

Abstract
Aging is associated with decreased antigen-specific immunity and increased chronic inflammation. While DNA-sensing pathways might be involved, the molecular factors underlying these age-related aberrancies in immune signaling are unclear. Here, we consider the potential role of aging-induced hypomethylated DNA as a putative stimulant of age-associated inflammation.
Aging is associated with immune system dysfunction
Emerging evidence from pathology, epidemiology, and animal studies conducted in mice, rats, and non-human primates alludes to a close relationship between aging and immune dysregulation [1,2]. However, the molecular underpinnings behind this relationship remain a mystery. A better understanding of how aging contributes to inflammation is needed to ideally improve the treatment and diagnosis of age-related diseases (ARDs) (see Glossary), such as cancer, rheumatoid arthritis, and Alzheimer’s disease (AD). While advances in DNA sequencing have shown that age-dependent epigenetic changes can impact gene regulation, there is little understanding of how these altered epigenetic patterns may impact cells of the mammalian immune system when nuclear DNA and chromatin are released into the extracellular space upon cellular demise. In this forum article, we provide a perspective on how hypomethylated DNA, arising during aging, can stimulate the immune system by activating immune receptors thought to be involved in detecting foreign (viral and bacterial) DNA. We discuss evidence supporting the hypothesis that aged hypomethylated DNA might be immunogenic and perhaps act as an overlooked, but key regulator of immune signaling during aging.
Misplaced self-molecules can activate immune responses following cell death
The mammalian immune system recognizes molecular patterns indicative of infection, injury, or tissue dysfunction through a common set of pattern recognition receptors (PRRs), which detect conserved molecular structures known as pathogen-associated molecular patterns (PAMPS) and damage-associated molecular patterns (DAMPs). Recently, the term DAMPs was expanded to include self-derived biomolecules that are damaged, misfolded, or displaced into the extracellular space, collectively termed ‘altered or misplaced self-molecules’ [3]. While cellular repair and protein degradation mechanisms exist to combat molecular damage to DNA, proteins, and lipids, a key feature of aging is the accumulation of altered and misplaced self-derived molecules due to an aging-dependent decline of such molecular pathways [3].
In addition, a decline in proteolytic activity is also observed in aged cells; this results in impaired autophagy and efferocytosis [1,3]. Defects in autophagy are linked to accelerated aging phenotypes and several ARDs, including AD and age-related retinal degeneration [3]. Inefficiently cleared apoptotic cells (ACs) arising during impaired efferocytosis undergo secondary necrosis, releasing intracellular DAMPs into the extracellular space [e.g., nuclear DNA, high mobility group-box 1 (HMGB1) protein, mitochondrial DNA (mtDNA), and chromatin] [4,5]. As mtDNA has been shown to be lowly methylated [6] and to prime the antiviral innate immune response in mice upon cytosolic escape [4], it is plausible that aged hypomethylated nuclear DNA might also be a key contributor to inflammation during aging. The disposal of cell debris and damaged organelles slows down during aging in tandem with the accumulation of high amounts of altered and misfolded proteins within aged cells [3]. In this scenario, the tissue-specific ‘education’ received by immune cells [7] following the release of endogenous material from young cells early in an organism’s life may not be compatible to carry out immune surveillance during advanced aging (Figure 1A). If the ‘rules’ for healthy maintenance of the organism grow outdated with age and the immune system fails to adapt, the result might resemble the chronic low-grade inflammatory state typically observed during aging [3].
Figure 1. Model for the potential contribution of global DNA hypomethylation to human aging.
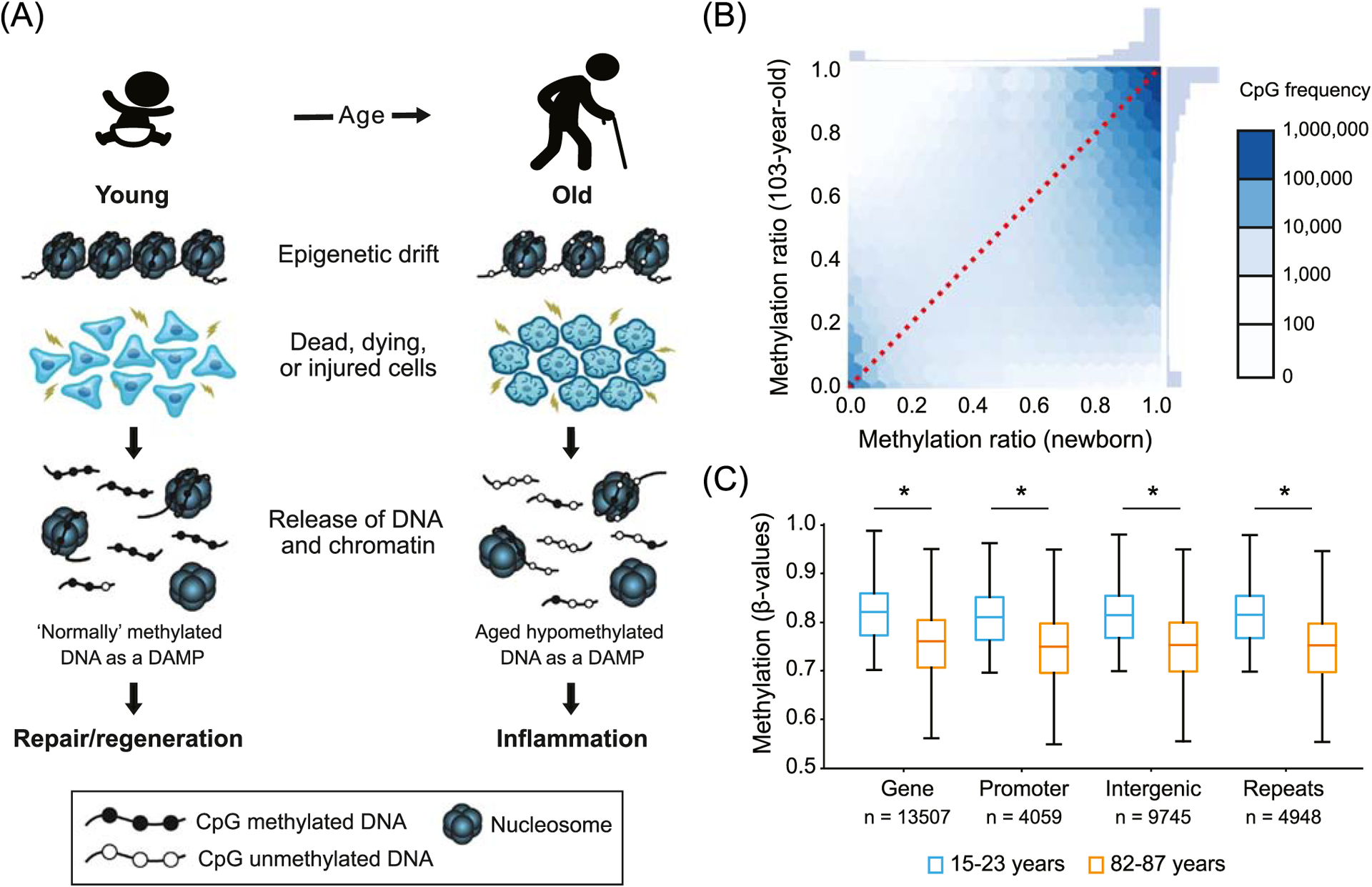
(A) Schematic illustrating the ‘altered self’ hypothesis with aging. DNA methylation patterns progressively diverge across the genome from highly conserved postnatal patterns over a lifetime due to a phenomenon known as epigenetic drift. The aged epigenome is characterized by deviations in global DNA methylation patterns and global DNA hypomethylation. These aging-associated epigenetic changes might generate ‘altered self’ molecules such as unmethylated DNA and chromatin fragments that are released into the extracellular space when cells die or are stressed. During aging, higher numbers of unmethylated genomic DNA fragments might stimulate PRRs and contribute to the low-grade chronic inflammation seen in elderly populations. (B) For illustrative purposes, an analysis of whole genome bisulfite sequencing data (from GEO accession GSE31438 [8]) for a newborn and a centenarian individual are shown. Illustrative CpG methylation (%) of 9 272 050 CpGs in the DNA from the newborn and the centenarian sample are depicted. (C) To illustrate the reproducibility of the observed trend in 1B (showing that highly methylated CpGs in a newborn are hypomethylated in a centenarian), we focused this illustration on changes in methylation between young and aged individuals from a larger cohort of blood sample donors. DNA methylation data at 476 366 sites throughout the genome of white blood cells that were collected from two cohorts ranging in age from 15 to 23 years old (n = 12) and 82–87 years old (n = 12) are shown. For these illustrative purposes, DNA methylation was displayed as ß-values (i.e., ratio of intensities of methylated to unmethylated alleles) only for CpG sites that were highly methylated in blood samples from young individuals (ß-value > 0.7). Boxplots show average ß-values across four genomic features (promoters, exons, introns, and intergenic regions). *P < 0.0001 (one-sided Welch’s t-test). DNA methylation array data was obtained from GEO Accession GSE87571 [9]. Abbreviation: DAMP, damage-associated molecular pattern.
Age-related epigenetic drift and the ‘altered self’ hypothesis
Epigenetics describes the set of reversible chemical modifications that govern the functional use of genetic information. Our cells’ ability to maintain epigenetic patterns also declines over a lifetime, resulting in epigenetic drift. Here, we propose a potential role for age-related DNA hypomethylation in generating altered self-DNA molecules that can stimulate PRRs when released during cell death.
In mammalian genomes, methylation commonly occurs in a CpG dinucleotide context. Several reports have found genome-wide losses in CpG methylation during aging (Figure 1B,C, shown for illustration purposes), most notably at repetitive DNA sequences [8,9]. As DNA methylation is known to contribute to key transposon-silencing mechanisms, hypomethylation at substantially CpG-dense transposable elements (TEs) can trigger reactivation of TEs and subsequent aberrant activation of immune signaling pathways [2,10]. Reactivation of TEs due to epigenetic drift was previously observed in a study reporting the reactivation of TEs in human fibroblasts passaged until cellular senescence, leading to the accumulation of cytoplasmic DNA, and the direct activation of type I interferon signaling in these cells [10].
Given the high occurrence of repetitive elements across mammalian genomes, changes in methylation in these regions would generate high numbers of unmethylated DNA fragments with highly conserved sequence motifs, potentially rendering them strong candidates for proinflammatory biomolecules during aging (Figure 1B,C, shown for illustration purposes) [8,9]. In the case of retrotransposons, it is possible that the amplification of transposable sequences might exacerbate the downstream effects of released unmethylated DNA on immune signaling, although this remains speculative. Indeed, it will be interesting to further explore this idea by comparing retrotransposon expression amounts between cells from young and aged individuals.
Widespread loss of cytosine methylation can increase DNA immunogenicity
CpG methylation, a common feature of mammalian genomes, is largely absent in immunogenic bacterial and viral DNA. During aging, the loss of CpG DNA methylation in genomic DNA may generate unmethylated DNA fragments that can trigger PRR activation in phagocytes during AC clearance, including Toll-like receptor 9 (TLR9) [11]. As discussed, phagocytes dispose of ACs via noninflammatory efferocytosis. One report indicated that DNA methylation regulated the suppressive versus inflammatory responses of dendritic cells (DCs) during AC efferocytosis [12]. It is known that phagocytes secrete wound-healing cytokines such as TGF-β during the disposal of ACs. However, upon treatment of ACs with the global demethylating agent 5-azacytidine, hypomethylated ACs induced inflammatory IL-6 signaling in mouse DCs. The authors restored the immunosuppressive properties of ACs by remethylating the hypomethylated DNA using a CpG methyltransferase [12]. Additionally, others have found that DNA from human DCs can become progressively more immunogenic with aging, as measured by IFN-α expression amounts, and this effect was dependent on DNA hypomethylation associated with aging [13]. These findings support the notion that DNA hypomethylation during aging can beget immunogenic self-derived DNA to trigger inflammatory signaling. To reinforce the notion that hypomethylated self-DNA might trigger immune responses, we elaborate on the connection of PRRs and DNA-sensing pathways with immune system dysfunction.
Crosstalk between DNA-sensing pathways and age-associated inflammation
Several cytosolic DNA-sensing (CDS) pathways activate an adaptor protein named stimulator of interferon genes (STING), which regulates the expression of type I interferons (IFNα and IFNβ) through activation of NF-кB and interferon regulatory factor (IRF3) (Table 1). IFN activation may synergistically enhance TLR-induced cytokine transcription by inducing chromatin remodeling (i.e., histone acetylation and transcription factor binding) at promoters and enhancers of target genes. In one of the largest multitissue analyses of aging in mice, researchers found that aging was associated with IFN activation, at both the transcriptional and chromatin levels, along with induction of CDS genes [14].
Table 1.
Characteristics of DNA sensorsa
| Ligand(s) | DNA sensors | Signaling pathway ➔ immune effects | Methylation sensitive |
|---|---|---|---|
| Unmethylated CpG motifs | TLR9 | MyD88-dependent IRF7 activation ➔ type I IFNs; MyD88-dependent NF-κB signaling via IKK ➔ IL-6, TNF-α |
Yes |
| Sugar-phosphate backbone of dsDNA >15 bp, CDNs, ssDNA duplex structures | cGAS | STING-dependent activation of TBK1/IRF3 and IKK/NF-κB ➔ type I IFNs, IL-6, and TNF-α | No |
| AT-rich B-DNA, viral, bacterial, and mammalian gDNA 500 >> 100 > 75 bp | DAI | NF-κB activation (via adaptor proteins RIP1 and RIP3) and STING-dependent TBK1-IRF3 activation ➔ type I IFNs and necrosis | Unknown |
| Poly (I:C), CpG-B | DHX9 | MyD88-dependent NF-κB signaling ➔ TNF-α, IL-6 | Yes |
| CpG-A | DHX36 | DHX36 forms a complex with TRIF, which activates NF-кB and IRF-3/7 ➔ type I IFN production | Yes |
| dsDNA, bacterial CDNs, cGAMP, poly (dA:dT), and poly(dG:dC) | DDX41 | STING-dependent activation of TBK1-IRF3 ➔ type I IFNs | Unknown |
| ssDNA and RNA, dsDNA sequence-independent, 150 >> 70 >> 50 bp | IFI16 | STING-dependent activation of TBK1-IRF3 and NF-κB ➔ type I IFNs. Viral DNA in nucleus activates inflammasome | No |
| Sugar-phosphate backbone of dsDNA, poly(dA:dT) | AIM2 | AIM2 bound to dsDNA recruits ASC to form inflammasome complex and activate caspase-1. Active caspase-1 ➔ IL-1β, IL-18, and pyroptosis | No |
| Viral and Alu-RNA, oxidized mtDNA, ATP, cardiolipin, MSU crystals | NLRP3 | NLRP3 oligomerizes with AIM2 and recruits ASC to form inflammasome, which activates pro-caspase-1. Active caspase-1 ➔ IL-1β and IL-18 | Unknown |
Abbreviations: AIM2, absent in melanoma 2; ASC, adaptor protein apoptosis speck-like protein; CDNs, cyclic dinucleotides; cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; cGAS, cyclic GMP-AMP synthase; CpG-A, class A CpG ODNs (characterized by a central, phosphodiester, CpG-containing palindromic motif and a 3′ phosphorothioate poly-G string); CpG-B, class B CpG ODNs (contain a full phosphorothioate backbone with one or more CpG dinucleotides); CpG ODN, CpG oligodeoxynucleotide; DEAD, aspartate-glutamate-alanine-aspartate; DDX41, DEAD-box helicase 41; DHX9, DEAH-box helicase 9; DHX36, DEAH-box helicase 36; dsDNA, double-stranded DNA; HIN-200, hematopoietic expression, interferon-inducible nature, and nuclear localization; IFI16, interferon gamma inducible protein 16; IFN, interferon; IKK, IкB kinase; IL-1β, interleukin-1 beta; IL-6, interleukin-6; IL-18, interleukin-18; IRF3, interferon regulator factor 3; IRF7, interferon regulator factor 7; MAPKs, mitogen-activated protein kinases; MSU, monosodium urate; mtDNA, mitochondrial DNA; MyD88, myeloid differentiation primary-response protein88; NF-кB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, nucleotide-binding oligomerization domain (NOD)-like receptor protein 3; poly(dA:dT), homopolymeric stretches of deoxyadenosine nucleotides (‘A’s) on one strand of double-stranded DNA; poly(I:C): mismatched double-stranded RNA with one strand being a polymer of inosinic acid, the other a polymer of cytidylyl acid; RIP1, receptor interacting proteins 1; RIP3, receptor interacting proteins 3; ssDNA, single-stranded DNA; STING, stimulator of interferon genes; TBK1, TANK binding kinase 1; TIR, Toll-IL receptor; TLR9, Toll-like receptor 9; TNF-α, tumor necrosis factor alpha; TRIF, TIR-domain-containing adapter-inducing interferon-β.
It is plausible that the crosstalk between DNA-sensing pathways and age-associated inflammation can be triggered by heightened aging-induced hypomethylated self-DNA. As immune cells take up extracellular material in phagosomes, there may be additional cofactors present during cellular demise that enhance DNA uptake and immunogenicity, such as HMGB1, a nuclear protein that binds DNA [3,5]. As unmethylated and hypomethylated DNA can be immunogenic, the increased amounts of ‘altered self’ molecules resulting from age-related methylation drift might potentially trigger continuous PRR activation and subsequent immune signaling crosstalk. This, in turn, might exacerbate immune dysfunction and inflammation in elderly populations, as observed in the aforementioned studies [3,14].
Concluding remarks
We propose that aging induces changes in DNA released upon cellular demise that originate from epigenetic drift in the chromatin landscape. We postulate that the resulting unmethylated DNA may act as an immunogenic, host-derived biomolecule, which should be further explored as a putative factor contributing to aging-induced chronic inflammation [3]. Notably, an inverse correlation has been observed between species lifespan (mouse, non-human primate, and human) and rate of epigenetic drift [15], which further strengthens the case that epigenetic drift might be one driver of aging. Even in species harboring significantly low levels of DNA methylation, such as Drosophila spp., epigenetic drift can still be observed in other chromatin marks (e.g., histone modifications) known to trigger PRR signaling [16]. We argue that a better understanding of aging-specific DAMPs and the role of autophagy in promoting longevity may reveal epigenetic drift as a potentially key contributor to species-specific lifespans [17]. Furthermore, this knowledge might help guide the development of personalized medicine based on a patient’s ‘epigenetic age’, the concentrations of inflammatory cytokines, as well as circulating unmethylated self-DNA. Taken together, aging-associated epigenetic changes might represent a new class of ‘immunogenic molecular patterns’ harboring potential as candidate pharmacological targets in the fight against aging and certain ARDs.
Acknowledgments
The authors thank Z. Smith and M. Blurton-Jones for their thoughtful feedback on prior drafts of this article. This work was supported by National Institutes of Health (NIH) National Institute of Biomedical Imaging and Bioengineering Grant R21EB027840-01, a NIH New Innovator Award (DP2) Grant DP2CA250382-01, National Science Foundation grants (DMS1763272 and EF2022182), and a grant from the Simons Foundation (594598 QN) to T.L.D.
Glossary
- Age-related disease (ARD)
pathology observed more frequently with aging (i.e., cancer, rheumatoid arthritis, Alzheimer’s disease).
- Autophagy
cellular process involving self-recycling and degradation of damaged organelles by the lysosome.
- Chronic low-grade inflammatory state
describes low-level inflammation that occurs persistently and globally, as determined from small increases in immune marker expression in blood or tissues.
- Cytosolic DNA-sensing (CDS) pathways
components of the innate immune system that detect DNA in the cytosol through the activity of pattern recognition receptors and activate inflammatory or defense mechanisms in response.
- Damage-associated molecular patterns (DAMPS)
molecules released from damaged or dying cells as part of the innate immune response.
- Efferocytosis
targeted removal and degradation of apoptotic cells by phagocytes such as macrophages and dendritic cells.
- Epigenetic age
estimate of an individual’s biological age through assessment of DNA methylation signatures within the genome.
- Epigenetic drift
process by which epigenetic modifications across the genome progressively diverge from conserved postnatal patterns over an organism’s lifespan.
- Hypomethylated DNA
DNA that has experienced a loss of methylation (i.e., of cytosine residues).
- Pathogen-associated molecular patterns (PAMPS)
molecular motifs that are conserved within groups of related microbes and that activate the host innate immune response.
- Pattern recognition receptors (PRRs)
immune receptors that recognize molecular patterns indicating infection, injury, or tissue dysfunction and that contribute to the activation of inflammatory signaling or wound healing responses; pattern recognition receptors can be grouped according to ligand specificity, localization, and function.
- Retrotransposon
genetic element that copies and pastes itself into various genomic locations via reverse transcription.
- Stimulator of interferon genes (STING)
adaptor protein in the endoplasmic reticulum that facilitates host defense mechanisms by regulating the expression of type I interferons IFNα and IFNβ.
- Toll-like receptor 9 (TLR9)
transmembrane protein that recognizes pathogen-associated molecular patterns and initiates cytokine production as part of the host innate immune response; usually activated by DNA lacking cytosine methylation.
- Type I interferons
signaling proteins released by infected cells; activate antimicrobial immune responses.
Footnotes
Declaration of interests
No interests are declared.
References
- 1.Aprahamian T et al. (2008) Ageing is associated with diminished apoptotic cell clearance in vivo. Clin. Exp. Immunol 152, 448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon M et al. (2019) LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab. 29, 871–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franceschi C et al. (2017) Inflammaging and ‘garb-aging’. Trends Endocrinol. Metab 28, 199–212 [DOI] [PubMed] [Google Scholar]
- 4.West AP et al. (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu L et al. (2014) HMGB1-DNA complex-induced autophagy limits AIM2 inflammasome activation through RAGE. Biochem. Biophys. Res. Commun 450, 851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu B et al. (2016) CpG methylation patterns of human mitochondrial DNA. Sci. Rep 6, 23421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matzinger P (2007) Friendly and dangerous signals: is the tissue in control? Nat. Immunol 8, 11–13 [DOI] [PubMed] [Google Scholar]
- 8.Heyn H et al. (2012) Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. U. S. A 109, 10522–10527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johansson A et al. (2013) Continuous aging of the human DNA methylome throughout the human lifespan. PLoS One 8, e67378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Cecco M et al. (2019) L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hemmi H et al. (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 [DOI] [PubMed] [Google Scholar]
- 12.Notley CA et al. (2017) DNA methylation governs the dynamic regulation of inflammation by apoptotic cells during efferocytosis. Sci. Rep 7, 42204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agrawal A et al. (2010) Age-associated epigenetic modifications in human DNA increase its immunogenicity. Aging (Albany NY) 2, 93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benayoun BA et al. (2019) Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 29, 697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maegawa S et al. (2017) Caloric restriction delays age-related methylation drift. Nat. Commun 8, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Z et al. (2018) Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. eLife 7, e35368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simonsen A et al. (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184 [DOI] [PubMed] [Google Scholar]