

Abstract
Polyfluoroarene moieties are of interest in medicinal chemistry, agrochemicals, and material sciences. Herein, we present the first polyfluoroarylation of unactivated alkyl halides via a halogen atom transfer process. This method converts primary, secondary, and tertiary alkyl halides into the respective polyfluoroaryl compounds in good yields in the presence of amide, carbamate, ester, aromatic, and sulfonamide moieties, including derivatives of complex bioactive molecules. Mechanistic work revealed that this transformation proceeds through an alkyl radical generated after the halogen atom transfer.
Graphical Abstract
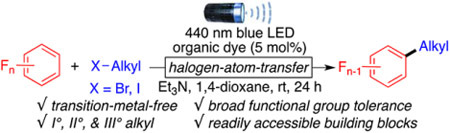
Organofluorine scaffolds are pivotal frameworks in various applications such as pharmaceuticals, material sciences, and pesticides and for positron emission tomography imaging (Scheme 1A).1 Fluorine atoms in a bioactive molecule can provide many beneficial properties such as increased membrane penetration and enhanced activity and can promote chemical or metabolic stability.2 Thus, the way to incorporate polyfluoroarenes into compounds has attracted considerable attention.3,4 Metal-catalyzed cross-couplings between ArF─H and aryl, alkenyl, or alkynyl groups have been explored.3-5 Strategies that proceed via nucleophilic aromatic substitution, such as the defluorinative functionalization of readily available polyfluoroarenes, often require the use of strong organometallic species such as Grignard, organolithium, or organozinc reagents.6 These harsh reaction conditions limit the functional group compatibility and reduce the complexity of possible molecular scaffolds.
Scheme 1. Molecules of Interest and Current Technologiesa.

a(A) Bioactive compounds of interest. (B) Photoredox methodologies to access alkylated polyfluoroarenes. (C) The presented halogen atom transfer (XAT) strategy.
Recently, photoredox-catalyzed polyfluoroarylation has been developed to enable mild and selective C─C bond formations, but the number of alkylation strategies remain limited.7 The Weaver group reported various photoredox functionalizations of C─F bonds in polyfluoroarenes, including a photocatalytic alkylation using alkenes as starting materials (Scheme 1B).7d In 2021, the Ritter group developed an alkylation of polyfluoroarenes via the radical decarboxylation of carboxylic acids.7e Finally, the Hu group reported a dual photo- and copper-catalyzed decarboxylative coupling of aliphatic N-hydroxyphthalimide (NHPI) esters with ArF─Zinc reagents.7f
Despite the progress made in the alkylation of polyfluoroarenes, the direct use of unactivated alkyl halides under metal-free conditions would significantly expand the scope and diversity of alkyl chains amenable for coupling compared to alkyl carboxylic acids, NHPI esters, and alkenes. Indeed, not only do alkyl halides represent one of the largest classes of building blocks in organic chemistry but they are also readily accessible from alcohols, another large chemical feedstock. Yet, access to alkyl radicals from alkyl halides remains challenging.8
As part of our interest in transition-metal-free cross-coupling reactions9 and inspired by the recent work of Leonori, Juliá, and Doyle,10 who demonstrated the ability of α-amino radicals to generate alkyl radicals from alkyl halides through halogen atom transfer processes (XAT), we present here the first example of a photoredox direct polyfluoarylation of unactivated alkyl halides via XAT (Scheme 1C). This metal-free transformation uses an α-aminoalkyl radical as the halogen abstracting reagent to obtain privileged alkyl polyfluoroarenes with wide functional group compatibilities, including esters, amides, carbamates, and sulfonamides.
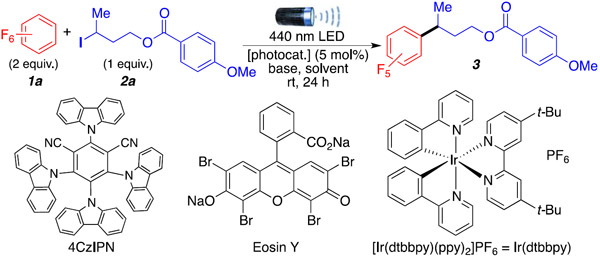
We started our investigation with perfluorobenzene (1a) and 3-iodobutyl 4-methoxybenzoate (2a) as model substrates (Table 1, entry 1) in the presence of 4CzIPN as the photocatalyst and triethylamine as the halogen abstracting agent (Table 1). Initial screens showed the formation of the desired product in various solvents (entries 1–3), with 1,4-dioxane providing the best results (44%) when the reaction was performed at higher concentrations (entry 4). Screening of bases showed that increasing the steric bulk around the nitrogen using diisopropylethyl amine (DIPEA) reduced the yield to 34% (entry 5). Using structurally rigid bases such as DABCO (entry 6) completely shut down the reactivity. Increasing the amount of Et3N (entries 7 and 8) led to small but noticeable increases in the yield. Similarly, increasing the amount of polyfluoroarene to 5 and 10 equiv (entries 9 and 10, respectively) further increased yields, affording the desired product in a 60% isolated yield (entry 10). Switching LED lamps from 440 to 427 nm (entry 11) reduced the yield to 38%. In the absence of either base (entry 12) or light (entry 13), the desired product was not observed. Finally, other common photocatalysts such as eosin Y (entry 14) or iridium-based catalysts (entry 15) provided the desired product in lower yields, 38% and 58%, respectively (see the Supporting Information for the full table of optimization on pages S7-S9).
Table 1.
Optimization of the Reaction and Its Conditions a
| ||||
|---|---|---|---|---|
| entry | base (equiv) | solvent (mL) | [photocatalyst] | yield (%)b |
| 1 | Et3N (2.0) | CH3CN (1.0) | 4CzIPN | 24 |
| 2 | Et3N (2.0) | DMSO (1.0) | 4CzIPN | 22 |
| 3 | Et3N (2.0) | 1,4-dioxane (1.0) | 4CzIPN | 26 |
| 4 | Et3N (2.0) | 1,4-dioxane (0.4) | 4CzIPN | 46 |
| 5 | DIPEA (2.0) | 1,4-dioxane (0.4) | 4CzIPN | 34 |
| 6 | DABCO (2.0) | 1,4-dioxane (0.4) | 4CzIPN | 0 |
| 7 | Et3N (3.0) | 1,4-dioxane (0.4) | 4CzIPN | 48 |
| 8 | Et3N (5.0) | 1,4-dioxane (0.4) | 4CzIPN | 50 |
| 9c | Et3N (5.0) | 1,4-dioxane (0.4) | 4CzIPN | 55 |
| 10d | Et3N (5.0) | 1,4-dioxane (0.4) | 4CzIPN | 62 (60)e |
| 11f | Et3N (5.0) | 1,4-dioxane (0.4) | 4CzIPN | 38 |
| 12 | – | 1,4-dioxane (0.4) | 4CzIPN | 0 |
| 13g | Et3N (5.0) | 1,4-dioxane (0.4) | 4CzIPN | 0 |
| 14 | Et3N (5.0) | 1,4-dioxane (0.4) | eosin Y | 38 |
| 15 | Et3N (5.0) | 1,4-dioxane (0.4) | Ir(dtbbpy) | 58 |
Optimal reaction conditions are as follows: 1 (2.0 mmol, 10 equiv), 2a (0.2 mmol, 1 equiv), base (1.0 mmol, 5 equiv), 1,4-dioxane (0.5 mL), 4CzIPN (5 mol %), 440 nm LED (40 W), room temperature (temperature around reaction flask was 35 °C due to heating caused by the LED lamp), reaction flask capped under argon, 24 h.
1H NMR yields using dibromomethane as internal standard.
1a (5 equiv) was used instead of 2 equiv.
1a (10 equiv) was used instead of 2 equiv.
Isolated yield.
A 427 nm LED (40W) was used instead of a 440 nm LED.
The reaction was performed in the dark.
We continued our investigations by exploring the scope of alkyl halides (Scheme 2). Ester moieties are well-tolerated in product 3 (60%). Cyclic and heterocyclic alkyl halides (4–6) afforded the desired products in good yields. Importantly, both alkyl iodides and alkyl bromides engage efficiently in this cross-coupling (42–70% yields), while alkyl chlorides remained unreactive. The reaction could also proceed with Boc-protected azetidines and piperidine motifs in 50% and 80% yields, respectively (7 and 8, respectively), which are important motifs in drug discovery.11 The ability of this method to tolerate Boc protecting groups further emphasizes the synthetic utility of this approach. Thus, we continued exploring the piperidine amide motif,4-iodo-1-benzoylpiperidine (9–14), bearing various substituents. Electron-donating and electron-withdrawing groups were well-tolerated (65–85%). Aromatic halogens in the para position were also compatible with this method, as well as halogens in the meta position, affording products 15 and 16 in 55% and 75% yields, respectively. Importantly, the orthogonal reactivity of these aryl halides through this coupling process may enable further functionalization of the products via Suzuki or Negishi cross-coupling reactions. Ortho substitutions and heteroaromatics such as thiophene afforded products 17 and 18 in moderate to good yields (55–65%). Less stable primary alkyl radicals afforded products in 19–21 in low but synthetically useful yields (20–45%).12 Notably, 1-adamantyl iodide, a tertiary alkyl iodide, also gave desired product 22 in a 30% yield. Finally, to highlight the late-stage functionalization potential of the method, compounds derived from commercially available bioactive molecules (ibuprofen, probenecid, and naproxen) were polyfluoroarylated to generate products 23–25, respectively, in good to excellent yields (50–89%). These functionalizations could be broadly applied to other bioactive compounds containing alcohol moieties that could be readily transformed into their halogen counter parts.13
Scheme 2. Alkyl Halide Scopea.

aReaction conditions are as follows: 1a (2.0 mmol, 10 equiv), 2 (0.2 mmol, 1 equiv), Et3N (1.0 mmol, 5 equiv), 1,4-dioxane (0.4 mL), 4CzIPN (5 mol %), room temperature (temperature around reaction flask was 35 °C due to heating caused by the LED lamp), 24 h. All yields are isolated. b1a (5 equiv). cReaction in DMSO (0.4 mL).
Next, we turned our attention to explore the scope of polyfluoroarenes 26–33 (Scheme 3). Substituted polyfluoroarenes generate inseparable mixtures of regioisomers due to unselective radical addition to different positions.14 However, the reaction tolerates bromo and chloro substitutions, affording products 26 and 27, respectively, in good to excellent yield (70–85%). It is worth indicating that alkylation occurred chemoselectively at the location of fluorine atoms with no observable substitution at the bromide or chloride, differentiating this method from previous methodologies.7d Polyfluoroheteroarenes such as pentafluoropyridine also gave product 28 in a moderate yield (60%). Electron-withdrawing CF3 (29), secondary amides (30 and 31), and sulfonamide 32 were well tolerated, affording the corresponding products in 85%, 75%, 65%, and 40% yields, respectively. Finally, 1,2,4,5-tetrafluoro-3-(trifluoromethyl)benzene also benefitted from the chemoselective alkylation, providing product 33 in a 66% yield with no observable substitution at the location of the hydrogen.
Scheme 3. Polyfluoroarene Scopea.

aReaction conditions are as follows : 1 (1.0 mmol, 5 equiv), 2a (0.2 mmol, 1 equiv), Et3N (1.0 mmol, 5 equiv), 1,4-dioxane (0.4 mL), 4CzIPN (5 mol %), room temperature (temperature around reaction flask was 35 °C due to heating caused by the LED lamp), overnight. All yields are isolated.
To explore the reaction mechanism, a series of control experiments were performed (Scheme 4). As expected, a carbon-centered radical generated from (3-iodopropyl)-benzene was confirmed by the formation of the cyclic byproduct 35 (Scheme 4A). Furthermore, the use of radical trapping agents such as 2,2,6,6-tetramethyl-peperidinylxoyl (TEMPO) only afforded a trace amount of product 4 (Scheme 4B). The use of other trapping agents (1,1-DPE, 1,4-DNB, and BHT) also produced similar results. Importantly, two radical intermediates were successfully trapped as TEMPO and 1,1-DPE adducts (36–38) and observed by GC-MS. The cyclohexane alkyl radical was trapped with both TEMPO (36) and 1,1-DPE (37). Additionally, the triethylamine radical at the α-position was also trapped using 1,1-DPE as product 38. These experiments indicate that alkyl and triethylamine radicals are involved in this transformation.
Scheme 4. Verification of the Presence of Radicalsa.

a (A) Cyclization byproduct experiment. (B) Radical trapping experiments. (C) Fluorescence spectra of 4CzIPN in 1,4-dioxane (0.01 mM) before and after the addition of different amounts of (1) C6F6, (2) iodocyclohexane, and (3) Et3N. (4) The resulting Stern–Volmer plot.
To further understand the mechanism of the reaction and determine the sequence of each step, we performed a series of fluorescence quenching experiments involving the photocatalyst (Scheme 4C and Supporting Information S27). The addition of polyfluoroarene 1a or alkyl halide 2a does not quench fluorescence of the excited state of the photocatalyst (Scheme 4C1 and C2). On the other hand, the addition of Et3N (Scheme 4C3) leads to a substrate-dependent quenching, indicating that the excited state of 4CzIPN initially reacts with Et3N via a single-electron transfer. This observation is in accordance with previous XAT reactions developed by Leonori and Juliá.10a,b
On the basis of the above obtained results and previous reports,7e,10 we propose a reaction mechanism in Scheme 5. First, photoexcited 4CzIPN oxidizes Et3N to form radical cation I. Fast deprotonation15 of I generates an α-aminoalkyl radical species II capable of performing the XAT with iodocyclohexane, which generates alkyl radical III and α-iodoamine IV (II and III can be trapped with 1,1-DPE and TEMPO; Scheme 4B). The irreversible dissociation of IV into iminium iodide V provides a XAT driving force. Radical species III then reacts with polyfluorobenzene to form the aryl radical intermediate VI via radical nucleophilic aromatic substitution. The reduction of VI by radical anion 4CzIPN•− affords the aryl anion species VII and regenerates the photocatalyst. Finally, the loss of fluoride from VII generates the desired final product.
Scheme 5.

Proposed Mechanism
In summary, this work presents the first polyfluoroarylation of alkyl halides via a XAT process. The reaction shows good functional group compatibility with synthetically useful moieties such a Boc protecting groups, esters, amides, and sulfonamides, all of which are of great interest to medicinal chemistry. The transformation proceeds in moderate to excellent yields with I°, II° and III° alkyl halides building blocks and alkyl halides derived from bioactive compounds. This process provides a complementary and metal-free approach to current decarboxylative and hydrofunctionalization approaches.
Supplementary Material
Funding
This publication was made possible with support from National Institute of Dental and Craniofacial Research Grant R21DE029156.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.1c04267.
1H and 13C{1H} NMR spectra, GS─MS spectra, and additional information on the substrate scope and its limitations (PDF)
REFERENCES
- (1).(a) Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in Medicinal Chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; (b) Lee E; Kamlet AS; Powers DC; Neumann CN; Boursalian GB; Furuya T; Choi DC; Hooker JM; Ritter T A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Babudri F; Farinola GM; Naso F; Ragni R Fluorinated Organic Materials for Electronic and Optoelectronic Applications: the Role of the Fluorine Atom. Chem. Commun 2007, 1003–1022. [DOI] [PubMed] [Google Scholar]; (d) Wang J; Sanchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]; (e) Jeschke P The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. Chem. Bio. Chem 2004, 5, 570–589. [DOI] [PubMed] [Google Scholar]
- (2).(a) Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]; (b) Ojima I; Awasthi D; Wei L; Haranahalli K Strategic Incorporation of Fluorine in the Drug Discovery of New-generation Antitubercular Agents Targeting Bacterial Cell Division Protein FtsZ. J. Fluor. Chem 2017, 196, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Liang T; Neumann CN; Ritter T Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem Int. Ed 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]; (b) Li X; He ST; Song QL Rapid Incorporation of a Difluoroacetate Radical into Para-Quinone Methides via Radical 1,6-Conjugate Addition. Chem. Commun 2021, 57, 6035–6038. [DOI] [PubMed] [Google Scholar]; (c) Weaver J; Senaweera S C─F Activation and Functionalization of Perfluoro- and Polyfluoroarenes. Tetrahedron 2014, 70, 7413–7428. [Google Scholar]
- (4).(a) Ahrens T; Kohlmann J; Ahrens M; Braun T Functionalization of Fluorinated Molecules by Transition-Metal-Mediated C─F Bond Activation To Access Fluorinated Building Blocks. Chem. Rev 2015, 115, 931–972. [DOI] [PubMed] [Google Scholar]; (b) Amii H; Uneyama K C─F bond Activation in Organic Synthesis. Chem. Rev 2009, 109, 2119–2183. [DOI] [PubMed] [Google Scholar]; (c) Kuehnel MF; Lentz D; Braun T Synthesis of Fluorinated Building Blocks by Transition-Metal-Mediated Hydrodefluorination Reactions. Angew. Chem., Int. Ed 2013, 52, 3328–3348. [DOI] [PubMed] [Google Scholar]; (d) Campbell MG; Ritter T Modern Carbon-Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev 2015, 115, 612–633. [DOI] [PubMed] [Google Scholar]
- (5).(a) Wu CZ; He CY; Huang Y; Zhang XG Thioether-Promoted Direct Olefination of Polyfluoroarenes Catalyzed by Palladium. Org. Lett 2013, 15, 5266–5269. [DOI] [PubMed] [Google Scholar]; (b) Liu HC; Li Y; Gong XP; Niu ZJ; Wang YZ; Li M; Shi WY; Zhang Z; Liang YM Cu-Catalyzed Direct C─H Alkylation of Polyfluoroarenes via Remote C(sp3)-H Functionalization in Carboxamides. Org. Lett 2021, 23, 2693–2698. [DOI] [PubMed] [Google Scholar]; (c) Nakamura Y; Yoshikai N; Ilies L; Nakamura E Nickel-Catalyzed Monosubstitution of Polyfluoroarenes with Organozinc Reagents using Alkoxydiphosphine Ligand. Org. Lett 2012, 14, 3316–3319. [DOI] [PubMed] [Google Scholar]; (d) Xu S; Wu GJ; Ye F; Wang X; Li H; Zhao X; Zhang Y; Wang JB Copper (I)-Catalyzed Alkylation of Polyfluoroarenes through Direct C-H Bond Functionalization. Angew. Chem., Int. Ed 2015, 54, 4669–4672. [DOI] [PubMed] [Google Scholar]; (e) Li XH; Fu B; Zhang Q; Yuan XP; Zhang Q; Xiong T; Zhang Q Copper-Catalyzed Defluorinative Hydroarylation of Alkenes with Polyfluoroarenes. Angew. Chem., Int. Ed 2020, 59, 23056–23060. [DOI] [PubMed] [Google Scholar]; (f) Dahiya A; Fricke C; Schoenebeck F Gold-Catalyzed Chemoselective Couplings of Polyfluoroarenes with Aryl Germanes and Downstream Diversification. J. Am. Chem. Soc 2020, 142, 7754–7759. [DOI] [PubMed] [Google Scholar]
- (6).(a) Fujii K; Ito S; Mikami K Synthetic Methodologies for Perfluoroaryl-Substituted (Diaryl)methylphosphonates, -Phosphinates via SNAr Reaction. J. Org. Chem 2019, 84, 12281–12291. [DOI] [PubMed] [Google Scholar]; (b) Sun YQ; Sun HJ; Jia J; Du AQ; Li XY Transition-Metal-Free Synthesis of Fluorinated Arenes from Perfluorinated Arenes Coupled with Grignard Reagents. Organometallics 2014, 33, 1079–1081. [Google Scholar]; (c) Kiplinger JL; Richmond TG; Osterberg CE Activation of Carbon-Fluorine Bonds by Metal Complexes. Chem. Rev 1994, 94, 373–431. [Google Scholar]; (d) Ong WJ; Swager TM Dynamic Self-Correcting Nucleophilic Aromatic Substitution. Nat. Chem 2018, 10, 1023–1030. [DOI] [PubMed] [Google Scholar]
- (7).(a) Xie J; Rudolph M; Rominger F; Hashmi ASK Photoredox-Controlled Mono- and Di-Multifluoroarylation of C-(Sp3)-H Bonds with Aryl Fluorides. Angew. Chem. Int. Ed 2017, 56, 7266–7270. [DOI] [PubMed] [Google Scholar]; (b) Senaweera SM; Singh A; Weaver JD Photocatalytic Hydrodefluorination: Facile Access to Partially Fluorinated Aromatics. J. Am. Chem. Soc 2014, 136, 3002–3005. [DOI] [PubMed] [Google Scholar]; (c) Singh A; Fennell CJ; Weaver JD Photocatalyst Size Controls Electron and Energy Transfer: Selectable E/Z Isomer Synthesis via C─F Alkenylation. Chem. Sci 2016, 7, 6796–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Singh A; Kubik JJ; Weaver JD Photocatalytic C─F Alkylation; Facile Access to Multifluorinated Arenes. Chem. Sci 2015, 6, 7206–7212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sun X; Ritter T Decarboxylative Polyfluoroarylation of Alkylcarboxylic Acids. Angew. Chem., Int. Ed 2021, 60, 10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yi XL; Mao RZ; Lavrencic L; Hu XL Angew. Chem., Int. Ed 2021, 60, 23557–23563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Chatgilialoglu C; Ferreri C; Landais Y; Timokhin VI Thirty Years of (TMS)3SiH: A Milestone in Radical-Based Synthetic Chemistry. Chem. Rev 2018, 118, 6516–6572. [DOI] [PubMed] [Google Scholar]; (b) Curran DP; McFadden TR Understanding Initiation with Triethylboron and Oxygen: The Differences between Low-Oxygen and High-Oxygen Regimes. J. Am. Chem. Soc 2016, 138, 7741–7752. [DOI] [PubMed] [Google Scholar]; (c) Neumann WP Tri-n-butyltin Hydride as Reagent in Organic Synthesis. Synthesis 1987, 1987, 665. [Google Scholar]; (e) Chatgilialoglu C Organosilanes as Radical-Based Reducing Agents in Synthesis. Acc. Chem. Res 1992, 25, 188–194. [Google Scholar]; (f) Ollivier C; Renaud P Organoboranes as a Source of Radicals. Chem. Rev 2001, 101, 3415–3434. [DOI] [PubMed] [Google Scholar]
- (9).(a) Pan L; Cooke MV; Spencer A; Laulhé S Dimsyl Anion Enables Visible-Light-Promoted Charge Transfer in Cross-Coupling Reactions of Aryl Halides. Adv. Synth. Cat 2021, DOI: 10.1002/adsc.202101052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Niu B; Blackburn BG; Sachidanandan K; Cooke MV; Laulhé S Metal-Free Visible-Light-Promoted C(sp3)─H Functionalization of Aliphatic Cyclic Ethers Using Trace O2. Green Chem. 2021, 23, 9454–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pan L; Elmasry J; Osccorima T; Cooke MV; Laulhé S Org. Lett 2021, 23, 3389–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pan L; Kelley AS; Cooke MV; Deckert MM; Laulhé S Transition-Metal-Free Photoredox Phosphonation of Aryl C─N and C─X Bonds in Aqueous Solvent Mixtures. ACS Sustainable Chem. Eng 2022, DOI: 10.1021/acssuschemeng.1c07394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Constantin T; Zanini M; Regni A; Sheikh NS; Juliá F; Leonori D Aminoalkyl Radicals as Halogen-atom Transfer Agents for Activation of Alkyl and Aryl Halides. Science 2020, 367, 1021–1026. [DOI] [PubMed] [Google Scholar]; (b) Constantin T; Juliá F; Sheikh NS; Leonori D A Case of Chain Propagation: α-Aminoalkyl Radicals as Initiators for Aryl Radical Chemistry. Chem. Sci 2020, 11, 12822–12828. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neff RK; Su Y-L; Liu S; Rosado M; Zhang X; Doyle MP Generation of Halomethyl Radicals by Halogen Atom Abstraction and Their Addition Reactions with Alkenes. J. Am. Chem. Soc 2019, 141, 16643–16650. [DOI] [PubMed] [Google Scholar]
- (11).(a) Mehra V; Lumb I; Anand A; Kumar V Recent Advances in Synthetic Facets of Immensely Reactive Azetidines. RSC Adv. 2017, 7, 45763–45783. [Google Scholar]; (b) Abdelshaheed MM; Fawzy IM; El-Subbagh HL; Youssef KM Piperidine nucleus in the field of drug discovery. Future J. Pharm. Sci 2021, 7, 188. [Google Scholar]; (c) Goel p.; Alam O; Naim MJ; Nawaz F; Iqbal M; Alam MI Recent Advancement of Piperidine Moiety in Treatment of Cancer- A review. Eur. J. Med. Chem 2018, 157, 480–502. [DOI] [PubMed] [Google Scholar]
- (12).(a) Romero KJ; Galliher MS; Pratt DA; Stephenson CRJ Radicals in Natural Product Synthesis. Chem. Soc. Rev 2018, 47, 7851–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hippler H; Viskolcz B Competition Between Alkyl Radical Addition to Carbonyl Bonds and H-Atom Abstraction Reactions. Phys. Chem. Chem. Phys 2002, 4, 4663–4668. [Google Scholar]
- (13).(a) Classon B; Liu Z; Samuelsson B New Halogenation Reagent Systems Useful for the Mild One-Step Conversion of Alcohols into Iodides or Bromides. J. Org. Chem 1988, 53, 6126–6130. [Google Scholar]; (b) Smeaton E; Smith MH; White MJ Science of Synthesis, Reagents: Halogenation; Georg Thieme Verlag KG: Stuttgart, NY, 2013. [Google Scholar]
- (14).(a) Nagib DA; MacMillan DWC Trifluoromethylation of Arenes and Heteroarenes by Means of Photoredox Catalysis. Nature 2011, 480, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ham WS; Hillenbrand J; Jacq J; Genicot C; Ritter T Divergent Late-Stage (Hetero)aryl C─H Amination by the Pyridinium Radical Cation. Angew. Chem., Int. Ed 2019, 58, 532–536. [DOI] [PubMed] [Google Scholar]
- (15).(a) Zhang X; Bordwell FG Acidities and Homolytic Bond Dissociation Energies of the Acidic C-H Bonds in Radical Cations. J. Org. Chem 1992, 57, 4163–4168. [Google Scholar]; (b) Shono T; Matsumura Y; Inoue K; Ohmizu H; Kashimura S Electroorganic chemistry. 62. Reaction of Iminium Ion with Nucleophile: a Versatile Synthesis of Tetrahydroquinolines and Julolidines. J. Am. Chem. Soc 1982, 104, 5753–5757. [Google Scholar]; (c) Hu J; Wang J; Nguyen TH; Zheng N The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem 2013, 9, 1977–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.