

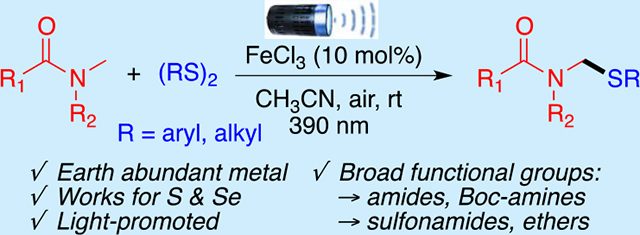
Abstract
A photoinduced, iron(III) chloride-catalyzed C–H activation of N-methyl amides and ethers leads to the formation of C–S and C–Se bonds via a ligand-to-metal charge transfer (LMCT) process. This methodology converts secondary and tertiary amides, sulfonamides, and carbamates into the corresponding amido-N,S-acetal derivatives in good yields. Mechanistic work revealed that this transformation proceeds through a hydrogen atom transfer (HAT) involving chlorine radical intermediates.
Graphical Abstract
Amines and amides are quintessential moieties in organic synthesis. They are found in a wide variety of natural products, pharmaceuticals, peptides, and fine chemicals.1 Similarly, sulfur-containing compounds represent over 20% of FDA-approved drugs, which is the third largest heteroatom after oxygen and nitrogen.2 A well-known bioactive scaffold in sulfur-containing drugs is the amido-N,S-acetal moiety found in natural products and antibacterials, such as penicillin, penicillin derivatives, and fusaperazine A (Scheme 1A).3
Scheme 1. Molecules of Interest & Current Technologiesa.
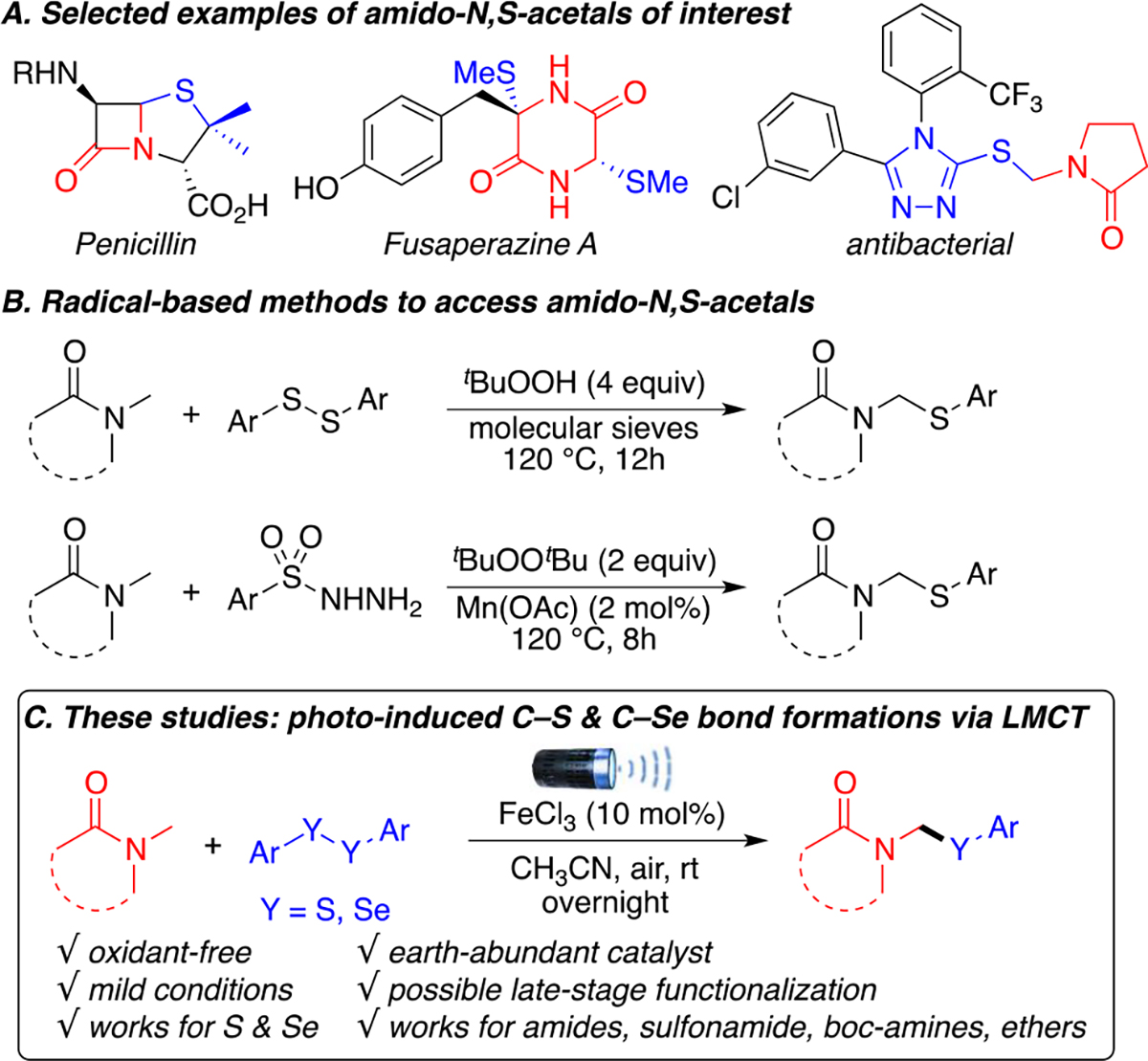
a(A) Selected bioactive compounds of interest. (B) Radical approaches to amido-N,S-acetals. (C) Proposed photoinduced method.
Traditional synthetic approaches to access these amido-N,S-acetals proceed through either the substitution reaction of halogenated amides at the amino carbon, or through the addition reaction to N-acyl imines by thiol nucleophiles. Unfortunately these prefunctionalized imines and halogenated intermediates are often unstable to moisture, not easily stored for prolonged periods of time, and require sophisticated reaction conditions.4 Direct C–H thiolation of amides represent a more attractive approach for late-stage diversification and functionalization, and bypasses the need for these unstable intermediates.
In the past decade, two radical-promoted site-selective C–H thiolations of amides at the amino carbon have been developed (Scheme 1B).5 Unfortunately, both these methods use peroxides at 120 °C to achieve the desired transformation, which poses safety concerns and limits scalability.6 In contrast, the development of a photoinduced methodology would enable such transformations to occur under mild conditions at room temperature and presumably without the use of unstable and potentially explosive peroxides. Among photoinduced transformations, ligand-to-metal charge transfer (LMCT) processes have recently gained increased attention7 because of the simplicity of the methods and the affordability of its catalysts, which often use earth abundant metals, such as copper and iron,8 as opposed to traditional iridium and ruthenium photoredox catalysts. C–H activation transformations promoted by LMCT processes have been shown to proceed through chlorine radical intermediates by Doyle, Rovis, Walsh, and Schelter among others.9 The high reactivity of the chlorine radical makes it an ideal intermediate to initiate hydrogen-atom-transfer (HAT) processes with all kinds of C(sp3)–H bonds10 from activated ethers and amides to unactivated alkanes.
Recently, our group has reported various site-selective C–H functionalization of amides and ethers under mild and photoinduced conditions,11a–c as well as photoinduced cross-couplings that generate C–S and C–Se bonds.11d Combining these interests to our interest in green photoinduced methodologies,12 we envisioned that chlorine radicals generated via LMCT processes could enable site-selective C(sp3)–H bond activation of the amino carbon of amides, sulfonamides, and carbamates to form stabilized alkyl radical and then react with disulfide to give the desired amido-N,S-acetals derivatives. Herein, we present the first example of a photoinduced C–H sulfenylation of amides, amide derivatives, and ethers using affordable and earth abundant FeCl3 via a LMCT process (Scheme 1C).
Our investigations began using diphenyl disulfide (1a) and N,N-dimethylacetamide (DMA) (2a) as model substrates (Table 1) in the presence of 10 mol % iron(III) chloride as catalyst under air and irradiation of 40 W 390 nm LED. An initial solvent screen comparing acetonitrile (CH3CN), DMSO, and THF (entries 1–3), revealed that the transformation only proceeds in acetonitrile (81%) and THF (26%). It is worth noting that neat conditions using DMA as solvent (entry 4) did not afford the desired product. This observation further differentiates this method with other radical approaches that require neat conditions to achieve the desired transformation, which limits the scope of the reaction to liquid amides.5 The use of other catalysts and iron sources (entries 5–8) did not improve the transformation (for full catalyst screen see Supporting Information S7). Lowering catalyst loading to 5 mol % (entry 9) had a deleterious effect on yield (59%). Similarly, using inert argon atmosphere (entry 10) lowered the yield to 61%. Switching LED lamps from 390 to 427 nm reduced the yield to 49% (entry 11). Control experiments in the absence of light (entry 12) or absence of catalyst (entry 13) did not generate the desired product. Additional optimization experiments detailing catalyst loading, reagent equivalencies, light sources, metal sources, solvents, and inert atmospheres are described in the Supporting Information S6–S10.
Table 1.
Optimization of the Reaction and Its Conditionsa

| |||
|---|---|---|---|
| entry | catalyst (mol %) | solvent (mL) | yield (%)b |
| 1 | FeCl3 (10%) | CH3CN (2.0) | 81 (78)c |
| 2 | FeCl3 (10%) | THF (2.0) | 26 |
| 3 | FeCl3 (10%) | DMSO (2.0) | NR |
| 4 | FeCl3 (10%) | DMA (2.0) | trace |
| 5 | FeCl3·6H2O (10%) | CH3CN (2.0) | 67 |
| 6 | FeCl2·4H2O (10%) | CH3CN (2.0) | 30 |
| 7 | FeBr3 (10%) | CH3CN (2.0) | NR |
| 8 | CeCl3 (10%) | CH3CN (2.0) | NR |
| 9 | FeCl3 (5%) | CH3CN (2.0) | 59 |
| 10d | FeCl3 (10%) | CH3CN (2.0) | 61 |
| 11e | FeCl3 (10%) | CH3CN (2.0) | 49 |
| 12f | FeCl3 (10%) | CH3CN (2.0) | NR |
| 13 | CH3CN (2.0) | NR | |
Optimal reaction conditions: 1a (0.2 mmol, 1 equiv), 2a (1.0 mmol, 5 equiv), solvent (2 mL), anhydrous FeCl3 (10 mol %), 390 nm LED (40 W), room temperature around reaction flask was 35 °C (heating caused by the LED lamp), reaction flask capped under air, 15 h.
1HNMR yields using dibromomethane as internal standard.
Isolated yield.
Under argon.
427 nm LED.
Reaction performed in the dark.
With optimal conditions in hand, we continued our investigations exploring the scope of aliphatic amides derivatives (Scheme 2). Using N,N-dimethylformamide (DMF) as amide source afforded the desired product 4 as a single regioisomer in good yield (65%), which differs from previous reports5a that obtain a mixture of regioisomers in 1:1 ratio. Linear amide substrates, such as N,N-dimethylbutyramide, N,N-dimethylpentanamide, N,N,3-trimethylbutanamide, and N,N-dimethylisobutyramide, afforded the desired corresponding products 5–8 in moderate to good yields (48–80%).
Scheme 2. Amides Scopea.
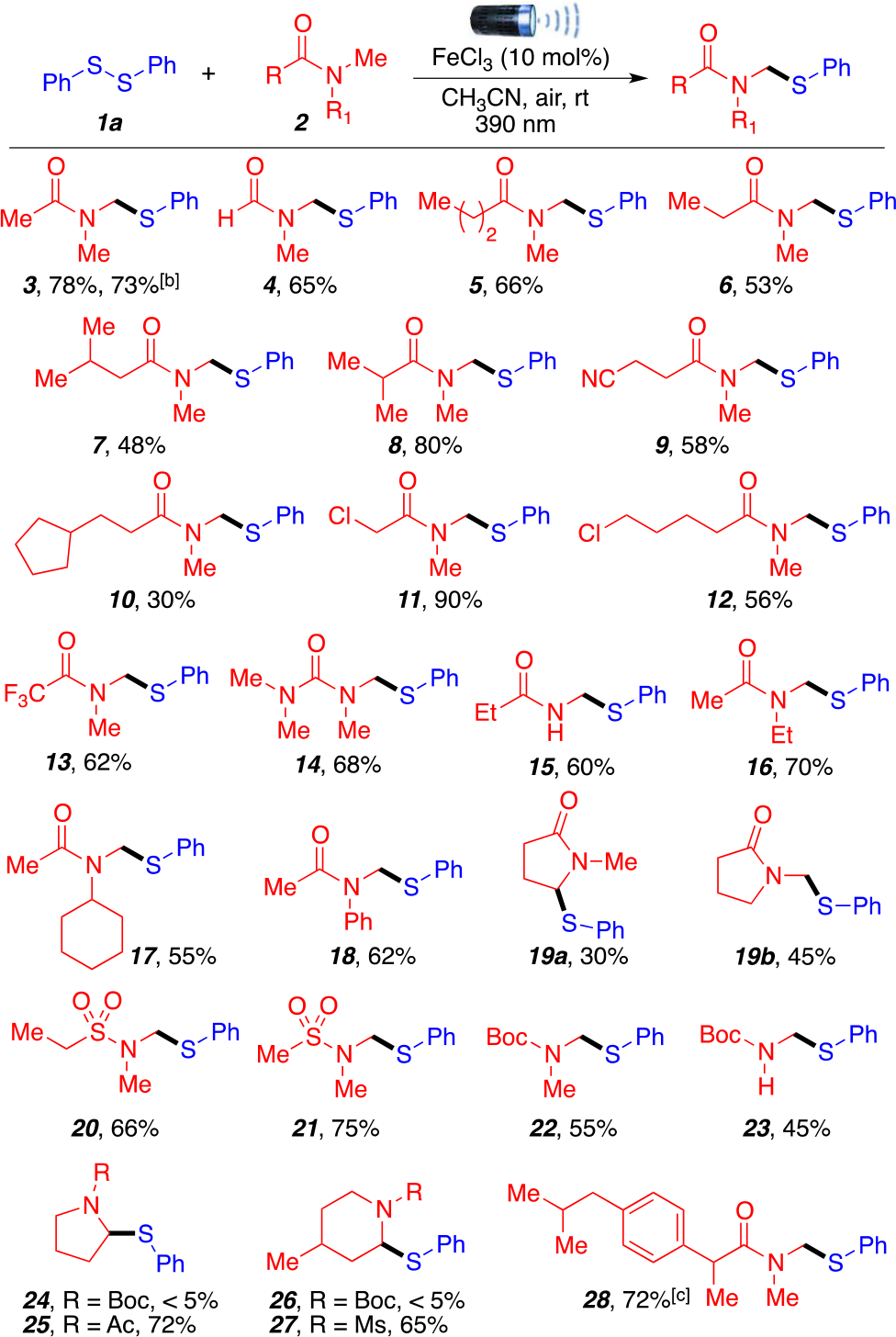
aReaction conditions: 1a (0.2 mmol, 1 equiv), 2 (1.0 mmol, 5 equiv), MeCN (2 mL), anhydrous FeCl3 (10 mol %), room temperature around reaction flask was 35 °C (heating caused by the LED lamp.), 15 h. All yields are isolated. b1 mmol scale reaction (see Supporting Information, page S19). cTwenty-four hours.
It is noteworthy that linear amides bearing various functional groups, nitriles, chloride, trifluoromethyl, and cyclopentane, were well tolerated and afforded products 9–13 in moderate to excellent yields (30–90%). Tetramethylurea afforded product 14 in good yield (68%). Secondary amides were also suitable substrates affording product 15 in 60% yield. Unsymmetrical aliphatic amides display excellent regioselectivity toward the methyl group over the methylene carbon affording products 16 and 17 in 70% and 55% yields, respectively. Aromatic moieties were also tolerated giving product 18 in moderate yield (62%). Importantly, when using N-methylpyrrolidone (NMP) as an amide source, two regioisomers were obtained (19a and 19b) in 75% overall yield with two regioisomers in a 3:2 ratio in favor of the primary carbon. As expected, this transformation could also proceed efficiently with sulfonamides and Boc-protected amine motifs (20–23) in moderate to good yields (45–75%), which are important frameworks in drug discovery and total synthesis.13 Other medicinally relevant motifs such as piperidine and pyrrolidine were also tested.14 While Boc-protected pyrrolidine and piperidine were not suitable substrates for this transformation (24 and 26), their acetyl-protected and mesyl-protected counterparts were well tolerated and afforded products 25 and 27 in 72% and 65% yield, respectively. Finally, to highlight the late-stage functionalization potential of our approach, an ibuprofen amide derivative was investigated to get the desired product 28 in good yield (72%), opening the door to the diversification of other bioactive compounds containing carboxylic acid moieties that can be readily transformed into their amide counter parts.
Next, we turned our attention to explore the scope of disulfides and other dichalcogenides (Scheme 3). Diaryl disulfides bearing electron donating and electron withdrawing groups on in the para position (29–31) were well tolerated (71–85%) with the exception of the nitro group, which afforded product 32 in moderate yield (45%). Sterics at the ortho position were also tolerated in good yields giving products 33 and 34 in 88% and 72% yield, respectively. Heteroaromatic substrates were also compatible and afforded products 35 and 36 in good yields (60–68%). Beyond aromatic disulfides, benzylic and alkyl disulfides were also investigated and afforded products 37 and 38 in low to moderate yields (45–63%). The lower yield obtained using di-tert-butyl disulfide is probably due to steric hindrance. Finally, other commercially available aryl dichalcogenides were investigated because of their potential bioactivity15 and synthetic interest.16 Lower yields were obtained when using diphenyl diselenide, but products 39–42 were obtained in synthetically useful yields for late-stage diversification studies (48–66%). It is worth mentioning that unreacted diselenide reagent was recovered in good yields. Unfortunately, diphenyl ditelluride was unsuitable in this reaction and only trace amounts of product 43 were formed. Previous methods have used FeCl3 to add selenides and tellurides to unsaturated systems17 but not to amides.
Scheme 3. Disulfides Scopea.
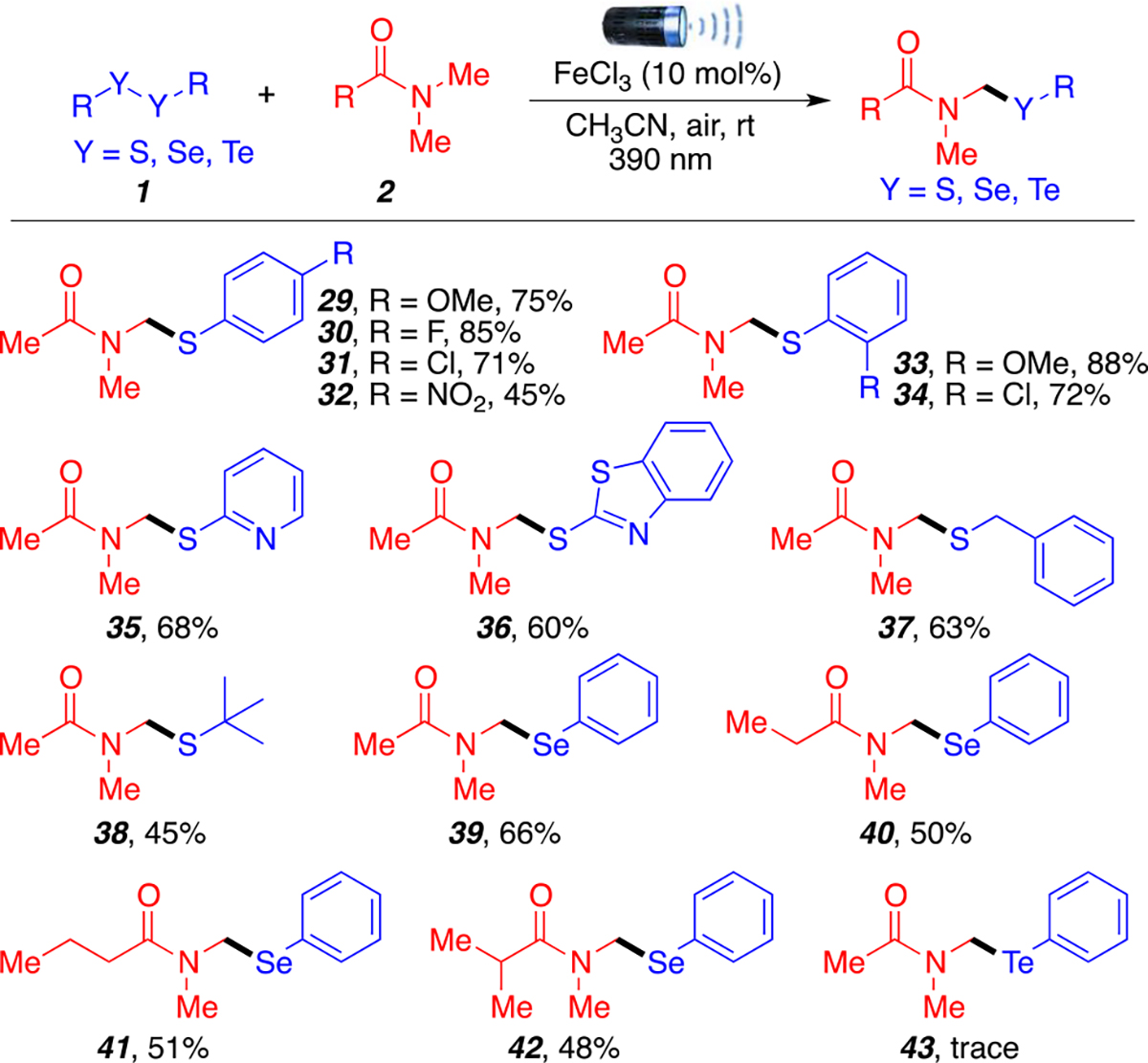
aReaction conditions: 1 (0.2 mmol, 1 equiv), 2a (1.0 mmol, 5 equiv), MeCN (2 mL), anhydrous FeCl3 (10 mol %), 390 nm LED (30 W), room temperature around reaction flask was 35 °C (heating caused by the LED lamp), 15 h. All yields are isolated.
Further exploration of the substrate scope revealed that this methodology could be extended to ethers, both cyclic and acyclic (Scheme 4). Commonly used ethers, such as THF, 1,4-dioxane, and diethyl ethers, afforded desired products 44–46 in moderate to good yields (66–75%). Tetrahydrothiophene was also evaluated and generated product 47 in good yield (80%), suggesting that this procedure may be expanded to other activated C(sp3)–H bonds alpha to heteroatoms. Finally, 2-methyl-THF afforded the desired product forming two regioisomers 48a (minor) and 48b (major) (1:3 ratio) as an inseparable mixture in 55% yield. Regioisomer 48b was generated as a mixture of diastereomers (d.r. = 3:1).
Scheme 4. Ethers Scopea.
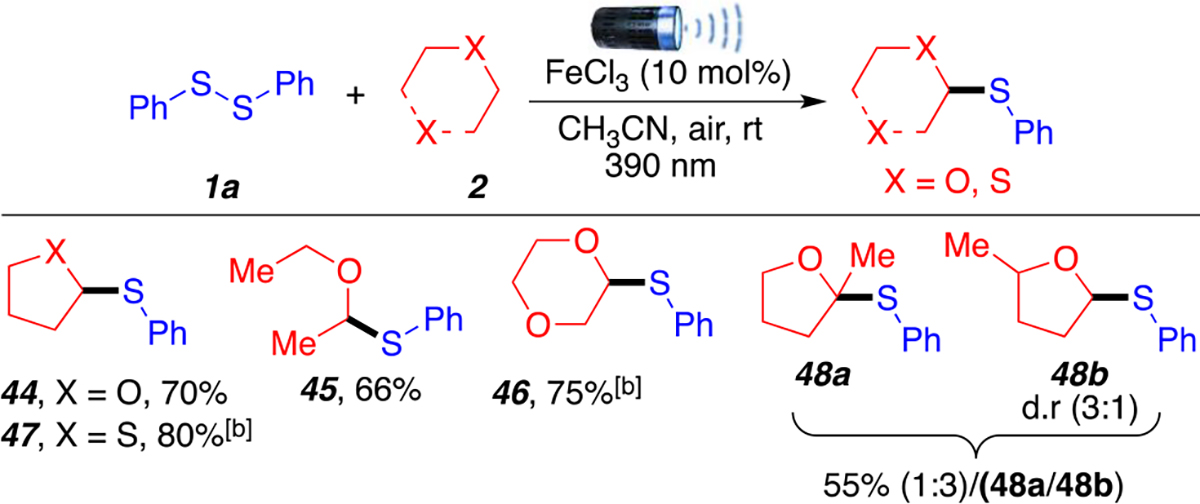
aReaction conditions: 1 (0.2 mmol, 1 equiv), ethers (2 mL), anhydrous FeCl3 (10 mol %), 390 nm LED (30 W), room temperature around reaction flask was 35 °C (heating caused by the LED lamp), 15 h. bMeCN (2 mL), ether (5.0 equiv, 1 mmol). All yields are isolated.
To expand our understanding of the reaction mechanism, a series of control experiments were performed (Scheme 5A). As expected, the addition of radical trapping agents, such as 2,2,6,6-tetramethyl-peperidinylxoyl (TEMPO), only afforded trace amount of product 3. The use of other trapping agents 1,1-diphenylethylene (1,1-DPE), 1,4-dinitrobenzene (1,4-DNB), and butylated hydroxytoluene (BHT) also produced similar results. Importantly, DMA radical intermediates were successfully trapped as TEMPO and 1,1-DPE adducts (49, 50) and observed by GC-MS. The chlorine radical was trapped with 1,1-DPE (51), while the thiyl radical was trapped as a 1,1-DPE and BHT adducts (52, 53) (Scheme 5A).
Scheme 5. Verification of the Presence of Radicals and Proposed Mechanisma.
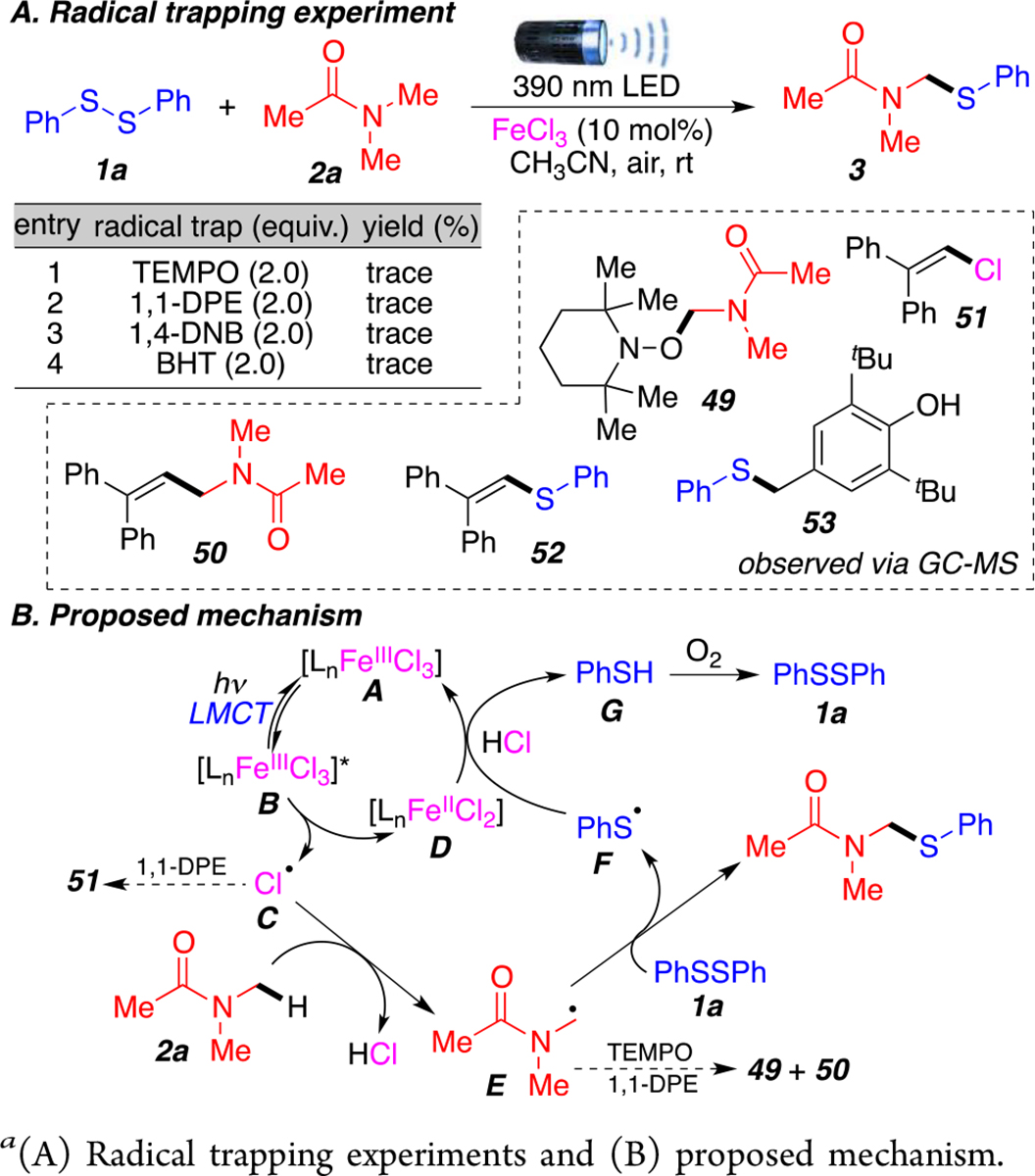
a(A) Radical trapping experiments and (B) proposed mechanism.
UV–vis experiments (Supporting Information, pages S11–S13) show an absorption band for FeCl3 with an absorption maximum at λ = 361 nm that tails off as far as 500 nm. On the other hand, neither diphenyldisulfide (1a) nor DMA (2a) absorb past 375 nm, strongly suggesting that FeCl3 in MeCN is the species capable of photoexcitation (Figure S1, page S11). Indeed, when FeCl3 absorbance was evaluated in different solvents (Figure S2, page S12), MeCN showed the most enhanced absorbance in the visible spectrum. Lastly, comparing different metal catalysts shows that FeCl3 had the strongest absorption, which may explain the lack of reactivity of the other metal chloride salts (Figures S3 and S4, pages S12–S13).
On the basis of these observations and reported literature,9,18 we propose that this transformation proceeds via a LMCT process between FeCl3 and acetonitrile as a ligand (Scheme 5B). Iron(III) complex A can undergo excitation to B under visible-light irradiation and subsequently generates highly reactive chlorine radical C and forms iron(II) species D. Chlorine radical intermediate C performs a HAT process with the C(sp3)–H bond alpha to nitrogen of the amide 2a to generate stabilized alkyl radical E. Reaction between radical intermediate E and diphenyl disulfide 1a forms the desired product and produces thiyl radical F. Radical intermediate F can then oxidize iron(II) D to regenerate iron catalyst A (see cyclic voltammetry experiments in Supporting Information, page S10) in the presence of HCl and forms thiol G, which can reform disulfide 1a in the presence of O2 from our atmospheric conditions (Scheme 5B). Although thermodynamically unfavorable,18d there is a possibility that O2 plays a minor role in the single electron oxidation of Fe(II) species D into Fe(III) catalyst A.
This work presents the first photoinduced C–H sulfenylation of amides, amide derivatives, and ethers using affordable and earth abundant FeCl3 via a LMCT process. Broad functional group compatibility with synthetically useful moieties has been observed using this protocol, such a Boc- and mesyl-protected amines, nitriles, halogens, and sulfonamides, all of which are of great interest to medicinal chemistry. The transformation proceeds with moderate to excellent yields with secondary and tertiary amides, providing a complementary approach to current thiolation of N-methyl amides and ethers.
Supplementary Material
Funding
This publication was made possible with support from National Institute of Dental & Craniofacial Research grant number 1R21DE029156–02.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c01505.
1H and 13C{1H} NMR spectra and characterization data for all products (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.orglett.2c01505
Contributor Information
Ben Niu, Department of Chemistry & Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
Krishnakumar Sachidanandan, Department of Chemistry & Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
Maria Victoria Cooke, Department of Chemistry & Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
Taylor E. Casey, Department of Chemistry & Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States
Sébastien Laulhé, Department of Chemistry & Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
REFERENCES
- (1).(a) Fischer J; Ganellin CR Analogue-Based Drug Discovery; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; p 457. [Google Scholar]; (b) Lawrence SA Amines: Synthesis, Properties and Applications, 1st ed..; Cambridge Univ. Press: Cambridge, 2004. [Google Scholar]
- (2).(a) Scott KA; Njardarson JT Analysis of USA FDA-Approved Drugs Containing Sulfur Atoms. Top Curr. Chem. 2018, 376, 5. [DOI] [PubMed] [Google Scholar]; (b) Ilardi EA; Vitaku E; Njardarson JT Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals To Reveal Opportunities for Drug Design and Discovery. J. Med. Chem. 2014, 57, 2832–2842. [DOI] [PubMed] [Google Scholar]; (c) Smith BR; Eastman CM; Njardarson JT Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem. 2014, 57, 9764–9773. [DOI] [PubMed] [Google Scholar]
- (3).(a) Parry RJ Biosynthesis of Some Sulfur-Containing Natural Products Investigations of the Mechanism of Carbon-Sulfur Bond Formation. Tetrahedron. 1983, 39, 1215–1238. [Google Scholar]; (b) Wu Y-J; Meanwell NA Geminal Diheteroatomic Motifs: Some Applications of Acetals, Ketals, and Their Sulfur and Nitrogen Homologues in Medicinal Chemistry and Drug Design. J. Med. Chem. 2021, 64, 9786–9874. [DOI] [PubMed] [Google Scholar]; (c) Sammes PG Recent Chemistry of the β-lactam Antibiotics. Chem. Rev. 1976, 76, 113–155. [Google Scholar]; (d) Usami Y; Aoki S; Hara T; Numata A New Dioxopiperazine Metabolites from a Fusarium Species Separated from a Marine Alga. J. Antibiot. 2002, 55, 655–659. [DOI] [PubMed] [Google Scholar]
- (4).(a) Hucher N; Pesquet A; Netchitaïlo P; Daïch A Exocyclic-Endocyclic N-Acyliminium Ion Equilibration via an Intramolecular α-Thioamidoalkylation in the Synthesis of Fused N, S-Heterocyclic Systems: Some New Parameters. Eur. J. Org. Chem. 2005, 2005, 2758–2770. [Google Scholar]; (b) Unsworth WP; Kitsiou C; Taylor RJK Direct Imine Acylation: Rapid Access to Diverse Heterocyclic Scaffolds. Org. Lett. 2013, 15, 258–261. [DOI] [PubMed] [Google Scholar]; (c) Ingle GK; Mormino MG; Wojtas L; Antilla JC Chiral Phosphoric Acid-Catalyzed Addition of Thiols to N-Acyl Imines: Access to Chiral N, S-Acetals. Org. Lett. 2011, 13, 4822–4825. [DOI] [PubMed] [Google Scholar]; (d) Wezeman T; Hu Y; McMurtrie J; Brase S; Masters KS Synthesis of Non-Symmetrical and Atropisomeric Dibenzo[1,3]-diazepines: Pd/CPhos-Catalysed Direct Arylation of Bis-Aryl Aminals. Aust. J. Chem. 2015, 68, 1859–1865. [Google Scholar]; (e) Rayner PJ; Gelardi G; O’Brien P; Horan RAJ; Blakemore DC On the synthesis of α-amino sulfoxides. Org. Biomol. Chem. 2014, 12, 3499–3512. [DOI] [PubMed] [Google Scholar]
- (5).(a) Tang R-Y; Xie Y-X; Xie Y-L; Xiang J-N; Li J-H TBHP mediated Oxidative Thiolation of a sp3 C-H Bond Adjacent to a Nitrogen Atom in an Amide. Chem. Commun. 2011, 47, 12867–12869. [DOI] [PubMed] [Google Scholar]; (b) Sun J; Wang Y; Pan Y Manganese Catalysed Sulfenylation of N-Methyl Amides with Arenesulfonyl Hydrazides. Org. Biomol. Chem. 2015, 13, 3878–3881. [DOI] [PubMed] [Google Scholar]
- (6).(a) Varjavandi J; Mageli OL Safe Handling of Organic Peroxides. J. Chem. Educ. 1971, 48, A451. [Google Scholar]; (b) Malow M; Wehrstedt KD Prediction of the Self-Accelerating Decomposition Temperature (SADT) for Liquid Organic Peroxides from Fifferential Scanning Calorimetry (DSC) Measurements. J. Hazard. Mater. 2005, 120, 21–24. [DOI] [PubMed] [Google Scholar]
- (7).(a) Zhao R; Shi L A Renaissance of Ligand-to-Metal Charge Transfer by Cerium Photocatalysis. Org. Chem. Front. 2018, 5, 3018–3021. [Google Scholar]; (b) Hu A; Guo JJ; Pan H; Tang H; Gao Z; Zuo Z δ-Selective Functionalization of Alkanols Enabled by Visible-Light Induced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc. 2018, 140, 1612–1616. [DOI] [PubMed] [Google Scholar]; (c) Chen Y; Wang X; He X; An Q; Zuo Z Photocatalytic Dehydroxymethylative Arylation by Synergistic Cerium and Nickel Catalysis. J. Am. Chem. Soc. 2021, 143, 4896–4902. [DOI] [PubMed] [Google Scholar]; (d) Ravetz BD; Wang JY; Ruhl KE; Rovis T Photoinduced Ligand-to-Metal Charge Transfer Enables Photocatalyst-Independent Light-Gated Activation of Co(II). ACS Catal. 2019, 9, 200–204. [Google Scholar]
- (8).(a) Yaroshevsky AA Abundances of Chemical Elements in the Earth’s Crust. Geochem. Int. 2006, 44, 48–55. [Google Scholar]; (b) Chirik P; Morris R Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals. Acc. Chem. Res. 2015, 48, 2495–2495. [DOI] [PubMed] [Google Scholar]
- (9).(a) Shields BJ; Doyle AG Direct C(sp3)-H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc. 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C-C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 14059–14063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Treacy SM; Rovis T Copper Catalyzed C(Sp3)-H Bond Alkylation via Photoinduced Ligand to Metal Charge Transfer. J. Am. Chem. Soc. 2021, 143, 2729–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kang YC; Treacy SM; Rovis T Iron-Catalyzed Photoinduced LMCT: A 1° C-H Abstraction Enables Skeletal Rearrangements and C(sp3)-H Alkylation. ACS Catal. 2021, 11, 7442–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kang YC; Treacy SM; Rovis T Iron-Catalyzed C(sp3)-H Alkylation through Ligand-to-Metal Charge Transfer. Synlett 2021, 32, 1767–1771. [Google Scholar]; (f) Yang Q; Wang Y-H; Qiao Y; Gau M; Carroll PJ; Walsh PJ; Schelter EJ Photocatalytic C-H Activation and the Subtle Role of Chlorine Radical Complexation in Reactivity. Science 2021, 372, 847–852. [DOI] [PubMed] [Google Scholar]; (g) An Q; Wang Z; Chen Y; Wang X; Zhang K; Pan H; Liu W; Zuo Z Cerium-Catalyzed C-H Functionalizations of Alkanes Utilizing Alcohols as Hydrogen Atom Transfer Agents. J. Am. Chem. Soc. 2020, 142, 6216–6226. [DOI] [PubMed] [Google Scholar]; (h) Xiong N; Li Y; Zeng R Iron-Catalyzed Photoinduced Remote C(sp3)-H Amination of Free Alcohols. Org. Lett. 2021, 23, 8968–8972. [DOI] [PubMed] [Google Scholar]; (i) Yamane M; Kanzaki Y; Mitsunuma H; Kanai M Titanium(IV) Chloride-Catalyzed Photoalkylation via C(sp3)-H Bond Activation of Alkanes. Org. Lett. 2022, 24, 1486–1490. [DOI] [PubMed] [Google Scholar]; (j) Dai Z-Y; Zhang S-Q; Hong X; Wang P-S; Gong L-Z A Practical FeCl3/HCl Photocatalyst for Versatile Aliphatic C-H Functionalization. Chem. Catalysis 2022, 2, 1211–1222. [Google Scholar]
- (10).(a) Gonzalez MI; Gygi D; Qin Y; Zhu Q; Johnson EJ; Chen YS; Nocera DG Taming the Chlorine Radical: Enforcing Steric Control over Chlorine-Radical Mediated C-H Activation. J. Am. Chem. Soc. 2022, 144, 1464–1472. [DOI] [PubMed] [Google Scholar]; (b) Fokin AA; Schreiner PR Selective Alkane Transformations via Radicals and Radical Cations: Insights into the Activation Step from Experiment and Theory. Chem. Rev. 2002, 102, 1551–1594. [DOI] [PubMed] [Google Scholar]
- (11).(a) Niu B; Blackburn BG; Sachidanandan K; Cooke MV; Laulhé S Metal-Free Visible-Light-Promoted C(sp3)-H Functionalization of Aliphatic Cyclic Ethers Using Trace O2. Green Chem. 2021, 23, 9454–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pan L; Elmasry J; Osccorima T; Cooke MV; Laulhé S Photochemical Regioselective C(sp3)-H Amination of Amides Using N-haloimides. Org. Lett. 2021, 23, 3389–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gasonoo M; Thom ZW; Laulhé S Regioselective α-Amination of Ethers Using Stable N-Chloroimides and Lithium tert-Butoxide. J. Org. Chem. 2019, 84, 8710–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pan L; Cooke MV; Spencer A; Laulhé S Dimsyl Anion Enables Visible-Light-Promoted Charge Transfer in Cross-Coupling Reactions of Aryl Halides. Adv. Synth. Catal. 2022, 364, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Pan L; Kelley AS; Cooke MV; Deckert MM; Laulhé S TransitionMetal-Free Photoredox Phosphonation of Aryl C-N and C-X Bonds in Aqueous Solvent Mixtures. ACS Sustainable Chem. Eng. 2022, 10, 691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Niu B; Sachidanandan K; Blackburn BG; Cooke MV; Laulhé S Photoredox Polyfluoroarylation of Alkyl Halides via Halogen Atom Transfer. Org. Lett. 2022, 24, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Mader MM; Shih C; Considine E; Dios AD; Grossman CS; Hipskind PA; Lin H-S; Lobb KL; Lopez B; Lopez JE; Cabrejas LMM; Richett ME; White WT; Cheung Y-Y; Huang Z; Reilly JE; Dinn SR Acyl Sulfonamide Antiproliferatives. Part 2: Activity of Heterocyclic Sulfonamide Derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 617–620. [DOI] [PubMed] [Google Scholar]; (b) Azevedo-Barbosa H; Dias DF; Franco LL; Hawkes JA; Carvalho DT From Antibacterial to Antitumour Agents: A Brief Review on The Chemical and Medicinal Aspects of Sulfonamides. Mini Rev. Med. Chem. 2020, 20, 2052–2066. [DOI] [PubMed] [Google Scholar]; (c) Ragnarsson U; Grehn L Dual Protection of Amino Functions Involving Boc. RSC Adv. 2013, 3, 18691–18697. [Google Scholar]
- (14).(a) Abdelshaheed MM; Fawzy IM; El-Subbagh HL; Youssef KM Piperidine Nucleus in the Field of Drug Discovery. Future J. Pharm. Sci. 2021, 7, 188. [Google Scholar]; (b) Goel p.; Alam O; Naim MJ; Nawaz F; Iqbal M; Alam MI Recent Advancement of Piperidine Moiety in Treatment of Cancer- A review. Eur. J. Med. Chem. 2018, 157, 480–502. [DOI] [PubMed] [Google Scholar]
- (15).(a) Mugesh G; du Mont WW; Sies H Chemistry of Biologically Important Synthetic Organoselenium Compounds. Chem. Rev. 2001, 101, 2125–2180. [DOI] [PubMed] [Google Scholar]; (b) Nogueira CW; Zeni G; Rocha JBT Organoselenium and Organotellurium Compounds: Toxicology and Pharmacology. Chem. Rev. 2004, 104, 6255–6286. [DOI] [PubMed] [Google Scholar]
- (16).(a) Prasad CD; Sattar M; Kumar S Transition Metal-Free Selective Oxidative C(sp3)-S/Se Coupling of Oxindoles, Tetralone, Arylacetamides: Synthesis of Unsymmetrical Organochalcogenides. Org. Lett. 2017, 19, 774–777. [DOI] [PubMed] [Google Scholar]; (b) Rathore V; Kumar S Visible-Light-Induced Metal and Reagent-Free Oxidative Coupling of sp2 C–H Bonds with Organodichalcogenides: Synthesis of 3-organochalcogenyl Indoles. Green Chem. 2019, 21, 2670–2676. [Google Scholar]
- (17).(a) Goulart TAC; Back DF; Moura E Silva S; Zeni G Diorganyl Diselenides and Iron(III) Chloride Drive the Regio- and Stereoselectivity in the Selenation of Ynamides. J. Org. Chem. 2021, 86, 980–994. [DOI] [PubMed] [Google Scholar]; (b) Recchi AMS; Back DF; Zeni G Sequential Carbon-Carbon/Carbon-Selenium Bond Formation Mediated by Iron(III) Chloride and Diorganyl Diselenides: Synthesis and Reactivity of 2-Organoselenyl-Naphthalenes. J. Org. Chem. 2017, 82, 2713–2723. [DOI] [PubMed] [Google Scholar]
- (18).(a) Jin Y; Zhang Q; Wang L; Wang X; Meng C; Duan C Convenient C(sp3)-H Bond Functionalisation of Light Alkanes and Other Compounds by Iron Photocatalysis. Green Chem. 2021, 23, 6984–6989. [Google Scholar]; (b) Takaki K; Yamamoto J; Matsushita Y; Morii H; Shishido T; Takehira K Oxidation of Alkanes with Dioxygen Induced by Visible Light and Cu(II) and Fe(III) Chlorides. Bull. Chem. Soc. Jpn. 2003, 76, 393–398. [Google Scholar]; (c) Takaki K; Yamamoto J; Komeyama K; Kawabata T; Takehira K Photocatalytic Oxidation of Alkanes with Dioxygen by Visible Light and Copper(II) and Iron(III) Chlorides: Preference Oxidation of Alkanes over Alcohols and Ketones. Bull. Chem. Soc. Jpn. 2004, 77, 2251–2255. [Google Scholar]; (d) Porsch K; Kappler A FeII Oxidation by Molecular O2 During HCl Extraction. Environ. Chem. 2011, 8, 190–197. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.