

In this issue of the Journal, Tompkins and colleagues (pp. 550–561) describe a novel pharmacological property of PD 102807 with potential clinical implications in obstructive lung diseases (1). Back in 1997, two different labs originally described PD 102807 as a selective M4 mAChR antagonist by combining radioligand binding assays in mAChR overexpressing Chinese hamster ovary cells and functional assays performed in various rodent tissues (2, 3). Since then, additional studies have indeed confirmed the M4 mAChR selectivity of PD 102807 (4). The current report by Tompkins and colleagues is unique as it is the first to demonstrate that PD 102807 can exert non-M4 activities by acting as a biased M3 mAChR ligand on the basis of a set of carefully designed experiments on HEK293 cells and hTERT-immortalized airway smooth muscle (ASM) cells. The choice of using immortalized ASM cells over the traditional primary cells could be called into question with respect to the physiological relevance. However, this cellular model represents the best option to study the function of M3 mAChR in ASM cells that otherwise would be impossible in primary cells because of the loss of the receptor expression (and hence function) (5). The authors have provided convincing arguments showing the ability of PD 102807 to trigger M3 mAChR-biased signaling in ASM cells by preferably activating M3-GRK2 and 3–β-arrestin–dependent pathways independently of the canonical M3-Gq/calcium signaling. Although these findings were derived from cell-based studies, there are a number of exciting lessons that can be drawn from their work (summarized in Figure 1).
Figure 1.
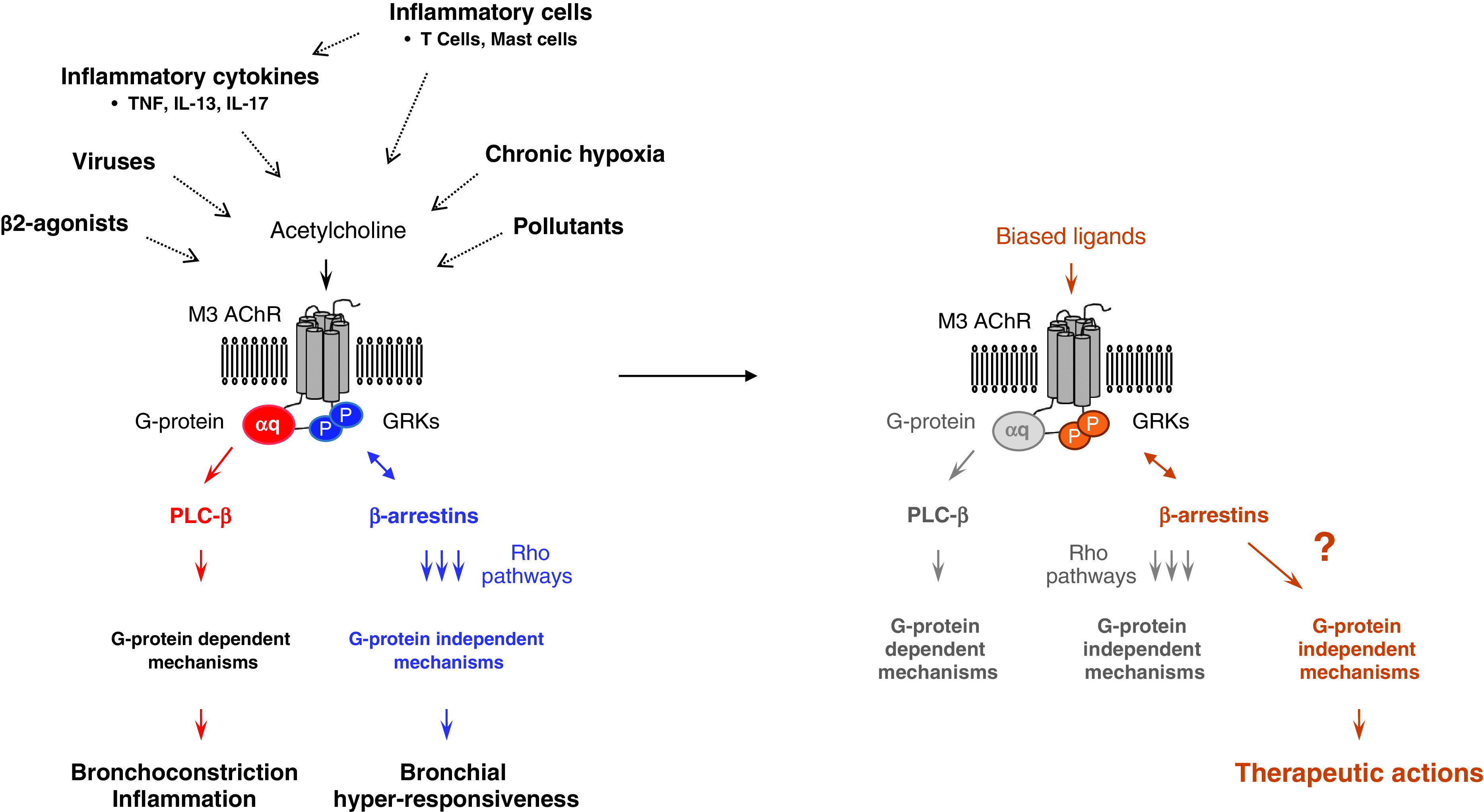
Biased M3 AChR agonism in the treatment of obstructive lung disease. Accumulating evidence suggests that M3 AChR-dependent pathways contribute to airway inflammation, airway remodeling, and abnormal lung function in asthma. The mechanisms underlying these proasthmatic responses appear to involve both canonical (G-protein dependent) and noncanonical (β-arrestin dependent) pathways, most likely acting in concert. Combined preclinical studies that have used engineered mice or receptor antagonists led to the conclusion that the pathological role of M3 AChR in allergic asthma is complex and determined by the type of activated downstream signaling pathway. Bronchial hyperresponsiveness is believed to result from muscarinic agonists activating preferably noncanonical pathways leading to Rho-dependent enhancement of airway smooth muscle contractility. In contrast, other asthmatic features such as airway inflammation (and possibly airway remodeling) are driven by G-protein–dependent mechanisms. The ability of various proasthmatic insults, including cytokines, respiratory viruses, inflammatory cells, and pollutants (among others), to alter M3 AChR receptor function may contribute to signaling bias associated with different clinical outcomes, although this hypothesis remains to be further investigated. With the biased M3 AChR ligands, the expectation is that they will only trigger G-protein–independent pathways that promote antiremodeling and bronchorelaxant actions via β-arrestin pathways that are distinct from those driving bronchial hyperresponsiveness. The study by Tompkins provides strong evidence for a therapeutic role of some of the M3 AChR-associated β-arrestin pathways that remain to be further investigated (?). GPCR = G-protein coupled receptor; GRKs = GPCR kinases; P = phosphorylation site; PLC = phospholipase.
From a clinical point of view, the first obvious one would be to suggest that biased M3 mAChR ligands might represent novel therapeutic approaches for the treatment of lung diseases. As stated by the authors, biased ligands are already being evaluated at various development stages for different human conditions ranging from preclinical to phase III studies, with carvedilol being the only biased drug approved for the treatment of congestive heart failure (6). In this study, Tompkins and colleagues demonstrate that PD 102807 not only antagonized M3-Gq–dependent procontractile pathways (i.e., calcium) but also suppressed TGFβ signaling and its procontractile function (contractility and αSMA) via Gq-independent pathways (i.e., GRK2 and 3–β-arrestin–AMPK). Interestingly, others using the same model of immortalized ASM cells have shown that M3 AChR engagement with methacholine led to an enhancement of the procontractile effects of TGFβ (7). Two main conclusions could be drawn from this latter study. The first is that PD 102807 is able to effectively use distinct signaling pathways to prevent TGFβ deleterious responses, and the second is that depending on the type of ligands (cognate vs. biased), engagement of M3 AChR in ASM cells has the potential to trigger two completely opposite outcomes (i.e., detrimental vs. therapeutic). One can therefore postulate that biased M3 mAChR ligands have the potential to alleviate excessive bronchoconstriction and/or prevent features of airway remodeling on the basis of their unique capacity to modulate TGFβ signaling pathways. These possibilities could be easily tested in animal models of allergic asthma treated with biased M3 mAChR ligands, similar to studies that have tested anticholinergics (8). The putative role of ASM in the therapeutic action of the most promising biased M3 mAChR ligands could then be examined using the precision cut lung slice model.
Both preclinical and clinical evidence using receptor antagonists/inverse agonists have indeed demonstrated a central role of M3 AChR in mediating key features of chronic obstructive pulmonary disease and asthma, including abnormal lung function, mucus hypersecretion, and airway inflammation and remodeling (8). For example, strong evidence for a role of M3 AChR in mediating allergen-induced airway remodeling and ASM mass came from the use of receptor knockout mice (9), so it would be sensible to examine the effect of biased ligands on these proasthmatic features. However, in contrast to the anticipated effects of receptor blockade (using receptor antagonism or knockdown strategies), predicting the impact of M3-biased signaling pathways in vivo may prove to be difficult. Indeed, we found that allergen-induced airway hyperresponsiveness was completely abrogated in sensitized mice engineered to express a G protein-biased M3-mAChR mutant receptor incapable of activating phosphorylation-dependent β-arrestin pathways (10). In contrast, allergen-induced pulmonary inflammation was not affected in these mutant mice. Together our study suggests that in experimental asthma, M3-Gq–independent pathways (i.e., noncanonical) exert a predominant role over Gq-dependent pathways in driving ASM abnormalities to muscarinic agonists.
So, even if suitable biased M3 mAChR ligands could be developed and trialed for lung diseases, one might wonder whether their therapeutic action, relying on Gq-independent pathways, would be altered in asthma. This is a legitimate question as a number of different insults such as cytokines, inflammatory cells (T cells), pollutants, respiratory viruses, mechanical strain, and anesthetics (to list a few) have been shown to modulate M3 AChR-associated procontractile responses, a consistent finding reported by many groups in both cultured ASM cells and isolated ASM tissues (reviewed in [11, 12]). Irrespective of the mechanisms leading to changes in M3 AChR responsiveness that are highly complex, often context- and stimulus-specific, all these studies point toward a defect occurring at different degrees of M3 AChR signaling. For example, TNFα (but the observation also applies to other cytokines) can increase carbachol-evoked ASM contractility by modulating both canonical (calcium signals) and noncanonical (β-arrestin and Rho-dependent calcium sensitization) (13). Those changes may result from alterations occurring at 1) receptor level (expression); 2) receptor coupling level (G-protein); and 3) signal transduction level (phospholipase and intracellular calcium pools). It is, therefore, essential to ensure that the beneficial responses of biased M3 ligands are still preserved in asthmatic airways.
Lastly, from a mechanistic point of view, it would be interesting to determine how PD 102807, described as a selective M4 mAChR antagonist more than 20 years ago, exerts its biased action on M3 mAChR. It is also unknown whether the beneficial outcomes of PD 102807 seen in ASM cells can be reproduced in other M3 AChR-expressing cells that contribute to lung diseases (e.g., lung epithelial cells). In addition to opening new areas of investigation, the present study has the merit of being the first to identify PD 102807 as a novel biased M3 mAChR ligand and to show the therapeutic value of studying G-protein coupled receptor (GPCR)-biased agonism in ASM cells. The best is probably yet to come.
Footnotes
This study was supported by the National Institute for Health Research Leicester Biomedical Research Centre Respiratory. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, and the Department of Health.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0335ED on September 1, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Tompkins E, Mimic B, Cuevas-Mora K, Schorsch H, Shah SD, Deshpande DA, et al. Pd 102807 induces m3 machr-dependent grk-/arrestin-biased signaling in airway smooth muscle cells. Am J Respir Cell Mol Biol . 2022;67:550–561. doi: 10.1165/rcmb.2021-0320OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gross J, Augelli-Szafran CE, Czeche S, Friebe T, Jaen JC, Penvose-Yi JR, et al. Functional characterisation of pd102807: a novel m4-selective muscarinic antagonist. Life Sci . 1997;60:1168. [Google Scholar]
- 3. Schwarz RD, Nelson CB, Augelli-Szafran CE, Penvose JR, Jaen JC, Wiley J, et al. Pharmacological characterization of pd102807: an m4 subtype selective muscarinic antagonist. Life Sci . 1997;60:1167. [Google Scholar]
- 4. Olianas MC, Onali P. PD 102807, a novel muscarinic M4 receptor antagonist, discriminates between striatal and cortical muscarinic receptors coupled to cyclic AMP. Life Sci . 1999;65:2233–2240. doi: 10.1016/s0024-3205(99)00488-9. [DOI] [PubMed] [Google Scholar]
- 5. Burgess JK, Ketheson A, Faiz A, Limbert Rempel KA, Oliver BG, Ward JPT, et al. Phenotype and functional features of human telomerase reverse transcriptase immortalized human airway smooth muscle cells from asthmatic and non-asthmatic donors. Sci Rep . 2018;8:805. doi: 10.1038/s41598-017-18429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov . 2018;17:243–260. doi: 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oenema TA, Smit M, Smedinga L, Racké K, Halayko AJ, Meurs H, et al. Muscarinic receptor stimulation augments TGF-β1-induced contractile protein expression by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol . 2012;303:L589–L597. doi: 10.1152/ajplung.00400.2011. [DOI] [PubMed] [Google Scholar]
- 8. Kistemaker LE, Gosens R. Acetylcholine beyond bronchoconstriction: roles in inflammation and remodeling. Trends Pharmacol Sci . 2015;36:164–171. doi: 10.1016/j.tips.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 9. Kistemaker LE, Bos ST, Mudde WM, Hylkema MN, Hiemstra PS, Wess J, et al. Muscarinic M3 receptors contribute to allergen-induced airway remodeling in mice. Am J Respir Cell Mol Biol . 2014;50:690–698. doi: 10.1165/rcmb.2013-0220OC. [DOI] [PubMed] [Google Scholar]
- 10. Bradley SJ, Wiegman CH, Iglesias MM, Kong KC, Butcher AJ, Plouffe B, et al. Mapping physiological G protein-coupled receptor signaling pathways reveals a role for receptor phosphorylation in airway contraction. Proc Natl Acad Sci USA . 2016;113:4524–4529. doi: 10.1073/pnas.1521706113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amrani Y, Panettieri RA., Jr Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr Opin Allergy Clin Immunol . 2002;2:39–45. doi: 10.1097/00130832-200202000-00007. [DOI] [PubMed] [Google Scholar]
- 12. Amrani Y. Airway smooth muscle modulation and airway hyper-responsiveness in asthma: new cellular and molecular paradigms. Expert Rev Clin Immunol . 2006;2:353–364. doi: 10.1586/1744666X.2.3.353. [DOI] [PubMed] [Google Scholar]
- 13. Brightling C, Berry M, Amrani Y. Targeting TNF-alpha: a novel therapeutic approach for asthma. J Allergy Clin Immunol . 2008;121:5–10, quiz 11–12. doi: 10.1016/j.jaci.2007.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]