

Abstract
Clear cell ovarian carcinoma (CCOC) is the second most common subtype of epithelial ovarian carcinoma. Late stage CCOC is not responsive to gold-standard chemotherapy and results in suboptimal outcomes for patients. In-depth molecular insight is urgently needed to stratify the disease and drive therapeutic development. We conducted global proteomics in 192 cases of CCOC compared to other epithelial ovarian carcinoma subtypes. Our results show distinct proteomic differences in CCOC compared to other epithelial ovarian cancer subtypes including alterations in lipid and purine metabolism pathways. Furthermore, we report potential clinically significant proteomic subgroups within CCOC, suggesting the biologic plausibility of stratified treatment for this cancer. Taken together, our results provide a comprehensive understanding of the CCOC proteomic landscape to facilitate future understanding and research of this disease.
Keywords: Clear cell ovarian carcinoma, proteomics, epithelial ovarian carcinoma
Introduction
Ovarian carcinoma is the fifth leading cause of death from cancer in North American women [1]. Epithelial ovarian cancer (EOC) is the most predominant and has molecularly and clinically distinct subtypes. High grade serous ovarian carcinoma (HGSC), the most common EOC, accounts for up to 60% of EOC cases [2, 3] and clear cell ovarian carcinoma (CCOC) is the second most common subtype accounting for 5–11% of EOC cases [4]. Each EOC subtype has distinct cellular origins, mutational and molecular profiles, and should be treated as different diseases. Despite this difference, gold-standard treatment for EOC subtypes is still taxane- and platinum-based chemotherapeutics. Late stage CCOC patients have worse outcome compared to other subtypes due to a lack of response to conventional chemotherapeutics [5, 6]. Understanding the CCOC molecular landscape is crucial to improve targeted therapeutic development.
Most “omics” studies have focused on HGSC, while CCOC omics characterizations have had limited case numbers. Genomic studies have indicated genomic mutation heterogeneity within the disease [7], without clear correlation between clinical outcome and mutational landscapes. Small-scale transcriptomic studies of EOC subtypes have provided a glimpse into CCOC biology [8, 9]. A recent study combining published microarray mRNA datasets and additional cases totaling 222 CCOC suggested molecular subgroups with different clinical outcomes [10]. However, clinical translation of these findings is hindered by the lack of robust outcome correlation, compounded by discordances between alterations in the genome, transcriptome, and proteome.
Proteomics analysis complements genomic and transcriptomic information with the quantification of the final gene product and provides easily translatable biomarker candidates for pathology applications. Global proteomics using archival clinical tissues have recently driven discoveries in many cancers. While large-scale proteomic characterization is available for HGSC, to our knowledge, such profiling has not been reported in CCOC. Herein, we describe the proteomic landscape of a large patient cohort of CCOC (n = 192) using SP3-clinical tissue proteomics (SP3-CTP), a global proteomic technique which allows high throughput proteomic characterization using archival tissue[11]. Furthermore, we studied the proteome of other EOC subtypes including HGSC (n = 34), endometrioid ovarian cancer (ENOC, n = 35), and low-grade serous cancers (LGSC, n = 31) to further facilitate biomarker development of ovarian cancer subtypes. We observed distinct proteomic subgroups within 192 cases of CCOC which could provide further understanding of the disease and its molecular landscape.
MATERIAL AND METHODS
Patient cohort and initial review
CCOC cases were obtained from the Canadian Ovarian Experimental Unified Resource (COEUR) [6], the Vancouver OVCARE tumour bank, and the Mayo Clinic. ENOC, HGSC, and LGSC cases were obtained from the OVCARE tumour bank and Vancouver general hospital archival tissue bank. For cases with multiple tumour blocks, the best block was chosen based on tumour content. Two 10μM scrolls were obtained from each case, followed by a hematoxylin/eosin stain (H/E). The fresh H/E slides were reviewed by experienced pathologists (DF, BTC, LNH) to confirm diagnosis, measure tumour size (cm2), and score for tumour content. Tumour content was defined as the percentage area occupied by tumour tissue on the slide. After histological review, only cases with confirmed diagnosis, having a tumour content of more than 20%, and a tumour area above 0.8 cm2 were used.
All studies using Vancouver patient tissues were conducted under the OVCARE tissue bank protocol approved by the research ethics board (H05–60119), use of Vancouver General Hospital archival tissues for proteomics was indicated under approved protocol H18–01652. Studies using tissues and clinical data from Mayo Clinic was approved by the local ethics board (18–005652). The COEUR cohort is a repository of tissues and clinical data from 2069 women diagnosed with ovarian cancer in Canada between 1992 and 2016 [6]. All Canadian biobanks received ethics approval from their local review boards to collect and share samples and clinical data. The COEUR program received local ethics approval from the Comité d’éthique de la recherche du Centre hospitalier de l’Université de Montréal and the CRCHUM ethics committee. All patients have signed a written informed consent at the local institution.
Global Proteomics and analysis
Two 10μM scrolls of the block with the highest tumour content as scored by experienced pathologists was used without macrodissection. Sample deparaffinization, protein isolation and Tandem Mass Tag (TMT) labeling was carried out as previously described [11, 12]. For detailed methods for proteomics data acquisition and data analysis, see supplemental methods.
Immunohistochemistry and interpretation
TMA and formalin-fixed paraffin embedded (FFPE) tissue blocks were sectioned at 4 μm onto charged glass slides, air-dried for 10 minutes, and baked for an hour at 60°C. All immunohistochemical stains were performed on the automated Ventana Benchmark and Discovery systems (Ventana Medical Systems) using cell conditioning solution CC1 (Ventana), heat induced antigen retrieval (37°C for 2 hours) and Ventana OptiView DAB detection kit. Primary antibodies used were GDA (HPA019352, Sigma, 1:500), PSAT1 (NBP1–32920, Novusbio, 1:1000), and LAMC1 (HPA001909, Sigma, 1:250). TMAs were scored by experienced pathologists (LNH, JH). Only staining in tumour areas were considered for histoscore evaluation. The histoscore was determined as the average of duplicate cores and was defined as the sum of the intensity of tumour staining (0–3) multiplied by the percentage of tumour cells staining (0 – 100%).
Statistical analysis
For survival analysis, we considered the univariable association between proteomics cluster group and clinicopathological parameters using a Chi-squared test for binary and categorical variables and a Kruskal-Wallis test for continuous variables. Cases with missing values were removed from analysis. We assessed the univariable effect of clinicopathological parameters and proteomics cluster group. Furthermore, multivariate survival models were fit to account for the effects of known prognostic factors. The Firth penalized maximum-likelihood bias-reduction method was used to estimate hazard ratios when the proportion of censored cases exceeded 80%. Statistical significance was set at 0.05 and no attempts were made to adjust for multiple comparisons. All statistical analyses were done using R project for statistical computing. For Kaplan-Meier curves, the associated log ranked pvalue was corrected with Benjamini-Hochberg when three or more conditions were compared.
Statistical survival analyses were performed using R statistical software using the “survminer” and “survival” packages.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [13] partner repository with the dataset identifier/accession code PXD032355.
Additional methods are available in supplementary materials.
RESULTS
Patient cohort characteristics
Thirty-eight CCOC cases were excluded due to insufficient tumour content. Four cases were reclassified as different subtypes and included in the proteomic analysis with their confirmed subtypes for a total case count of 192 CCOC, 34 HGSC, 35 ENOC, and 31 LGSC (Figure S1A). The average tumour content of ENOC, HGSC, and LGSC cases were high (76%, 85%, and 81%, respectively). In contrast, the CCOC cohort contained more cases with lower tumour content, however 76% of cases had tumour contents above 60%.
The median age at diagnosis for CCOC, ENOC, HGSC, and LGSC were 56 (37 −82), 54 (32 – 89), 58 (42 – 88) and 57 (26 – 78) respectively (Figure S1B). While both CCOC and ENOC contained mainly stage 1 and 2 diseases (70.5% and 71.4%), most cases of HGSC and LGSC were diagnosed at late stage (76.5% and 65.6% respectively) (Figure S1C). HGSC and LGSC had median overall survival (OS) of 58.1 and 62.6 months, and a median progression-free survival (PFS) of 24.7 and 25.3 months respectively. CCOC had a median OS of 51.6 months and PFS of 30.4 months, where ENOC had the best OS and PFS at 66.7 and 63.8 months (Figure S1D–E). The clinical characteristics are representatie of previously reported data [6, 14–16].
Within the CCOC cohort, later stage at diagnosis was associated with worse outcome. Compared to stage 1, patients diagnosed with stages 2– 4 had worse OS with a hazard ratio of 3.85 (2.3 −6.46, LRT pvalue = 0.000), and PFS with a hazard ratio of 3.96 (2.36 – 6.63, LRT pvalue = 0.000) (Table 1). Furthermore, while age at diagnosis was not prognostic, disease optimal debulking at surgery was significantly associated with better OS and PFS (Table 1). Both stage and residual disease have been previously reported as prognostic factors[16].
Table 1.
Univariate survival analysis of clinicopathological parameters of CCOC
| Age at diagnosis | # of events/n | Comparison | Hazard Ratio (95% CI) | P value |
|---|---|---|---|---|
| OS | 74 / 191 | 1.01 (0.98–1.03) | 0.6573 | |
| PFS | 75 / 155 | 1.00 (0.97–1.02) | 0.7746 | |
|
| ||||
| Stage (reference: 1) | ||||
| OS | 72 / 187 | 2–4 | 3.85 (2.30–6.46) | 0 |
| PFS | 74 / 153 | 2–4 | 3.96 (2.36–6.63) | 0 |
|
| ||||
| Residual disease (reference: non-optimal) | ||||
| OS | 56 / 129 | optimal | 0.28 (0.17–0.48) | 0 |
| PFS | 60 / 125 | optimal | 0.21 (0.12–0.35) | 0 |
|
| ||||
|
ARID1A IHC (reference: 0) |
||||
| OS | 1/2 | 1.56 (0.92–2.65) | 0.0911 | |
| PFS | 1/2 | 1.34 (0.80–2.25) | 0.2538 | |
Using an IHC assay for ARID1A, we showed that of 154 cases with interpretable ARID1A staining, 69 cases exhibited a complete loss (44.8%), and 6 had ARID1A subclonal loss. In a univariate survival analysis, ARID1A protein loss was not prognostic (Table 1). Overall, our proteomic cohort characteristics are similar to previously reported data, suggesting that the subtypes were well-represented.
Global proteomics identify CCOC as a distinct disease compared to other ovarian cancer subtypes
The global proteomic analysis identified 7468 proteins, of which 3069 were quantified in all samples. Around 90% of the proteins identified were quantified with at least 3 peptides and 80% by 4 or more (Figure S2A, B). In total, 84 proteins out of the 86 proteins targeted by isoDoping were detected with 2 or more peptides. IsoDoped proteins were quantified more consistently compared to endogenous proteins, with 90% of the isoDoped proteins quantified in all samples (Figure S2C).
In a principal component analysis (PCA), CCOC displays a distinct proteomic landscape compared to other EOC subtypes (Figure 1A). Interestingly, the CCOC cohort was separated into two primary clades by hierarchical clustering (Figure 1B). We investigated confounding factors contributing to proteomic differences including batch effect, the biobank location, and tumour content. While there was no contribution of batch and biobank location to proteomic clusters, tumour content was associated with some hierarchical clusters within the CCOC cohort (Figure 1B, Figure S2D–F).
Figure 1.
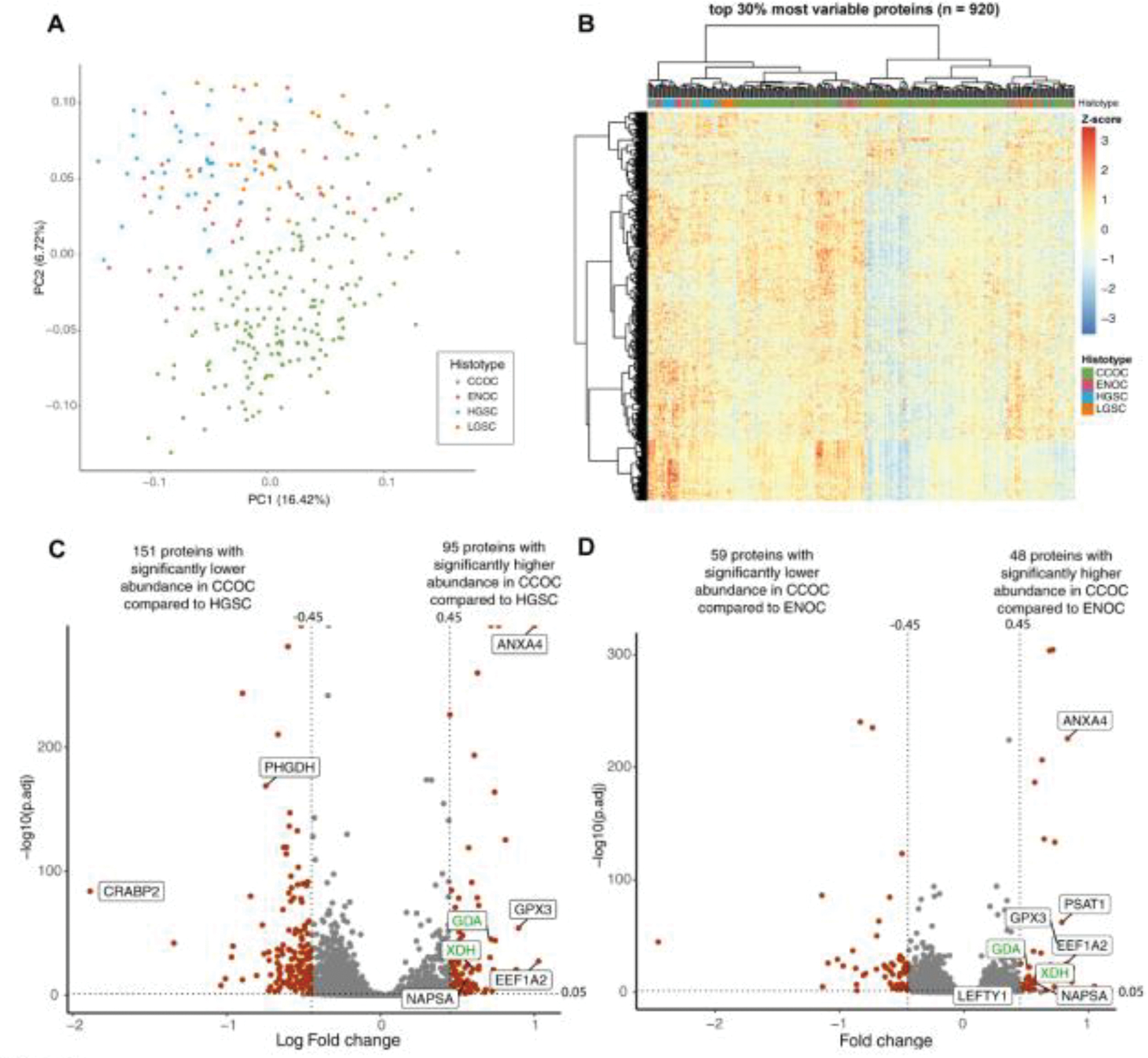
CCOC has a distinct proteome. (A) Principal component analysis and (B) hierarchical heatmap representing unsupervised clustering of the top 30% most differentially abundant proteins separate CCOC from other EOC subtypes. Volcano plot showing PECA results comparing (C) CCOC to HGSC and (D) CCOC to ENOC. Grey indicates proteins not having significantly abundance variation, whereas red are significantly differentially abundant proteins between the subtypes. Proteins labeled in black represent notable targets consistent with previous publications. Proteins labeled in green represent some novel biomarker targets.
Significantly differentially abundant proteins were defined as proteins with more than 1 peptide, padj <0.05, and log2 fold change of > 0.45 or < −0.45 (Figure 1C, D). Differential abundance analysis identified clinical CCOC markers (NAPSA), previously characterized IHC CCOC markers (CTH, LEFTY1) [17, 18], and highly expressed in CCOC in previous protein and transcriptome analysis (PSAT1, ANXA4, GPX3, LAMC1, SOD2) [8, 19]. Moreover, previously described HGSC markers CRABP2 and L1RE1 were reported as highly abundant in HGSC compared to CCOC (Figure 1C, 1D) [19, 20].
Notably, many metabolic enzymes had higher abundance in CCOC compared to other subtypes, including purine metabolism pathway proteins (XDH, GDA), and serine/cysteine associated pathway proteins (PSAT1, CTH, GPX3). The laminin subunits (LAMC1, LAMB1, LAMA4) were highly abundant in CCOC. Using IHC, we confirmed that LAMC1 was expressed in CCOC tumour cells, and at a higher abundance compared to HGSC and ENOC (Figure S3A, S3B). Tables S2 and S3 details the differentially abundant proteins between the EOC subtypes.
We performed gene set enrichment analysis (GSEA) to identify pathways enriched in each EOC subtype. When comparing to ENOC and HGSC, CCOC was enriched in processes related to homeostasis, extracellular matrix, and lipid metabolism. Similar pathways were enriched in ENOC and HGSC compared to CCOC, including pathways in protein metabolism and DNA replication (Figure 2A, 2B, Table S4, S5).
Figure 2.

Enriched pathways in a GSEA analysis comparing CCOC to ENOC and HGSC. GO annotation representing the top 15 enriched pathways in (A) CCOC and HGSC, and (B) CCOC and ENOC. All enriched pathways have an adjusted p value of less than 0.05. Red: coagulation related pathways; Blue: metabolism related pathways; Green: protein metabolism pathways; and orange: DNA synthesis pathways.
Distinct clinically relevant subgroups are present within CCOCs
To investigate the heterogeneity of CCOC, we used consensus clustering k-means clustering algorithm to identify 4 robust clusters within CCOC (Figure S4A–B). Based on morphological and proteomic features, we separated CCOC into 4 defined clusters: metabolically active, coagulative, fibrotic, and necrotic (Figure 3A).
Figure 3.

CCOC has distinct subgroups. (A) heatmap depicting four distinct clusters within CCOC. (B) GSEA depicting enriched pathways in each CCOC proteomic subgroup, and Kaplan-Meier plot describing the(C) overall survival and (D) progression free survival of patients in each cluster.
There were no predominant morphological architecture or cytoplasmic features correlated to the clusters (Figure S4C, S4D), suggesting that morphological features in CCOC does not reflect molecular changes. The fibrotic cluster was enriched for fibrous tissue (p<0.001), whereas the necrotic cluster had more tumour necrosis compared to the other clusters (p<0.001) (Figure S4E, S4F). There were no significant correlations between host immune infiltrates or mitotic counts and the proteomic clusters (FigureS4G, S4H).
Through GSEA, the fibrotic cluster showed enrichment for proteins in the extracellular matrix pathways, consistent with the abundance of fibrotic areas. Furthermore, the necrotic cluster showed an enrichment in coagulation and platelet activation pathways, suggesting acute inflammatory responses to necrosis. In contrast, the metabolically active cluster was enriched for protein turnover control, cell cycle control, tricarboxylic acid cycle, electron transport chain, amino acid, and fatty acid metabolic processes. The coagulative cluster was enriched in lipoprotein transport and breakdown, as well as complement activation and coagulation pathways despite the lack of overt necrosis on morphology (Figure 3B, Figure S4E).
As the metabolic and coagulative clusters were the most distinct in outcome and least affected by fibrotic and necrotic tissues, we performed differential protein expression analysis comparing these two subgroups. Differentially expression analysis identified inflammatory processes (Mucin 5B, S100A9, S100A8, SERPINA3) and fibrinogen subunits (FGG, FGB, FGA) to be significantly accumulated in the coagulative cluster compared to the metabolic cluster (Table S6). Furthermore, fibrinogen subunits expression did not correlate with the amount of necrotic tissues in the coagulative cluster (Figure S4I–K).
While the fibrotic and necrotic clusters had similar distributions of stage, the metabolic cluster contained more stage 1 and 2 patients (58.6% and 23%, respectively). In contrast, coagulative cluster had the highest percentage of stage 3 and 4 patients (56%). The clinicopathological parameters of each cluster is described in Table S7. Survival analysis showed no significant difference of OS between the clusters (Figure 3C), but the coagulative cluster had a significantly worse PFS (Figure 3D). Univariate analysis indicated metabolic cluster was associated with better PFS but not OS where coagulative cluster had significantly worse PFS (Table 2). In a multivariate survival analysis adjusting for age, grade, stage, residual disease, ARID1A IHC, and adjuvant treatment, the metabolic cluster was associated with significant better OS and PFS, whereas coagulative cluster was a worse prognosticator (Table 3).
Table 2.
Univariate survival analysis of CCOC proteomic clusters.
| # of events/n | Comparison cluster | Hazard Ratio (95% CI) | LRT p-value | |
|---|---|---|---|---|
|
| ||||
| Proteomic cluster (Reference: Metabolic) | ||||
| OS | 74 / 191 | Fibrotic | 1.50 (0.81–2.80) | 0.194 |
| Coagulative | 1.83 (1.05–3.21) | |||
| Necrotic | 1.43 (0.58–3.51) | |||
| PFS | 75 / 155 | Fibrotic | 1.79 (0.96–3.36) | 0.0365 |
| Coagulative | 2.21 (1.27–3.85) | |||
| Necrotic | 1.63 (0.66–4.01) | |||
|
| ||||
| Proteomics cluster (Reference: Metabolic) | ||||
| OS | 74 / 191 | F,C,N* | 1.65 (1.01–2.69) | 0.0413 |
| PFS | 75 / 155 | F,C,N* | 1.97 (1.20–3.24) | 0.0053 |
|
| ||||
| Proteomics cluster (Reference: Coagulative) | ||||
| OS | 74 / 191 | M,F,N* | 0.65 (0.40–1.06) | 0.0889 |
| PFS | 75 / 155 | M,F,N* | 0.58 (0.36–0.93) | 0.0261 |
Proteomic Cluster M: Metabolic active cluster; F: Fibrotic cluster; C: Coagulative cluster; N: Necrotic cluster
Table 3.
Multivariate analysis of CCOC protein clusters and other clinicopathological parameters.
| # of events/n | comparison | Hazard Ratio (95% CI) | LRT p-value | |
|---|---|---|---|---|
|
| ||||
| Overall survival | 34 / 80 | |||
| Cluster (reference: Metabolic) | F,C,N* | 3.02 (1.44–6.69)F | 0.0027 | |
| Cluster (reference: Coagulative) | M,F,N* | 0.46 (0.22–0.96)F | 0.0364 | |
| Age and diagnosis | 1.00 (0.96–1.04)F | 0.8952 | ||
| Stage (reference: Stage I) | II-IV | 7.98 (3.22–23.55)F | 0 | |
| Residual disease (reference: non-optimal) | optimal | 0.29 (0.14–0.61)F | 0.0015 | |
| ARID1A (IHC) (reference: 0) | 1/2 | 1.03 (0.49–2.26)F | 0.8882 | |
| Adjuvant treatment (reference: no) | yes | 0.06 (0.01–0.39)F | 0.013 | |
|
| ||||
| Progression-free survival | 37 / 78 | |||
| Cluster (reference: Metabolic) | F,C,N* | 3.02 (1.46–6.65)F | 0.002 | |
| Cluster (reference: Coagulative) | M,F,N* | 0.44 (0.21–0.92)F | 0.0274 | |
| Age and diagnosis | 0.98 (0.94–1.02)F | 0.3481 | ||
| Stage (reference: Stage I) | II-IV | 6.10 (2.79–14.80)F | 0 | |
| Residual disease (reference: non-optimal) | optimal | 0.24 (0.12–0.50)F | 0.0002 | |
| ARID1A (IHC) (reference: 0) | 1/2* | 1.07 (0.54–2.15)F | 0.8024 | |
| Adjuvant treatment (reference: no) | yes | 0.31 (0.06–3.09)F | 0.4782 | |
indicates that the Firth’s penalized maximum likelihood bias reduction method was used to estimate the hazard ratio.
Proteomic Cluster M: Metabolic active cluster; F: Fibrotic cluster; C: Coagulative cluster; N: Necrotic cluster
We next investigated whether any of the proteomic clusters showed enrichment of recurrent CCOC gene mutations ARID1A and PIK3CA. While the ARID1A mutations resulted in the loss of ARID1A protein expression[21, 22], the PIK3CA mutations are activating mutations[23]. In our cohort, ARID1A protein loss had a similar prevalence in the metabolic, fibrotic, and necrotic clusters (33%, 39%, 40% respectively), but was enriched in the coagulative cluster (66.7%, Pearson Chi-square 13.1, p = 0 .043) (Table S7). PIK3CA mutation status was determined using targeted exome sequencing (manuscript in preparation), the metabolic, coagulative, and necrotic clusters exhibited similar percentages of cases with PIK3CA mutations (61%, 51.3%, and 58.3%), whereas the fibrotic cluster had the lowest proportion of PIK3CA mutated cases (21.8%). Furthermore, we did not observe significant correlation between protein clusters and the top 10 most altered genes on exome sequencing panel (Figure S4L).
Applying previous signatures in CCOC cohort
Due to the scarcity of CCOC, limited previous knowledge was available to study disease heterogeneity. Recently, Tan et al evaluated the mRNA landscape using microarray data for 222 CCOC cases [10]. They identified two distinct mRNA expression subtypes based on unsupervised clustering – a mesenchymal-like (MesCC-like), and an epithelial-like subgroup (EpiCC-like). While MesCC-like subgroup was enriched for later stage and worse outcome, EpiCC-like subgroup contained earlier stage patients and better clinical outcome. We applied the gene signature from Tan et al containing 1052 genes in the EpiCC group, and 1145 in the MesCC groups to our proteomic cohort. 605 proteins corresponding to the gene signatures were present in the proteome data and were used in an unsupervised clustering (Figure S5A). Cases classified as MesCC were enriched for low tumour content, and largely comprised of the fibrotic and necrotic clusters from our cohort. This was expected as stromal cells are more likely to exhibit a mesenchymal expression signature. A detailed morphological review was not available for Tan et al due to the use of frozen tissue and publicly deposited microarray data, raising the possibility that the MesCC signature was derived from stromal tissues. To lessen the contribution of cases with poor tissue content, we repeated the unsupervised clustering using only the intrinsic metabolic and coagulative subgroups. While the clustering showed good separation, the heatmap did not suggest they were driven by the MesCC and EpiCC gene signatures (Figure S5B).
Clear cell carcinomas of different anatomical sites exhibit morphological and clinical similarities. One of the most prevalent CCOC counterpart is clear cell renal cell carcinomas (ccRCC), which clinicopathological landscapes we have compared in a previous review article [24]. Clark et al characterized the proteogenomic landscapes of ccRCC with matched normal kidney tissue[25]. We applied the Clark et al ccRCC proteomic signature on our CCOC cohort using hierarchical clustering (Figure S5C). We show our CCOC proteomic clusters were predominately preserved using the ccRCC proteomic signatures. This intriguing finding further solidifies the similarities between the two clear cell cancers, suggesting that the clear cell phenotype may be driven by similar biological processes despite distinct cellular origins and driver mutations.
Target validation using immunohistochemistry
We noticed many metabolic enzymes to be differentially expressed in CCOC. Notable elevated enzymes included cystathionine gamma lyase (CTH), which has been previously studied [17], and phosphoserine aminotransferase (PSAT1) previously reported to have a higher mRNA expression in CCOC compared to other subtypes, which we confirmed by IHC (Figure S6A – B). PSAT1 expression was found to have no significant association with outcome in CCOC (Figure S6C–D). Moreover, we discovered multiple enzymes involved in purine metabolism had a higher abundance in CCOC compared to other subtypes, including guanine deaminase (GDA) and xanthine deaminase (XDH) (Table S1, S2). GDA and XDH are enzymes involved in the sequential breakdown of guanine into uric acid, suggesting aberrations in this pathway.
We used a combination of the proteomic cohort and an independent confirmation cohort from a local gynecological bank to validate the proteomic findings. GDA exhibited tumour-specific cytoplasmic staining in CCOC and ENOC, and was null in HGSC (Figure 4A). IHC analysis of the proteomic cohort confirmed the findings, with CCOC having the highest median histoscore compared to ENOC and HGSC (275, 130, 100 respectively, p < 0.001) (Figure 4B). The confirmation cohort GDA IHC data had a similar result with HGSC and ENOC having a GDA median histoscore of 70 and 152, where CCOC had a median histoscore of 240 (Figure 4C). Interestingly, high GDA expression in CCOC correlated to a better disease specific survival (log-rank p = 0.01), but no differences were seen in overall or progression-free survival (Figure 4D–E). This survival advantage also reflected in the proteomic results (Figure S6 E,F). Significantly better disease specific survival was maintained on multivariate survival analysis (Table 4). Our results suggest that the activation of purine metabolism through the increased expression of GDA is a unique feature in CCOC.
Figure 4.

Guanine deaminase (GDA) immunohistochemical validation. (A) Representative immunohistochemistry staining of GDA in different ovarian cancer subtypes, and boxplots representing (B) GDA histoscores in proteomic cases, and (C) an independent confirmation cohort. (D) Disease specific survival and (E), progression-free survival in CCOC with high GDA and low GDA expression. The statistical significance in multiple group comparisons is calculated with a Kruskal-Wallis test with a post-hoc Dunn’s test with Benjamini-Hochberg correction.
Table 4.
Multivariable analysis of GDA expression in clear cell ovarian carcinoma
| # of events / n | Comparison | Hazard Ratio (95% CI) | LRT P-value | |
|---|---|---|---|---|
|
| ||||
| Overall survival | 38 / 67 | |||
| GDA (IHC: h-score) - optimal cut point (clear cell) (reference: <230) | >=230 | 0.53 (0.27–1.05)F | 0.0751 | |
| Age at surgery | 1.03 (0.99–1.07)F | 0.0984 | ||
| Grade (reference: grade 1/2) | grade 3 | 0.25 (0.05–2.47)F | 0.3803 | |
| Stage (reference: I) | II-IV | 3.62 (1.69–8.73)F | 0.0005 | |
| Adjuvant treatment (reference: none) | chemo | 1.02 (0.24–9.5)F | 0.7144 | |
|
| ||||
| Diseases specific survival | 30 / 66 | |||
| GDA (IHC: h-score) - optimal cut point (clear cell) (reference: <230) | >=230 | 0.44 (0.21–0.92)F | 0.0350 | |
| Age at surgery | 1.01 (0.97–1.05)F | 0.5669 | ||
| Grade reference: grade 1/2) | grade 3 | 0.46 (0.05–61.63)F | 0.5288 | |
| Stage (reference: I) | II-IV | 3.72 (1.6–10.11)F | 0.0012 | |
| Adjuvant treatment (reference: none) | chemo | 0.97 (0.23–9.06)F | 0.7499 | |
|
| ||||
| Progression-free survival | 32 / 37 | |||
| GDA (IHC: h-score) - optimal cut point (clear cell) (reference: <230) | >=230 | 0.85 (0.33–2.21)F | 0.7553 | |
| Age at surgery | 0.98 (0.94–1.03)F | 0.4511 | ||
| Grade (reference: grade 1/2) | grade 3 | 3 (0.28–412.92)F | 0.1419 | |
| Stage (reference: I) | II-IV | 1.86 (0.76–5.43)F | 0.1458 | |
| Adjuvant treatment (reference: none) | chemo | 0.87 (0.2–8.22)F | 0.8462 | |
indicates that the Firth’s penalized maximum likelihood bias reduction method was used to estimate the hazard ratio.
Discussion
Epithelial ovarian carcinoma subtypes are being increasingly recognized as different diseases due to their distinct mutational landscape and cells of origin [26]. While CCOC and ENOC share similar mutations and are derived from either the ciliated or secretory cells of ovarian endometriotic cysts [27], HGSC originates from serous tubal intraepithelial carcinoma in the fallopian tubes [28].
Principal component analysis showed CCOC has a distinct proteomic landscape. Pathway analysis comparing CCOC to ENOC and HGSC recognized distinct differences in molecular processes such as coagulation and lipid metabolism. Increased lipid metabolism processes may suggest that the clear cytoplasm of CCOC could contain fatty droplets in addition to glycogen, potentially acting as an alternative fuel for tumour growth. Metabolic enzymes in the oxidative response pathways were highly expressed in CCOC (CTH, GPX3, SOD2), suggesting an active response to oxidative damage. Previous literature has postulated the role of reactive oxygen species (ROS) damage caused by menstrual cycling in the carcinogenesis of endometriosis-associated ovarian cancers [29]. A proteomic signature reflecting increased adaptation to oxidative stress suggests that ameliorating ROS damage could be a crucial step in the malignant transformation and maintenance of CCOC, especially in a background of florid inflammation and coagulation in endometriotic cysts. This finely balanced redox state could serve as a potential therapeutic option where ROS-generating therapeutics could tip the disease towards increased cell death. This could also explain CCOC’s increased response to radiation therapy.
Obtaining the proteomic landscape of a large cohort of CCOC afforded us the ability to study the disease heterogeneity. Since we did not macrodissect the tumour tissue, stromal components contributed to the proteomic complexity in a small number of cases. While this presents a drawback in stratifying the disease based on the tumour cell proteome, the abundance of stromal components may represent tumour biology. Among the cases with lower tumour content, we observed fibrosis-rich and necrosis-rich clusters with no associated outcome differences, suggesting that tumour morphology may not play a prognostic role.
Cases with high tumour content were separated into metabolically-active and coagulative clusters. Despite the lack of necrosis on histology, the coagulative cluster exhibited high expressions of coagulative proteins. Clinically, CCOC is associated with increased risk of venous thromboembolism (VTE) [30], reported in as high as 42% of patients [31]. The accumulation of fibrinogen subunits in tumours in the coagulative cluster could reflect VTE risk, therefore serving as a potential prognostic biomarker guiding clinical decision on VTE prophylaxis. A large case series with outcome and VTE information would be an optimum strategy to study this association. The metabolically active cluster was enriched in cell cycle processes, suggesting a more proliferative state and potentially a better response to cytotoxic chemotherapeutics. Moreover, the proteomic signature suggests the metabolic CCOC subgroup is more dependent on using carbohydrate and fatty acids as cellular fuels, providing possible targets for metabolic therapeutics.
Interestingly, the CCOC proteomic clusters did not correlate with PIK3CA mutation, there was only weak association between ARID1A protein loss and the coagulative cluster. In the literature, the consequences of these mutations have not been elucidated in the appropriate cell of origin. Alterations in ARID1A and PIK3CA frequently co-occur, and studies in adjacent endometriosis suggests that ARID1A protein loss is an early event in CCOC tumorigenesis [32, 33]. This lack of correlation suggests redundant pathway alternations in CCOC precursor cells, followed by convergent tumour evolution as the malignancy matures in a background of microenvironment stresses. This finding is consistent with previous studies showing ARID1A expression was not a prognostic factor [34]. Moreover, we noticed similarities in the proteomic landscapes of CCOC and ccRCC, suggesting a decreased contribution of driver mutations in tumour maintenance. Studying the roles of these mutations in the correct cell of origin and cellular cntext is crucial to deciphering the oncogenesis of CCOC.
In a nanostring immune profiling in CCOC, Heong et al discovered four clinically relevant immune subtypes – PD1 high, CTLA4 high, antigen-presentation, and pro-angiogenic [35]. Our proteomic analysis did not identify immune protein differences between the clusters, suggesting immune related processes are not a primary driver of the molecular differences between the subgroups. However, the importance of the tumour immune response should not be overlooked especially given the optimistic response to immunotherapy in CCOC. Further correlation between the subtypes found by Heong et al and the proteome characteristics can be studied through PD1 and CTLA4 IHC, particularly in its relation to metabolic and inflammatory protein expression.
A recent study identified two clinically relevant DNA methylation clusters among 271 cases of CCOC [36]. Methylation cluster 1 was associated with advanced stage and TP53 mutations, whereas methylation cluster 2 was found to represent earlier stage cases and PIK3CA mutations. Transcriptomic analysis completed by Cunningham et al identified a difference in expression related to inflammation and immune pathways between the two clusters. Thirty-eight cases were common between our proteomic cohort and the methylation study, in which 18 were methylation cluster 1, and 20 cases were methylation cluster 2. Methylation cluster 2 was enriched for cases in the metabolically active proteomic cluster (10/20, 50%), while methylation cluster 1 had 7 metabolically active cases (7/18, 39%). Although there was not enough power to conclude statistical significance, it is suggestive that an active metabolism could contribute to a unique methylation profile by increasing the availability of methyl donor metabolites.
We observed many metabolic alterations in CCOC, including increased XDH and GDA expression, suggesting an active purine metabolism pathway. Higher abundance of GDA in CCOC is correlative of a significantly improved disease specific survival in univariate and multivariate analysis. This may suggest a loss in GDA activity as a route of chemotherapy or radiotherapy resistance. The role of the purine pathway and its mechanism of activity could be studied in CCOC and its precursors. If GDA expression is an early event in CCOC oncogenesis, its decreased expression could represent a late metabolic adaptation giving rise to subpopulations that are more resistant to treatments.
In this study, we profiled the global proteomic landscape of CCOC compared to other EOC subtypes. Our study identified CCOC markers previously described in transcriptome and small-scale proteomic studies[8, 19], but also provided new insights into CCOC biology. Furthermore, we show that CCOC is a heterogenous disease with subtypes having distinct metabolic enzyme alterations. The results of this study provide many targetable pathway candidates, beyond those we preliminarily validated, for further prognostic and therapeutic development and understanding CCOC biology.
Supplementary Material
Table S1 Isodoping library protein list
Table S6 differential protein expression PECA analysis results of the metabolically active cluster compared to the coagulative cluster
Table S7 Clinical and pathological parameters of CCOC proteomic clusters
Figure S1 Clinical and pathological parameters of the proteomic cohort
Figure S2 Proteomic analysis parameters
Figure S3 Imunohistochemistry stain of LAMC1
Figure S4 CCOC contain diverse proteomic subgroups
Figure S5 Previous transcriptomic and proteomic signatures applied to CCOC.
Figure S6 Immunohistochemistry validation of proteomic targets
Table S2 differential protein expression PECA analysis results of CCOC compared to HGSC
Table S3 differential protein expression PECA analysis results of CCOC compared to ENOC
Table S4 Gene Set Enrichment Analysis of CCOC compared to HGSC
Table S5 Gene Set Enrichment Analysis of CCOC compared to ENOC
Acknowledgements
This study was supported by grants from the Canadian Cancer Society (Impact grant #705647) and the Canadian Institutes of Health Research (Foundation grant #154290). The VGH & UBC Hospital Foundation and the BC Cancer Foundation provided funding to OVCARE: British Columbia’s Ovarian Cancer Research Team. This study uses resources provided by the Canadian Ovarian Cancer Research Consortium’s COEUR biobank funded by the Terry Fox Research Institute and Ovarian Cancer Canada, and managed by the Centre de recherche du Centre hospitalier de l’Université de Montréal (CRCHUM), the COEUR biobank recognizes the contribution of institutions across Canada (for a full list see https://www.tfri.ca/coeur) and is affiliated with the Canadian Tissue Repository Network. We thank the molecular pathology core facility of the CRCHUM, and in particular Liliane Meunier, for sample processing. J.X. Ji is supported by a Vanier Canada graduate scholarship and UBC-BCCA MD/PhD studentship. D. Huntsman is supported by the Dr. Chew Wei Memorial Professorship in Gynecologic Oncology and the Canada Research Chairs program (Research Chair in Molecular and Genomic Pathology). We would like to thank all patients who have donated samples to the tumour banks to support gynecological cancer research. We would like to thank Dr. Nissreen Mohammad for help with scoring TMAs.
Footnotes
Conflict of interest statement
The authors declare no conflict of interest
Ethics approval statement
All studies using Vancouver patient tissues were conducted under the OVCARE tissue bank protocol approved by the research ethics board (H05–60119), use of Vancouver General Hospital archival tissues for proteomics was indicated under approved protocol H18–01652. Studies using tissues and clinical data from Mayo Clinic was approved by the local ethics board (18–005652). The COEUR cohort is a repository of tissues and clinical data from 2069 women diagnosed with ovarian cancer in Canada between 1992 and 2016. All Canadian biobanks received ethics approval from their local review boards to collect and share samples and clinical data. The COEUR program received local ethics approval from the Comité d’éthique de la recherche du Centre hospitalier de l’Universié de Montréal and the CRCHUM ethics committee.
Patient consent statement
All patients have signed a written informed consent at the local institution.
Data Availability statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier/accession code PXD032355.
References
- 1.American Cancer Society. Cancer Statistics Center. 2017. [cited 2017 22 June]; Available from: https://cancerstatisticscenter.cancer.org/#/
- 2.Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA: A Cancer Journal for Clinicians 2018; 68: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park HK, Ruterbusch JJ, Cote ML. Recent Trends in Ovarian Cancer Incidence and Relative Survival in the United States by Race/Ethnicity and Histologic Subtypes. Cancer Epidemiol Biomarkers Prev 2017; 26: 1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.del Carmen MG, Birrer M, Schorge JO. Clear cell carcinoma of the ovary: a review of the literature. Gynecol Oncol 2012; 126: 481–490. [DOI] [PubMed] [Google Scholar]
- 5.Chan JK, Teoh D, Hu JM, et al. Do clear cell ovarian carcinomas have poorer prognosis compared to other epithelial cell types? A study of 1411 clear cell ovarian cancers. Gynecol Oncol 2008; 109: 370–376. [DOI] [PubMed] [Google Scholar]
- 6.Le Page C, Rahimi K, Kobel M, et al. Characteristics and outcome of the COEUR Canadian validation cohort for ovarian cancer biomarkers. BMC Cancer 2018; 18: 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang YK, Bashashati A, Anglesio MS, et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat Genet 2017; 49: 856–865. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz DR, Kardia SL, Shedden KA, et al. Gene expression in ovarian cancer reflects both morphology and biological behavior, distinguishing clear cell from other poor-prognosis ovarian carcinomas. Cancer Res 2002; 62: 4722–4729. [PubMed] [Google Scholar]
- 9.Stany MP, Vathipadiekal V, Ozbun L, et al. Identification of novel therapeutic targets in microdissected clear cell ovarian cancers. PLoS One 2011; 6: e21121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan TZ, Ye J, Yee CV, et al. Analysis of gene expression signatures identifies prognostic and functionally distinct ovarian clear cell carcinoma subtypes. EBioMedicine 2019, 10.1016/j.ebiom.2019.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes CS, McConechy MK, Cochrane DR, et al. Quantitative Profiling of Single Formalin Fixed Tumour Sections: proteomics for translational research. Sci Rep 2016; 6: 34949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asleh K, Negri GL, Spencer Miko SE, et al. Proteomic analysis of archival breast cancer clinical specimens identifies biological subtypes with distinct survival outcomes. Nat Commun 2022; 13: 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez-Riverol Y, Csordas A, Bai J, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research 2018; 47: D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plaxe SC. Epidemiology of low-grade serous ovarian cancer. Am J Obstet Gynecol 2008; 198: 459 e451–458; discussion 459 e458–459. [DOI] [PubMed] [Google Scholar]
- 15.Wentzensen N, Poole EM, Trabert B, et al. Ovarian Cancer Risk Factors by Histologic Subtype: An Analysis From the Ovarian Cancer Cohort Consortium. J Clin Oncol 2016; 34: 2888–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugiyama T, Kamura T, Kigawa J, et al. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer 2000; 88: 2584–2589. [PubMed] [Google Scholar]
- 17.Hughes CS, McConechy MK, Cochrane DR, et al. Quantitative Profiling of Single Formalin Fixed Tumour Sections: proteomics for translational research. Scientific Reports 2016; 6: 34949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akiya M, Yamazaki M, Matsumoto T, et al. Identification of LEFTY as a molecular marker for ovarian clear cell carcinoma. Oncotarget 2017; 8: 63646–63664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toyama A, Suzuki A, Shimada T, et al. Proteomic characterization of ovarian cancers identifying annexin-A4, phosphoserine aminotransferase, cellular retinoic acid-binding protein 2, and serpin B5 as histology-specific biomarkers. Cancer Sci 2012; 103: 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia Z, Cochrane DR, Tessier-Cloutier B, et al. Expression of L1 retrotransposon open reading frame protein 1 in gynecologic cancers. Hum Pathol 2019; 92: 39–47. [DOI] [PubMed] [Google Scholar]
- 21.Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 2010; 363: 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maeda D, Mao TL, Fukayama M, et al. Clinicopathological significance of loss of ARID1A immunoreactivity in ovarian clear cell carcinoma. Int J Mol Sci 2010; 11: 5120–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo KT, Mao TL, Jones S, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol 2009; 174: 1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji JX, Wang YK, Cochrane DR, et al. Clear cell carcinomas of the ovary and kidney: clarity through genomics. J Pathol 2018; 244: 550–564. [DOI] [PubMed] [Google Scholar]
- 25.Clark DJ, Dhanasekaran SM, Petralia F, et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019; 179: 964–983 e931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobel M, Kalloger SE, Boyd N, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med 2008; 5: e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cochrane DR, Tessier-Cloutier B, Lawrence KM, et al. Clear cell and endometrioid carcinomas: are their differences attributable to distinct cells of origin? J Pathol 2017; 243: 26–36. [DOI] [PubMed] [Google Scholar]
- 28.Karnezis AN, Cho KR, Gilks CB, et al. The disparate origins of ovarian cancers: pathogenesis and prevention strategies. Nat Rev Cancer 2017; 17: 65–74. [DOI] [PubMed] [Google Scholar]
- 29.Shigetomi H, Tsunemi T, Haruta S, et al. Molecular mechanisms linking endometriosis under oxidative stress with ovarian tumorigenesis and therapeutic modalities. Cancer Invest 2012; 30: 473–480. [DOI] [PubMed] [Google Scholar]
- 30.Abu Saadeh F, Norris L, O’Toole S, et al. Venous thromboembolism in ovarian cancer: incidence, risk factors and impact on survival. Eur J Obstet Gynecol Reprod Biol 2013; 170: 214–218. [DOI] [PubMed] [Google Scholar]
- 31.Duska LR, Garrett L, Henretta M, et al. When ‘never-events’ occur despite adherence to clinical guidelines: the case of venous thromboembolism in clear cell cancer of the ovary compared with other epithelial histologic subtypes. Gynecol Oncol 2010; 116: 374–377. [DOI] [PubMed] [Google Scholar]
- 32.Ayhan A, Mao TL, Seckin T, et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int J Gynecol Cancer 2012; 22: 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamamoto S, Tsuda H, Takano M, et al. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol 2012; 25: 615–624. [DOI] [PubMed] [Google Scholar]
- 34.Heinze K, Nazeran TM, Lee S, et al. Validated biomarker assays confirm that ARID1A loss is confounded with MMR deficiency, CD8(+) TIL infiltration, and provides no independent prognostic value in endometriosis-associated ovarian carcinomas. J Pathol 2021, 10.1002/path.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heong V, Tan TZ, Miwa M, et al. A multi-ethnic analysis of immune-related gene expression signatures in patients with ovarian clear cell carcinoma. J Pathol 2021; 255: 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunningham JM, Winham SJ, Wang C, et al. DNA Methylation Profiles of Ovarian Clear Cell Carcinoma. Cancer Epidemiol Biomarkers Prev 2022; 31: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Isodoping library protein list
Table S6 differential protein expression PECA analysis results of the metabolically active cluster compared to the coagulative cluster
Table S7 Clinical and pathological parameters of CCOC proteomic clusters
Figure S1 Clinical and pathological parameters of the proteomic cohort
Figure S2 Proteomic analysis parameters
Figure S3 Imunohistochemistry stain of LAMC1
Figure S4 CCOC contain diverse proteomic subgroups
Figure S5 Previous transcriptomic and proteomic signatures applied to CCOC.
Figure S6 Immunohistochemistry validation of proteomic targets
Table S2 differential protein expression PECA analysis results of CCOC compared to HGSC
Table S3 differential protein expression PECA analysis results of CCOC compared to ENOC
Table S4 Gene Set Enrichment Analysis of CCOC compared to HGSC
Table S5 Gene Set Enrichment Analysis of CCOC compared to ENOC
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [13] partner repository with the dataset identifier/accession code PXD032355.
Additional methods are available in supplementary materials.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier/accession code PXD032355.