

Abstract
Coronavirus disease (COVID-19) begins with upper airway symptoms but proceeds in a significant proportion of patients to life-threatening infection of the lower respiratory tract, where an exuberant inflammatory response, edema, and adverse parenchymal remodeling impair gas exchange. Respiratory failure is caused initially by flooding of the airspaces with plasma exudate, sloughed epithelium, and inflammatory cells. For many patients with COVID-19, this acute phase has been observed to give way to a prolonged course of acute respiratory distress syndrome, and a significant proportion of patients go on to develop fibroproliferative remodeling of the lung parenchyma, which lengthens the duration of respiratory impairment and mechanical ventilation. Monocyte-derived macrophages have previously been implicated in the fibrotic phase of lung injury in multiple models. From several recent studies that used single-cell genomic techniques, a profile of the transcriptomic state of COVID-19 lung macrophages has emerged. Linkages have been made between these macrophages, which are monocyte-derived and CD163+, and profibrotic macrophages found in other contexts, including animal models of fibrosis and idiopathic pulmonary fibrosis. Here, emerging concepts of macrophage profibrotic function in COVID-19 are highlighted with a focus on gaps in knowledge to be addressed by future research.
Keywords: COVID-19, ARDS, fibrosis, monocyte-derived macrophage, CD163
Elie Metchnikoff first described macrophages nearly 150 years ago, observing phagocytic cells that could engulf either dead host cells or pathogens such as bacteria. Studies since that time have revealed a wide variety of roles for macrophages beyond phagocytosis, including paracrine interactions with other cells in specific microenvironments (1). These functions are highly dependent on tissue type and context.
In the lung, mechanistic studies have defined disparate functions for macrophages according to disease, location, and ontogeny. At steady state, the lung has two major types of macrophages defined by their anatomic niche: alveolar and interstitial. Alveolar macrophages reside within the alveolar air sacs of the lower respiratory tract and are embryonically derived, self-renewing throughout the lifespan (2–4). They support a number of homeostatic functions, including clearance of surfactant and detection of pathogen- and damage-associated molecular patterns (5–7). A second relevant anatomic space is the lung interstitium, which lies between the luminal and vascular spaces throughout the organ, and macrophage diversity within this compartment has been increasingly recognized by single-cell transcriptomic studies (8, 9). Disparate microenvironments within the lung, from airway to bronchovascular cuff to the alveolar interstitium, define various functions and immunoregulatory roles (10), although in most cases the context-specific functions are yet to be fully worked out. Here I review the role of lung macrophages in coronavirus disease (COVID-19)–related fibrotic disease.
Macrophages in Pulmonary Fibrosis
For context, it is helpful to consider the profile of profibrotic macrophages found in the most common fibrosing disease of the lung, idiopathic pulmonary fibrosis (IPF), a chronic and progressive disorder of the lung for which there are no curative therapies (11). In IPF, patients present with shortness of breath attributable pathologically to multiple regions of activated fibroblasts that form clusters and deposit collagens as well as other extracellular matrix proteins. These fibroblastic foci are progressive and are also associated with parenchymal remodeling, including grossly dilated adjacent airspaces in the peripheral lung known as regions of honeycombing.
The root cause of fibroblastic activation in IPF is not known, although some consensus exists around the view that the process begins with alveolar epithelial dysfunction (12). Alveolar epithelial cells have been noted to develop features of cellular senescence (13–15), a DNA damage response associated with replicative arrest and a secretory state known as the senescence-associated secretory profile, which has been shown to be profibrotic in animal models (13, 16). The causes of induction of senescence itself may be variable and multifactorial, ranging from telomere dysfunction associated with genetic predisposition or aging to injury, infection, or inflammation. Why this senescence persists remains a mystery in most cases, although in a minority of patients there is a hereditary cause, such as a genetic predisposition to telomere dysfunction, as in certain familial variants of IPF (17). Senescent or otherwise dysfunctional epithelial cells recruit profibrotic immune cells, including macrophages, and also express integrin αvβ6, which activates TGF-β (transforming growth factor-β) and thereby induces the profibrotic state of fibroblasts (18–20).
Two antifibrotics approved by several international drug-regulatory agencies, nintedanib and pirfenidone, slow the progression of IPF (21, 22). However, transplant remains necessary for many patients because of progressive obliteration of the gas-exchanging volume of the lung. Thus, identifying therapeutic targets is an urgent need, and targeting the profibrotic function of macrophages has emerged as a candidate approach. In the setting of acute lung injury or inflammation, mouse models have clearly demonstrated that, after an initial period of neutrophilic inflammation, monocyte-derived macrophages (moMacs) are recruited to and are the predominant inflammatory cell type in the lung (23–25). Among lung fibrosis models, a single intratracheal instillation of the DNA-damaging agent bleomycin is the most commonly used for testing of pathways relevant to lung fibrosis (26). In the bleomycin model, bone marrow–derived cells have been found to predominate in the fibrotic phase of injury, with characteristic gene expression programs. As distinct from embryonically derived alveolar macrophages, recruited moMacs localize to sites of fibroblast and collagen accumulation (Figure 1) (25). Several groups have shown that these moMacs are profibrotic, exerting their fibrotic function by paracrine signaling to fibroblasts—that is, by secreting mediators that induce production by fibroblasts of collagens and other matrix molecules that comprise fibrotic scar. Among the many mediators that have been identified are TNF-α, TGF-β, IL1β, PDGF (platelet-derived growth factor), and Wnt ligands (24, 25, 27, 28). In IPF, confirming clinical relevance of the mouse model, moMac markers have likewise been identified in single-cell RNA sequencing (scRNAseq) studies (20, 29, 30)—for example, the moMac marker MAF BZIP transcription factor B (MAFB), which is not expressed in alveolar macrophages (31). Fluorescence microscopy using second harmonic imaging to identify areas of collagen accumulation localized MAFB-expressing moMacs to areas of dense fibrotic scar, not to spared regions of the lung (Figure 2) (25).
Figure 1.

Mouse lung immunofluorescence. Cx3cr1-CreERT2: R26-loxp-STOP-loxp-TdTomato (Tandem dimer Tomato) mice were treated with tamoxifen for Cre activation and intratracheal bleomycin to induce lung injury, and lung sections were prepared in the fibrotic period, at 14 days. The two photos show the same microscopic field, with (right) and without (left) SiglecF (Sialic acid binding Ig-like lectin F) fluorescence. TdTomato+ monocyte-derived macrophages (moMacs) localize to Pdgfrb+ (Platelet-derived growth factor receptor beta) fibroblasts in the fibrotic niche, expressing some level of the resident macrophage marker SiglecF. Alveolar macrophages, which express SiglecF but not the Cx3cr1 marker TdTomato, lie outside the fibrotic niche. Scale bar, 50 mm. Reprinted by permission from Reference 25.
Figure 2.

Idiopathic pulmonary fibrosis (IPF) lung immunofluorescence for healthy and fibrotic regions indicated by second harmonic signal for collagen (SH) from the same sample. The transcription factor and moMac marker MAF BZIP transcription factor B (MAFB) was detected in CD68+ macrophages in fibrotic regions but not in healthy lung. Scale bar, 50 mm. Reprinted by permission from Reference 25.
Fibrosis in COVID-19
Patients who develop COVID-19 pneumonia and require mechanical ventilation for hypoxemia nearly all have acute respiratory distress syndrome (ARDS), a syndromic descriptor for hypoxemia and respiratory failure from widespread lung edema after disruption of the alveolocapillary barrier (32). Since the early phase of the pandemic, a remarkable feature of COVID-19 has been the prolonged duration of mechanical ventilation compared with patients with non–COVID-19 causes of ARDS (33). Many patients with severe ARDS from COVID-19 develop nonresolving impairment in lung function over the course of weeks to months, and some require lung transplant (34). Computed tomography (CT) scans have revealed parenchymal evolution through the course of the illness, with reticular opacifications consistent with fibrosis found in up to 21% of patients after >3 weeks of illness (35, 36). Autopsy studies have provided confirmation of fibroproliferative remodeling in lethal COVID-19. Several series have reported a high prevalence of diffuse or focal organizing pneumonia, with clusters of activated fibroblasts alongside regions of diffuse alveolar damage, a finding that correlated with duration of illness (37–39).
Patients discharged from the hospital continue to have lung dysfunction and CT findings consistent with fibrotic lung disease for months (40, 41). Fibrotic changes resolve in the majority of patients by 1 year. For example, in one study, a single-center cohort of 61 patients followed after discharge after COVID pneumonia requiring mechanical ventilation (42), CT scans were performed for 36 patients at 1 year: only 4 had fibrotic changes. However, of the 36, 29 also had CT scans earlier in their course, at 3 months, with 8 showing fibrotic change. This pattern of resolution across time is consistent with a gradual recovery of normal lung architecture as part of the tissue wounding and ensuing healing response. Nonetheless, it indicates a significant burden of fibrotic disease in the acute to subacute phase, lasting several months after infection, which contributes to the prolonged requirement for mechanical ventilation and ICU stay, known risk factors for life-threatening complications, including ventilator-associated pneumonia, ICU delirium, and mortality (43–45).
The fibrotic reaction in COVID-19 ARDS can be viewed as analogous to the wound-healing response of sterile injury induced by bleomycin. The pathophysiology of the fibrosis observed in the bleomycin model is in fact similar enough to IPF that the model has been used for development of antifibrotic therapies (26), although its natural history differs from IPF in that it naturally resolves over time (similar to lung fibroproliferative responses induced by COVID-19). Recent analyses at the single-cell level have revealed common parenchymal and immune cellular populations across COVID, IPF, and murine lung fibrosis. For example, the epithelial senescence phenotype appears to be a common dysfunctional cellular state present in both bleomycin injury and IPF—in particular, Krt8+ (Keratin 8–positive) cells with expression of senescence markers (16). These cells are the predominant population expressing αvβ6, an integrin that is essential for TGF-β activation and for fibrosis (18). Interestingly, comparative analyses of scRNAseq data have also revealed a similarity of transcriptomic profiles between lung fibrosis and COVID-19 pneumonia. Bharat and colleagues (46) performed scRNAseq on explanted COVID-19 and IPF lungs acquired at the time of transplantation and found that both IPF and COVID-19 lungs were notable for a marked increase in a Krt8+ epithelial senescent cell population compared with healthy controls, and the epithelial senescence phenotype was confirmed by a subsequent study (39).
Macrophage Transcriptomic Identity in COVID-19
With respect to the immune compartment within the lung, early on in the pandemic Liao and colleagues found that patients with severe COVID-19 had a higher proportion of myeloid cells in the lung lavage than mild cases (47). Subsequently, longitudinal sampling of the airway in intubated patients by Szabo and colleagues revealed an association between airway aspirate myeloid cells and death from COVID-19 (48). An intriguing result from this paper was that the proportion of myeloid cells was markedly higher in aged patients—a potential clue to worse outcomes in the elderly. Several scRNAseq studies of lung cells from COVID-19 found a marked heterogeneity in the macrophage compartment, with multiple clusters detected (47–51). Notably, these reports revealed a predominant moMac ontogeny in the expanded lung myeloid compartment, similar to the bleomycin model and to IPF, whether cells were isolated by lavage or by tissue dissociation postmortem.
CD163 was a consistent marker of moMacs in these studies, and coexpression of a wide range of inflammatory chemokines was also a common feature. Trajectory analysis suggested that lung cells expressing CD163 had differentiated from the monocyte pool (51). Furthermore, monocyte progenitors expressing CD163 were increased in the peripheral blood in patients with COVID-19 (48), and the presence of these CD163+ peripheral blood monocytes was associated with severe disease (52). In a recent breakthrough that shed light on a common profibrotic moMac profile, direct transcriptomic comparison by Wendisch and colleagues revealed that this CD163+ moMac compartment overlapped with similar clusters detected in multiple studies of IPF lungs and not with macrophages found in healthy lungs (53). Furthermore, compared with IPF, much less overlap was detected between COVID-19 lung macrophages and lung macrophages from patients with chronic obstructive pulmonary disease. In animal models of both sterile injury–induced fibrosis and of severe acute respiratory syndrome (SARS) and SARS–coronavirus 2 (SARS-CoV-2) infection, CD163+ macrophages have likewise been prominent (54–56). In several studies, COVID-19 macrophages were found to express genes associated with fibrosis, including TGFβ1, Secreted Phosphoprotein 1 (SPP1), and CCL18; importantly, CD163+ cells colocalized with activated fibroblasts, reminiscent of the fibrosis models and IPF samples discussed above (Figure 3), and had greater proximity to areas of collagen accumulation than CD163− macrophages (53). Furthermore, analysis of a mass cytometry comparing COVID-19 and control lung monocytes and macrophages (57, 58) revealed higher concentrations in COVID-19 samples of IL1β, a known profibrotic and prosenescence factor in lung injury (59, 60).
Figure 3.
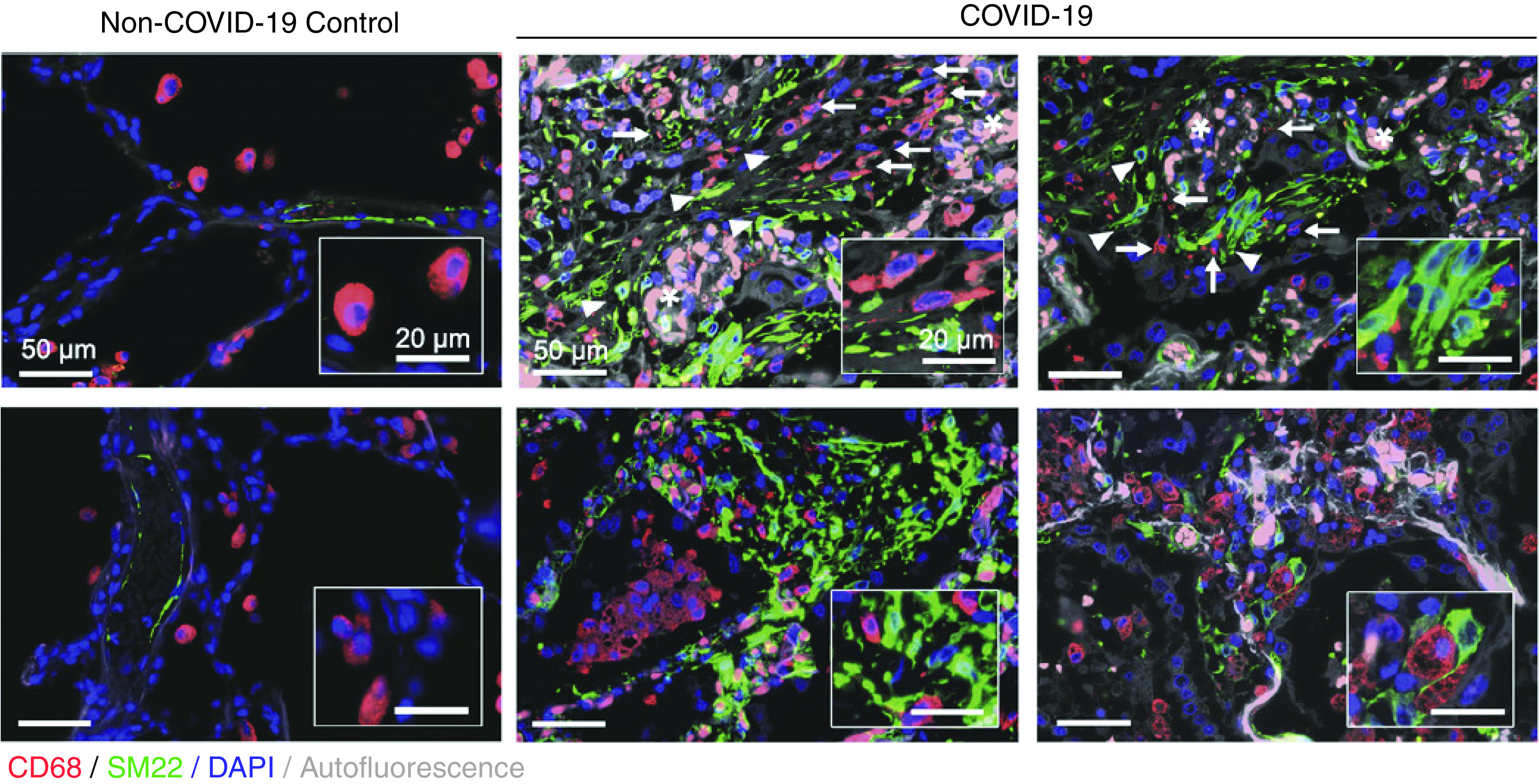
Coronavirus disease (COVID-19) pneumonia samples acquired at autopsy showing localization of macrophages (CD68) in the fibrotic niche in proximity to mesenchymal cells (SM22), likely activated fibroblasts or myofibroblasts, akin to noninfectious causes of lung fibrosis such as bleomycin-induced fibrosis and IPF. Arrows indicate macrophages, arrowheads indicate expanded SM22 foci, and asterisks denote erythrocyte-filled capillaries in alveolar septa. Reprinted by permission from Reference 53.
Taken together, these results suggest that CD163+ cells, by localizing to the fibrotic niche and expressing profibrotic factors, may directly activate the mesenchyme and induce the fibroproliferative response seen in COVID-19 infection (Figure 4).
Figure 4.
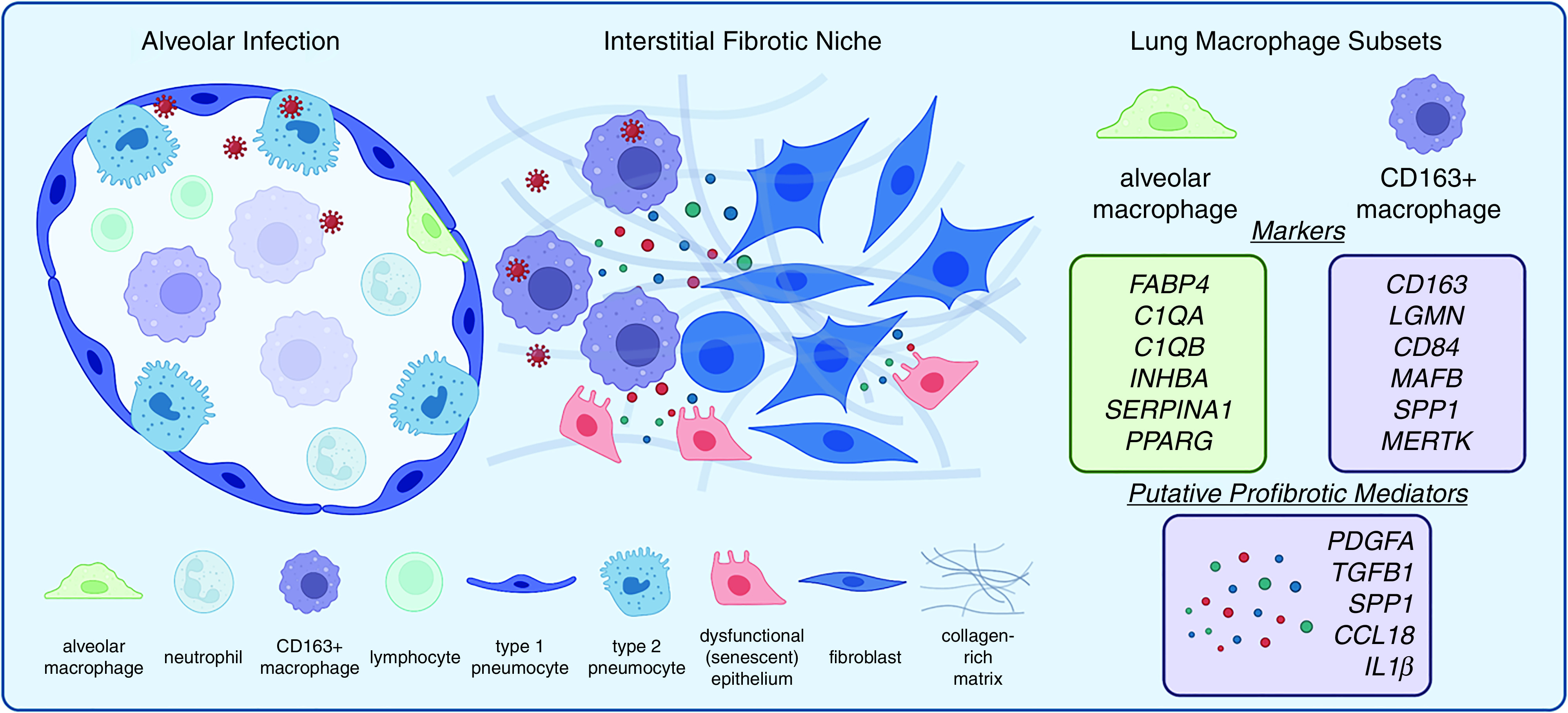
Fibrotic progression in COVID-19. Left: Alveolitis due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is notable for infection of both myeloid and epithelial cells. Both compartments evolve in response to infection, with a senescence response in epithelial cells and with macrophage polarization including CD163+ expression, resulting in profibrotic cell-to-cell interactions in an expanding interstitial fibrotic niche. Right: Markers of two major lung macrophage subtypes in health and fibrotic diseases (20, 29, 30, 47–51, 53) and COVID-19 CD163+ moMac-associated secreted factors with fibrogenic potential based on experimental and clinical studies of idiopathic pulmonary fibrosis (IPF) (25, 27, 30, 57, 74, 77).
Drivers of Macrophage Polarization
The growing literature on CD163+ moMacs is highly suggestive of a profibrotic polarization, but how conditions within the COVID-19–infected lung might induce this transcriptomic polarization remains incompletely understood. Traditionally, an M1–M2 paradigm has been used to characterize macrophage polarization states, with M2 macrophages being implicated in the profibrotic state observed in the context of fibrosis at many tissular sites (61). However, with the advent of single-cell sequencing and the ability to characterize transcriptomes at a more granular level, a much greater heterogeneity has been appreciated, and the common transcriptomic profile observed for moMacs in both COVID-19 and IPF provides a detailed, marker-based fingerprint. Therefore, the question arises, could viral infection itself induce the gene expression profile observed? Alternatively, one possibility is that, rather than an effect of viral infection, monocytic ontogeny (i.e., the derivation from monocytes) may itself render an inherent profibrotic transcriptomic state. This possibility would be difficult to rule out. However, local factors in the tissue can also play an important role in driving the functional phenotype of macrophages in fibrosis, depending on the model. For example, helminthic infections induce macrophage-mediated maintenance of type 2 immunity and recruitment of IL13-secreting T cells, a sequence that has been found to be necessary for fibrosis (62). In fact, this pathobiology may be relevant to COVID-19, where type 2 cytokines including IL13 have been found to be upregulated, and retrospective analyses have shown protection in patients who received IL13 blockade with dupilumab (63). Whether these phenomena are macrophage dependent is as yet unknown.
Nonetheless, viral pneumonias present a circumstance where host cell sensing of the virus itself could provide a mechanism for transcriptomic polarization of macrophages, given that viral nucleic acids induce host responses though the action of innate sensing pathways, including the TLRs (Toll-like receptors) TLR3, TLR7/8, TLR9, and other nucleic acid sensors such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5). Therefore, understanding how macrophage gene expression is modified by SARS-CoV-2 infection is an important direction for ongoing and future research. Interestingly, TLR expression has been found to increase in samples from patients with IPF (64), and TLR3 polymorphism has been associated with disease progression (65, 66). Whether these findings reflect an interaction between aberrant responses to viral infection and disease progression in IPF represents an intriguing possibility but is unknown.
Viral entry in most cells is dependent on the expression of the cell membrane receptor angiotensin-converting enzyme 2 (ACE2) and is enhanced by the transmembrane protease transmembrane serine protease 2 (TMRPSS2) (67). scRNAseq analysis enables detection of viral transcripts as well as their correlation to specific cell types. Unsurprisingly, both the native positive strand and the replication-intermediate negative strand mRNA have been found abundantly in epithelial cells of both the upper and lower respiratory tract in multiple studies. Reanalyzing BAL samples sequenced by scRNAseq (47) with a focus on viral reads, Bost and colleagues found that SARS-CoV-2 transcripts were also detectable within macrophages (68); in a subsequent study, analysis of autopsy lung samples from patients with COVID-19, with comprehensive profiling of all cell types, revealed that myeloid cells bore the largest burden of SARS-CoV-2 reads (69). Moreover, negative strand mRNA reads, indicating some level of replication, have been detected within some subclusters of macrophages, including both tissue resident macrophages and moMacs (49). Entry mechanisms may differ by macrophage ontogeny. In monocyte-derived cells, a recently described Fc gamma receptor (FcγR)-mediated internalization of spike antibody–opsonized virus was found to be important (70); on the other hand, alveolar macrophages were infected in vitro in a partially ACE2-dependent manner (71). Whether infected lung macrophages produce newly synthesized viral particles—so-called productive versus abortive infection—is a matter of some debate and may relate to macrophage lineage. Several in vitro studies with monocytes or moMacs demonstrated infection in vitro without being able to find evidence of productive infection (70, 72, 73). However, alveolar macrophages isolated by lavage demonstrated productive infection (71).
In any case, infection of macrophages is likely to be important for clinical outcomes because of the polarization effect on macrophages—the skewing of gene expression toward an inflammatory and profibrotic profile. Remarkably, scRNAseq analysis supported this idea: patients with severe disease not only had a higher proportion of the SPP1+ moMacs discussed above but these cells also had the highest number of SARS-CoV-2 mRNA reads (68). Importantly, viral read–positive SPP1+ cells, compared with read-negative SPP1+ cells, had a higher number of inflammatory chemokines such as CCL7, CCL8, and CCL18. Notably, lung macrophage CCL18 has been associated with disease progression in lung fibrosis (74). To directly address the question of whether viral sensing by infected myeloid cells induces the CD163+ moMac gene expression profile detected in patient samples, Wendisch and colleagues measured gene expression by RNAseq after in vitro infection with SARS-CoV-2 (53).
In their experiment, CD14+ CD16− classical monocytes were isolated from peripheral blood of healthy human donors and stimulated with SARS-CoV-2 or with agonists for multiple nucleic acid sensors, including RIG-I and MDA5 (3p-hpRNA) and TLR7/8 (R848). Remarkably, compared with the agonist-treated and unstimulated control cells, SARS-CoV-2 infection increased genes discussed above that were detected in the profibrotic macrophage subcluster common to both IPF and COVID-19 lungs, including CD163, MRC1, TGFBI, MMP9, MERTK, and LGMN; at the genome-wide level, there was a statistically significant increase for the SARS-CoV-2–treated cells relative to the controls of overlap with a recently reported IPF macrophage profile (75). In a further proteomic validation of these RNAseq data, influenza A virus was used as the comparator for SARS-CoV-2 infection. Comparisons of proteins detected with infection with SARS-CoV-2 but not influenza A virus bore statistically significant similarity to published IPF macrophage expression profiles; interestingly, phospho-proteomic analysis confirmed phosphorylation (indicative of activation) of CCAAT Enhancer Binding Protein Beta (CEBPB), a transcription factor associated with the profibrotic function of macrophages in the bleomycin model (24). Taken together, these experiments revealed a profibrotic polarizing effect of direct infection of monocytes with SARS-CoV-2 and clear the path for future work to determine mechanisms of induction of the profibrotic state by the virus, to reveal potentially druggable pathways.
Conclusions and Perspectives on Future Research
Interactions between macrophages and fibroblasts have received increasing attention as a fundamental feature of tissue patterning in both health and disease. This crosstalk is notable for a paracrine interdependence, wherein macrophages and fibroblasts achieve a numerical, 1:1 steady-state ratio based on mutual trophism, with colony stimulating factor 1 (CSF1) secretion by fibroblasts supporting macrophage growth and PDGFs secreted by macrophages supporting fibroblasts (76). These specific factors have been shown to be relevant to macrophage–fibroblast interactions in the fibrotic niche, where disruption of normal anatomic boundaries of the alveolar and interstitial spaces and expansion of a morphologically simple wound bed expand the opportunity for unrestricted paracrine signaling–based interdependence (25, 77).
Although studies with animal models focused on this mechanistic role of macrophage–fibroblast crosstalk have so far not been reported for SARS-CoV-2 as they have for bleomycin and other sterile injury models, given the similarity in gene expression profile of moMacs in patients with IPF and COVID-19, a working hypothesis has now distinctly emerged that the fibrotic reaction seen in COVID-19 is due at least in part to the effects of CD163+ moMacs on the mesenchyme. Furthermore, the results of Wendisch and colleagues (53), together with multiple reports finding SARS-CoV-2 mRNA in macrophages in samples from patients, indicate that the profibrotic polarization of moMacs in COVID-19 is directly induced by the response to viral internalization in these cells. Future studies in experimental models of gene function focused on CD163+ moMacs (and adjacent fibroblasts) should be able to test individual factors, their upstream regulators, and effector pathways in regard to the profibrotic effects observed. In this respect, the recent model of COVID-19 infection reported by Sefik and colleagues in which hematopoietically humanized mice recapitulated both moMac infiltration and a prominent late fibrotic phase holds promise for testing specific macrophage-dependent pathways (78).
Much progress in recent years has been made in understanding the myeloid and specifically moMac contribution to fibrosis in multiple settings. In fibrosing lung injury models, deletion or reconstitution of moMacs has proven their profibrotic role. Importantly, patient samples have revealed a similar profile of gene expression between IPF and severe COVID-19 pneumonia. In COVID-19, studies specifically deleting macrophage-derived factors or macrophages themselves in experimental models are much needed. The translational relevance is that the fibroproliferative response to severe SARS-CoV-2 infection impairs gas exchange and prolongs mechanical ventilation, putting patients at risk for death; thus, approaches that seek to reverse fibrotic remodeling induced by the observed profibrotic polarizing effect of SARS-CoV-2 infection on macrophages are urgently needed. These insights could lead to the development of novel therapeutics. For example, recently, a drug screening approach was used in cultured lung slices for IPF and focused on the profibrotic function of macrophages (75). A similar approach could be taken for COVID-19. Questions that remain unaddressed and will be illumined by future work include the role of macrophage heterogeneity in the fibrotic process, including, specifically, non-CD163+ cells, and how aging of the lung influences the recruitment and profibrotic function of moMacs.
Acknowledgments
Acknowledgment
Figure 4 was created under license with BioRender.com.
Footnotes
Supported by the UCSF Division of Pulmonary, Critical Care, Allergy, and Sleep Medicine and by the UCSF Sandler Asthma Basic Research Center.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0107TR on June 8, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity . 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tan SY, Krasnow MA. Developmental origin of lung macrophage diversity. Development . 2016;143:1318–1327. doi: 10.1242/dev.129122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med . 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature . 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, et al. Pulmonary macrophage transplantation therapy. Nature . 2014;514:450–454. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature . 2014;506:503–506. doi: 10.1038/nature12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neupane AS, Willson M, Chojnacki AK, Vargas E Silva Castanheira F, Morehouse C, Carestia A, et al. Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell . 2020;183:110–125.e11. doi: 10.1016/j.cell.2020.08.020. [DOI] [PubMed] [Google Scholar]
- 8. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol . 2017;57:66–76. doi: 10.1165/rcmb.2016-0361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science . 2019;363:eaau0964. doi: 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 10. Bain CC, MacDonald AS. The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol . 2022;15:223–234. doi: 10.1038/s41385-021-00480-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis An official ATS/ERS/JRS/ALAT statement. Idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med . 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moss BJ, Ryter SW, Rosas IO. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev Pathol . 2022;17:515–546. doi: 10.1146/annurev-pathol-042320-030240. [DOI] [PubMed] [Google Scholar]
- 13. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun . 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med . 2021;203:707–717. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schuliga M, Kanwal A, Read J, Blokland KEC, Burgess JK, Prêle CM, et al. A cGAS-dependent response links DNA damage and senescence in alveolar epithelial cells: a potential drug target in IPF. Am J Physiol Lung Cell Mol Physiol . 2021;321:L859–L871. doi: 10.1152/ajplung.00574.2020. [DOI] [PubMed] [Google Scholar]
- 16. Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun . 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Courtwright AM, El-Chemaly S. Telomeres in interstitial lung disease: the short and the long of it. Ann Am Thorac Soc . 2019;16:175–181. doi: 10.1513/AnnalsATS.201808-508CME. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell . 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 19. John AE, Graves RH, Pun KT, Vitulli G, Forty EJ, Mercer PF, et al. Translational pharmacology of an inhaled small molecule αvβ6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat Commun . 2020;11:4659. doi: 10.1038/s41467-020-18397-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv . 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nathan SD, Albera C, Bradford WZ, Costabel U, Glaspole I, Glassberg MK, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med . 2017;5:33–41. doi: 10.1016/S2213-2600(16)30326-5. [DOI] [PubMed] [Google Scholar]
- 22. Richeldi L, Cottin V, du Bois RM, Selman M, Kimura T, Bailes Z, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS trials. Respir Med . 2016;113:74–79. doi: 10.1016/j.rmed.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 23. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med . 2017;214:2387–2404. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Satoh T, Nakagawa K, Sugihara F, Kuwahara R, Ashihara M, Yamane F, et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature . 2017;541:96–101. doi: 10.1038/nature20611. [DOI] [PubMed] [Google Scholar]
- 25. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol . 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Königshoff M, Kolb M, et al. ATS Assembly on Respiratory Cell and Molecular Biology An official American Thoracic Society workshop report: use of animal models for the preclinical assessment of potential therapies for pulmonary fibrosis. Am J Respir Cell Mol Biol . 2017;56:667–679. doi: 10.1165/rcmb.2017-0096ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity . 2016;44:582–596. doi: 10.1016/j.immuni.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cosin-Roger J, Ortiz-Masià MD, Barrachina MD. Macrophages as an emerging source of Wnt ligands: relevance in mucosal integrity. Front Immunol . 2019;10:2297. doi: 10.3389/fimmu.2019.02297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med . 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J . 2019;54:1802441. doi: 10.1183/13993003.02441-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soucie EL, Weng Z, Geirsdóttir L, Molawi K, Maurizio J, Fenouil R, et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science . 2016;351:aad5510. doi: 10.1126/science.aad5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med . 2017;377:562–572. doi: 10.1056/NEJMra1608077. [DOI] [PubMed] [Google Scholar]
- 33. Bain W, Yang H, Shah FA, Suber T, Drohan C, Al-Yousif N, et al. COVID-19 versus non-COVID-19 acute respiratory distress syndrome: comparison of demographics, physiologic parameters, inflammatory biomarkers, and clinical outcomes. Ann Am Thorac Soc . 2021;18:1202–1210. doi: 10.1513/AnnalsATS.202008-1026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roach A, Chikwe J, Catarino P, Rampolla R, Noble PW, Megna D, et al. Lung transplantation for Covid-19-related respiratory failure in the United States. N Engl J Med . 2022;386:1187–1188. doi: 10.1056/NEJMc2117024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y, Dong C, Hu Y, Li C, Ren Q, Zhang X, et al. Temporal changes of CT findings in 90 patients with COVID-19 pneumonia: a longitudinal study. Radiology . 2020;296:E55–E64. doi: 10.1148/radiol.2020200843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jalaber C, Lapotre T, Morcet-Delattre T, Ribet F, Jouneau S, Lederlin M. Chest CT in COVID-19 pneumonia: a review of current knowledge. Diagn Interv Imaging . 2020;101:431–437. doi: 10.1016/j.diii.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Valdebenito S, Bessis S, Annane D, Lorin de la Grandmaison G, Cramer-Bordé E, Prideaux B, et al. COVID-19 lung pathogenesis in SARS-CoV-2 autopsy cases. Front Immunol . 2021;12:735922. doi: 10.3389/fimmu.2021.735922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borczuk AC, Salvatore SP, Seshan SV, Patel SS, Bussel JB, Mostyka M, et al. COVID-19 pulmonary pathology: a multi-institutional autopsy cohort from Italy and New York City. Mod Pathol . 2020;33:2156–2168. doi: 10.1038/s41379-020-00661-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. D’Agnillo F, Walters KA, Xiao Y, Sheng ZM, Scherler K, Park J, et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci Transl Med . 2021;13:eabj7790. doi: 10.1126/scitranslmed.abj7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mumoli N, Bonaventura A, Colombo A, Vecchié A, Cei M, Vitale J, et al. Lung function and symptoms in post-COVID-19 patients: a single-center experience. Mayo Clin Proc Innov Qual Outcomes . 2021;5:907–915. doi: 10.1016/j.mayocpiqo.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stockley JA, Alhuthail EA, Coney AM, Parekh D, Geberhiwot T, Gautum N, et al. Lung function and breathing patterns in hospitalised COVID-19 survivors: a review of post-COVID-19 Clinics. Respir Res . 2021;22:255. doi: 10.1186/s12931-021-01834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zangrillo A, Belletti A, Palumbo D, Calvi MR, Guzzo F, Fominskiy EV, et al. COVID-BioB Study Group One-year multidisciplinary follow-up of patients with COVID-19 requiring invasive mechanical ventilation. J Cardiothorac Vasc Anesth . 2022;36:1354–1363. doi: 10.1053/j.jvca.2021.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moitra VK, Guerra C, Linde-Zwirble WT, Wunsch H. Relationship between ICU length of stay and long-term mortality for elderly ICU survivors. Crit Care Med . 2016;44:655–662. doi: 10.1097/CCM.0000000000001480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fiest KM, Soo A, Hee Lee C, Niven DJ, Ely EW, Doig CJ, et al. Long-term outcomes in ICU patients with delirium: a population-based cohort study. Am J Respir Crit Care Med . 2021;204:412–420. doi: 10.1164/rccm.202002-0320OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu D, Wu C, Zhang S, Zhong Y. Risk factors of ventilator-associated pneumonia in critically iii patients. Front Pharmacol . 2019;10:482. doi: 10.3389/fphar.2019.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bharat A, Querrey M, Markov NS, Kim S, Kurihara C, Garza-Castillon R, et al. Lung transplantation for patients with severe COVID-19. Sci Transl Med . 2020;12:eabe4282. doi: 10.1126/scitranslmed.abe4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med . 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 48. Szabo PA, Dogra P, Gray JI, Wells SB, Connors TJ, Weisberg SP, et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity . 2021;54:797–814.e6. doi: 10.1016/j.immuni.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, et al. NU SCRIPT Study Investigators Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature . 2021;590:635–641. doi: 10.1038/s41586-020-03148-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol . 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 51. Wauters E, Van Mol P, Garg AD, Jansen S, Van Herck Y, Vanderbeke L, et al. CONTAGIOUS collaborators Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res . 2021;31:272–290. doi: 10.1038/s41422-020-00455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell . 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wendisch D, Dietrich O, Mari T, von Stillfried S, Ibarra IL, Mittermaier M, et al. Deutsche COVID-19 OMICS Initiative (DeCOI) SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell . 2021;184:6243–6261.e27. doi: 10.1016/j.cell.2021.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Speranza E, Williamson BN, Feldmann F, Sturdevant GL, Pérez-Pérez L, Meade-White K, et al. Single-cell RNA sequencing reveals SARS-CoV-2 infection dynamics in lungs of African green monkeys. Sci Transl Med . 2021;13:eabe8146. doi: 10.1126/scitranslmed.abe8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gu H, Chen Q, Yang G, He L, Fan H, Deng YQ, et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science . 2020;369:1603–1607. doi: 10.1126/science.abc4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rawle DJ, Le TT, Dumenil T, Yan K, Tang B, Nguyen W, et al. ACE2-lentiviral transduction enables mouse SARS-CoV-2 infection and mapping of receptor interactions. PLoS Pathog . 2021;17:e1009723. doi: 10.1371/journal.ppat.1009723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, et al. A molecular single-cell lung atlas of lethal COVID-19. Nature . 2021;595:114–119. doi: 10.1038/s41586-021-03569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rendeiro AF, Ravichandran H, Bram Y, Chandar V, Kim J, Meydan C, et al. The spatial landscape of lung pathology during COVID-19 progression. Nature . 2021;593:564–569. doi: 10.1038/s41586-021-03475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, et al. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell . 2020;27:366–382.e7. doi: 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest . 2007;117:3786–3799. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity . 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Borthwick LA, Barron L, Hart KM, Vannella KM, Thompson RW, Oland S, et al. Macrophages are critical to the maintenance of IL-13-dependent lung inflammation and fibrosis. Mucosal Immunol . 2016;9:38–55. doi: 10.1038/mi.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Donlan AN, Sutherland TE, Marie C, Preissner S, Bradley BT, Carpenter RM, et al. IL-13 is a driver of COVID-19 severity. JCI Insight . 2021;6:e150107. doi: 10.1172/jci.insight.150107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Margaritopoulos GA, Antoniou KM, Karagiannis K, Samara KD, Lasithiotaki I, Vassalou E, et al. Investigation of Toll-like receptors in the pathogenesis of fibrotic and granulomatous disorders: a bronchoalveolar lavage study. Fibrogenesis Tissue Repair . 2010;3:20. doi: 10.1186/1755-1536-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McElroy AN, Invernizzi R, Laskowska JW, O’Neill A, Doroudian M, Moghoofei M, et al. Candidate role for Toll-like receptor 3 L412F polymorphism and infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med . 2022;205:550–562. doi: 10.1164/rccm.202010-3880OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O’Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, et al. The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med . 2013;188:1442–1450. doi: 10.1164/rccm.201304-0760OC. [DOI] [PubMed] [Google Scholar]
- 67. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol . 2022;23:3–20. doi: 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell . 2020;181:1475–1488.e12. doi: 10.1016/j.cell.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature . 2021;595:107–113. doi: 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Junqueira C, Crespo Â, Ranjbar S, de Lacerda LB, Lewandrowski M, Ingber J, et al. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature . 2022 doi: 10.1038/s41586-022-04702-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Magnen M, You R, Rao AA, Davis RT, Rodriguez L, Simoneau CR, et al. Immediate myeloid depot for SARS-CoV-2 in the human lung [preprint] bioRxiv . 2022 doi: 10.1126/sciadv.adm8836. https://www.biorxiv.org/content/10.1101/2022.04.28.489942v2 [DOI] [PMC free article] [PubMed]
- 72. Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC, et al. Attenuated interferon and proinflammatory response in SARS-CoV-2-infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis . 2020;222:734–745. doi: 10.1093/infdis/jiaa356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Boumaza A, Gay L, Mezouar S, Bestion E, Diallo AB, Michel M, et al. Monocytes and macrophages, targets of severe acute respiratory syndrome coronavirus 2: the clue for coronavirus disease 2019 immunoparalysis. J Infect Dis . 2021;224:395–406. doi: 10.1093/infdis/jiab044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med . 2006;173:781–792. doi: 10.1164/rccm.200509-1518OC. [DOI] [PubMed] [Google Scholar]
- 75.Ayaub E, Poli S, Ng J, Adams T, Schupp J, Quesada-Arias L, et al. Single cell RNA-seq and mass cytometry reveals a novel and a targetable population of macrophages in idiopathic pulmonary fibrosis [preprint] bioRxiv 2021https://www.biorxiv.org/content/10.1101/2021.01.04.425268v1.
- 76. Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, et al. Circuit design features of a stable two-cell system. Cell . 2018;172:744–757.e17. doi: 10.1016/j.cell.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bhattacharyya A, Boostanpour K, Bouzidi M, Magee L, Chen TY, Wolters R, et al. IL10 trains macrophage profibrotic function after lung injury. Am J Physiol Lung Cell Mol Physiol . 2022;322:L495–L502. doi: 10.1152/ajplung.00458.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sefik E, Israelow B, Mirza H, Zhao J, Qu R, Kaffe E, et al. A humanized mouse model of chronic COVID-19. Nat Biotechnol . 2021 doi: 10.1038/s41587-021-01155-4. [DOI] [PMC free article] [PubMed] [Google Scholar]