

Abstract
Glioblastoma (GBM) is an aggressive primary brain cancer; with a 5-year survival of ~5%. Challenges that hamper GBM therapeutic efficacy include: (i) tumor heterogeneity, (ii) treatment resistance, (iii) immunosuppressive tumor microenvironment (TME) and (iv) the blood-brain barrier (BBB). The C-X-C Motif Chemokine Ligand-12/ C-X-C Motif Chemokine Receptor-4 (CXCL12/CXCR4) signaling pathway is activated in GBM and is associated with tumor progression. Although the CXCR4 antagonist (AMD3100) has been proposed as an attractive anti-GBM therapeutic target, it’s poor pharmacokinetic properties, and unfavorable bioavailability have hampered its clinical implementation. Thus, we developed synthetic protein nanoparticles (SPNPs) coated with the transcytotic peptide iRGD (AMD3100-SPNPs) to target the CXCL2/CXCR4 pathway in GBM via systemic delivery. We showed that AMD3100-SPNPs block CXCL12/CXCR4 signaling in three mouse and human GBM cell cultures in vitro and in a GBM mouse model in vivo. This results in (i) inhibition of GBM proliferation, (ii) reduced infiltration of CXCR4+ monocytic myeloid derived suppressor cells (M-MDSCs) into the TME, (iii) restoration of BBB integrity, and (iv) induction of immunogenic cell death (ICD), sensitizing the tumor to radiotherapy, and leading to anti-GBM immunity. Additionally, we showed that combining AMD3100-SPNPs with radiation led to long term survival; with ~60% of GBM tumor bearing mice remaining tumor free, after rechallenging with a second GBM in the contralateral hemisphere. This was due to a sustained anti-GBM immunological memory response that prevented tumor recurrence without additional treatment. In view of the potent ICD induction and reprogrammed tumor microenvironment, this SPNP-mediated strategy has a significant clinical translation applicability.
Keywords: glioma, chemo-immunotherapy, systemic delivery, tumor microenvironment
Graphical Abstract
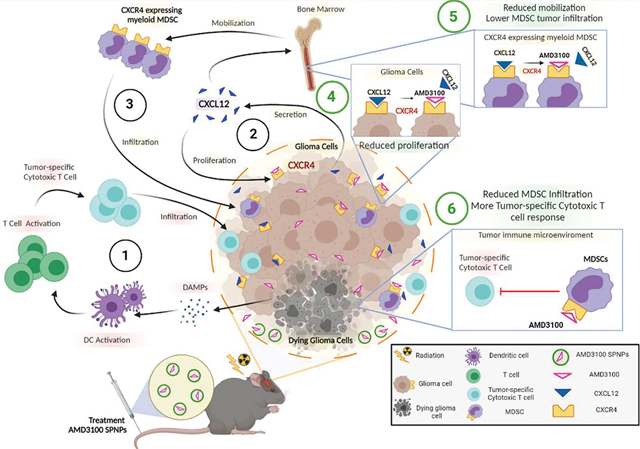
Immunological mechanism targeting Glioblastoma (GBM) upon blocking CXCR4 signaling pathway with AMD3100-conjugated nanoparticles (SPNPs). (1) Radiotherapy induces glioma cell death, followed by release of Damage-associated molecular patterns (DAMPs) release. Dendritic cells (DC) are activated by DAMPs and migrate to the regional lymph node where they prime cytotoxic T lymphocyte immune response. Tumor-specific cytotoxic T cells infiltrate the tumor and target glioma cells. (2) Glioma cells express CXCR4, as well its ligand CXCL12. CXCL12 induces glioma cell proliferation and, (3) CXCL12 mediates mobilization of CXCR4 expressing MDSC from bone marrow, which infiltrate the tumor, and inhibit tumor-specific cytotoxic T cells activity. GEMM of glioma were treated systemically with SPNPs AMD3100-SPNPs plus radiation. AMD3100-SPNPs inhibit the interaction between CXCR4 and CXCL12, thus (4) inhibiting glioma cell proliferation and (5) reducing mobilization in the bone marrow of CXCR4 expressing myeloid MDSC, (6) leading a reduced MDSC tumor infiltration and enhancing tumor specific cytotoxic T cell response.
Introduction
Glioblastoma (GBM) is an aggressive brain tumor with poor prognosis, characterized by a high level of cellular and molecular heterogeneity, high proliferative capacity, and invasive borders, making GBM challenging to treat.1 The invasive characteristics of GBM lead to infiltration of tumor cells into the normal brain tissue, making GBM difficult to completely resect and increasing the likelihood of tumor recurrence. This is compounded by the presence of immunosuppressive immune cells which hinder the effectiveness of immunotherapies.2 Moreover, the presence of the blood-brain barrier (BBB) provides both physical and biochemical barriers to drug delivery into the brain.3 This limits the brain permeability of many chemotherapeutic drugs, including monoclonal antibodies, antibody-drug conjugates, and hydrophilic molecules that do not readily cross lipid bilayers.4
The CXCL12/CXCR4 signaling pathway is involved in multiple physiological processes including hematopoiesis,5 retention of hematopoietic stem cells (HSCs) in the bone marrow, and central nervous system (CNS) development.6, 7 Several studies have illustrated the involvement of activated CXCL12/ CXCR4 signaling in solid cancers in promoting survival, growth, and metastasis.8 In GBM, it has been previously demonstrated the CXCL12/CXCR4 signaling pathway is important for sustained invasion,9 enhanced angiogenesis,10 and maintenance of glioma stem-cell migration and therapeutic resistance.11 This pathway is particularly upregulated under hypoxic conditions, a feature that is associated with worse prognosis in GBM.12
CXCR4 is expressed by many cells, including hematological progenitor cells, myeloid cells, stromal fibroblasts, endothelial cells, epithelial cells, and tumor cells.13, 14, 15 Most importantly, CXCR4 expression on myeloid cells promotes trafficking of myeloid-derived suppressor cells (MDSCs) in several cancers such as osteosarcoma,16 ovarian cancer,17 colorectal cancer,18 metastatic liver,19 and leukemia.20 Enhanced MDSCs infiltration promotes an immunosuppressive tumor microenvironment and further contributes to immunotherapeutic resistance. Thus, blocking CXCR4 provides an attractive target to promote effective immunotherapy. However, the impact of CXCL12/CXCR4 signaling on immune-mediated therapeutic outcomes in GBM and its impact on reprograming the immunosuppressive tumor immune microenvironment (TIME) has not yet been elucidated.
The presence of the BBB limits the efficacy of both conventional and novel therapies for GBM. Thus, the development of nano-based therapies that have the ability to cross the BBB is of great interest to improve GBM clinical outcomes. A wide variety of nanoparticles have been tested in preclinical settings to facilitate effective drug delivery into brain tumors.21 Remaining challenges in nanoparticle safety, include the fact that most of the widely used NPs are composed of non-organic materials that tend to accumulate in the liver.22 Another compounding factor, especially for GBM therapeutics, relates to their limited BBB penetration capacity.20 We recently developed biologically compatible NPs comprised of polymerized human serum albumin (HSA) and oligo ethylene glycol (OEG), equipped with the transcytotic peptide iRGD.23 HSA was used as the primary component because of its biological compatibility, and well-studied kinetic, and interactions with other cellular proteins and receptors.24 The incorporation of the tumor targeting transcytotic peptide, iRGD, results in the ability of the SPNPs to target the tumor mass, after systemic delivery.23 We previously demonstrated that these SPNPs loaded with siRNA against signal transducer and activator of transcription 3 (STAT3i) are able to silence STAT3 within the GBM cells in vivo.23 The main advantage of the SPNPs is that they can deliver their therapeutic cargo to the target tissue (GBM) after systemic delivery.
Here, we used SPNPs loaded with the CXCR4 inhibitor, AMD3100, to block CXCR4 signaling in an aggressive intracranial GBM model. We demonstrate that blocking CXCR4 results in reduced MDSCs trans-endothelial migration in vitro and reduced infiltration of CXCR4+ M-MDSCs to the GBM TIME in vivo. Interestingly, we showed that blocking CXCR4 sensitizes GBM cells to radiation-induced immunogenic cell death (ICD) which triggers an anti-GBM adaptive immune response. With the potent ICD induction and reprogrammed immunosuppressive microenvironment, SPNPs- elicited antigen presentation, immune priming, and GBM specific immunological T-cell mediated immunity.
Results
Infiltration of CXCR4+ M-MDSCs within the tumor immune microenvironment in genetically engineered mouse GBM models
To study the impact of the CXCL12/CXCR4 signaling pathway on the infiltration of immature myeloid cells to the tumor immune microenvironment (TIME), we used three different implantable glioma models. Two models were aggressive, fast-growing tumors, developed using the SB-generated neurospheres, i.e. OL61, and RPA as described before (Figure 1A).25, 26 The third model was a less aggressive, slower-growing GBM model developed using RCAS-TVA system (Arf−/−) (Figure 1A).25 When comparing the median survival (MS) between the three groups, we found that both OL61 and RPA had a median survival (MS= 18dpi and 14dpi, respectively). Whereas the Arf−/− tumor-bearing mice had a longer MS compared to the other groups (Arf−/− MS= 69dpi, P< 0.001) (Figure 1B). Next, we investigated the frequencies and the subsets of CXCR4+ myeloid derived suppressor cells (MDSCs) infiltrating into the TIME of each GBM model. MDSCs infiltrating the TIME could be of granulocytic (PMN-MDSCs) (CD45high/CD11b+/Ly6G+/Ly6Clow) or monocytic (M-MDSCs) (CD45high/CD11b+/Ly6G−/Ly6Chigh) origin. First, we characterized the GBM infiltrating MDSCs subsets in each GBM model and found that in contrast to PMN-MDSCs which showed no differences in frequencies between the three groups, the frequencies of M-MDSCs infiltrating the tumors in the aggressive GBM models (i.e. OL61 and RPA) were significantly higher compared to the Arf−/− model (36% and 35% vs 19%, respectively, (P<0.01, Figure 1C–E)). Next, we analyzed the expression of CXCR4 on both PMN-MDSCs and M-MDSCs. Consistent with their abundance, we found that the majority of M-MDSCs infiltrating the OL61 and RPA tumor models express higher level of CXCR4, with no difference in the frequency of CXCR4+ PMN-MDSCs between the three tumor models (Figure 1F– I). This suggests that the frequency of CXCR4+ M-MDSCs which infiltrate GBM correlates with tumor aggressiveness.
Figure 1. Aggressive genetically engineered GBM models are associated with activated CXCR4/CXCL12 signaling and infiltration of immunosuppressive myeloid cells.
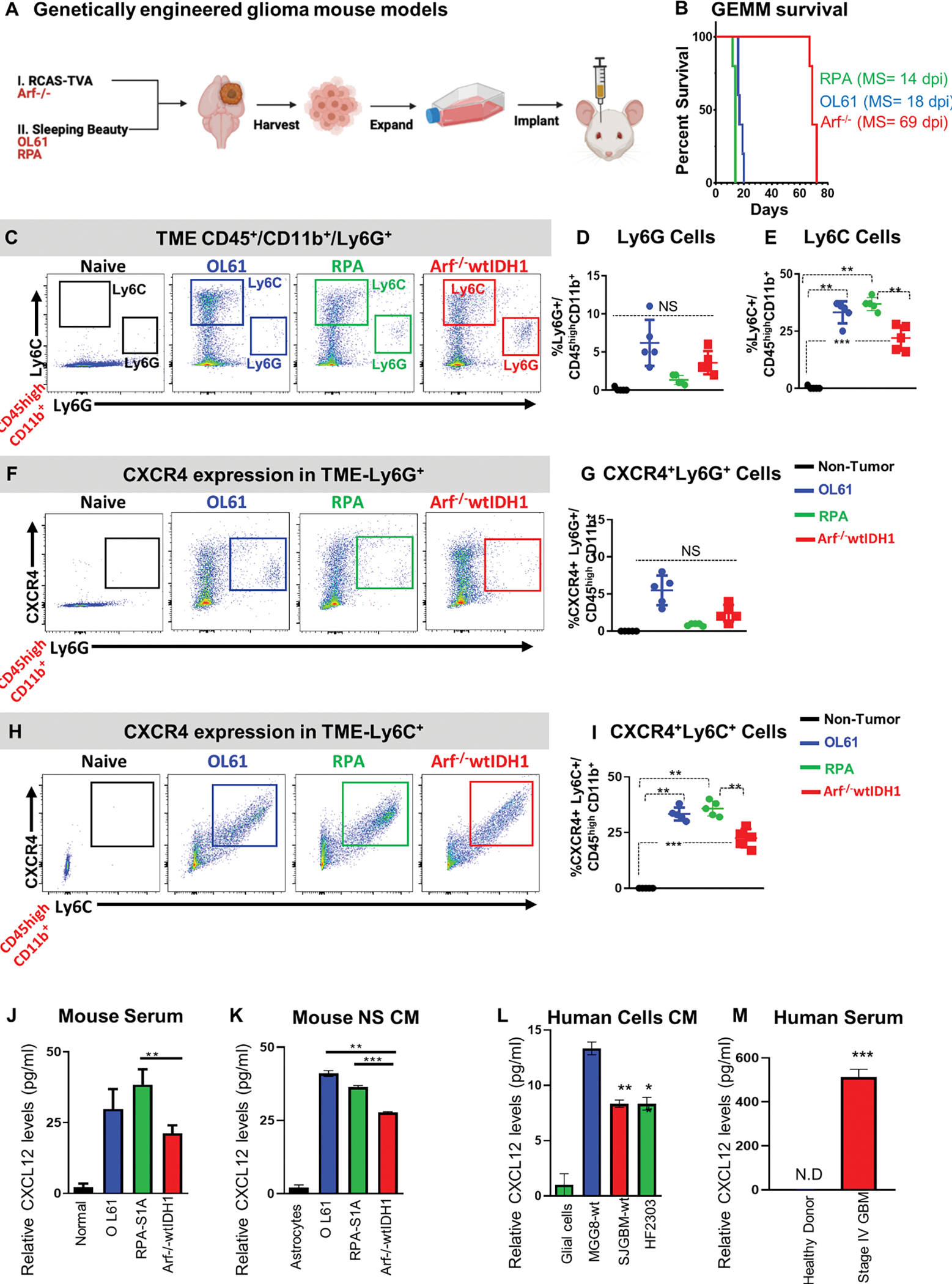
(A) Experimental design of the generation of GBM models using the sleeping beauty (SB, rapidly growing, RPA and OL61) and RCAS-TVA technology (slow-growing, Arf−/−). Neurospheres from each model were harvested, cultured and used for intracranial implantation in animals. (B) Kaplan-Meier survival curves of mice bearing Arf−/−, OL61, or RPA (MS: median survival). (C-E) Characterization of granulocytic and monocytic myeloid cell populations (Ly6G vs Ly6C respectively) in normal brain or OL61, RPA, and Arf−/− tumor bearing mice. Arf−/− wtIDH1 tumor bearing mice display lower percentage of monocytic myeloid cells (M-MDSCS; Ly6C+) compared to OL61 and RPA tumor bearing mice. (F, G) Flow analysis of the Ly6G+ CXCR4+ myeloid cells in normal brain or in the tumor from OL61, RPA, and Arf−/− implanted mice. (H-I) Flow analysis of the Ly6C+ CXCR4+ myeloid cells in normal brain or in the tumor from OL61, RPA, and Arf−/− implanted mice. Quantitative ELISA of the CXCL12 levels in mouse serum of tumor-bearing animals (J), conditioned media from cultured mouse, (K) conditioned media from human cells (L), and serum from control and GBM patients (M). p<0.05, ** p<0.01, *** p<0.001. Student’s t test, (n=5/group).
CXCL12 is the primary ligand that binds to CXCR4 receptors expressed by GBM cells, and its expression is also responsible for the homeostasis of HSCs in the bone marrow (BM).7, 11, 27 We hypothesized that the enhanced infiltration of M-MDSCs in aggressive GBM models results from activation of CXCL12/CXCR4 signaling. Quantitative ELISA analysis showed that the level of CXCL12 was lower in the Arf−/− GBM model in both mouse serum from implanted animals as well as IN conditioned media of cultured GBM cells (P<0.01, Figure 1J, K). We also observed that CXCL12 is expressed at higher levels in patient derived glioma cells (MGG8, SJGBM2, and HF2303) when compared to media from normal glial cells (Figure 1L). We also found significant increase in CXCL12 level in serum from stage IV GBM patients compared to serum from healthy donors (Figure 1M). Overall, these results suggest that CXCL12/CXCR4 signaling is activated in aggressive GBM and is associated with enhanced infiltration of immunosuppressive M-MDSCs.
CXCR4 is expressed primarily by monocytic MDSCs in the spleen and blood from GBM mouse models and is associated with poor prognosis in GBM patients.
We next asked if the change we observed in the frequency of CXCR4+ MDSCs in the TIME from different GBM models was associated with changes in CXCR4 expressing myeloid cells in the blood, spleen, and BM. We found that in both the blood and spleen, PMN-MDSCs were the dominant population with no difference in the frequencies between the three glioma models (Figure 2A, B, D, E). However, the frequency of M-MDSCs was significantly higher in the blood from OL61 and RPA tumor-bearing mice compared to the frequency observed in Arf−/− glioma-bearing mice (Figure 2A, B). Like the TIME, we found that there was increased expression of CXCR4 by the M-MDSC population in the blood from OL61 tumor bearing mice compared to the two other models (Figure 2C). There were no differences in the frequencies of M-MDSC and PMN-MDSC populations in the spleen of the three GBM models, however, M-MDSCs were the major population expressing CXCR4 (Figure 2D–F).
Figure 2. CXCR4 is expressed primarily by monocytic MDSCs (CD45high/CD11b+/Ly6Chigh) and is associated with poor prognosis.
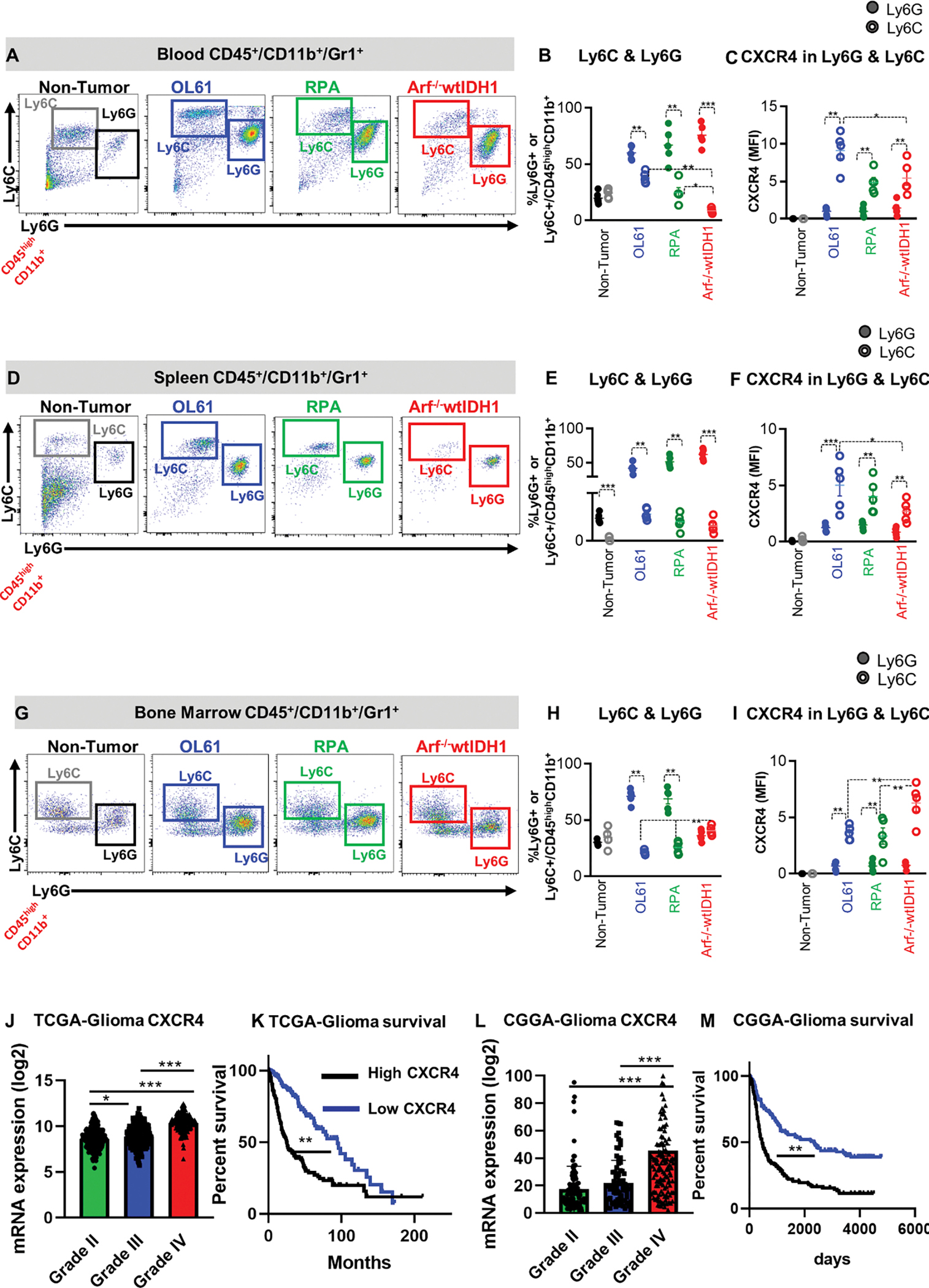
(A, B) Representative flow cytometry plots and quantification of the percentage of PMN-MDSCs (CD45high/CD11b+/Ly6G+/Ly6Clow) or M-MDSCs (CD45high/CD11b+/ Ly6Chigh) in bone marrow (BM) from normal mice (N), and mice implanted with OL61, RPA, or Arf−/− wtIDH1 neurospheres. (C) Quantitative analysis of CXCR4 expression in conditions from (B). (D, E) Representative flow cytometry plots and quantification of the percentage of PMN-MDSCs (CD45high/CD11b+/Ly6G+/Ly6Clow) or M-MDSCs (CD45high/CD11b+/ Ly6Chigh) in blood from normal mice (N), and mice implanted with OL61, RPA, or Arf−/− wtIDH1 neurospheres. (F) Quantitative analysis of CXCR4 expression in conditions from (E). (G, H) Representative flow cytometry plots and quantification of the percentage of PMN-MDSCs (CD45high/CD11b+/Ly6G+/Ly6Clow) or M-MDSCs (CD45high/CD11b+/ Ly6Chigh) in spleen from normal mice (N), and mice implanted with OL61, RPA, or Arf−/− wtIDH1 neurospheres. (I) Quantitative analysis of CXCR4 expression in conditions from (H). (J) Analysis of CXCR4 gene expression for glioma patients according to their grade, Grade II (n=226), Grade III (n= 244), and Grade IV (n=150). (K) Kaplan-Meier survival analysis of TCGA glioma patients with high vs low level of CXCR4 expression. (L) Analysis of CXCR4 gene expression for glioma patients in CGGA database according to their grade, Grade II (n=103), Grade III (n= 79), and Grade IV (n=139). (M) Kaplan-Meier survival analysis of CGGA glioma patients with high vs low level of CXCR4 expression. * p<0.05, ** p<0.01, *** p<0.005, One-way ANOVA, (n=5/group).
We also found that the frequency of M-MDSCs was higher in the BM from the Arf−/− GBM mouse model when compared to Ol61 and RPA GBM models (Figure 2G, H). This corresponded to higher expression of CXCR4 in the M-MDSCs residing in the BM of the Arf−/− GBM model (Figure 2I). There were no significant differences in the frequencies of CD8+ or CD4+ T cells in the BM between the three GBM mouse models (Figure S1A–C), suggesting that CXCL12-CXCR4 signaling does not mediate T cell sequestration. These results suggest that there is increased trafficking of CXCR4+ M-MDSCs from the bone morrow into circulation, and to the TIME, in the OL61 and RPA GBM models, that promote an immunosuppressive glioma TIME.
To evaluate if CXCR4 expression is associated with GBM tumor progression in the clinical setting, we queried the TCGA data and analyzed CXCR4 expression in three different glioma grades (Grade II, III, and IV). We also examined the median survival (MS) of glioma patients in the context of CXCR4 (CXCR4) expression (i.e. MS of patients expressing high vs low levels of CXCR4). TCGA-Glioma analysis revealed that CXCR4 gene expression level is correlated with tumor grade, and its expression is associated with a worse prognosis in glioma patients (Figure 2J, K). Similar results were found when analyzing data from the Chinese Glioma Genome Atlas (CGGA) (Figure 2L, M). These results suggest that CXCR4 expression is associated with poor prognosis in glioma patients.
CXCR4 signaling disrupts brain endothelial cells’ barrier permeability and promotes myeloid cells’ infiltration
To assess the effect of CXCR4 signaling on blood-brain barrier (BBB) integrity and permeability, we used an in vitro model established in a transwell dual-chamber system. In this model, brain microvascular endothelial cells (BECs) were grown to confluence in the upper chamber, on collagen type IV coated filters of the trans-well dual-chamber system. To mimic the BBB biology, pericytes were grown on the opposite side (basolateral) of the filter (Figure 3A). First, we tested the effect of CXCR4 inhibition on myeloid cells’ transmigration from the apical-basolateral side. Conditioned media (CM) collected from OL61, Arf−/− and RPA cultures treated with either vehicle or the CXCR4 inhibitor AMD3100 (according to the IC50 (Figure S2)), were placed in the basolateral side (Figure 3A). MDSCs were placed on the apical side of the BBB transwell system. Results showed that treatment with AMD3100 significantly reduced the number of myeloid cells which had transmigrated through the basolateral chamber with all three GBM cells CM tested (Figure 3B).
Figure 3. CXCR4 signaling enhances myeloid cells transmigration and increases brain endothelial cell (BEC) barrier permeability.
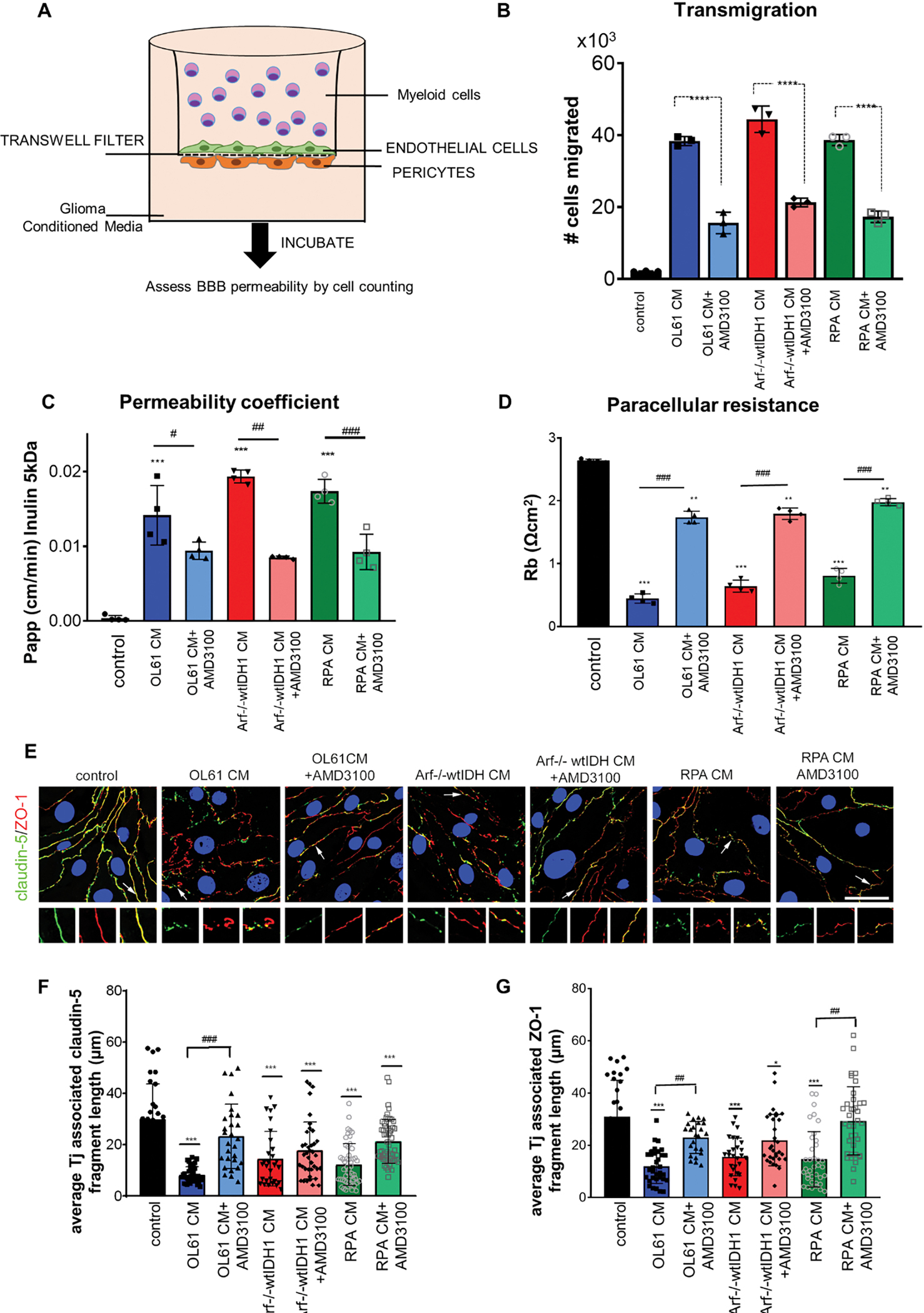
(A) Diagram of transwell dual-chamber system used for cell migration assay. (B) Bar graph represents the number of the myeloid cells migrated through the endothelial-pericytes transmembrane. (C) Permeability coefficient (Papp) for FITC-inulin in mBMEC monolayers exposed to condition media collected from OL61, Arf−/−wtIDH and RPA cells with or without CXCR4 inhibitor AMD3100 for 24 hrs. (D) Bar graph represents paracellular resistance (Rb) value at 24 hrs for all analyzed groups. (E) Immunofluorescence staining for tight junction (Tj) proteins claudin-5 and ZO-1 in control and cells exposed to OL61, OL61+AMD3100, Arf−/−wtIDH, Arf−/−wtIDH+AMD3100, and RPA and RPA+AMD3100 for 24 hrs. Arrow and magnified images indicate pattern and colocalization of claudin-5 and ZO-1 on the cell border. Scale bar 50mm. Quantitation of the average TJ-associated (F) claudin-5 and (G) ZO-1 fragment length in claudin-5/ZO-1 costained immunofluorescent images in control and cells exposed to OL61, OL61+AMD3100, Arf−/−wtIDH, Arf−/−wtIDH+AMD3100, and RPA and RPA+AMD3100 for 24 hrs. Data are shown as means ± SD. n = 3–5; ***p<0.0001 and **p<0.001 comparing to control. ###p<0.0001 comparing experimental groups with and without inhibitor AMD3100.
We also analyzed the effects of conditioned media (CM) collected from glioma cells on the barrier properties of mouse brain endothelial cells (mBECs). Results showed that CM collected from OL61, RPA and Arf−/− GBM cells increased brain endothelial barrier permeability for the small molecular weight tracers, i.e., Cadaverine (1kDa), Dextran (3kDa), and Inulin (5kDa), but not for high molecular weight Dextran (10kDa) (Figure S3, and Figure 3C). Our results also show that blocking CXCR4 with AMD3100 rescued the impaired barrier integrity (Figure S3, and Figure 3C). This was validated in multi-frequency measurements of barrier properties (resistance, capacitance, and impedance) showing that AMD3100 treatment caused a two-fold increase in the paracellular resistance (Figure 3D). Morphologically, the effects of GBM CM on brain endothelial barrier integrity were associated with alteration in tight junction (Tj) protein expression, localization, and complex organization (Figure 3E). Because of their role in preserving the integrity of the BBB,28 the major occlusion Tj protein claudin-5 and ZO-1 were analyzed by immunofluorescence and confocal microscopy. Results showed that CM collected from the three cultured GBM cells caused defragmented, and punctate staining of claudin-5 and ZO-1 in mBECs (Figure 3E–G). This was associated with dislocation of both Tj proteins from mBECs and significant reduction of claudin-5 and ZO-1 associated Tj length (Figure 3E–G). Then, we analyzed the interactions between the ZO-1 and claudin-5 by performing a proximal ligation assay (PLA) to detect protein-protein interactions at higher specificity and sensitivity. Consistent with the immunofluorescence, PLA analysis showed decreased interactions between claudin-5 and ZO-1 in mBECs treated with CM only (P<0.01, Figure S4). Blocking CXCR4 resulted in more than 2-fold increase in the interactions between claudin-5 and ZO-1 (P<0.01, Figure S4). This functional rescue of the barrier upon CXCR4 blockade was accompanied by re-establishing the Tj complex, shown by restoration of claudin-5 and ZO-1 localization on the cell border mired in increased length of the Tj fragment and increased the number of interactions between claudin and ZO-1 (P<0.01, Figure 3F, G, Figure S4, Figure S5). We further validated this effect using human brain endothelial cells (hBECs) incubated with CM collected from two patient derived GBM cells (SJGBM2 and HF2303) in the presence or absence of either vehicle or AMD3100 (according to the IC50 (Figure S2B)). Consistent with the mouse data, our results showed that blocking CXCR4 expression reversed the effect of CM-mediated disruption of ZO-1 and claudin-5 (P<0.01, Figure S4). These results verified the protective effects of the CXCR4 blockade from GBM-mediated derangement of the BBB integrity.
Blocking of CXCR4 sensitizes mouse and human glioma cells to radiotherapy and induces immunogenic cell death
CXCR4 signaling is crucial for GBM cell survival and proliferation.11, 27 We next sought to determine whether CXCR4 inhibition would induce radio-sensitization and also induce immunogenic cell death (ICD). First, we performed cell viability (Figure 4A) and real-time cell proliferation assay (Figure S6) in three mouse glioma (i.e. RPA, OL61, Arf−/−) and three human glioma cell cultures (i.e. MGG8, SJGBM2, HF2303) treated with either saline, AMD3100, IR, or AMD3100+ IR. Results showed that treatment with either AMD3100 or IR alone markedly inhibited cell proliferation in all mouse (Figure 4B–D) and human (Figure 4E–G) glioma cells. Interestingly, combination treatment of AMD3100 with IR showed a greater decrease in cell viability in all GBM cells tested, compared to AMD3100 or IR alone (Figure 4B–G).
Figure 4. CXCR4 blockade enhances radio-sensitivity and immunogenic cell death in mouse and human glioma cells.
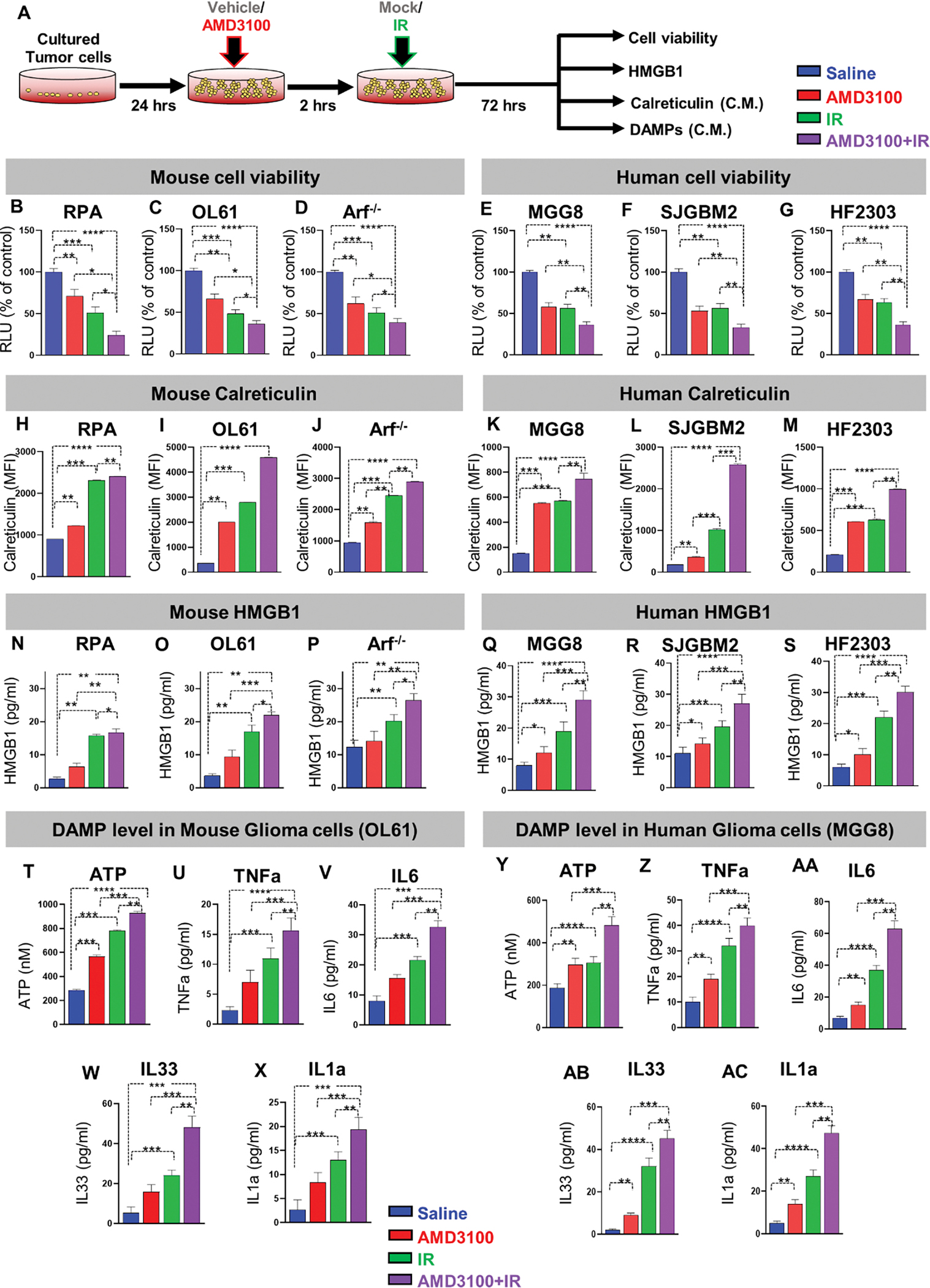
(A) Schematic shows the in vitro application of AMD3100 and/ or radiation in mouse and human cell cultures. Mouse and patient-derived glioma cells were treated with either free-AMD3100 or in combination with radiation at their respective IC50 doses for 72h. All mouse and human glioma cells were pre-treated with AMD3100 2h prior to irradiation with 3Gy and 10Gy of radiation respectively. (B-D) Bar plot shows the % viable mouse glioma cells (RPA, OL61, or Arf−/−) after treatment with saline, AMD3100, IR (3Gy), or AMD3100+IR. (E-G) Bar plot shows the % viable human glioma cells (MGG8, SJGBM2, or HF2303) after treatment with saline, AMD3100, IR (3Gy), or AMD3100+IR. (H-J) Bar graphs represent levels of immunogenic cell death (ICD) marker Calreticulin in mouse glioma cells (RPA, OL61, or Arf−/−) after treatment with saline, AMD3100, IR (3Gy), or AMD3100+IR. (K-M) Bar graphs represent levels of immunogenic cell death (ICD) marker Calreticulin in human glioma cells (MGG8, SJGBM2, or HF2303) after treatment with saline, AMD3100, IR (3Gy), or AMD3100+IR. (N-P) Bar graphs represent levels of immunogenic cell death (ICD) marker HMGB1 in mouse glioma cells (RPA, OL61, or Arf−/−) after treatment with saline, AMD3100, IR (3Gy), or AMD3100+IR. (Q-S) Bar graphs represent levels of immunogenic cell death (ICD) marker HMGB1 in human glioma cells (MGG8, SJGBM2, or HF2303) after treatment with saline, AMD3100, IR (3Gy), or AMD3100+IR. (T-AC) Quantitative ELISA show the levels of DAMPs (ATP, TNFα, IL6, IL33, IL1α), as markers for ICD in the mouse glioma cells OL61 and the human glioma cells MGG8 after treatment with saline, AMD3100, IR (3Gy), or AMD3100+IR. All AMD3100 treatment were done at IC50 values of the corresponding cell line alone or in combination with 3Gy of IR. (Blue= Saline red= AMD3100 alone, green= IR alone, violet= AMD3100 + IR). MFI= mean fluorescence intensity. ns= non-significant, *p< 0.05 **p< 0.01, ***p< 0.0001, ****p< 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 3 biological replicates).
We hypothesized that combining CXCR4 antagonist AMD3100 with radiation would induce immunogenic cell death (ICD) in glioma cells. This unique mechanism of cell death results in an increased secretion of Damage-Associated Molecular Patterns (DAMPs) molecules. Therefore, we measured the levels of HMGB1, Calreticulin (CRT), as well as ATP, IL1α, IL6, IL33, and TNFα in multiple GBM mouse and human cells upon treatment with saline, AMD3100, IR, or combination therapy. Mouse-GBM cells treated with AMD3100 displayed a 1.3, 4.3, and 1.6-fold (p ≤ 0.0001) increase in CRT expression relative to vehicle control group for RPA (Figure 4H), OL61 (Figure 4I), and Arf−/− (Figure 4J) cells respectively. This response was further increased by approximately 2, 2.5, and 2-fold (P<0.01) with AMD3100+IR treatment for RPA, OL61, and Arf−/− cells, respectively (Figure 4H–J). A similar response was observed in human-glioma cells treated with AMD3100 alone or AMD3100+IR (Figure 4K–M). We also tested the release of HMGB1 in the supernatants of mouse and human-glioma cells in response to AMD3100, IR, or AMD3100+IR treatments. Upon treatment with AMD3100, we observed a 1.5, 2, and 1.3-fold increase in HMGB1 release in the CM from RPA (Figure 4N), OL61 (Figure 4O), and Arf−/− (Figure 4P) (p ≤ 0.0001) mouse glioma cells, respectively. We also observed that, combination therapy resulted in ~2-fold increase in the extracellular HMGB1 release in the supernatant of all mouse glioma cells (Figure 4N–P). A similar response was observed in human-glioma cells treated with AMD3100 alone or in combination with IR (Figure 4Q–S). Next, we assessed DAMPs released from all glioma cells which received mono or dual therapy as described. Consistent with HMGB1 and Calreticulin expression, the levels of ATP, TNFα, IL6, IL33, and IL1α in all three mouse glioma cells (OL61, RPA, and Arf−/−) was significantly higher in the combination treatment (Figure 4T–X Figure S7A, B) and in MGG8 human-glioma cells (Figure 4Y–AC, Figure S7C, D). Taken together, these results demonstrated that AMD3100 in combination with IR enhances the radio-sensitivity and induces ICD in both mouse and human-glioma cells.
Synthetic protein nanoparticles’ (SPNPs) design, synthesis, and characterization
AMD3100 has poor pharmacokinetic and BBB penetration, and therefore, its delivery to the GBM TIME is limited.29 Thus, we designed SPNPs to facilitate systemic delivery of AMD3100 to GBM. SPNPs were formulated following the order of components depicted in Figure 5A followed by electrohydrodynamic (EHD) jetting of the formulation to form well-defined nanoparticles (Figure 5B).30,31 In particular, a 7.5w/v% of human serum albumin was dissolved in a solvent system of 80% water and 20% ethylene glycol, by volume. Thereafter, bovine serum albumin conjugated Alexa Fluor 647 was introduced into the solution at a 0.50 wt% relative to HSA. The transcytotic peptide, iRGD, was added, followed by the addition of AMD3100 at 3.75mg/mL of the total solution. Lastly, the bifunctional OEG macromer (NHS-OEG-NHS, 2kDa) was added at 10% by weight relative to the HSA. After jetting, the SPNPs were incubated at 37 °C to allow the OEG macromers to react with the albumin molecules resulting in water stable SPNPs.
Figure 5. Preparation of electrohydrodynamic (EHD)-jetting and characterization of AMD3100-SPNPs.
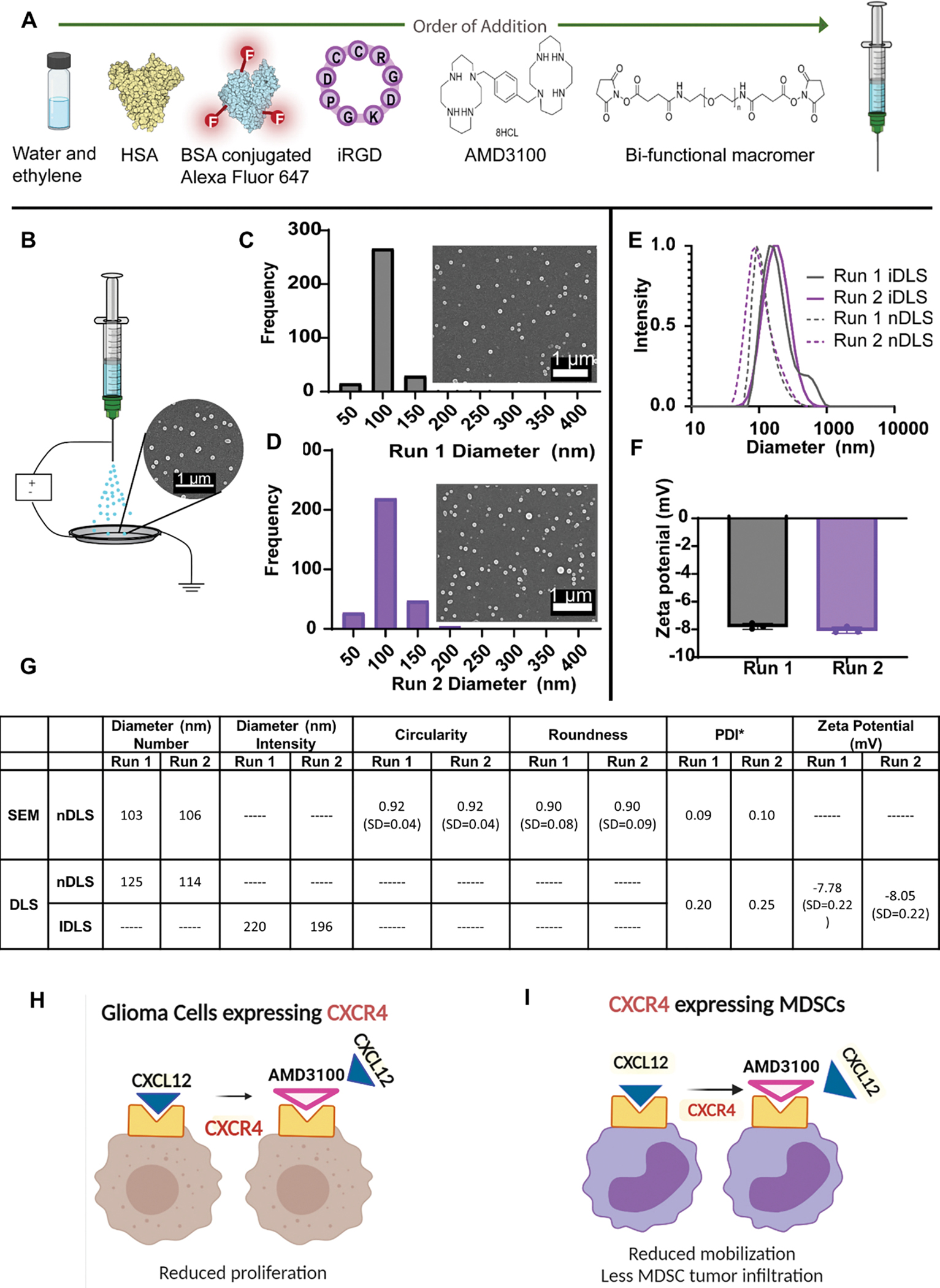
(A) Formulation of AMD3100-SPNPs indicating the order of addition of different components. (B) Schematic of the jetting process for AMD3100-SPNPs depicting a Scanning Electron Microscopy (SEM) image of the SPNPs jetted atop of the collection plate (scale bar = 1μm). (C) Size distribution of SPNPs of an independent run, Run 1, in their dry state characterized via SEM and ImageJ analysis. Average diameter, 103 ± 20 nm (PDI = 0.09). Scale bar = 1 μm. (D) Size distribution of SPNPs of a second independent run, Run 2, in their dry state characterized via SEM and ImageJ analysis. Average diameter, 106 ± 25 nm (PDI = 0.10). (E) Numbers based Dynamic Light Scattering (nDLS) size distribution (dashed) and Intensity based DLS (IDLS) of SPNPs in PBS comparing Run 1 and Run 2. (F) Zeta potential of Run 1 and Run 2. (G) Summary table of SPNP characterization of size, shape and charge. (H, I) Schematics represent the therapeutic advantages of blocking CXCR4 in glioma cells. (H) and myeloid cells (I).
To test the batch-to-batch variation during SPNPs formulation, we ran quality control comparing two batches of the nanoparticles (i.e. Run1 and Run2, Fig 5C–G). Results showed that the dry-state SPNPs after jetting had an average diameter ~100nm in both batches (Figure 5C, D). Moreover, there were no differences between the two batches in all quality control parameters tested (i.e. size, zeta potential, and PDI; Figure 5E–G), highlighting the narrow dispersity within and between batches. Furthermore, the AMD3100-SPNPs were highly spherical, as indicated by a circularity of 0.92 ± 0.04 and roundness of 0.89 ± 0.08 (Figure 5G). Once fully hydrated, the average diameter in intensity based dynamic light scattering (DLS) measurements of SPNPs in Run 1 and Run 2 were 220 nm and 196 nm, respectively (Figure 5G). The nDLS diameter for Run 1 and Run 2 were 125nm (PDI=0.2) and 114nm (PDI =0.25), respectively (Figure 5E, G). The zeta potential was −7.78 mV for Run 1 and −8.05 mV for Run 2.
To investigate whether the jetting process and the crosslinking have any significant effects on the secondary structure of albumin, we performed circular dichroism spectroscopy on SPNPs, SPNPs loaded with HSA before and after jetting, and naïve HSA (Figure S8). The secondary structures of each spectra were analyzed and categorized according to alpha helix (regular and distorted), beta sheet (regular and distorted), turns, and unordered. Results showed that neither the EHD jetting process, nor the crosslinking have a significant effect on HSA secondary structure compared to native HSA structure (Fig S9A, B).
We next tested the stability of the formulated SPNPs ex vivo by incubating equal number of SPNPs with mouse or human serum at 37˚ C. Results showed that the SPNPs particle size were similar between the SPNPs incubated with serum when compared to control (PBS) (Figure S9) after 24 hrs. Next, we tested the release behavior of AMD3100 SPNPs by performing in vitro drug release profile over a period of 9 days (Figure S10). We noticed an initial burst of AMD3100 release of 13% within the first two hours for a rate of 6.7%/hour (Figure S10). This was followed by a slower release rate (~2% per hour) during the13 hours and 17 hours timepoints (cumulative release of 22% and 35%, respectively). Thereafter, the release rate further decreased for the remainder of time <1% per hour (Figure S10). This indicates that the maximum AMD3100 release occurs at the early times after SPNPs administration (up to ~24 hours).
We hypothesize that the designed AMD3100 SPNPs will facilitate AMD3100 entry to the GBM microenvironment. This would result in a dual anti-tumor effect by inhibiting GBM cell proliferation, induction of immunogenic dell death, enhanced efficacy of radiotherapy, and reduce the infiltration of immunosuppressive CXCR4+ M-MDSCs in the TIME (Figure 5H, I).
AMD3100 activity is preserved when packaged in the SPNP formulation
To examine if the pharmacological activity of AMD3100 was retained in the nanoparticle formulation, we tested the efficacy of AMD3100-SPNPs to induce radio-sensitization and ICD in vitro using both mouse (OL61) and human (SJGBM2) glioma cells as described above (Figure S11, A). Monotherapy with either AMD3100-SPNPs or IR inhibit cellular proliferation, which was markedly decreased upon combination (Figure S11 B, C). We also observed that AMD3100-SPNPs combined with IR further enhanced calreticulin, HMGB1, and DAMPs release from both mouse and human glioma cells (Figure S11, D–L). These results indicate that the in vitro pharmacological activity of AMD3100 is not affected by its formulation as AMD3100-SPNPs.
SPNPs-mediate AMD3100 delivery to brain in vivo
In order to assess whether synthetic protein nanoparticles encapsulation improved the access of AMD3100 into the brain, we administered free AMD3100 or AMD3100-SPNPs systemically in brain tumor-bearing mice and measured the content of the drug in brain and plasma 8hr later using Ultra Performance Liquid Chromatography mass spectroscopy (UPLC-MS) (Figure S12A). While the total levels of AMD3100 in plasma were similar in mice treated with free AMD3100 (21.7 ± 1.92 ng) or AMD3100-SPNP (24.2 ±2.1 ng) (Figure S12B), the total levels of AMD3100 found in brain tissue were higher when mice received AMD3100-SPNPs (12.875 ng ± 2.89) than when they received free AMD3100 (4.175 ng±1.62) (Figure S12B, p<0.05, Student’s t test). In addition, the brain/plasma ratio (B/P) for AMD3100 was found to be ~3 times higher when AMD3100 was delivered encapsulated in SPNP (0.53 ± 0.11) than when it was delivered as free drug (0.19 ±0.07) (p<0.05, Student’s t test) (Figure S12C).
AMD3100 encapsulated in SPNPs when combined with radiation elicit enhanced therapeutic efficacy and anti-GBM immune response
Previous data indicated that blocking CXCR4 signaling prevents glioma progression.7, 12, 16, 27 To test the efficacy of AMD3100-SPNPs in vivo, GBM-bearing mice were treated intravenously with a total of 10 doses of AMD3100-SPNPs over the course of a three-week treatment regimen (Figure 6A). Since radiation therapy is the current standard of care for GBM, we tested the efficacy of AMD3100-SPNPs alone or in combination with two cycles of radiotherapy on the survival of tumor-bearing mice. We established a treatment modality which included a multi-dosage regimen of systemic nanoparticles administration combined with 10-days of localized radiotherapy in the GBM tumor model.23, 25, 32 Results showed that administration of empty SPNPs did not affect median survival compared to saline treated group (Figure 6B). Administration of AMD3100 or IR alone significantly enhanced MS of tumor-bearing mice (MS= 45 dpi for AMD3100, MS= 28dpi for IR; P<0.05, Figure 6B). However, AMD3100-SPNPs significantly enhanced survival compared to control or AMD3100 alone, underscoring the role of SPNPs in improving AMD3100 delivery to the GBM (MS= 61 dpi for AMD3100-SPNPs, MS= 45 dpi for AMD3100; P<0.05, Figure 6B). Interestingly, combining AMD3100-SPNPs with radiation resulted in a greater survival benefit with 67% of mice surviving long-term (P<0.05, Figure 6B). These mice remained tumor-free even after they were rechallenged with OL-61 in the contralateral hemisphere indicating the presence of sustained anti-GBM memory response. On the other hand, control mice succumbed due to tumor burden (MS= 19 days; P ≤ 0.0001, Figure 6C). Microscopic examination showed no evidence of intracranial tumor even after 60 days of tumor rechallenge (Figure 6D). In contrast, hematoxylin and eosin staining revealed the presence of a tumor in the hemisphere region of the saline and IR treated mice (Figure 6D). No apparent regions of hemorrhages, necrosis, or invasion were present in the long-term survivors after receiving the combination therapy (Figure 6D). To assess whether the combination treatment may have affected the surrounding brain architecture, we performed IHC staining using glial fibrillary acidic protein (GFAP) and myelin basic protein (MBP) as markers for myelin sheaths integrity. Result showed that there were no apparent changes in brain architecture in mice received the combined AMD3100-SPNPs + IR treatment when compared to the saline-treated control group (Figure 6D, middle images).
Figure 6. Combining AMD3100-SPNPs with IR prolong survival of GBM tumor bearing mice.
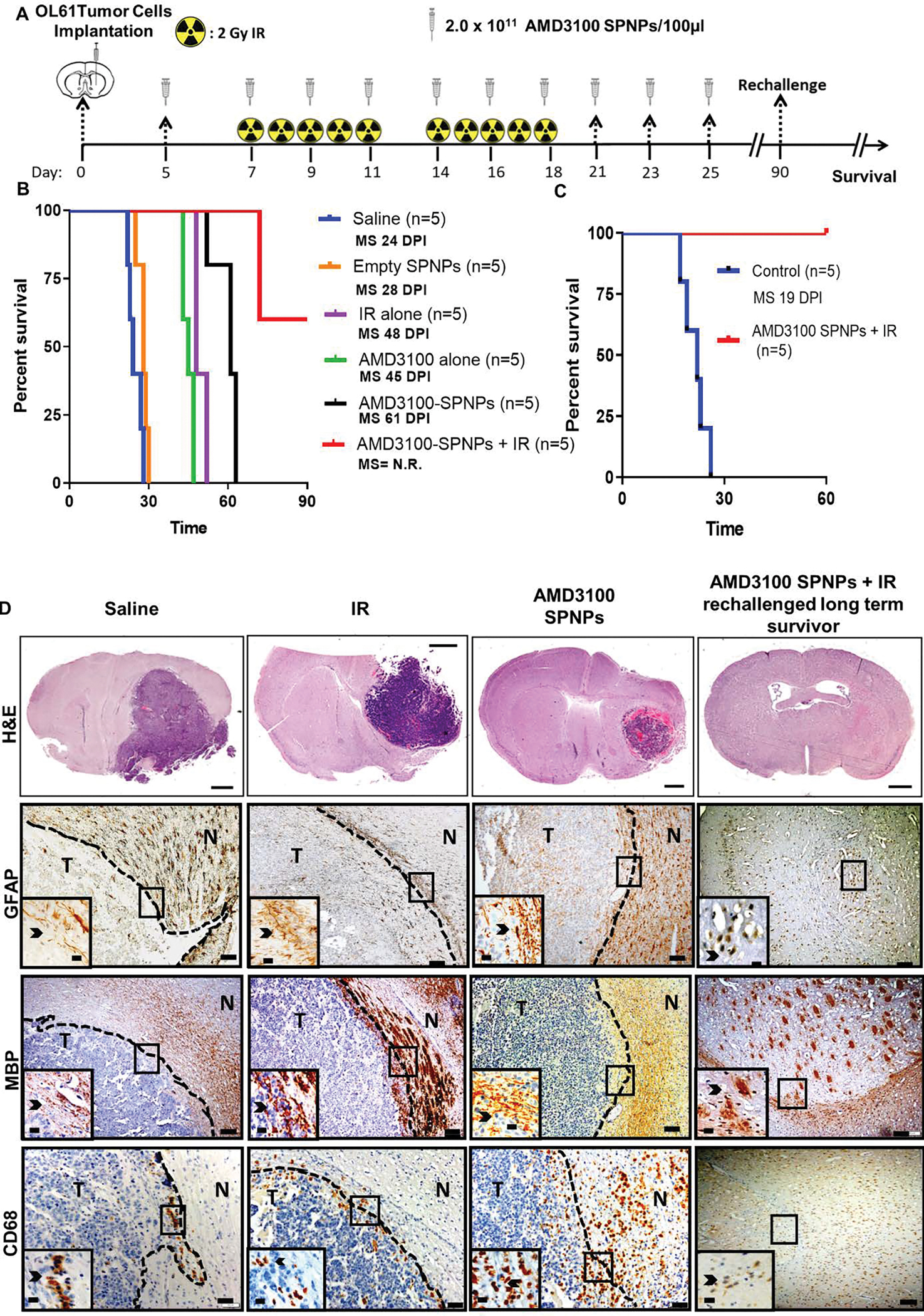
(A) Timeline of treatment for the combined AMD3100-SPNPs+ IR survival study. (B) Kaplan–Meier survival curve. Significant increase in median survival is observed in all groups receiving AMD3100 alone (i.p.) or IR (p<0.01). Mice (n=5) treated with AMD3100-SPNPs (i.v.) + IR reach long-term survival timepoint (100 dpi) with no signs of residual tumor (C) Kaplan-Meier survival plot for re-challenged long-term survivors from AMD3100-SPNPs+IR (n=5), or control (OL61 Untreated) (n=5). Data were analyzed using the log-rank (Mantel-Cox) test. Days post implantation= dpi. NS= Not significant. **p<0.01, ***p<0.005. (D) H&E staining of 5μm paraffin embedded brain sections from saline (24 dpi), IR (48 dpi), AMD3100-SPNPs alone (45 dpi) and long-term survivors from AMD3100-SPNPs + IR treatment groups (60 dpi after rechallenging with OL61 cells) (scale bar = 1mm). Paraffin embedded 5μm brain sections for each treatment groups were stained for CD68, myeline basic protein (MBP) and glial fibrillary acidic protein (GFAP). Low magnification (10X) panels show normal brain (N) and tumor (T) tissue (black scale bar = 100μm). Black arrows in the high magnification (40X) panels (black scale bar = 20μm) indicate positive staining for the areas delineated in the low-magnification panels. Representative images from an experiment consisting of 3 independent biological replicates are displayed.
To assess whether the tumor aggressiveness was due to both proliferating tumor cells and invasion of the normal brain tissue, we carried out an experiment to determine the growth of intracranially implanted tumors at 6 days and 10 days post-implantation. We performed hematoxylin and eosin (H&E) staining and immunohistochemistry using anti-Vimentin, anti-Ki67, and anti-CD31 antibodies to identify tumor cells, and to assess proliferation and micro-vessel density, respectively, at different time points of tumor development. Although the images show a clear distinction between the tumor mass and the surrounding brain tissue, there are tumor cell clusters infiltrating the brain beyond the apparent borders of the tumors (Fig S13). In addition, we observed abundant neovascularization (Fig S14A), high expression of vimentin and abundance of Ki67+ cells within the tumor at day 6 and day 10, indicative of the highly proliferative nature of these tumors (Fig S14B, C).
Furthermore, recognizing that a significant proportion of SPNPs would be cleared via the liver,23 we performed serum biochemistry analysis of liver and kidney metabolites, complete blood cell counts (CBCs), and histopathological analysis of liver sections to inspect potential toxicity of the combined therapy. Results showed that there were no significant differences in the CBCs for animals receiving SPNPs, AMD3100-SPNPs, or AMD3100-SPNPs + IR treatment compared with animals in the saline treatment group (Figure S15). Moreover, no difference was observed in the serum level of kidney (creatinine, blood urea nitrogen) and liver (aminotransferase, aspartate aminotransferase) metabolites tested in animals receiving SPNPs, AMD3100, AMD3100-SPNPs, or AMD3100- SPNPs + IR treatment compared to saline treatment group (Figure S15).
Furthermore, pathological examination of liver tissue sections revealed no differences in the hepatocytes, stromal central, and portal areas between control saline and different treatment groups (Figure S16A–F). These results suggest that radiotherapy and AMD3100-SPNPs is a safe and effective combination for GBM tumor eradication with no cytotoxicity.
To assess the level of immune cellular infiltrates, brain tissue sections of mice from the same experimental groups were evaluated by immunohistochemistry using markers for macrophages (CD68), T-cells (CD8 and CD4), and microglia (IBA1) (Figure 6D, Figure S17). Visual inspection showed increased infiltration of macrophages (CD68+ cells) within the TIME and the adjacent brain parenchyma in the group that received monotherapy of either AMD3100-SPNPs, or IR compared to saline treated group (Figure 6D, bottom). In contrast, the long-term survivors from the AMD3100-SPNPs + IR group have a reduced number of CD68+ macrophages compared to the other treated groups (Figure 6D, bottom). We also observed IBA1+ microglia, CD8+ and CD4+ T cells within the tumor and the surrounding brain parenchyma in AMD3100-SPNPs or IR alone treated groups compared to the control group. However, there was a reduced level of IBA1+, CD8+ and CD4+ populations in AMD3100-SPNPs + IR long-term survivors compared to other treatment groups (Figure S17 1st to 3rd row). These data indicate that there was no overt inflammation in long-term survivors. Thus, our findings suggest that AMD3100-SPNPs in combination with IR is capable of eliciting an antitumor response leading to long-term survival and immunological memory.
Nanoparticle-mediated CXCR4 blockade combined with radiation therapy primed an adaptive immune response and enhanced T-cell mediated cytotoxicity
To examine the role of combining AMD3100-SPNPs with IR treatment in adaptive immunity, we assessed the frequency of tumor specific T cells within the TIME by flow cytometry. Using glioma implantable mouse model that harbors the surrogate tumor antigen ovalbumin (OVA), we compared the frequencies of OVA specific CD8+ T cells infiltrating the TIME after different treatment modalities (Figure 7A). Tumor-specific T cells were characterized by staining for the SIINFEKL-H2Kb-OVA tetramer, an OVA cognate antigen within the CD8 T cell population. Results showed that the tumor-specific CD8 T cells (CD3+/CD8+/SIINFEKL-H2Kb tetramer) within the AMD3100-SPNPs + IR group were increased by two-fold compared to AMD3100-SPNPs alone and saline groups (P<0.01, Figure 7B). In addition, CD8 T cells infiltrating the tumor of the combination therapy have enhanced expression of the T-cell effector molecules interferon-γ (IFN-γ) and granzyme B (GzmB) (P<0.001, Figure 7C, D). Taken together with the enhanced survival observed in the combination therapy group (Figure 6B), these results suggest that a robust anti-GBM immune response was elicited by the combined AMD3100-SPNPs + IR therapy (Figure 7E). To functionally test if combining AMD3100-SPNPs with radiotherapy results in priming an anti-tumor T-cell response, we co-cultured splenocytes derived from mice with the treatment formulations shown in Figure 7A, with OL61-OVA tumor cells in a ratio 20:1 (T-cell to tumor cells) ex vivo (Figure 7F). Our data showed that splenocytes derived from combination therapy elicited enhanced cytotoxicity against tumor cells compared to AMD3100-SPNPs, and saline groups (P<0.05, Figure 7G). Additionally, we assessed the frequency of infiltrating T-cells elicited by the combination therapy using OVA-independent OL61 model (Figure S18). Using this model, we found that combining AMD3100-SPNPs with radiation elicits enhanced infiltration of CD3+ and CD8+ T cells (Figure S18B, C). Consistent with previous results, T-cells from the combination therapy group exhibited higher expression levels of effector molecules (i.e., GzmB, and IFN-γ) compared to control (saline or AMD3100-SPNPs alone). No differences were observed between these effector molecules’ expression in the combination therapy and IR groups (Figure S18D, E). These results further validated that AMD3100-SPNPs, in combination with IR, elicit an antitumor immune response leading to long-term survival and immunological memory. Involvement of this adaptive immune response protects against secondary tumors, which is essential for successful glioma therapy that results in long-term eradication of resistant and recurrent GBM.
Figure 7. Combining AMD3100-SPNPs + IR enhances the adaptive antitumor immune response.
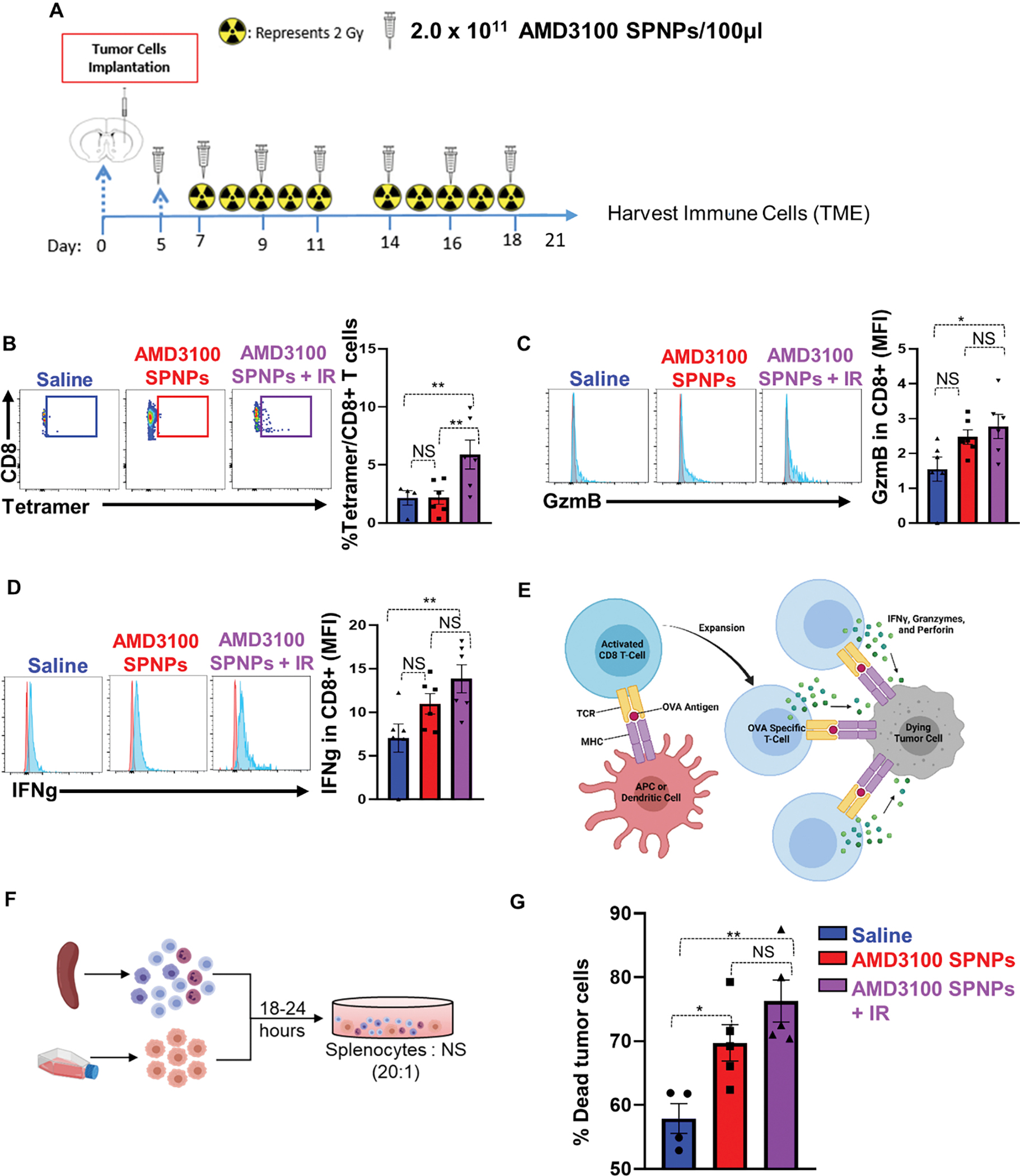
(A) Experimental design represents the timeline for the combination treatment of AMD3100-SPNPs + IR to assess the efficacy of GBM-infiltrating T cell function. (B) Representative flow cytometry plots and analysis represents the frequency of tumor specific CD8+ T cells within the TIME in saline, AMD3100-SPNPs, or AMD3100-SPNPs+ IR group. OL61-OVA tumors were analyzed by staining for the SIINFEKL-Kb tetramer. (C, D) Representative flow cytometry plots and analysis represent the expression of effector T cells molecules Granzyme B (GzmB) (C) and Interferon-g (IFN-γ) (D) in CD8 T cells in filtrating the TIME of each group. (E) Schematic represents the process of priming and expansion of OVA specific CD8 T cells which target OL61-OVA cells and triggers tumor cell death. (F) Schematic represents the killing assay of tumor cells co-cultured with splenocytes from each treatment group. (G) Quantitative analysis of the percentage of tumor cells death in co-culturing condition of OL61-OVA tumor cells with Splenocytes from OL61-OVA implanted mice treated with saline, AMD3100-SPNPs, or AMD3100-SPNPs+IR. Red histogram= isotype control, blue histogram= representative sample GzmB or IFN-γ expression. *p< 0.05 **p< 0.01, ***p< 0.0001, ****p<0.0001; One way ANOVA. Bars represent mean ± SEM. (n=4–5/group).
Discussion
The current standard of care for primary GBM involves surgical resection followed by concurrent radiation and adjuvant chemotherapy with temozolomide (TMZ).33 Despite therapeutic advances and intense research efforts to develop effective therapeutic modalities, the median survival of GBM patients remains poor ~18–24 months.34 GBM patients succumb to their disease ~9–12 months post-tumor recurrence, highlighting the urgent need for developing more effective and safer anti-GBM therapies that can elicit an anti-GBM immunological memory response. We developed synthetic protein nanoparticles loaded with the CXCR4 inhibitor, AMD3100. Our data demonstrate that systemic delivery of SPNPs loaded with AMD3100, inhibits GBM progression; and when used in combination with radiotherapy it leads to GBM immunogenic cell death and an effective anti-GBM adaptive immune response. Furthermore, we showed that systemic administration of AMD3100-SPNPs resulted in a less suppressive TIME by inhibiting the infiltration of inhibitory CXCR4+ M-MDSCs.
Myeloid-derived suppressor cells (MDSCs) are major component of the TIME hindering effective anti-GBM immunotherapy.35, 36 Studies showed that the number of circulating MDSCs is positively correlated with the stage of the tumor and with tumor burden, in several cancer types including glioma.37 We previously demonstrated, that depletion of immunosuppressive MDSCs significantly enhanced the efficacy of an immune-mediated gene therapy strategy 37, 38. In the present study, we showed that CXCR4 is mainly expressed by GBM infiltrating, immunosuppressive monocytic MDSCs (M-MDSCs). Using multiple genetically engineered GBM mouse models, we showed that the frequency of CXCR4+ M-MDSCs infiltrating the GBM is correlated with GBM aggressiveness. This is consistent with the fact that CXCR4 expression in human patients was correlated with the glioma grade and associated with poor prognosis using both TCGA and CGGA databases.
Immunogenic cell death (ICD) represents a functionally unique response elicited by dying tumor cells which results in induction of adaptive anti-tumor immunity.39, 40 Our results show that blocking CXCR4 using either free AMD3100 or ADM3100 encapsulated in SPNPs, in mouse and human GBM leads to inhibition of GBM proliferation and increases in the expression of HMGB1 and Calreticulin as well as the secretion of and Damage-Associated Molecular Patterns (DAMPS) including ATP, TNFa, IL33, and IL-6. These DAMPs directly activate macrophages and DCs by binding to cell surface receptors such as CD91, TLR2, TLR4, and the Receptor for Advanced Glycation End-products (RAGE).41, 42 Activation of these receptors leads to priming of both the innate and adaptive immunity which promotes the engulfing of dying cells, enhances antigen presentation, and increases the production of IL-1β.43, 44 All these signals are responsible for eliciting immune responses targeted to tumor cells and the establishment of antitumor immunological memory.39, 42, 44 To summarize, the activity of AMD3100 was not affected by its encapsulation into SPNPs.
We demonstrated that CXCR4 is an important receptor for the maintenance of BBB integrity. Our data show that blocking CXCR4 on both human and mouse GBM cells, reduced GBM-mediated BBB disruption as well as decreased myeloid cells transmigration across brain microvascular endothelial cells (mBECs) in vitro. This was validated by intact tight junctions (Claudin-5/Zo-1) and increased paracellular resistance between mBECs monolayer upon blocking CXCR4 signaling. This illustrates that CXCL12/CXCR4 is an important signaling pathway that is hijacked by the GBM to compromise the BBB integrity, and that systemic administration of CXCR4 blocker may counteract the GBM-induced alterations in BBB permeability.
Altered BBB permeability was evident by the leakage of molecular weight tracers such as cadaverine (1kDa), inulin (5kDa) and Dextran (5kDa) into the bottom chamber in the presence of GBM cells’ CM, which contained secreted factors produced by the glioma cells (Figure S3). Although the 10kDa dextran was not permeable, we observed that there were alterations in the tight junction proteins in the endothelial barrier after incubation with GBM CM (Figure 3E). Albeit the migration of the myeloid cells and other immune cells through the BBB barrier occurs via diapedesis.45 Collectively, our results indicate that monocytic cells’ migration into the tumor microenvironment is reduced by the blockade of CXCR4.
Previous studies showed that nanoparticles can selectively target the GBM TIME.46, 47 The SPNPs used in this study make use of a biologically compatible material albumin-based and the ability to target the GBM using iRGD, which enhances the efficiency of the SPNPs to deliver AMD3100 to the TME. These SPNPs have been shown to facilitate delivery of therapeutics to the GBM TME, after systemic delivery, without toxicity.23 In addition, the properties of AMD3100-SPNPs were reproducible in independent preparations as shown in Figure 5 and HSA’s secondary structure was mostly preserved after being exposed to the EHD jetting and crosslinking (Figure S8). The SPNPs were shown to be stable in both human and mouse serum at 37 °C when compared to being suspended in PBS. The diameter of the SPNPs in serum and PBS were not statistically different (P>0.05).
Several studies have shown that the transcytotic peptide (iRGD) enhanced the delivery of small molecules and nanoparticles to the target tissue when co-delivered systemically.48, 49 iRGD triggers a stepwise transcytotic delivery machinery responsible for the target tissue (tumor) penetration properties. The process of iRGD-mediated delivery is triggered by binding to αvβ3 and αvβ5 integrins. Therefore, the tissue selectivity for iRGD-mediated drug delivery depends mainly on the expression of these two integrins in the target tissue.50 We have observed increased levels of both αvβ3 and αvβ5 integrins in GBM bearing mice and a corresponding increase in tumor accumulation of SPNPs.23 We showed that when iRGD-loaded SPNPs were incubated with glioma cells, particle uptake was increased by 5-fold compared to nanoparticles without iRGD.23 Therefore, the adaptation of iRGD to the HSA nanoparticle formulation promoted nanoparticle transport and CNS penetration, specifically the entry and diffusion of the cargo to the tumor microenvironment. This strategy is leveraged here to deliver AMD3100 in vivo (Figure 6).
CXCL12/CXCR4 promotes cancer growth and angiogenesis in multiple solid tumors. CXCR4 inhibitors are in clinical trials for the treatment of multiple solid tumors.51, 52 Structurally, AMD3100 is a bicyclam, composed of two cyclam rings joined by an aromatic bridge. However, AMD3100 has poor pharmacokinetic and BBB penetration, therefore, its delivery to the GBM microenvironment is limited.29 Our approach using SPNPs loaded with AMD3100 facilitates delivery of AMD3100 to the brain via systemic administration with no overt side effects or cytotoxicity.23, 48
Since radiation therapy is the current standard of care for GBM, we tested the efficacy of SPNPs loaded with AMD3100 in combination with radiation therapy. Our results showed that blocking CXCR4 significantly enhanced the efficacy of radiotherapy-mediated tumor cell killing both in vitro and in vivo. This was accompanied with enhanced release of DAMPs associated with ICD which elicits activation of antigen-presenting DCs to prime cytotoxic CD8+ T cells and leads to a robust anti–GBM response in our model. Blocking CXCR4 sensitizes GBM to ionization radiation causing enhanced GBM cell death resulting in tumor regression, long-term survival, anti-GBM immune response, and long-term immunological memory. Because of the robust anti-GBM memory response, this strategy can effectively inhibit recurrent GBM. Finally, we observed no signs of pathophysiology in the liver nor significant differences in CBCs or serum chemistry biomarkers for kidney or liver dysfunction, suggesting no toxicity occurred in response to the treatment.
In addition, in order to demonstrate the generation of tumor-antigen specific T cells in the GBM TIME, we used a GBM mouse glioma model harboring the surrogate tumor antigen (Ag) ovalbumin (OVA). Using this model, we monitored the generation of glioma Ag-specific T cells; our results demonstrate an increase in the frequency of glioma–specific T cells in the TIME of GBM–bearing mice treated with AMD3100 encapsulated in SPNPs when used in combination with IR. In addition, we found that the majority of T-cell infiltrating tumors with combination AMD3100-SPNPs + IR therapy express higher levels of effector molecules, i.e., GzmB and IFN-γ. These data strongly suggest that inhibiting CXCR4 in combination with IR induces an effective GBM–specific immune response.
In summary, our study brings a new mechanistic insight of the role of the CXCL12/CXCR4 signaling pathway in glioma. The use of SPNPs provides effective and minimally invasive delivery of AMD3100 to GBM bearing mice. We show that AMD3100 encapsulated in SPNPs when used in combination with radiation elicits an effective ant-GBM response by (1) Inducing potent ICD, (2) reprogramming the immunosuppressive glioma microenvironment and (3) inhibiting the migration of MDSCs into the GBM. The sensitization of GBM to radiotherapy in an orthotopic in GBM model, promoted anti-glioma CD8+ T cells and elicited anti-GBM immunological memory responses. This strategy can be harnessed to elicit beneficial immunotherapeutic outcomes in GBM patients.
Materials and Methods
Reagents
DMEM-F12 (11330057), DMEM (12430054), RPMI-1640 (11875119), FBS (10437028), PBS (14190250), were acquired from GIBCO, Life Technologies. Epidermal growth factor (EGF) and fibroblast growth factor (FGF) were obtained from Peprotech, Supplements N2 (17502048), and B27 (17504044) and penicillin-streptomycin (15240062) were bought from GIBCO, Life Technologies. Specific anti- mouse CD8 (100706), anti-mouse CD45 (147716), CD3 (100218), Gr1 (108430), CD11b (101226), Ly6G (127648), Ly6C (128028), CXCR4 (146508), GzmB (372216), and IFN-γ (505826) antibodies for flow cytometry analysis were obtained from Biolegend. SIINFEKL tetramers were obtained from MBL International (TB-5001–1). Viability dye (LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit, NC0180395) was purchased from Fisher Scientific. For immunohistochemistry, anti-mouse MBP (MAB386) and GFAP (AB5541) primary antibody was purchased from Millipore Sigma; anti-Nestin antibody (NB100–1604) was purchased from Novus biologicals; anti-mouse Iba1 (ab178846) and CD68 (ab125212) antibodies were purchased from Abcam; anti-mouse CD8a [(HS-361003(SY)] antibody was purchased from Cedarlane; anti-mouse CD4 (48274S) antibody was purchased from Cell Signaling. Alexa Fluor 647 anti-Calreticulin antibody (ab196159) was purchased from Abcam. AMD3100 was purchased from Selleck Chemical LLC (Cat# S8030). All fluorescence-conjugated antibodies used in this study are listed in Table S1.
Synthesis of AMD3100 synthetic protein nanoparticles (AMD3100-SPNPs)
AMD3100-SPNPs were fabricated via the electrohydrodynamic (EHD) jetting process previously established in our group.1–4 Briefly, human serum albumin (HSA) was dispersed in a solvent mixture (80:20 v/v) of ultrapure water and ethylene glycol; final concentration of 7.5 w/v%. Bovine serum albumin (BSA) labeled with Alexa Fluor 647 was added at 0.5 w/w% relative to HSA to generate fluorescently labeled SPNPs. Next, the cyclic nine amino acid transcytotic peptide, iRGD, was added at 355 ng per mg of albumin. Subsequently, 3.75mg of AMD3100 per mL of jetting solution was incorporated after first dissolving the drug in a small aliquot of ultrapure H2O. Lastly, a bifunctional OEG (NHS-OEG-NHS, 2kDa) was added at 10.0 w/w% relative to HAS. This was done to crosslink the SPNPs. To prepare empty SPNPs, we followed the same formulation steps except that ultrapure water was added, in lieu of AMD3100. The complete jetting solutions were pumped through a syringe outfitted with a stainless steel 26-gauge blunt tip stainless needle, the flow rate was kept constant at 0.2 mL h−1; while being subjected to a constant voltage (ranging from 7.5 to 10.2 kV) to form a stable Taylor cone at the tip of the needle. Particles were collected in grounded aluminum pans at a needle to collector distance of 15 cm. Particles were then incubated at 37 °C during 7d to achieve the polymerization process, yielding water-stable protein nanoparticles. SPNPs were then kept in their dry state, at room temperature and in dark until used for experimentation.
Collection and purification of albumin nanoparticles
SPNPs were collected according to a standard protocol developed in our group 1–4. In brief, a small volume (5–10 mL) of PBS + 0.5% Tween 20, was added to the aluminum pans containing crosslinked, water-stable AMD3100-SPNPs. Gentle mechanical scraping of the pans using a plastic razor blade was used to transfer the SPNPs from the solid surface into the collection solution. To disperse any aggregates, the NP suspension was briefly sonicated and passed through a 40 μm cell strainer filter. To pellet and remove remaining large protein aggregates, the solution was centrifuged 4 min at 4,000 rpm (3,220 xg). The supernatant containing SPNPs was aliquoted into 2 mL Eppendorf tubes and centrifuged at 15,000 rpm (21,500 xg) to pellet the SPNPs. Finally, the pelleted particles were combined into a single concentrated sample. SPNPs were used within 1 week after collection, they were continually stored at 4°C in sterile PBS in the interim.
Characterization of SPNP size, shape, and concentration
SPNPs were extensively characterized as previously described to establish reasonable tolerances against which each batch could be compared. Prior to their use in any experiments, collected particles were similarly measured to ensure they met specifications and to confirm batch-to-batch properties were consistently maintained. Physical characterization comprised the measurement of particle size in both their dry and hydrated state. To determine particle size, shape, and examine their morphology, small silicon wafers were positioned on the grounded aluminum collection surface and were subjected to the same seven days incubation period required to complete the step-growth polymerization. Samples were gold sputter coated and afterwards imaged via Scanning Electron Microscopy (SEM) using a FEI NOVA 200 SEM/FIB instrument using a current of 0.14nA and a voltage of 17kV. SEM images of SPNPs (n>300) were characterized using ImageJ software according to protocols previously developed in our lab31. To investigate SPNP properties in after hydration, SPNPs were collected and purified as described above. The stock SPNP solution was diluted 100-fold in 0.22 μm filtered 10 mM KNO3 for subsequent DLS (dynamic light scattering) and zeta potential measurements. To further investigate size and solution concentration, nanoparticle tracking analysis (NTA) was used. DLS, zeta potential, and NTA analyses were performed using the Malvern Nano ZSP and NanoSight NS300 instruments. The concentration of the AMD3100-SPNP solution was assessed employing the BCA (bicinchoninic acid) assay. To analyze the shape and size of the SPNPs, a SEM PDI is presented. This value doesn’t have a commonly accepted definition, nevertheless we define it as the SDSEM/DiameterSEM,AVG. This is done to provide an easily reference number for quantitative comparison between SEM derived data sets. This value can also be compared, qualitatively, to the SDDLS/DiameterDLS,AVG for number average DLS data. While one could square these values for comparison to intensity DLS PDI values, this approach is not recommended due to the non-linear relationship between number/count-based scores and intensity (direct spectral deconvolution) data.
Serum stability of AMD3100-SPNPs
SPNPs samples were distributed into mouse or human serum (n=3) and rotated at 37˚ C. SPNPs samples in PBS at 37˚ C were included as controls. After 24 hours, aliquots of the samples were removed for determination of particle size distribution. Three instrumental determinations were averaged for each sample.
Circular Dichroism (CD) Spectroscopy and estimation of secondary structures
CD spectroscopy measurements were taken of HSA, the pre-jetting solution, the post jetting solution, and the crosslinked SPNPs. All samples’ concentrations were measured via a BCA Assay then diluted in ultrapure water to achieve a final concentration of 0.05mg/mL. Samples were recorded using CD spectrometer (Jasco J-815) in a rectangular quartz cuvette of 1mm pathlength. Measurement parameters were set to a wavelength range of 260 to 190nm, with a step size of 0.2nm, data integration time of 2 seconds, slit width of 1.00nm, and a scanning speed of 100nm/min. Each spectrum was an average of 3 measurements and was subsequently baseline corrected using an ultrapure water spectrum before analysis. Dichroweb was used to further analyze the CD spectroscopic data.53 CDDSTR was chosen as the analysis program with the reference Set 7.54, 55 Output data included fractional contributions from Helix 1 (alpha helix, regular), Helix 2 (alpha helix, distorted), Sheet 1 (Beta sheet, regular), Sheet 2 (Beta sheet, distorted), turns and unordered.55
AMD3100-SPNPs in vivo dose preparations
AMD3100-SPNPs were collected and purified as previously described prior to diluting the resulting solution to a concentration of 2.7 mg mL−1 in sterile PBS. Aliquots of the stock solution were diluted and standard DLS, zeta potential, and NanoSight measurements as detailed above were completed to ensure batch-to-batch consistency. Once validated, 100 μL of the stock SPNP solution was allocated per dose, per mouse for tail vein injection. Control SPNPs, encapsulating no AMD3100, were collected, purified, characterized, and dosed identically to AMD3100-SPNPs on a particle mass basis.
Cell Line and Cell Culture Conditions
Genetically engineered mouse glioma models: RPA (PDGFRA D842V/ shTP53-GFP/ shATRX), and OL61 (shp53/PDGFB/NRAS), OL61-OVA, were developed by the sleeping beauty model as described before.27, 56 Arf−/− wtIDH1 (PDGFB/shP53/shATRX/Ink4a/Arf−/−). Tumors were generated by injection of DF-1 (ATCC, CRL-12203) cells transfected with a combination of RCAS plasmids (RCAS PDGFB-HA, RCAS shp53-RFP) using the FuGENE 6 transfection kit (Roche, 11814443001) according to the manufacturer’s protocol and as previously described.57
Human glioma cells: HF2303 glioblastoma cells were grown in Dulbecco’s modified eagle (DMEM) media supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 0.3 mg/mL L-glutamine. SJGBM2 pediatric glioma cells were a kind gift by the Children’s Oncology Group (COG) Repository, Health Science Center, Texas Tech University. These cells were cultured in IMDM medium with L-glutamine (0.3 mg/mL) (Gibco,12440–053), 20% FBS (Gibco,10437–028), and antibiotic-antimycotic (1X) (Gibco, 15240–062) at 37 °C, 5% CO2. Cells were maintained in a humidified incubator at 95% air/5% CO2 at 37°C and passaged every 2–4 days.
Generation of Ovalbumin Expressing Glioma Cells
Wild type OL61 glioma cells (5.0 × 105) were cultured in a 6-well plate in 1mL of media (DMEM, Thermofisher Cat# 12430054; 10% FBS, FisherSci Cat# 10437028; 1x Anti-Anti, Thermofisher Cat# 15240062) with 8ug of polybrene and 100uL of 10x pLVX-cOVA-Puro (Addgene Cat#135073) lentiviral particles generated by the University of Michigan Vector Core; 1mL of media was added to the cells at 24- and 72-hours post-incubation. Once the cells became confluent, they were transferred to a larger T-25 cm2 flask to expand. Cells were then selected with 10ug/mL of puromycin added directly to the culture media. Cells were then passaged two times before assessing ovalbumin expression via western blot.
Animal Strains
Six- to eight-week-old female C57BL/6 mice were acquired from Jackson Laboratory (Bar Harbor, ME). Mice housed in a pathogen free environment in our vivarium. All studies involving live mine were performed in accordance with the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan.
Intracranial GBM Models
Tumor implantation was done as described before.58 Briefly, mice are anesthetized using ketamine and dexmedetomidine prior to stereotactic implantation with 50,000; 100,000 and 200,000 cells for OL61, Arf−/− and RPA S8 in the right striatum respectively. The coordinates for implantation are 0.5 mm anterior and 2.0 mm lateral from the bregma and 3.0 mm ventral from the dura. Neurospheres were injected at a rate of 1 μL/min. Mice were given a combination of buprenorphine (0.1mg/kg) and carprofen (5mg/kg) for analgesia. At symptomatic stage, tumors were isolated and immune cells were characterized as described in the flow cytometry section (see below).
Mouse brain endothelial cells
Microvascular endothelial cells from mouse brain (mBMECs) were prepared using a protocol described previously.59 Briefly, brains were collected from 6–8-week-old C57BL/6 mice, minced in Hanks balanced salt solution (HBSS; Invitrogen), and homogenized using a Dounce-type homogenizer. Myelin was removed by resuspending homogenates in an 18% suspension of dextran (molecular weight, 60,000 to 90,000; USB) and centrifuging. Red blood cells were removed by centrifuging isolated microvessels in a Percoll gradient (ThermoFisher) for 10min at 2,700 rpm. The isolated microvessels were digested in HBSS solution containing 10 U/ml DNase I (Sigma-Aldrich), 1 μg/ml collagenase/dispase (Roche), and 1 μg/ml Na-p-tosyl-l-lysine chloromethyl ketone (TLCK) at 37°C for 20 min. They were then precipitated with CD31-coated magnet beads (Dynabeads; ThermoFisher). The microvessles were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen) with the addition of 10% inactivated fetal calf serum, 2.5 μg/ml heparin (Sigma-Aldrich), 20 mM HEPES, 2 mM glutamine, 1× antibiotic/antimycotic (ThermoFisher), and endothelial cell growth supplement (BD Bioscience). They were grown in 6-well plates which had been previously coated with collagen type IV (BD Bioscience). This protocol yields primary endothelial cell cultures, with a 99% purity. To assess this, we performed immunocytochemistry using a marker for anti-platelet endothelial cellular adhesion molecule 1 [anti-PECAM-1] antibody (BD Bioscience).
Microvascular endothelial cells from human brain (HBMEC) were obtained from iXCells Biotechnologies. HBMECs were plated on flasks coated with fibronectin and maintained in endothelial cell medium, containing 5% fetal bovine serum, 1% endothelial cells growth supplement and 1% penicillin/streptomycin solution, at 37°C in 5% CO2/95% air. HBMECs were used at 1st or 2nd passage for all experiments.
Tumor cell apoptosis assay
OL61-OVA cells were dispersed using Accutase (Gibco, Life Technologies) washed, counted, and plated (50,000 cells) in a 96 wells plate. Splenocytes from each group were washed, counted and co-cultured with OL61-OVA cells in the ratio of 1:20 (i.e., 50,000 OL61-OVA + 1,000,000 Sp). As a control condition OL61-OVA were cultured alone. The final volume in each well was 200 ul. The co-cultures were incubated at 37˚C 5% CO2 overnight. At the end of the incubation period, all the wells were transferred into a V-bottom plate for staining. The staining procedure was done as described in the flow cytometry staining. Cells were washed with PBS and stained with viability dye (LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit, Cat# NC0180395). Cells were then stained with CD45 to distinguish the tumor cells from splenocytes. For AnnexinV staining, cells were washed 1x with flow buffer resuspended in 100 ul of APC-AnnexinV 1/50 diluted in AnnexinV Binding buffer. Incubated for 10 min at RT protected from light. Cells were then passed to a flow tube and 100 ul of AnnexinV binding buffer was added. Data acquisition was performed using FACS ARIA II (BD Biosciences) and analyzed using Flow Jo version 10 (Treestar).
In vivo radiation
Ten days’ post OL61-OVA tumor cells (2 × 104) implantation (bioluminescence signal= 106), mice were subjected to Irradiation (IR) dose of 2 Gy for 5 days a week for two weeks for a total of 20gy of ionizing radiation. The procedure was performed as described before.25, 32 Briefly, mice were lightly anaesthetized with isoflurane. Mice were then placed under a copper Orthovoltage source, with the irradiation beam directed to the brain and body shielded by iron collimators. Irradiation treatment was given to mice at the University of Michigan Radiation Oncology Core.
Cell treatment
Brain endothelial cells were cultured for 5 days to established characteristics of the brain endothelial barrier (e.g., TJ complexes and transport molecules). The mBMEC (mouse brain microvascular endothelial cells) were incubated with tumor conditioned media collected from OL61, Arf−/−, or RPA cells in presence or absence of AMD3100 inhibitors, while HBMEC (human brain microvascular endothelial cells) were incubated with tumor conditioned media collected from cultured HF2303 or SJGBM2 cells in presence or absence of AMD3100 inhibitors. For each condition, cells were seeded at density of 1 × 106 cells into 6-well plate. Cells were then incubated with vehicle or AMD3100 (Plerixafor) at their respective IC50 doses for 72h in triplicate wells per condition. For irradiation, mouse and patient-derived glioma cells were irradiated with 3Gy and 10Gy, 2hrs after AMD3100 treatment, respectively. Conditioned media were then collected, centrifuged at 1000 × g for 10mins and filtered through 0.22μm syringe filter before they were incubated with mBMEC or HBMEC.
Collection of tumor conditioned media for transmigration and cellular permeability assay
To analyze the transmigration assay and measure the cellular permeability in presence of tumor conditioned media, mouse neurospheres and human glioma cells were seeded at density of 1 × 106 cells into 6-well plate. Cells were then incubated with either free-AMD3100 (Plerixafor) or in combination with radiation at their respective IC50 doses for 72h in triplicate wells per condition. Conditioned media were then collected, centrifuged at 1000 × g for 10mins and filtered through 0.22μm syringe filter for use.
Cell survival analysis
Mouse OL61 (shp53/NRAS/PDGFβ), Arf−/− wtIDH1(shp53/PDGFβ/Arf−/−) and RPA (shp53/PDGFRα/shATRX) cells and patient-derived glioblastoma cells (MGG8, HF2303 and SJ-GBM2 wtIDH1) were plated at a density of 1000 cells per well in a 96-well plate (Fisher, 12–566-00) 24h prior to treatment. Cells were then incubated with either free-AMD3100 (Plerixafor, Ontario Chemicals, Guelph, Ontario) or in combination with radiation at their respective IC50 doses for 72h in triplicate wells per condition. All the mouse and patient-derived glioma cells were pre-treated with AMD3100 2h prior to irradiation with 3Gy and 10Gy of radiation respectively. Cell viability was determined with CellTiter-Glo® Luminescent Cell Viability Assay (Promega, G7570) following manufacturer’s protocol. Resulting luminescence was read with the Enspire Multimodal Plate Reader (Perkin Elmer). Data was represented graphically using the GraphPad Prism software and statistical significance was determined by one-way ANOVA followed by Tukey’s test for multiple comparisons.
DAMPs release measurement
To analyze the DAMPs in the tumor conditioned media and on tumor cells, mouse neurospheres and human glioma cells were seeded at density of 1 × 106 cells into 6-well plate. Cells were then incubated with either free-AMD3100 or in combination with radiation at their respective IC50 doses for 72h in triplicate wells per condition. All the mouse and patient-derived glioma cells were pre-treated with AMD3100 2h prior to irradiation with 3Gy and 10Gy of radiation respectively. Release of DAMPs was assessed 72h post radiation. For assessing the level of Calreticulin (CRT), cells were harvested, dispersed with accutase and stained with anti-Calreticulin Ab (1:200) in PBS supplemented with 2% FCS (flow buffer) for 30 min at 4°C. Cells were rinsed twice using flow buffer. The samples were then analyzed using a BD FACS ARIA SORP using 647 lasers. Data was analyzed with Flowjo v.10 software. Concentrations of HMGB1, IL33, IL6, IL1α, TNF-α in the culture supernatants was measured by ELISA according to manufacturer’s protocol (R&D), Immunology Core at the Rogel Cancer Center, Michigan Medical School. ATP levels in the cell cultures’ supernatants were determined by ENLITEN ATP Assay following the manufacturer’s instructions (Promega).
Real-time cell proliferation assay
Real-time cell analysis (RTCA) of cell proliferation was monitored using the xCELLigence DP system (Agilent). Before cell seeding, E-plates were coated with 0.5% laminin and equilibrated for 1 hour at 37°C, 5% CO2 with 100μL of respective media per well. A total of 5 × 103 cells for HF2303 and 3 × 103 cells for OL61 (shp53/NRAS/PDGFβ) and Arf−/− wtIDH1(shp53/PDGFβ/Arf−/−) were plated to a final volume of 200μL per well. E-plates were then equilibrated for 30 minutes. Cells were treated with either free-AMD3100 or in combination with radiation at their respective IC50 doses in triplicate wells per condition before starting the experiment. All the mouse and patient-derived glioma cells were pre-treated with AMD3100 2h prior to irradiation with 3Gy and 10Gy of radiation respectively.
Immunofluorescence analysis
Cell samples to be processed for immunofluorescence staining were preincubated in blocking solution which contained 5% normal horse serum and 0.05% Triton 100X (Sigma-Aldrich) in PBS. Samples were incubated overnight at 4°C with the primary antibodies as follows: anti-claudin-5 Alexa-Fluor 488 conjugated, and ZO-1-Alexa Fluor 594 conjugated (Thermo Fisher). We used a confocal laser scanning microscope to view and analyze the samples (Nikon A1, HSD, Japan). Quantitation of TJ-associated fragments for claudin-5 and ZO-1 were performed on 12 images obtained from three independent slides using Fiji software.
Proximity Ligation Assay
Mouse BMEC grown as monolayers were washed, fixed and preincubated with permeabilization solution (DPBS and 0.5% Triton X-100) for 5 min. This was followed by incubation in blocking solution (DPBS++ containing 0.5% (v/v) Triton X-100 and 5% goat serum) for 2 hrs. After, samples were incubated with primary antibody pairs (mouse anti-ZO-1 and rabbit anti-claudin-5 antibodies) overnight. The mBMEC monolayers were then rinsed with DPBS containing 5% goat serum with gentle agitation, treated with rabbit Plus (DUO92002) and anti-mouse Minus (DUO92004) and incubated in a humidified incubator at 37 °C, 5% CO2 for 1 hr. Protein-protein interactions were assessed using the Detection Kit Red (DUO92008) following the manufacturer’s (Sigma) specifications. Samples were incubated with ZO-1-Alexa488 antibody for labeling lateral cells border and mounted on slides using Duolink In Situ Mounting Medium with 4,6-diamidino-2-phenylindole (DUO82040), prior to imaging. For quantification, Fiji software was used for the automatic quantification of claudin-5-ZO-1 interaction (positive dots). Five random images were taken and analyzed to provide a representative sampling of the tissue. The number of positive dots is expressed as average Claudin-5-Zo-1 positive dots/positive cellular counts relative to the number of nuclei present in each imaging field.
Permeability assays
The permeability of brain endothelial cell monolayers to cadaverin-569 (1kDa), Dextran (Cascade blue 3kDa, Thermo Fisher, D7132), Dextran Texas Red (10kDa) (Thermo Fisher, D1828), and FITC-inulin (5 kDa, Sigma Aldrich, I3661), were measured as described in Kazakoff et al., and modified in our laboratory.60 Briefly, hBMEC or mBMEC were plated on a Transwell Dual chamber system at a density of 1×105. Monolayer permeability (transendothelial electrical resistance) was measured daily for one week. Experiments to assess permeability were started by adding the tracers’ cocktail (1 μg/ml), in phenol red free DMEM (Thermo Fisher) to the apical chamber. After 30 min incubation, media was sampled from the receiving chamber. A fluorescent reader (Tecam) was employed to determine fluorescence intensity. The concentration was assessed from a standard curve. The permeability (Papp; cm/min) of the monolayer for each time point (T) was calculated using the formula: P=(C(B)T−C(B))×V(B)×2 /(C(A)+C(A)T)×A; where C(B) and C (B)T are, the concentrations of tracer in the basal chamber at the start, and at the end of the time interval of 30 min (in μg/ml), respectively. V(B) is the volume of the basal chamber (ml). C(A) and C(A)T are, the tracer concentrations in the apical (donor) chamber at the start, and at the end of the 30 min time interval (μg/ml), respectively. The area of the filter is A (cm2).
Complete serum chemistry
Blood was taken from the submandibular vein from OL61 OVA glioma bearing mice, and transferred to serum separation tubes (Biotang). Samples were incubated at room temperature for 60mins to allow for blood coagulation. Tubes were then centrifuged at 2000 rpm (400 × g). Complete serum chemistry for all samples was assessed by the Veterinary Core Facility at the Medical School.
Immunohistochemistry
For neuropathological analysis, brains were fixed using 4% paraformaldehyde (PFA). Fixed brains were paraffin embedded and 5μm thick sections were generated utilizing a Leica RM2165 microtome. To perform immunohistochemistry on brain sections they were permeabilized during 20 min using a solution of TBS-0.5% Triton-X (TBS-Tx). Then antigen retrieval was performed by incubating the sections for an additional 20 min at 96 °C with 10 mM sodium citrate (pH 6). After this step, the sections were cooled at room temperature (RT) and washed with TBS-Tx (five times; 5 min/each wash). Sections were then blocked for 1 h at RT using 10% goat serum in TBS-Tx. Incubation with the following primary antibodies overnight at RT, i.e., CD4 (Cell Signaling, 48274S, 1:1000), GFAP (Millipore Sigma, AB5541, 1:200), MBP (Millipore Sigma, MAB386, 1:200), IBA1 (Abcam, ab178846, 1:1000), CD8 (Cedarlane, [(HS-361003(SY)], 1:100),CD68 (Abcam, ab125212 1:1000) or Nestin (Novus Biologicals, (NB100–1604), 1:1000), which were diluted in 1% goat serum TBS-Tx. The next day sections were washed 5 times using TBS-Tx. Sections stained with CD4, GFAP and IBA1 primary antibodies, were incubated using a biotinylated secondary antibody; whilst sections stained with CD8, CD68, MBP and Nestin were incubated with HRP secondary antibody. Secondary antibodies were used at 1:1000 dilution in 1% goat serum TBS-Tx, sections were incubated in the dark for 4h. HRP and biotin-labeled sections were developed using to 3, 3′-diaminobenzidine (DAB) (Biocare Medical) with nickel sulfate precipitation. Sodium azide soluntion (10%) was used to stop the reaction. Brain sections were then washed 3 times in 0.1 M sodium acetate, followed by dehydration using xylene, and were subsequently coverslipped using DePeX Mounting Medium (Electron Microscopy Sciences). Images were obtained using a brightfield microscope (Olympus BX53) at 10X and 40X magnification.
For tumor size quantification, paraffin embedded 5μm thick brain sections from each experimental groups were stained with H&E as described previously.27 Sections comprising of tumor or from rechallenged brains (~12 sections per mouse braon) were imaged using the brightfield (Olympus BX53) setting. Brain tumor size was quantified using ImageJ’s Otsu threshold to determine the glioma mass in pixels.
Livers were embedded in paraffin for histological analysis, 5μm thick sections were generated using the microtome system and sections were stained using H&E as described by our team previously.27 Brightfield images were acquired employing an Olympus MA BX53 microscope.
Determination of AMD3100 brain/plasma ratio
Ten days post-tumor implantation (3 × 104 OL61 cells), mice harboring intracranial gliomas were divided into 3 groups (n= 4 mice/group) and injected with: 1) Saline, 2) Free AMD3100 (delivered i.p) and 3) AMD3100-SPNP (delivered i.v). On day 11, mice were anesthetized using ketamine and dexmedetomidine, blood was collected, and brains were harvested after clearing the blood by perfusing with 300 ml of Tyrode solution.
Detection of AMD3100 in plasma
AMD3100 (Selleckchem lot#: S8030) and internal standard (I.S., AMD3100-d4, Cayman Cat No. 26490) were weighed and dissolved in methanol. A series of AMD3100 standard solutions (200, 160, 120, 80, 40, 20, 10 and 5 ng/mL) were prepared using methanol. 250 ng/mL of AMD3100-d4 was used as internal standard (IS). Plasma samples (20 μL) were either spiked with 20 μL internal standard (IS) or not spiked; and then they were extracted with 240 μL methanol containing 1% TFA. The mixture was vortexed and centrifuged for 5 min at 12,000 rpm. Supernatants were blown dry with a stream of Nitrogen at 45°C. We reconstituted the residue using 80 μL of 50% methanol in water containing 0.1% formic acid and was then centrifuged for 5 mins at 12000 rpm. We used Ultra Performance Liquid Chromatography mass spectroscopy (UPLC-MS) to analyze the supernatants. Supernatants for each sample (3ul volume) were analyzed by UPLC-MS. Analysis was carried out through Xselect HSS T3 column (4.6 mm×100 mm, 3.5 μm, Waters, Milford, MA, USA). Mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in methanol) were operated with an isocratic elution using a flow rate of 1.0 mL/min. The column and sample temperature were 40 °C and 20 °C respectively. The injection volume was 3 μL. Analysis of AMD3100 was performed using the Waters Acquity UPLC system (Waters Corp., Milford, MA, USA) consisting of a quaternary solvent manager, sample manager, heated column compartment, ESI mass detector (QDa), and a cooling autosampler.
Detection of AMD3100 in brain
Brain tissue was dispersed into a single cell suspension and passed through a 70μm cell strainer with 3 mL PBS. The suspension was frozen using liquid nitrogen and thawed in a 37°C-water bath 3 times to generate brain tissue lysate. A series of AMD3100 standard solutions (200, 160, 120, 80, 40, 20, 10 and 5 ng/mL) were prepared using methanol. 250 ng/mL AMD3100-d4 was used as internal standard (IS). Brain tissue lysates (600ul) with or without spiking with 20 μL internal standard (IS) solution, were extracted using 1.2 mL methanol containing 1% TFA. The samples were vortexed and centrifuged for 5 min at 12,000 rpm. Supernatants were subsequently dried using a stream of nitrogen at 45°C. The residue was reconstituted with 40μL of 50% methanol in water containing 0.1% formic acid and centrifuged at 12000 rpm for 5 min. Supernatants (3ul volume) were analyzed by UPLC-MS. Analysis was carried out using Acquity UPLC BEH C18 column (2.1 mm×100 mm, 1.7 μm, Waters, Milford, MA, USA). Mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) were operated using a gradient elution at a flow rate of 0.3 mL/min2. The column and sample temperature were 40 °C and 20 °C respectively. The injection volume was 3 μL. Analysis of AMD3100 was performed as described above.
Drug release in vitro
AMD3100-SPNPs were manufactured and characterized (SEM, DLS, BCA assay) as described before, and resuspended in 1.3 mL of DPBS. The SPNPs were placed on ice and tip sonicated during 30 seconds, using 1 second on, followed by 2 seconds off. Samples were then transferred to a dialysis device (Float-a-Lyzer®, MWCO 8–10kDa) prepared following the manufacturer’s instructions. The device was placed in 30 mL of DI water and was constantly stirred at 37 ˚C. The dialysis device was removed at predetermined time points and placed in 30 mL of DI water. After transferring from the dialysis device, the sample was frozen and freeze-dried over 2 days. After freeze-drying, the SPNP samples were solubilized in 1 mL of 50% (v/v) HPLC grade methanol and 50% (v/v) ultrapure water. They were later evaluated using UPLC-MS.
Flow Cytometry
For flow cytometry analysis, cells within the TIME, spleen and blood from tumor bearing mice were processed as described before.61 Brain tumor tissue, spleen, and blood were collected and homogenized using Tenbroeck (Corning) homogenizer in DMEM supplemented with 10% FBS. Tumor infiltrating immune cells were purified using 30%70% Percoll (GE Lifesciences) density gradient. Cells were resuspended in PBS supplemented with 2% FBS and non-specific antibody binding was blocked with FC block (CD16/CD32). PMN-MDSCs were labelled as CD45high/CD11b+/Ly6G+/Ly6Clow, whereas M-MDSCs were gated as CD45high/ CD11b+/Ly6G−/Ly6Chigh. Tumor-specific T-cells were labeled with CD45, CD3, CD8 and SIINFEKL-H2Kb-tetramer. Activated T cells were labeled with CD45, CD3, CD8 and GzmB antibodies. Granzyme B and IFN-γ were stained using BD kit for intracellular staining following the manufacturer’s specifications. During the staining process, all cells were kept at 4°C, stained first with a viability dye to remove dead cells. Then cells were washed 2X with flow buffer. Following the washing step, all cells were incubated with FC block for 10 min, followed by incubation with the antibodies mix for 30 min. Finally, all cells were fixed using 4% PFA. In case of intracellular staining, cells were first premetallized and incubated with intercellular antibodies for 30 min. Flow data was acquired using a FACSAria flow cytometer (BD Bioscience) and analyzed using Flow Jo version 10 (Treestar).
TCGA and CGGA survival analysis
Glioma TCGA dataset were downloaded from Gliovis portal http://gliovis.bioinfo.cnio.es/glioma patients expressing high vs low CXCR4 were stratified based on the median cut off expression value. All CGGA data were downloaded from the Chinese Glioma Genome Atlas http://www.cgga.org.cn/ and stratified in a way similar to TCGA data.
Quantitative ELISA
Conditioned media from mouse and human glioma cells were harvested after culturing of 2 × 105 cells/ 1mL for 48 hours in appropriate culture media. Quantitation of CXCL12 was determined by ELISA (Duosets, R&D Systems, Minneapolis, MN) using the manufacturer’s suggested protocol with few modifications. Briefly, diluted coating Ab was added to ELISA microplates (Greiner Bio-One, Monroe, NC) and incubated overnight. Assay plates were then washed, blocked, and samples and standards added and incubated overnight at 4° C. Diluted secondary Ab was added after washing and incubated for 3 hours at room temperature, followed by washing and HRP incubation for 90 minutes. Following a final series of washes, plates were developed with TMBX substrate (Surmodics, Eden Prairie, MN) and stopped by the addition of an equal volume of 0.4% NaF. Absorbances were obtained at 620 nm and sample concentrations were determined by comparison to the CXCL12 standards using a 4-parameter curve fit (Synergy HT & Gen5 Software, BioTek Instruments, Winooski, VT). Serum level of CXCL12 was assessed using undiluted serum isolated from the blood of tumor-bearing mice or stage IV glioma patients.
Statistical Analysis
Sample sizes were selected based on pilot data from experiments done in our labs and published results from the literature. Animal experiments were performed after randomization. Data were analyzed by one- or two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons post-test. Kaplan Meier survival curves were assessed using the log-rank (Mantel-Cox) test with Prism 8.1 (GraphPad Software). Data were normally distributed and variance between groups was similar. P values less than 0.05 were considered statistically significant. All data was represented as means ± SEM, sample size is indicated for all experiments. No experimental values were excluded from the analysis.
Supplementary Material
Acknowledgments
This work was supported by “National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) Grants R37-NS094804, R01-NS105556, R01-NS122536, R01-NS124167, R21- NS123879–01 and Rogel Cancer Center Scholar Award to M.G.C.; NIH/NINDS Grants R01-NS076991, R01-NS082311, R01-NS096756, R01NS122234; and NIH/NCI R01-CA243916 to P.R.L.; the Department of Neurosurgery; the Pediatric Brain Tumor Foundation, Leah’s Happy Hearts Foundation, Ian’s Friends Foundation (IFF), Chad Tough Foundation, and Smiles for Sophie Forever Foundation to [M.G.C. and P.R.L]. NIH/NCI T32- CA009676 Post-Doctoral Fellowship to M.S.A. NIH/NCI F31CA247104 to AAA, and NIH/NCI F31CA247104 to JAJ. Agencia Nacional de Promoción Científica y Tecnológica, Argentina, PICT-2018–3088 and PICT-2019–00117 and Instituto Nacional del Cancer Argentina, Asistencia Financiera IV to MC. to M.C.
Footnotes
Associated Content
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website
Table S1: Fluorescence-conjugated antibodies used in the study, Figure S1: CXCL12/CXCR4 signaling does not affect T cells frequencies in the bone marrow, Figure S2: Dose-response curves for mouse and human glioma cells treated with AMD3100, Figure S3: AMD3100 preserves the trans-endothelial impermeability of the brain endothelial cells (BEC), Figure S4: CXCR4 signaling causes derangement in tight junction molecules, Figure S5: CXCR4 signaling disrupts human brain endothelial cell (hBEC) barrier, Figure S6: Real-time cell proliferation analysis of mouse neurospheres and human glioma cells following AMD3100 + IR treatment, Figure S7: CXCR4 antagonist, AMD3100 in combination with radiation increases the release of DAMPs from both human and mouse glioma cells, Figure S8: Preservation of secondary structures of HSA during AMD3100-SPNP production, Figure S9: AMD3100-SPNPs are stable in vivo, Figure S10: AMD3100 is released at a faster rate at early timepoints. Figure S11. CXCR4 blockade by AMD3100-SPNPs enhances radio-sensitivity and immunogenic cell death in mouse and human glioma cells, Figure S12: Brain vs. plasma ratios of AMD3100 delivered as free small molecule or encapsulated in synthetic protein nanoparticles (SPNPs), Figure S13: Tumor progression and invasion into surrounding brain parenchyma, Figure S14: Neuropathological analysis of mouse brain post-glioma implantation, Figure S15: Mouse serum biochemical analysis following intravenous AMD3100-SPNP in combination with IR treatment, Figure S16: Histopathological assessment of livers from tumor bearing mice treated with AMD3100-SPNPs + IR, Figure S17: AMD3100-SPNPs treatment in combination with radiation enhances immune infiltration of glioma bearing mice, Figure S18: combining AMD3100-SPNPs + IR enhances the adaptive antitumor immune response.
References
- (1).Ramirez YP; Weatherbee JL; Wheelhouse RT; Ross AH Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals (Basel) 2013, 6 (12), 1475–1506. DOI: 10.3390/ph6121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Himes BT; Geiger PA; Ayasoufi K; Bhargav AG; Brown DA; Parney IF Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front Oncol. 2021, 11, 770561. DOI: 10.3389/fonc.2021.770561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Arvanitis CD; Ferraro GB; Jain RK The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat Rev Cancer. 2020, 20 (1), 26–41. DOI: 10.1038/s41568-019-0205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Parrish KE; Sarkaria JN; Elmquist WF Improving drug delivery to primary and metastatic brain tumors: strategies to overcome the blood-brain barrier. Clin Pharmacol Ther. 2015, 97 (4), 336–346. DOI: 10.1002/cpt.71. [DOI] [PubMed] [Google Scholar]
- (5).Ma Q; Jones D; Borghesani PR; Segal RA; Nagasawa T; Kishimoto T; Bronson RT; Springer TA Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci U S A. 1998, 95 (16), 9448–9453. DOI: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zou YR; Kottmann AH; Kuroda M; Taniuchi I; Littman DR Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393 (6685), 595–599. DOI: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- (7).Teicher BA; Fricker SP CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010, 16 (11), 2927–2931. DOI: 10.1158/1078-0432.ccr-09-2329. [DOI] [PubMed] [Google Scholar]
- (8).Lombardi L; Tavano F; Morelli F; Latiano TP; Di Sebastiano P; Maiello E Chemokine receptor CXCR4: role in gastrointestinal cancer. Crit Rev Oncol Hematol. 2013, 88 (3), 696–705. DOI: 10.1016/j.critrevonc.2013.08.005. [DOI] [PubMed] [Google Scholar]
- (9).Rubin JB; Kung AL; Klein RS; Chan JA; Sun Y; Schmidt K; Kieran MW; Luster AD; Segal RA A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A. 2003, 100 (23), 13513–13518. DOI: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kioi M; Vogel H; Schultz G; Hoffman RM; Harsh GR; Brown JM Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010, 120 (3), 694–705. DOI: 10.1172/jci40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Goffart N; Kroonen J; Di Valentin E; Dedobbeleer M; Denne A; Martinive P; Rogister B Adult mouse subventricular zones stimulate glioblastoma stem cells specific invasion through CXCL12/CXCR4 signaling. Neuro Oncol. 2015, 17 (1), 81–94. DOI: 10.1093/neuonc/nou144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Eckert F; Schilbach K; Klumpp L; Bardoscia L; Sezgin EC; Schwab M; Zips D; Huber SM Potential Role of CXCR4 Targeting in the Context of Radiotherapy and Immunotherapy of Cancer. Front Immunol 2018, 9, 3018. DOI: 10.3389/fimmu.2018.03018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gupta SK; Lysko PG; Pillarisetti K; Ohlstein E; Stadel JM Chemokine receptors in human endothelial cells. Functional expression of CXCR4 and its transcriptional regulation by inflammatory cytokines. J Biol Chem. 1998, 273 (7), 4282–4287. DOI: 10.1074/jbc.273.7.4282. [DOI] [PubMed] [Google Scholar]
- (14).Murdoch C; Monk PN; Finn A Functional expression of chemokine receptor CXCR4 on human epithelial cells. Immunology 1999, 98 (1), 36–41. DOI: 10.1046/j.1365-2567.1999.00848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Guyon A CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front Cell Neurosci. 2014, 8, 65. DOI: 10.3389/fncel.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jiang K; Li J; Zhang J; Wang L; Zhang Q; Ge J; Guo Y; Wang B; Huang Y; Yang T; et al. SDF-1/CXCR4 axis facilitates myeloid-derived suppressor cells accumulation in osteosarcoma microenvironment and blunts the response to anti-PD-1 therapy. Int Immunopharmacol. 2019, 75, 105818. DOI: 10.1016/j.intimp.2019.105818. [DOI] [PubMed] [Google Scholar]
- (17).Obermajer N; Muthuswamy R; Odunsi K; Edwards RP; Kalinski P PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011, 71 (24), 7463–7470. DOI: 10.1158/0008-5472.can-11-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chang SC; Lin PC; Yang SH; Wang HS; Li AF; Lin JK SDF-1alpha G801A polymorphism predicts lymph node metastasis in stage T3 colorectal cancer. Ann Surg Oncol. 2009, 16 (8), 2323–2330. DOI: 10.1245/s10434-009-0501-x. [DOI] [PubMed] [Google Scholar]
- (19).Benedicto A; Romayor I; Arteta B CXCR4 receptor blockage reduces the contribution of tumor and stromal cells to the metastatic growth in the liver. Oncol Rep. 2018, 39 (4), 2022–2030. DOI: 10.3892/or.2018.6254. [DOI] [PubMed] [Google Scholar]
- (20).Huang X; Xiong M; Jin Y; Deng C; Xu H; An C; Hao L; Yang X; Deng X; Tu Z; et al. Evidence that high-migration drug-surviving MOLT4 leukemia cells exhibit cancer stem cell-like properties. Int J Oncol. 2016, 49 (1), 343–351. DOI: 10.3892/ijo.2016.3526. [DOI] [PubMed] [Google Scholar]
- (21).Zhou J; Patel TR; Sirianni RW; Strohbehn G; Zheng M-Q; Duong N; Schafbauer T; Huttner AJ; Huang Y; Carson RE; et al. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc Natl Acad Sci U S A. 2013, 110 (29), 11751. DOI: 10.1073/pnas.1304504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zottel A; Videtič Paska A; Jovčevska I Nanotechnology Meets Oncology: Nanomaterials in Brain Cancer Research, Diagnosis and Therapy. Materials (Basel) 2019, 12 (10), 1588. DOI: 10.3390/ma12101588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gregory JV; Kadiyala P; Doherty R; Cadena M; Habeel S; Ruoslahti E; Lowenstein PR; Castro MG; Lahann J Systemic brain tumor delivery of synthetic protein nanoparticles for glioblastoma therapy. Nature Commun. 2020, 11 (1), 5687. DOI: 10.1038/s41467-020-19225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lin T; Zhao P; Jiang Y; Tang Y; Jin H; Pan Z; He H; Yang VC; Huang Y Blood–Brain-Barrier-Penetrating Albumin Nanoparticles for Biomimetic Drug Delivery via Albumin-Binding Protein Pathways for Antiglioma Therapy. ACS Nano 2016, 10 (11), 9999–10012. DOI: 10.1021/acsnano.6b04268. [DOI] [PubMed] [Google Scholar]
- (25).Nunez FJ; Mendez FM; Kadiyala P; Alghamri MS; Savelieff MG; Garcia-Fabiani MB; Haase S; Koschmann C; Calinescu AA; Kamran N; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci Transl Med. 2019, 11 (479). DOI: 10.1126/scitranslmed.aaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Koschmann C; Calinescu AA; Nunez FJ; Mackay A; Fazal-Salom J; Thomas D; Mendez F; Kamran N; Dzaman M; Mulpuri L; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med. 2016, 8 (328), 328ra328. DOI: 10.1126/scitranslmed.aac8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Calinescu AA; Yadav VN; Carballo E; Kadiyala P; Tran D; Zamler DB; Doherty R; Srikanth M; Lowenstein PR; Castro MG Survival and Proliferation of Neural Progenitor-Derived Glioblastomas Under Hypoxic Stress is controlled by a CXCL12/CXCR4 Autocrine-Positive Feedback Mechanism. Clin Cancer Res. 2017, 23 (5), 1250–1262. DOI: 10.1158/1078-0432.CCR-15-2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Jiao H; Wang Z; Liu Y; Wang P; Xue Y Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 2011, 44 (2), 130–139. DOI: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- (29).De Clercq E Mozobil® (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir Chem Chemother. 2019, 27, 2040206619829382. DOI: 10.1177/2040206619829382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gregory JV; Vogus DR; Barajas A; Cadena MA; Mitragotri S; Lahann J Programmable Delivery of Synergistic Cancer Drug Combinations Using Bicompartmental Nanoparticles. Adv Healthc Mater. 2020, 9 (21), e2000564. DOI: 10.1002/adhm.202000564. [DOI] [PubMed] [Google Scholar]
- (31).Quevedo DF; Habibi N; Gregory JV; Hernandez Y; Brown TD; Miki R; Plummer BN; Rahmani S; Raymond JE; Mitragotri S; et al. Multifunctional Synthetic Protein Nanoparticles via Reactive Electrojetting. Macromol Rapid Commun. 2020, 41 (23), 2000425. DOI: 10.1002/marc.202000425. [DOI] [PubMed] [Google Scholar]
- (32).Kadiyala P; Carney SV; Gauss JC; Garcia-Fabiani MB; Haase S; Alghamri MS; Nunez FJ; Liu Y; Yu M; Taher A; et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J Clin Invest. 2021, 131 (4). DOI: 10.1172/JCI139542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ellison DW; Aldape KD; Capper D; Fouladi M; Gilbert MR; Gilbertson RJ; Hawkins C; Merchant TE; Pajtler K; Venneti S; et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 2020, 30 (5), 863–866. DOI: 10.1111/bpa.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nava F; Tramacere I; Fittipaldo A; Bruzzone MG; DiMeco F; Fariselli L; Finocchiaro G; Pollo B; Salmaggi A; Silvani A; et al. Survival effect of first- and second-line treatments for patients with primary glioblastoma: a cohort study from a prospective registry, 1997–2010. Neuro Oncol. 2014, 16 (5), 719–727. DOI: 10.1093/neuonc/not316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kamran N; Kadiyala P; Saxena M; Candolfi M; Li Y; Moreno-Ayala MA; Raja N; Shah D; Lowenstein PR; Castro MG Immunosuppressive Myeloid Cells’ Blockade in the Glioma Microenvironment Enhances the Efficacy of Immune-Stimulatory Gene Therapy. Mol Ther. 2017, 25 (1), 232–248. DOI: 10.1016/j.ymthe.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mirghorbani M; Van Gool S; Rezaei N Myeloid-derived suppressor cells in glioma. Expert Rev Neurother. 2013, 13 (12), 1395–1406. DOI: 10.1586/14737175.2013.857603. [DOI] [PubMed] [Google Scholar]
- (37).Gielen PR; Schulte BM; Kers-Rebel ED; Verrijp K; Petersen-Baltussen HMJM; ter Laan M; Wesseling P; Adema GJ Increase in Both CD14-Positive and CD15-Positive Myeloid-Derived Suppressor Cell Subpopulations in the Blood of Patients With Glioma But Predominance of CD15-Positive Myeloid-Derived Suppressor Cells in Glioma Tissue. J Neuropathol Exp Neurol. 2015, 74 (5), 390–400. DOI: 10.1097/NEN.0000000000000183. [DOI] [PubMed] [Google Scholar]
- (38).Alghamri MS; McClellan B; Avvari RP; Thalla R; Kamran N; Zhang L; Ventosa M; Taher A; Faisal SM; Núñez FJ; et al. G-CSF Secreted by mutant IDH1 glioma stem cells abolishes myeloid cell immunosuppression and enhances the efficacy of immunotherapy. Sci Adv. 2021, 7(40), eabh3243. DOI: 10.1126/sciadv.abh3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kroemer G; Galluzzi L; Kepp O; Zitvogel L Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013, 31, 51–72. DOI: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- (40).Moriya T; Kitagawa K; Hayakawa Y; Hemmi H; Kaisho T; Ueha S; Ikebuchi R; Yasuda I; Nakanishi Y; Honda T; et al. Immunogenic tumor cell death promotes dendritic cell migration and inhibits tumor growth via enhanced T cell immunity. iScience 2021, 24 (5), 102424. DOI: 10.1016/j.isci.2021.102424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kepp O; Tesniere A; Zitvogel L; Kroemer G The immunogenicity of tumor cell death. Curr Opin Oncol. 2009, 21 (1), 71–76. DOI: 10.1097/CCO.0b013e32831bc375. [DOI] [PubMed] [Google Scholar]
- (42).Obeid M; Panaretakis T; Joza N; Tufi R; Tesniere A; van Endert P; Zitvogel L; Kroemer G Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007, 14 (10), 1848–1850. DOI: 10.1038/sj.cdd.4402201. [DOI] [PubMed] [Google Scholar]
- (43).Kepp O; Senovilla L; Vitale I; Vacchelli E; Adjemian S; Agostinis P; Apetoh L; Aranda F; Barnaba V; Bloy N; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3 (9), e955691. DOI: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Solari JIG; Filippi-Chiela E; Pilar ES; Nunes V; Gonzalez EA; Figueiró F; Andrade CF; Klamt F Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC Cancer 2020, 20 (1), 474. DOI: 10.1186/s12885-020-06964-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mapunda JA; Tibar H; Regragui W; Engelhardt B How Does the Immune System Enter the Brain?. Front Immunol. 2022, 13, 805657. DOI: 10.3389/fimmu.2022.805657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jain KK Use of nanoparticles for drug delivery in glioblastoma multiforme. Expert Rev Neurother. 2007, 7 (4), 363–372. DOI: 10.1586/14737175.7.4.363. [DOI] [PubMed] [Google Scholar]
- (47).Wegscheid ML; Morshed RA; Cheng Y; Lesniak MS The art of attraction: applications of multifunctional magnetic nanomaterials for malignant glioma. Expert Opin Drug Deliv. 2014, 11 (6), 957–975. DOI: 10.1517/17425247.2014.912629. [DOI] [PubMed] [Google Scholar]
- (48).Sugahara KN; Teesalu T; Karmali PP; Kotamraju VR; Agemy L; Greenwald DR; Ruoslahti E Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010, 328 (5981), 1031–1035. DOI: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sugahara KN; Scodeller P; Braun GB; de Mendoza TH; Yamazaki CM; Kluger MD; Kitayama J; Alvarez E; Howell SB; Teesalu T; et al. A tumor-penetrating peptide enhances circulation-independent targeting of peritoneal carcinomatosis. J Control Release 2015, 212, 59–69. DOI: 10.1016/j.jconrel.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sugahara KN; Teesalu T; Karmali PP; Kotamraju VR; Agemy L; Girard OM; Hanahan D; Mattrey RF; Ruoslahti E Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16 (6), 510–520. DOI: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Biasci D; Smoragiewicz M; Connell CM; Wang Z; Gao Y; Thaventhiran JED; Basu B; Magiera L; Johnson TI; Bax L; et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc Natl Acad Sci U S A. 2020, 117 (46), 28960. DOI: 10.1073/pnas.2013644117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Pan Q; Cao X; Luo Y; Li J; Feng J; Li F Chemokine receptor-4 targeted PET/CT with (68)Ga-Pentixafor in assessment of newly diagnosed multiple myeloma: comparison to (18)F-FDG PET/CT. Eur J Nucl Med Mol Imaging 2020, 47 (3), 537–546. DOI: 10.1007/s00259-019-04605-z. [DOI] [PubMed] [Google Scholar]
- (53).Miles AJ; Ramalli SG; Wallace BA DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2022, 31 (1), 37–46. DOI: 10.1002/pro.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Sreerama N; Woody RW Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000, 287 (2), 252–260. DOI: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- (55).Sreerama N; Venyaminov SY; Woody RW Estimation of the number of alpha-helical and beta-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 1999, 8 (2), 370–380. DOI: 10.1110/ps.8.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Calinescu A-A; Núñez FJ; Koschmann C; Kolb BL; Lowenstein PR; Castro MG Transposon mediated integration of plasmid DNA into the subventricular zone of neonatal mice to generate novel models of glioblastoma. J Vis Exp. 2015, (96). DOI: 10.3791/52443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Hambardzumyan D; Amankulor NM; Helmy KY; Becher OJ; Holland EC Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol. 2009, 2 (2), 89–95. DOI: 10.1593/tlo.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Baker GJ; Castro MG; Lowenstein PR Isolation and Flow Cytometric Analysis of Glioma-infiltrating Peripheral Blood Mononuclear Cells. J Vis Exp. 2015, (105). DOI: 10.3791/53676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Johnson AM; Roach JP; Hu A; Stamatovic SM; Zochowski MR; Keep RF; Andjelkovic AV Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 2018, 32 (5), 2615–2629. DOI: 10.1096/fj.201700699R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Kazakoff PW; McGuire TR; Hoie EB; Cano M; Iversen PL An In Vitro model for endothelial permeability: assessment of monolayer integrity. In Vitro Cell Dev Biol Anim. 1995, 31 (11), 846–852. DOI: 10.1007/BF02634568. [DOI] [PubMed] [Google Scholar]
- (61).Kamran N; Li Y; Sierra M; Alghamri MS; Kadiyala P; Appelman HD; Edwards M; Lowenstein PR; Castro MG Melanoma induced immunosuppression is mediated by hematopoietic dysregulation. Oncoimmunology 2018, 7 (3), e1408750. DOI: 10.1080/2162402x.2017.1408750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.