biosimilar, CT-P16 might help to alleviate some of the pharmacoeconomic challenges faced by healthcare systems aiming to deliver effective therapies for patients with nsNSCLC [16]. In addition, bevacizumab-based regimens may be associated with a reduction in brain metastasis, with lower incidence reported for patients with nsNSCLC treated with a bevacizumab- and atezolizumab-based regimen versus an atezolizumab-based regimen [41].
While the current article reports data after 1 year of follow-up for the last patient enrolled, the study is ongoing and full data for secondary endpoints due to be assessed during the whole study period, as well as longer-term safety data, will become available upon study completion. This will allow further understanding of the comparability of the efficacy, safety, and immunogenicity of CT-P16 to EU-bevacizumab. Another future consideration is that EMA and FDA approval pathways permit extrapolation of regulatory approval for biosimilars to all indications licensed for the reference product, provided this is scientifically justified and supported by the findings of the comprehensive comparability exercise with the reference product [17, 18]. Considerations for extrapolation of regulatory approval for bevacizumab biosimilars have been discussed in detail in a recent review article [42]; indeed, the extrapolation of results was mentioned for a phase III study of the bevacizumab biosimilar FKB238 in patients with nsNSCLC [34]. As such, data from the CT-P16 development program, including the CT-P16 3.1 study reported herein, are anticipated to support extrapolation of regulatory approval for CT-P16 to all indications approved for reference bevacizumab, if biosimilarity is established by the regulatory authorities.
Conclusion
The initial findings from this ongoing, phase III trial demonstrate the equivalent efficacy of CT-P16 to EU-bevacizumab in patients with metastatic or recurrent nsNSCLC. Safety, immunogenicity, and pharmacokinetics were also comparable between groups.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank all patients and investigators involved in this study, as well as the members of the Data Safety Monitoring Board. Yuriy Ostapenko (Clinic of National Institute of Cancer, Kyiv, Ukraine) made substantial contributions to the acquisition of study data and provided feedback on early drafts of the manuscript; however, due to the ongoing exceptional circumstances in Ukraine, they were unable to be contacted to provide approval of the manuscript version submitted for publication. Nonetheless, the authors considered it important to transparently recognize the role of Yuriy Ostapenko in the development of this work. The authors would also like to thank Igor Bondarenko (Municipal Non-profit Enterprise, City Clinical Hospital #4 Dnipro City Council—PPDS, Dnipro, Ukraine) for their substantial contributions to the acquisition of data for the work, and Tudor Ciuleanu (Prof Dr I. Chiricuta Institute of Oncology, Cluj-Napoca, Romania) for their substantial contributions to the acquisition of data for the work and manuscript development. Medical writing support, including development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking, and referencing, was provided by Emma Evans, PhD, CMPP, and Beatrice Tyrrell, DPhil, CMPP, at Aspire Scientific Limited (Bollington, UK). Funding for medical writing support for this article was provided by Celltrion, Inc. (Incheon, Republic of Korea). Selected results from the current study were presented at the annual meeting of the American Association for Cancer Research (8–13 April 2022).
Authors’ contributions
Claire Verschraegen, Zoran Andric, Fedor Moiseenko, Tamta Makharadze, Alona Oleksiienko, Eduardo Yañez Ruiz, TaeHong Park, Sijin Park, Hana Ju, and Yuichiro Ohe: Investigation; data curation; writing—original draft; writing—review and editing. Sergii Shevnya: Writing—investigation; data curation; writing—original draft; writing—review and editing. SungHyun Kim and KeumYoung Ahn: Conceptualization; methodology; formal analysis; writing—original draft; writing—review and editing.
Declarations
Funding
This work was supported by Celltrion, Inc. (Incheon, Republic of Korea). The study sponsor played a role in the study design, data collection and analysis, decision to publish, and preparation of the manuscript.
Conflicts of interest
Fedor Moiseenko has received support for travel to meetings from AstraZeneca, Biocad, MSD, Novartis, Pfizer, and Roche; payment for lectures, including service on speakers’ bureaus, from AstraZeneca, Biocad, Eli Lilly, MSD, Novartis, Pfizer, and Roche; and has provided expert testimony for AstraZeneca, BMS, Eli Lilly, and Roche. SungHyun Kim, KeumYoung Ahn, and TaeHong Park are employees of and hold stocks in Celltrion, Inc. Sijin Park and Hana Ju are employees of Celltrion, Inc. Yuichiro Ohe has received grants from AstraZeneca, BMS, Chugai, Daiichi-Sankyo, Dainippon-Sumitomo, Janssen, Kissei, Kyorin, Lilly, LOXO, Novartis, ONO, Pfizer, Taiho, and Takeda; consulting fees or honoraria from AstraZeneca, Bayer, BMS, Boehringer Ingelheim, Chugai, Eli Lilly, Kyowa Hakko Kirin, MSD, Nippon Kayaku, ONO, Pfizer, and Taiho; fees for participation in review activities such as data monitoring boards from Amgen, AnHeart Therapeutics Inc., AstraZeneca, BMS, Boehringer Ingelheim, Celltrion, Chugai, Kyorin, Nippon Kayaku, and ONO; payment for the writing or reviewing of manuscripts from Eisai; and payment for lectures including service on speakers’ bureaus from AstraZeneca, Bayer, BMS, Boehringer Ingelheim, Chugai, Eli Lilly, Kyowa Hakko Kirin, MSD, Nippon Kayaku, ONO, Pfizer, and Taiho. Claire Verschraegen, Zoran Andric, Tamta Makharadze, Sergii Shevnya, Alona Oleksiienko, and Eduardo Yañez Ruiz have no conflicts of interest to declare.
Data availability statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Code availability
Not applicable.
Ethics approval
This study was conducted in accordance with the ethical principles originating in the Declaration of Helsinki, International Conference on Harmonisation Good Clinical Practice guidelines, and all applicable regulations. Prior to study initiation, the protocol was approved by the relevant Institutional Review Board or independent Ethics Committee at each center.
Consent to participate
Informed written consent to participate in this study was obtained from all participants.
Consent to publish
All participants provided consent for their data to be published.
Contributor Information
Claire Verschraegen, Email: claire.verschraegen@osumc.edu.
Zoran Andric, Email: drzoranandric@gmail.com.
Fedor Moiseenko, Email: moiseenkofv@gmail.com.
Tamta Makharadze, Email: tamta.makharadze@gmail.com.
Sergii Shevnya, Email: shevnia1969@gmail.com.
Alona Oleksiienko, Email: aliona.aleksandrovna92@gmail.com.
Eduardo Yañez Ruiz, Email: eduardoyanez.sim@gmail.com.
SungHyun Kim, Email: sunghyun.kim@celltrion.com.
KeumYoung Ahn, Email: keumyoung.ahn@celltrion.com.
TaeHong Park, Email: taehong.park@celltrion.com.
Sijin Park, Email: spark141008@gmail.com.
Hana Ju, Email: hana.ju@celltrion.com.
Yuichiro Ohe, Email: yohe@ncc.go.jp.
References
- 1.Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) plus carboplatin and paclitaxel as first-line treatment of advanced/metastatic recurrent nonsquamous non-small cell lung cancer. Oncologist. 2007;12(6):713–718. doi: 10.1634/theoncologist.12-6-713. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 3.European Medicines Agency. Avastin. Summary of product characteristics. 2022. https://www.ema.europa.eu/en/documents/product-information/avastin-epar-product-information_en.pdf. Accessed 15 Jul 2022.
- 4.US Food and Drug Administration. Highlights of prescribing information, Avastin (bevacizumab). 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125085s337lbl.pdf. Accessed 15 Jul 2022.
- 5.Keating GM. Bevacizumab: a review of its use in advanced cancer. Drugs. 2014;74(16):1891–1925. doi: 10.1007/s40265-014-0302-9. [DOI] [PubMed] [Google Scholar]
- 6.Amit L, Ben-Aharon I, Vidal L, Leibovici L, Stemmer S. The impact of bevacizumab (Avastin) on survival in metastatic solid tumors—a meta-analysis and systematic review. PLoS ONE. 2013;8(1):e51780. doi: 10.1371/journal.pone.0051780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lauro S, Onesti CE, Righini R, Marchetti P. The use of bevacizumab in non-small cell lung cancer: an update. Anticancer Res. 2014;34(4):1537–1545. [PubMed] [Google Scholar]
- 8.Janning M, Loges S. Anti-angiogenics: their value in lung cancer therapy. Oncol Res Treat. 2018;41(4):172–180. doi: 10.1159/000488119. [DOI] [PubMed] [Google Scholar]
- 9.Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, et al. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86:102017. doi: 10.1016/j.ctrv.2020.102017. [DOI] [PubMed] [Google Scholar]
- 10.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 11.Reck M, von Pawel J, Zatloukal P, Ramlau R, Gorbounova V, Hirsh V, et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non–small-cell lung cancer: AVAiL. J Clin Oncol. 2009;27(8):1227–1234. doi: 10.1200/JCO.2007.14.5466. [DOI] [PubMed] [Google Scholar]
- 12.Goulart B, Ramsey S. A trial-based assessment of the cost-utility of bevacizumab and chemotherapy versus chemotherapy alone for advanced non-small cell lung cancer. Value Health. 2011;14(6):836–845. doi: 10.1016/j.jval.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Zheng H, Xie L, Zhan M, Wen F, Xu T, Li Q. Cost-effectiveness analysis of the addition of bevacizumab to chemotherapy as induction and maintenance therapy for metastatic non-squamous non-small-cell lung cancer. Clin Transl Oncol. 2018;20(3):286–293. doi: 10.1007/s12094-017-1715-1. [DOI] [PubMed] [Google Scholar]
- 14.Wellcome and IAVI. Expanding access to monoclonal antibody-based products: a global call to action. 2020. https://www.iavi.org/phocadownload/expanding/Expanding%20access%20to%20monoclonal%20antibody-based%20products.pdf. Accessed 15 Jul 2022.
- 15.Buske C, Ogura M, Kwon HC, Yoon SW. An introduction to biosimilar cancer therapeutics: definitions, rationale for development and regulatory requirements. Future Oncol. 2017;13(15s):5–16. doi: 10.2217/fon-2017-0153. [DOI] [PubMed] [Google Scholar]
- 16.Monk BJ, Lammers PE, Cartwright T, Jacobs I. Barriers to the access of bevacizumab in patients with solid tumors and the potential impact of biosimilars: a physician survey. Pharmaceuticals. 2017;10(1):19. doi: 10.3390/ph10010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. https://www.fda.gov/media/82647/download. Accessed 15 Jul 2022.
- 18.European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf. Accessed 15 Jul 2022.
- 19.European Medicines Agency. Abevmy. Summary of product characteristics. 2022. https://www.ema.europa.eu/en/documents/product-information/abevmy-epar-product-information_en.pdf. Accessed 15 Jul 2022.
- 20.European Medicines Agency. Alymsys. Summary of product characteristics. 2022. https://www.ema.europa.eu/en/documents/product-information/alymsys-epar-product-information_en.pdf. Accessed 15 Jul 2022.
- 21.European Medicines Agency. Aybintio. Summary of product characteristics. 2021. https://www.ema.europa.eu/en/documents/product-information/aybintio-epar-product-information_en.pdf. Accessed 15 Jul 2022.
- 22.European Medicines Agency. Mvasi. Summary of product characteristics. 2022. https://www.ema.europa.eu/en/documents/product-information/mvasi-epar-product-information_en.pdf. Accessed 15 Jul 2022.
- 23.European Medicines Agency. Onbevzi. Summary of product characteristics. 2021. https://www.ema.europa.eu/en/documents/product-information/onbevzi-epar-product-information_en.pdf. Accessed 15 Jul 2022.
- 24.European Medicines Agency. Oyavas. Summary of product characteristics. 2022. https://www.ema.europa.eu/en/documents/product-information/oyavas-epar-product-information_en.pdf. Accessed 15 Jul 2022.
- 25.European Medicines Agency. Zirabev. Summary of product characteristics. 2022. https://www.ema.europa.eu/en/documents/product-information/zirabev-epar-product-information_en.pdf. Accessed 15 Jul 2022.
- 26.US Food and Drug Administration. Highlights of prescribing information, Mvasi (bevacizumab-awwb). 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761028s008lbl.pdf. Accessed 15 Jul 2022.
- 27.US Food and Drug Administration. Highlights of prescribing information, Zirabev (bevacizumab-bvzr). 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761099s006lbl.pdf. Accessed 15 Jul 2022.
- 28.US Food and Drug Administration. Highlights of prescribing information, Alymsys (bevacizumab-maly). 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761231s000lbl.pdf. Accessed 15 Jul 2022.
- 29.Cho SH, Han S, Ghim JL, Nam MS, Yu S, Park T, et al. A randomized, double-blind trial comparing the pharmacokinetics of CT-P16, a candidate bevacizumab biosimilar, with its reference product in healthy adult males. BioDrugs. 2019;33(2):173–181. doi: 10.1007/s40259-019-00340-x. [DOI] [PubMed] [Google Scholar]
- 30.Civoli F, Kasinath A, Cai X-Y, Wadhwa M, Exley A, Oldfield P, et al. Recommendations for the development and validation of immunogenicity assays in support of biosimilar programs. AAPS J. 2019;22(1):7. doi: 10.1208/s12248-019-0386-y. [DOI] [PubMed] [Google Scholar]
- 31.Thatcher N, Goldschmidt JH, Thomas M, Schenker M, Pan Z, Paz-Ares Rodriguez L, et al. Efficacy and safety of the biosimilar ABP 215 compared with bevacizumab in patients with advanced nonsquamous non-small cell lung cancer (MAPLE): a randomized, double-blind. Phase III study. Clin Cancer Res. 2019;25(7):2088–2095. doi: 10.1158/1078-0432.CCR-18-2702. [DOI] [PubMed] [Google Scholar]
- 32.Reck M, Luft A, Bondarenko I, Shevnia S, Trukhin D, Kovalenko NV, et al. A phase III, randomized, double-blind, multicenter study to compare the efficacy, safety, pharmacokinetics, and immunogenicity between SB8 (proposed bevacizumab biosimilar) and reference bevacizumab in patients with metastatic or recurrent nonsquamous non-small cell lung cancer. Lung Cancer. 2020;146:12–18. doi: 10.1016/j.lungcan.2020.05.027. [DOI] [PubMed] [Google Scholar]
- 33.Reinmuth N, Bryl M, Bondarenko I, Syrigos K, Vladimirov V, Zereu M, et al. PF-06439535 (a bevacizumab biosimilar) compared with reference bevacizumab (Avastin®), both plus paclitaxel and carboplatin, as first-line treatment for advanced non-squamous non-small-cell lung cancer: a randomized, double-blind study. BioDrugs. 2019;33(5):555–570. doi: 10.1007/s40259-019-00363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Syrigos K, Abert I, Andric Z, Bondarenko IN, Dvorkin M, Galic K, et al. Efficacy and safety of bevacizumab biosimilar FKB238 versus originator bevacizumab: results from AVANA, a Phase III trial in patients with non-squamous non-small-cell lung cancer (non-sq-NSCLC) BioDrugs. 2021;35(4):417–428. doi: 10.1007/s40259-021-00489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trukhin D, Poddubskaya E, Andric Z, Makharadze T, Bellala RS, Charoentum C, et al. Efficacy, safety and immunogenicity of MB02 (bevacizumab biosimilar) versus reference bevacizumab in advanced non-small cell lung cancer: a randomized, double-blind, Phase III study (STELLA) BioDrugs. 2021;35(4):429–444. doi: 10.1007/s40259-021-00483-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv192–iv237. 10.1093/annonc/mdy275. [DOI] [PubMed]
- 37.Hanna NH, Schneider BJ, Temin S, Baker S, Brahmer J, Ellis PM, et al. Therapy for stage IV non–small-cell lung cancer without driver alterations: ASCO and OH (CCO) joint guideline update. J Clin Oncol. 2020;38(14):1608–1632. doi: 10.1200/JCO.19.03022. [DOI] [PubMed] [Google Scholar]
- 38.Socinski MA, Waller CF, Idris T, Bondarenko I, Luft A, Beckmann K, et al. Phase III double-blind study comparing the efficacy and safety of proposed biosimilar MYL-1402O and reference bevacizumab in stage IV non-small-cell lung cancer. Ther Adv Med Oncol. 2021;13:17588359211045845. doi: 10.1177/17588359211045845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aguiar P, Jr, Giglio AD, Perry LA, Penny-Dimri J, Babiker H, Tadokoro H, et al. Cost-effectiveness and budget impact of lung cancer immunotherapy in South America: strategies to improve access. Immunotherapy. 2018;10(10):887–897. doi: 10.2217/imt-2017-0183. [DOI] [PubMed] [Google Scholar]
- 40.Liao W, Huang J, Hutton D, Li Q. Cost-effectiveness analysis of first-line pembrolizumab treatment for PD-L1 positive, non-small cell lung cancer in China. J Med Econ. 2019;22(4):344–349. doi: 10.1080/13696998.2019.1570221. [DOI] [PubMed] [Google Scholar]
- 41.Nogami N, Barlesi F, Socinski MA, Reck M, Thomas CA, Cappuzzo F, et al. IMpower150 final exploratory analyses for atezolizumab plus bevacizumab and chemotherapy in key NSCLC patient subgroups with EGFR mutations or metastases in the liver or brain. J Thorac Oncol. 2022;17(2):309–323. doi: 10.1016/j.jtho.2021.09.014. [DOI] [PubMed] [Google Scholar]
- 42.Melosky B, Reardon DA, Nixon AB, Subramanian J, Bair AH, Jacobs I. Bevacizumab biosimilars: scientific justification for extrapolation of indications. Future Oncol. 2018;14(24):2507–2520. doi: 10.2217/fon-2018-0051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.