

Abstract
G protein–coupled receptors (GPCRs) not only are turned on or off to control canonical G protein signaling but also may be fine-tuned to promote qualitative/biased signaling. Qualitative signaling by M3 muscarinic acetylcholine receptors (mAChRs) has been proposed, but its impact on physiologic systems remains unclear, and currently no biased M3 mAChR ligands have been described. Herein, we identify PD 102807 as a biased M3 ligand and delineate its signaling and function in human airway smooth muscle (ASM) cells. PD 102807 induced M3-mediated β-arrestin recruitment but not calcium mobilization. PD 102807 inhibited methacholine (MCh)-induced calcium mobilization in (M3-expressing) ASM cells. PD 102807 induced phosphorylation of AMP-activated protein kinase (AMPK) and the downstream effector acetyl–coenzyme A carboxylase (ACC). PD 102807– induced phosphorylated (p)-AMPK levels were greatly reduced in ASM cells with minimal M3 expression and were not inhibited by the Gq inhibitor YM-254890. Induction of p-AMPK and p-ACC was inhibited by β-arrestin 1 or GRK2/3 knockdown. Similarly, MCh induced phosphorylation of AMPK/ACC, but these effects were Gq dependent and unaffected by GRK2/3 knockdown. Consistent with the known ability of AMPK to inhibit transforming growth factor β (TGF-β)–mediated functions, PD 102807 inhibited TGF-β–induced SMAD-Luc activity, sm-α-actin expression, actin stress fiber formation, and ASM cell hypercontractility. These findings reveal that PD 102807 is a biased M3 ligand that inhibits M3-transduced Gq signaling but promotes Gq protein-independent, GRK-/arrestin-dependent, M3-mediated AMPK signaling, which in turn regulates ASM phenotype and contractile function. Consequently, biased M3 ligands hold significant promise as therapeutic agents capable of exploiting the pleiotropic nature of M3 signaling.
Keywords: acetylcholine, asthma, COPD, biased signaling
Early G protein–coupled receptor (GPCR) dogma posits that the receptor active state varies only in the degree of receptor activation and generation of its second messenger. It is now appreciated that GPCRs may undergo many different conformations to promote various forms of signaling, sometimes not related to the traditional G protein coupling and second messenger generation. Qualitative signaling/biased agonism concepts hold that many receptor conformations exist. Rather than just being turned on or off, GPCRs can be fine-tuned to achieve various signaling outcomes as opposed to simply effecting a different magnitude of a canonical second messenger signal (1–4). In this context, other proteins, such as arrestins, have been proposed to act as signaling molecules.
GPCR kinases (GRKs) and arrestins were originally identified as mediators of receptor desensitization and internalization (5, 6). Briefly, the active (ligand-bound) form of a GPCR is phosphorylated by GRKs, which allows arrestins to bind to the receptor and subsequently desensitize the receptor by means of steric hindrance of G protein binding and internalization of the receptor by means of clathrin-coated pits (7). GRK-mediated phosphorylation of GPCRs is not only important for arrestin recruitment but may also impart further signaling specificity by means of a “barcode” of phosphoresidues in the C terminus of GPCRs (8). More recently, the GRK-arrestin axis has been proposed as a mediator of biased signaling by GPCRs, with arrestins serving as scaffolds for initiating G protein–independent signaling (9, 10).
Biased signaling has been most widely studied for β-adrenoceptors (β-ARs). In the cardiac system, β-AR–mediated arrestin signaling improves heart function (11, 12). Arrestin-biased β-2 adrenoreceptor signaling has been identified in the lung, where it promotes asthma pathology, including airway hyperresponsiveness and mucus production (13–16). Furthermore, biased signaling has now been established for a growing number of receptors, including angiotensin II type 1 receptor, mu and kappa opioid receptors, dopamine 2 receptors, serotonergic receptors (5-HT2BRs), adenosine receptors, and others (17). The emerging breadth of biased GPCR signaling suggests potentially wide-ranging effects on the regulation of (patho-)physiological processes. However, to date, many ligands have only been screened with respect to their ability to stimulate or inhibit second-messenger induction; thus, the full extent of their signaling capabilities—including whether such signaling is skewed toward a given pathway or whether it is G protein dependent or independent—is unknown.
M3 muscarinic acetylcholine receptors (mAChRs) are key regulators of airway physiology; they are the major mediators of (physiologic and pathophysiologic) airway smooth muscle (ASM) contraction and airway mucus production. In addition, M3 mAChRs have been identified as major drivers of pathology in asthma and chronic obstructive pulmonary disease, contributing to airway obstruction (18), structural changes in the airway wall (airway remodeling) (19–21) and inflammation (19, 21, 22). M3 mAChRs also exhibit cooperativity with growth factors, including transforming growth factor β (TGF-β), to promote airway pathology (23, 24). The importance of M3 mAChRs is evidenced by the effectiveness of inhaled mAChR antagonists (anticholinergics) as treatment for airway obstruction in chronic obstructive pulmonary disease (25) and asthma (26). M3 mAChRs are Gq-coupled GPCRs whose canonical signaling leads to calcium mobilization, resulting in ASM contraction and airway obstruction. Currently, studies into potential Gq-independent M3 mAChR signaling have been hampered by the lack of biased M3 ligands (27–29). In this study, we identify PD 102807—a previously described muscarinic antagonist with selectivity for the M4 mAChR—as a biased M3 mAChR ligand that induces arrestin recruitment and activates AMP-activated protein kinase (AMPK) signaling in a GRK-arrestin– dependent manner while inhibiting Gq protein–mediated signaling in ASM cells. Some of the results of these studies have been previously reported in the form of an abstract (30).
Methods
Reagents
A list of reagents used is available (see data supplement).
Cell Culture
Human ASM cells stably expressing telomerase reverse transcriptase (hTERT) with physiological/high expression of M3 mAChR (two cell lines) and with low M3 mAChR expression (two cell lines) were provided by Dr. W. T. Gerthoffer and have been previously described in detail (31). Detailed methods are available in the data supplement.
siRNA-Mediated Knockdown
siRNA ON-TARGETplus SMARTpool oligos directed against β-arrestin-1 (L-011971) or β-arrestin-2 (L-007292) were purchased from Dharmacon. GRK2/3 (5′-GAT CTT CGA CTC ATA CAT CTT-3′) siRNA oligos were annealed at 95°C for 5 min and allowed to cool; ASM cells were transfected using Dharmafect 1 (Dharmacon). Mock-transfected cells were treated with Dharmafect 1 without oligos. Detailed methods are available in the data supplement.
β-Arrestin Recruitment
β-arrestin recruitment was determined as described previously by Carr and colleagues (32). HEK293 cells were transfected with pcDNA3-M3-RLucII and pcDNA3-βarrestin2-GFP10 using Fugene (Promega). Cells were incubated with coelenterazine 400a and stimulated with mAChR ligands. Bioluminescence resonance energy transfer (BRET) was measured using a Tecan Infinite F500 microplate reader. Detailed methods are available in the data supplement.
Intracellular Calcium Measurements, Immunoblotting, and M3 mAChR Heterologous Expression
Intracellular calcium measurements were performed using Fluo-4 AM and Flexstation, as described previously (33). Immunoblotting was performed using standard methods. For heterologous M3 mAChR expression, low M3–expressing cells were transfected with lentiviral particles from Origene (control: PS100064V; M3 mAChR: RC212436L1V), according to the manufacturer’s instructions. Detailed methods are available in the data supplement.
Luciferase Reporter Assay
Stable expression of a reporter construct for SMAD-Luc was established in hTERT ASM cells using Cignal Lenti luciferase reporter viral particles (SA Biosciences). Cells were stimulated for 6 hours and then harvested in passive lysis buffer. Luminescence was determined after adding firefly luciferase substrate reagent using a microplate luminometer. Detailed methods are available in the data supplement.
Immunofluorescence Staining and Imaging
Immunocytochemical methods were used to stain for smooth muscle α-actin (α-SMA), phalloidin, and DAPI. Detailed methods are available in the data supplement.
Gel Contraction
For collagen gel contraction assays, cells were plated onto 10-cm dishes and cultured for 3 days. Then the cells were treated with 1 ng/ml TGF-β with or without PD 102807 (10 μM). After 72 hours of treatment, cells were trypsinized and plated in collagen solution onto a 96-well plate at a density of 100,000 cells per well. After acquiring baseline images, collagen gels containing cells were stimulated with histamine (1 μM) for 10 min, and images were obtained. The collagen gel area was quantified, and percent reduction in gel area was calculated. Detailed methods are available in the data supplement.
Statistical Analysis
Data analysis was performed using GraphPad Prism 8 (GraphPad Software, La Jolla, CA); data are expressed as means ± SEM. Group comparisons were performed using a one-way ANOVA followed by a Dunnett’s test compared with the vehicle condition or Bonferroni’s multiple comparison test. A value of P < 0.05 was considered significant to reject the null hypothesis.
Results
PD 102807 Promotes M3 mAChR Arrestin Recruitment and Inhibits Gq Signaling
To assess the ability of various mAChR ligands to induce arrestin recruitment to M3 mAChR (M3), we heterologously expressed M3-RLuc and GFP-β-arrestin 2 in HEK293 cells and assessed the ability of mAChR ligands to induce 1) arrestin recruitment and 2) calcium mobilization (Figure 1). We found that the degree of arrestin recruitment (Figures 1A–1D) was proportional to the degree of calcium mobilization (Figures 1E and 1F) for all but one of the ligands tested: PD 102807. PD 102807 is a known characterized muscarinic receptor antagonist with selectivity for the M4 mAChR (half maximal inhibitory concentration: 91 nM for M4 and 950 nM for M3) (34, 35). In our M3-expressing hTERT ASM cells, PD 102807 inhibited M3-mediated, Gq-dependent calcium mobilization (36) in a competitive manner, as evidenced by a rightward shift of the methacholine (MCh) concentration-response curve (Figure 1C). Full agonists induced a robust increase in both arrestin recruitment and calcium mobilization, whereas partial agonists displayed low efficacy for both (Figures 1D–1F). However, PD 102807 did not fit this pattern, as it induced arrestin recruitment without concomitant calcium mobilization. These findings suggest that PD 102807 is an arrestin-biased M3 ligand.
Figure 1.
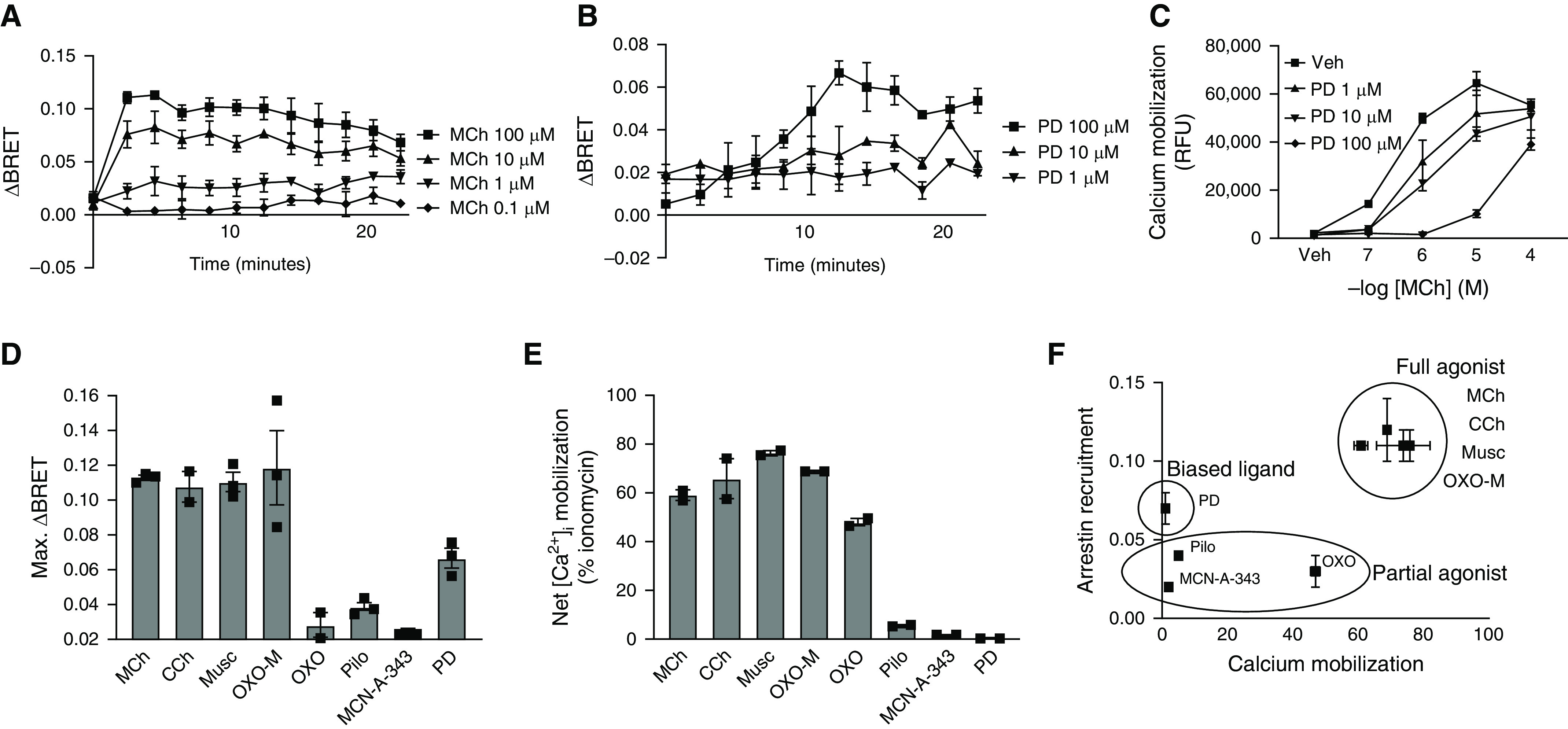
β-arrestin recruitment and calcium mobilization by muscarinic ligands. (A) Methacholine (MCh) (0.1, 1, 10, or 100 μM) and (B) PD 102807 (1, 10, or 100 μM) induce M3-mediated β-arrestin recruitment as assessed by bioluminescence resonance energy transfer (BRET) signal in HEK293 cells. (C) PD 102807 is a competitive antagonist of the M3 mAChR. Calcium mobilization in airway smooth muscle (ASM) cells by different concentrations of MCh in the presence or absence of PD 102807 (1, 10, or 100 μM; 15 min pretreatment) using Flexstation. (D) Maximal increase in β-arrestin recruitment BRET signal by mAChR ligands (100 μM) in HEK293 cells. (E) Net maximal calcium mobilization by mAChR ligands (100 μM) in human ASM cells stably expressing telomerase reverse transcriptase (hTERT) ASM cells. (F) Relationship between net maximal calcium mobilization and β-arrestin recruitment for each of the characterized mAChR ligands. Data are means ± SEM from two to four experiments. CCh = carbachol; MCh = methacholine; Musc = muscarine; OXO = oxotremorine sesquifumarate; OXO-M = oxotremorine-M; Pilo = pilocarpine; PD = PD 102807; Veh = vehicle.
M3-mediated AMPK Phosphorylation Is Modulated by Arrestins; PD 102807 Induced AMPK Phosphorylation Is M3 Dependent
In addition to identifying potential biased ligands as defined by arrestin recruitment, we also sought to identify associated signaling pathways activated by PD 102807. Using antibodies directed against known phosphopeptide residues, we aimed to identify aspects of M3-mediated signaling modulated by arrestins. For these initial screening studies, we knocked down expression of both β-arrestin isoforms (β-arrestin 1/2) in hTERT ASM cells by siRNA transfection, stimulated the cells with MCh (100 μM) for 5 min, and subjected cell lysates to the phosphoscreen using the ProteinSimple Wes automated capillary electrophoresis system. We observed a decrease in signal of MCh-induced AMPK-specific phosphoresidues in cells transfected with arrestin siRNA, as indicated by the overall decrease in the area under the curve (Figure E1A). This indicates a decrease in phosphorylation of AMPK consensus sequence peptides and suggests a decrease of MCh-induced AMPK activity. We then confirmed, using specific anti-phosphorylated (p)-AMPK antibodies, that M3-mediated AMPK phosphorylation is inhibited by arrestin knockdown (Figure E1B). Next, we determined time and concentration dependence for MCh- and PD 102807–induced AMPK signaling. PD 102807 reached a peak AMPK phosphorylation at 20 min and maintained elevated p-AMPK over the next 4 hours (Figure 2A). MCh-induced p-AMPK peaked at 5 min but waned over the next 4 hours (Figure 2B). MCh showed similar kinetics in inducing p-ERK (Figures E1D and E1E), whereas PD 102807 did not induce p-ERK (Figure E1F). MCh and PD 102807 both induced a concentration-dependent (1–100 μM) increase in p-AMPK, at 5 and 20 min, respectively (Figures 2C and 2D). In addition, we determined the phosphorylation status of acetyl-coenzyme A-carboxylase (ACC), a downstream target of AMPK whose phosphorylation is a proxy for AMPK activity. We found that both MCh and PD 102807 induced p-ACC in a concentration-dependent manner (Figures 2E and 2F). To determine the M3 dependence of MCh- and PD 102807–mediated p-AMPK induction, we evaluated responses to MCh and PD 102807 (100 μM; 5 and 20 min, respectively) in additional hTERT ASM cell lines (31) that we have determined to express very low levels of M3 mAChR (33, 37, 38). MCh- and PD 102807–induced p-AMPK was almost fully blunted in these cells (Figure 2G). We then used lentivirus to heterologously express M3 mAChR in these cells; PD 102807 strongly increased AMPK phosphorylation under these conditions. Finally, in the presence of the clinically relevant muscarinic receptor antagonist tiotropium bromide (100 nM), PD 102807– induced p-AMPK was almost fully inhibited (Figure 2I). These data indicate that PD 102807–induced AMPK signaling is M3 dependent.
Figure 2.
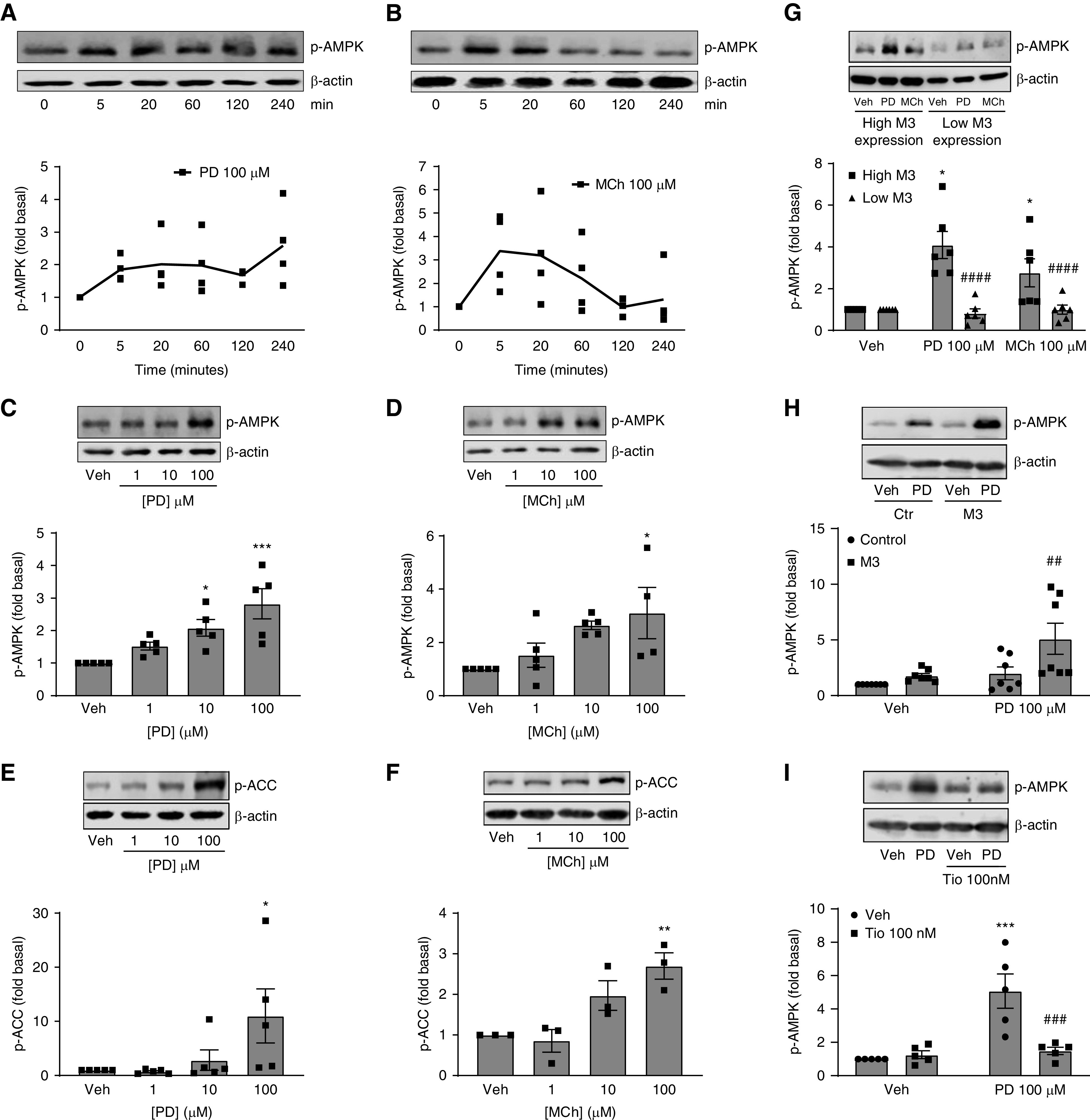
Methacholine and PD 102807 induce phosphorylated (p)-AMPK and p-ACC in a time- and concentration-dependent manner in human ASM cells; PD 102807–induced p-AMPK is M3 dependent. (A and B) Human hTERT ASM cell lines expressing M3 mAChRs were stimulated with (A) PD 102807 (100 μM) or (B) MCh (100 μM) for 0–240 min. (C–F) Cells were stimulated with increasing concentrations (1, 10, or 100 μM) of (D, F) MCh for 5 min or (C, E) PD 102807 for 20 min. *P < 0.05, **P < 0.01, and ***P < 0.001; one-way ANOVA followed by Dunnett’s multiple comparison test for differences from vehicle. (G) Cell lines expressing either high or low levels of M3 mAChR were stimulated with MCh (100 μM; 5 min) or PD 102807 (100 μM; 20 min). (H) In cell lines expressing low M3 mAChR, heterologous expression of M3 was achieved using lentiviral transfection. Transfected cells (control vector vs. M3 mAChR vector) were then stimulated with PD 102807 (100 μM; 20 min). (I) Cell lines expressing M3 mAChR were stimulated with PD 102807 (100 μM; 20 min) in the presence or absence of tiotropium bromide (100 nM; 30 min pretreatment). *P < 0.05, ***P < 0.001 versus vehicle, ##P < 0.01, ###P < 0.001, and ####P < 0.0001 versus own control; one-way ANOVA followed by Bonferroni multiple comparison test. Immunoblotting was performed using specific antibodies against p-AMPK, p-ERK, and p-ACC, as described in Methods. Representative blots are shown; loading was corrected for β-actin. Data are means ± SEM from three to seven experiments. Veh = vehicle.
MCh-Induced, but Not PD 102807–Induced, p-AMPK Is Gq Dependent
Because canonical M3 signaling is mediated by the Gq protein in most cell types, we clarified the role of Gq protein in M3 signaling in ASM cells by using an inhibitor of Gq signaling, YM-254890 (10 μM). Gq inhibition did not affect PD 102807–induced p-AMPK (Figure 3A). However, MCh-induced p-AMPK was strongly inhibited (88%) by YM-254890 (Figure 3B). Similarly, YM-254890 inhibited MCh-induced p-ERK by 82% (Figure 3C). These data indicate that, although both MCh-induced AMPK signaling and PD 102807–induced AMPK signaling are M3 dependent, MCh-induced p-AMPK is driven by canonical M3-Gq signaling, whereas the PD 102807–induced p-AMPK is M3 mediated but Gq independent.
Figure 3.
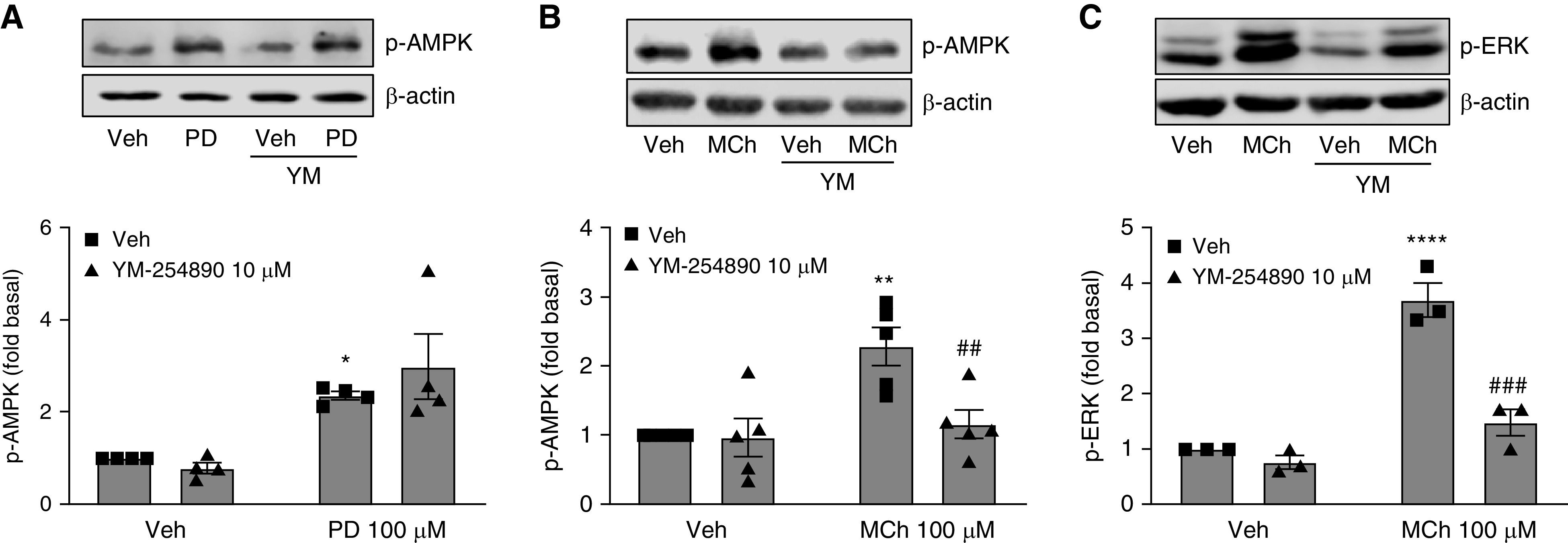
Gq inhibitor YM-254890 abrogates MCh-induced, but not PD 102807–induced, p-AMPK in human ASM cells expressing M3 mAChR. Human hTERT ASM cell lines were preincubated with YM-254890 (10 μM) for 15 min and then stimulated with (A) PD 102807 (100 μM; 20 min) or (B and C) MCh (100 μM; 5 min), and cell lysates were used for immunoblotting. Representative immunoblots for (A and B) p-AMPK and (C) p-ERK are shown; loading was corrected for β-actin. Data are means ± SEM from three to five experiments. *P < 0.05, **P < 0.01, and ****P < 0.0001 versus vehicle stimulation; ##P < 0.01, and ###P < 0.001 versus vehicle-pretreated, MCh-stimulated condition; one-way ANOVA followed by Bonferroni multiple comparison test. Veh = vehicle.
PD 102807–Induced AMPK Signaling Is GRK2/3 Dependent; Arrestins Modulate M3-Mediated AMPK Signaling
To further elucidate the signaling pathways involved in M3-mediated AMPK activation by PD 102807, we focused on the potential role of arrestins and GRK2/3, given the substantial body of literature showing that arrestins mediate signaling bias for various receptors, and given our data demonstrating that PD 102807 induces arrestin recruitment. To determine how the different β-arrestin isoforms and GRK2/3 regulate signaling, we selectively knocked down β-arrestin 1 and β-arrestin 2, as well as GRK2/3, using siRNA and determined phosphorylation of AMPK. Knockdown of β-arrestin 1 expression resulted in 64% inhibition of PD 102807–induced p-AMPK, whereas β-arrestin 2 knockdown augmented PD 102807–induced p-AMPK (Figure 4A). On the other hand, MCh-induced p-AMPK was decreased by 46% for β-arrestin 1 knockdown and 53% for β-arrestin 2 knockdown (Figure 4D). We determined that GRK2/3 knockdown completely abrogated PD 102807–induced p-AMPK, whereas it did not affect MCh-induced p-AMPK (Figures 4B and 4E). Similarly, GRK2/3 knockdown abolished the PD 102807–induced p-ACC but did not affect MCh-induced responses (Figures 4C and 4F). Collectively, these results indicate that GRK2/3 and arrestins modulate PD 102807– induced, M3-mediated AMPK signaling in ASM cells.
Figure 4.
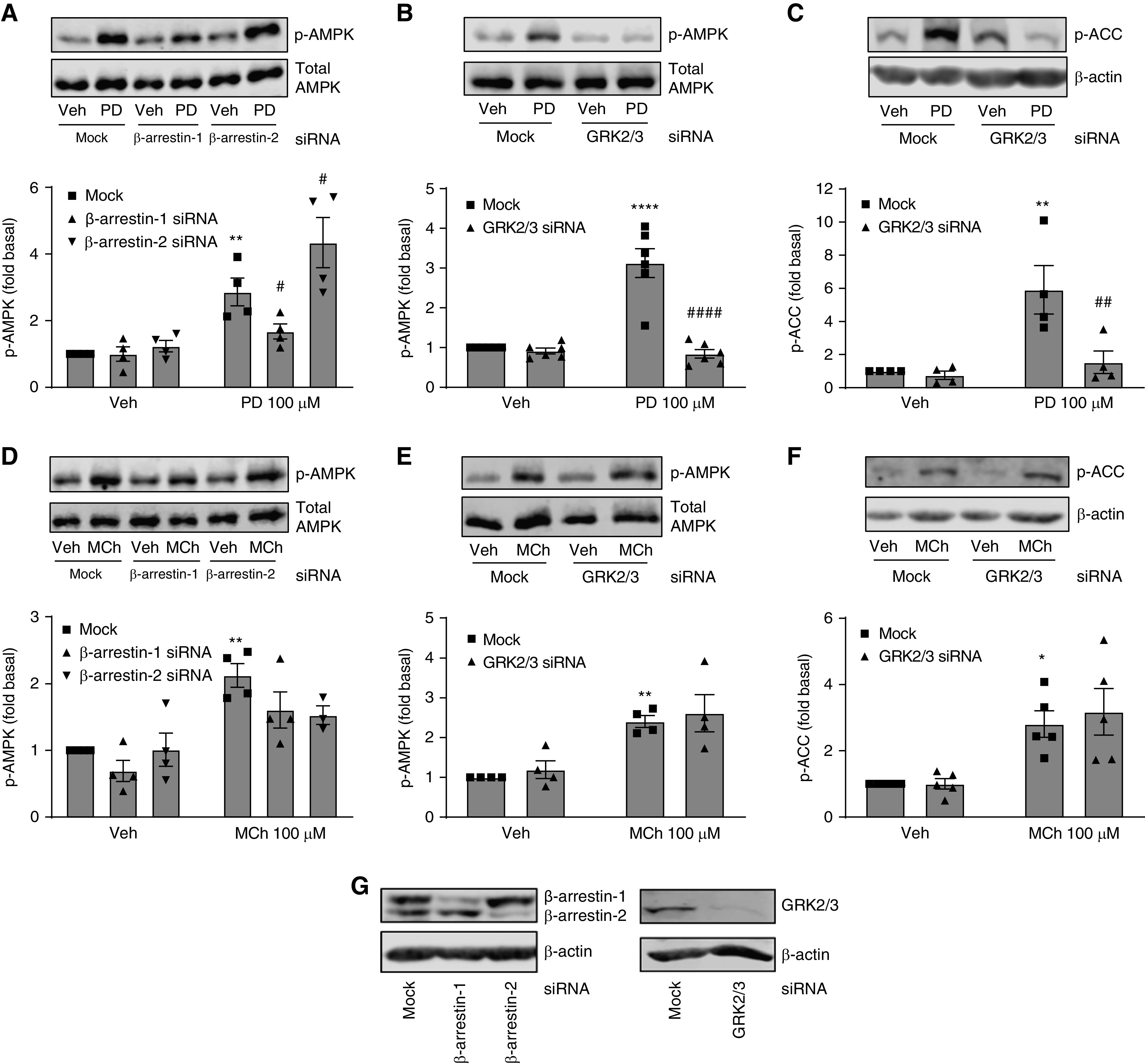
Role of β-arrestin and GRK2/3 in PD 102807–induced or MCh-induced p-AMPK and p-ACC. After mock, β-arrestin-1, β-arrestin-2, or GRK2/3 siRNA transfection, cells were stimulated with (A–C) PD 102807 (100 μM; 20 min), or (D–F) MCh (100 μM; 5 min), and (A, B, D, and E) p-AMPK and (C and F) p-ACC were assessed by immunoblotting. (G) β-arrestin-1, β-arrestin-2, and GRK2/3 knockdowns were confirmed by immunoblotting. Representative immunoblots for p-AMPK and p-ACC are shown; loading was corrected for total AMPK or β-actin. Data are means ± SEM from three to six experiments. *P < 0.05, **P < 0.01, and ****P < 0.0001 versus vehicle stimulation; #P < 0.05, ##P < 0.01, and ####P < 0.0001 versus mock-transfected PD 102807–stimulated condition; one-way ANOVA followed by Bonferroni multiple comparison test. Veh = vehicle.
PD 102807 Inhibits TGF-β Signaling and Hypercontractility in ASM Cells
AMPK is an established antagonist of TGF-β–induced signaling and pathology (39–41), a fact that is highly relevant to ASM cell biology, as M3 mAChRs and TGF-β cooperate to drive ASM cell phenotype switching to a hypercontractile phenotype characterized by increased contractile protein expression and contractility (23, 24). TGF-β is known to activate transcription factor SMAD in a variety of cell types. Using a luciferase reporter assay, we determined that PD 102807 inhibited TGF-β–induced SMAD-Luc activity by 58% in hTERT ASM cells with high M3 receptor expression (Figure 5A).
Figure 5.
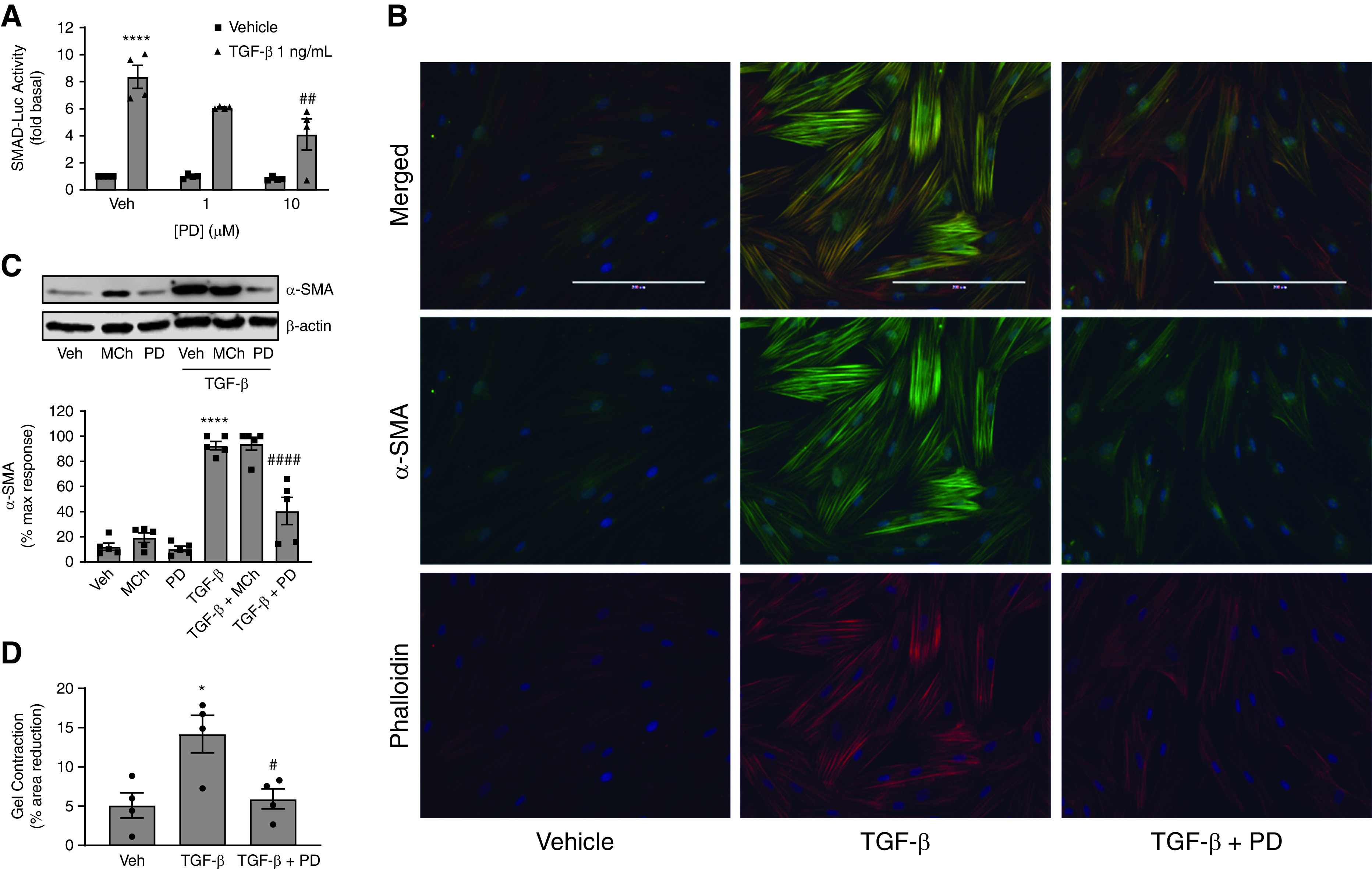
PD 102807 inhibits TGF-β–induced signaling and function in ASM cells in an M3-dependent manner. (A) SMAD-Luc transfected human hTERT ASM cell lines were preincubated with PD 102807 (1 or 10 μM) for 20 min and then stimulated with TGF-β (1 ng/mL) for 6 hours. Luciferase activity was determined as described in Methods. (B) hTERT ASM cell lines were treated with TGF-β (1 ng/mL) ± PD 102807 (10 μM) for 3 days. Cells were then fixed and permeabilized, and smooth muscle α-actin (α-SMA) (488 nm; green) and phalloidin (594 nm; red) expression was determined by immunofluorescence. Cells were counterstained with DAPI (blue) as described in Methods. Representative images are shown. (C) hTERT ASM cells were stimulated with TGF-β (1 ng/mL) with or without MCh (10 μM) or PD 102807 (10 μM) for 3 days, and α-SMA expression was determined by immunoblotting. Representative immunoblots for α-SMA are shown; loading was corrected for β-actin. (D) ASM cell contraction was after cells were treated with TGF-β (1 ng/mL) with or without PD 102807 (10 μM) for 3 days. Collagen gel contraction data are shown as percent reduction in gel area after histamine stimulation (1 μM; 10 min). Data are means ± SEM from four to five experiments. *P < 0.05 and ****P < 0.0001 versus vehicle-stimulated condition; #P < 0.05, ##P < 0.01, and ####P < 0.0001 versus vehicle-pretreated TGF-β condition; one-way ANOVA followed by Bonferroni multiple comparison test. Veh = vehicle.
Next, we focused on the functional significance of TGF-β signaling inhibition by M3 biased signaling. TGF-β is known to increase expression of contractile proteins in ASM; this is one of several ways in which TGF-β contributes to ASM cell phenotype plasticity and airway hypercontractility, important features of airway pathology in obstructive airway disease. We stimulated M3-expressing ASM cells with TGF-β (1 ng/ml) for 3 days in the absence or presence of PD 102807 (10 μM). Using immunocytochemistry, we determined that the TGF-β–induced increase in the expression of α-SMA, as well as the formation of actin stress fibers (phalloidin staining), is inhibited by PD 102807 (Figure 5B). We then used immunoblotting to quantify the effects of PD 102807 and MCh on TGF-β–induced α-SMA expression. In cells with high M3 expression, PD 102807 inhibited TGF-β–induced α-SMA expression by 65% (Figure 5C). In cells with low M3 expression, PD 102807 did not significantly affect TGF-β–induced SMAD-Luc activity or α-SMA expression (Figures E1H and E1I).
Consistent with the effect of PD 102807 on TGF-β–induced modulation of ASM structural phenotype, PD 102807 inhibited TGF-β–induced ASM cell hypercontractility as assessed in a collagen gel contraction assay (Figure 5D). TGF-β treatment increased histamine-induced ASM cell contraction by 2.8-fold, which was effectively inhibited by PD 102807 pretreatment. Collectively, these data indicate that PD 102807 inhibits TGF-β–induced signaling, α-SMA expression, and hypercontractility in human ASM cells. To further implicate the role of GRK-arrestin–biased M3 signaling in PD 102807-mediated regulation of ASM phenotype, we used GRK2/3 knockdown, given that GRK2/3 knockdown can be sustained for high levels in ASM over the course of a chronic experiment (Figures 6A and 6B). GRK2/3 knockdown reversed the ability of PD 102807 to inhibit TGF-β–induced α-SMA expression by 58% (Figure 6A).
Figure 6.

GRK2/3 knockdown attenuates PD 102807–mediated inhibition of TGF-β–induced α-SMA expression. Following (A) GRK2/3 siRNA transfection, ASM cells were stimulated with TGF-β (1 ng/ml) for 3 days in the presence or absence of PD 102807 (10 μM), and α-SMA expression was determined by immunoblotting. (B) GRK2/3 knockdown after the 3-day treatment (Day 6 post–siRNA transfection) was confirmed by immunoblotting. Representative immunoblots for α-SMA are shown; loading was corrected for β-actin. Data are means ± SEM from four experiments. *P < 0.05; ***P < 0.001 versus vehicle pretreated TGF-β condition; #P < 0.05 versus mock-transfected, PD 102807–pretreated TGF-β–stimulated condition; one-way ANOVA followed by Bonferroni multiple comparison test. Veh = vehicle.
Discussion
The present study identifies PD 102807 as a biased M3 ligand, which induces GRK2/3-mediated, arrestin-mediated, and Gq-independent activation of AMPK signaling in ASM cells. Our reductionist model—HEK293 cells heterologously expressing an M3-arrestin BRET pair—allowed us to screen for the ability of mAChR ligands to recruit arrestins to M3 mAChRs. Using this model, we observed that, for most mAChR ligands, the ability to recruit arrestins correlates with the ability to induce Gq signaling (calcium mobilization). When ligand-induced calcium mobilization is plotted against ligand-induced arrestin recruitment, the various ligands cluster together on the basis of their intrinsic ability to activate the M3 receptor (i.e., full agonists vs. partial agonists). The PD 102807 signaling profile—antagonism of M3-Gq–stimulated calcium and agonism of arrestin recruitment— indicates that this compound acts as a biased M3 ligand capable of arrestin recruitment independent of promoting Gq signaling.
In concert with the aforementioned studies, we focused on identifying potential biased arrestin-dependent signaling by M3 mAChR in human ASM cells. We used antibodies against specific kinase phosphoresidue sequences in conjunction with automated capillary electrophoresis (using the ProteinSimple Wes system) to reveal that MCh increased the phosphorylation of residues specific to AMPK in an arrestin-dependent manner. Using a specific phospho-AMPK antibody, we then confirmed that MCh-induced AMPK phosphorylation is impaired in ASM cells in which expression of β-arrestins is knocked down. We also determined that PD 102807 induces phosphorylation of AMPK; this is the first indication of a biased ligand inducing discrete signaling by the M3 mAChR. Indeed, we show that PD 102807 inhibits M3-mediated Gq signaling (calcium mobilization) yet promotes arrestin recruitment to M3 mAChR and induces phosphorylation of AMPK and the downstream AMPK target ACC. Using several experimental approaches, we determined that both MCh and PD 102807 require M3 expression to induce p-AMPK and that PD 102807 induction of p-AMPK is inhibited by the specific, clinically used muscarinic antagonist tiotropium, thereby demonstrating that this is an M3-dependent signal.
On the basis of our initial findings (Figures E1A–E1C), we expected that MCh-induced p-AMPK would be a Gq-independent/biased signal. However, even though we show that knocking down arrestins decreases the MCh-induced p-AMPK, this MCh-induced signal appears to be strongly dependent on Gq activation. Indeed, M3 mAChR-mediated, Gq- and calcium-dependent phosphorylation of AMPK has previously been reported in skeletal muscle cells (42). However, PD 102807–induced activation of AMPK signaling is not dependent on Gq activation; the PD 102807–induced signaling is modulated by arrestins and is strongly dependent on GRK2/3, further establishing signaling bias by PD 102807. GRK2/3 does not appear to be required for MCh-induced AMPK signaling.
The GRK-arrestin axis has been widely studied in the context of receptor desensitization. On the other hand, regulation of signaling by GRK/arrestin, whether canonical G protein or alternative biased/qualitative, is a relatively new concept, and more research is needed to elucidate the underlying mechanisms. With respect to the arrestin regulation of canonical Gq protein signaling, in our previously published studies, we determined that, in ASM cells, β-arrestin 1 expression is required for M3-mediated calcium mobilization as well as contraction both ex vivo and in vivo (38). Conversely, we have previously determined that GRK2/3 knockdown does not affect M3-mediated calcium mobilization in ASM cells (33). Our current findings show that MCh-induced p-AMPK is fully inhibited by Gq inhibition, whereas PD 102807-induced p-AMPK is not affected. The ability to use the Gq inhibitor YM-254890 to effectively distinguish between Gq and non-Gq (GRK-arrestin–dependent) signaling is important, given that at least one study (43) suggested that some arrestin-dependent signals may still require G protein activation. However, both MCh- and PD 102807– induced p-AMPK are partially inhibited by β-arrestin 1 knockdown. These data, indicating that knockdown of β-arrestin 1 diminishes both canonical Gq and noncanonical biased M3 signaling, suggest that β-arrestin 1 may regulate both Gq-dependent and -independent signaling. Under β-arrestin 2 knockdown conditions, the PD 102807–induced p-AMPK was increased; this suggests that the PD 102807– induced biased signaling is not mediated by β-arrestin 2 but that β-arrestin 2 may have a limiting effect on noncanonical biased M3 signaling.
Our data show that GRK2/3 is a crucial mediator of PD 102807–induced, noncanonical biased M3 signaling. Knockdown of GRK2/3 fully inhibits PD 102807–induced p-AMPK and p-ACC, whereas the MCh-induced p-AMPK and p-ACC are not affected. Our previous studies showing no role for GRK2/3 in regulating MCh-induced Gq signaling (33), as well as our present data, support a role for GRK2/3 as a specific regulator of biased M3 signaling. Furthermore, these findings bolster the evidence that PD 102807 induces a Gq-independent, biased M3-mediated p-AMPK signal, whereas MCh-induced p-AMPK is mediated by canonical Gq signaling.
With respect to regulation of ASM physiology, we identified a role for PD 102807–induced M3-mediated GRK-arrestin biased signaling in inhibiting TGF-β signaling in ASM cells (Figure 7). This is a highly relevant observation in the context of obstructive airway diseases, because TGF-β is an important driver of airway hypercontractility mediated by increased ASM contractile protein expression and airway remodeling, resulting in thickened airway wall (23, 44, 45). We found PD 102807 to inhibit TGF-β–induced SMAD-Luc activity, as well as α-SMA expression, in cells that express M3R but not in cells with low M3 mAChR (M3R) expression, indicating M3 dependence. Similarly, we found that PD 102807 inhibits TGF-β–induced increase in the formation of α-SMA fibers as well as actin stress fibers. Furthermore, we determined that TGF-β–induced increase in contractile protein expression also results in increased contractility of ASM cells. Consistent with our contractile protein expression data, PD 102807 inhibits the TGF-β–induced hypercontractility. In an attempt to further demonstrate M3 dependence, we tested the effect of the nonselective mAChR antagonist tiotropium on the effect of PD 102807–mediated regulation of the TGF-β–induced increase in α-SMA expression. We found that tiotropium had limited ability to inhibit the effects of PD 102807 over the 3-day stimulation (data not shown). Despite some reversal of PD 102807–induced AMPK phosphorylation at several time points, the inhibition is not complete over the course of 3 days, likely because of PD 102807 sufficiently competing with tiotropium for receptor binding over time. This is consistent with competitive behavior and not surprising, given the relatively higher concentration of PD 102807 versus tiotropium. Even under optimal circumstances, experiments in which an effect is induced (e.g., by TGF-β), is then inhibited by a compound (e.g., PD 102807), and then needs to be inhibited by a second compound (e.g., tiotropium) remain challenging, especially over the course of multiple days. However, we confirmed that the effects of PD102807 are GRK dependent, as we show that PD 102807 is less effective at inhibiting the TGF-β–induced expression of α-SMA in ASM cells when GRK2/3 is knocked down. These data indicate that PD 102807–induced GRK-arrestin–biased M3 signaling may be a promising pathway to attenuate airway hypercontractility, with obvious therapeutic implications for obstructive airway diseases.
Figure 7.

PD 102807 is an M3 mAChR biased ligand that inhibits M3-mediated Gq signaling and promotes (Gq-independent) GRK2/3-arrestin–dependent signaling. M3 mAChR agonists, such as MCh, promote Gq-mediated calcium mobilization and ASM contraction (left side). In addition, MCh induces a transient phosphorylation of AMPK that is Gq calcium dependent. The biased M3 ligand PD 102807, on the other hand, acts as an orthosteric antagonist for M3-Gq–dependent signaling and promotes GRK2/3-arrestin–dependent sustained AMPK signaling (right side). TGF-β–induced signaling, mediated by TGF-β receptors II and I (TβRII/I), promotes phenotype switching to a hypercontractile ASM phenotype. PD 102807–induced GRK2/3-arrestin–dependent AMPK signaling inhibits TGF-β signaling and thereby attenuates TGF-β–induced ASM hypercontractility.
AMPK is a heterotrimeric serine/threonine complex comprising an α catalytic, and β and γ regulatory subunits. AMPK was initially identified as a crucial regulator of cellular energy homeostasis, detecting changes in cytosolic AMP/ATP ratio and steering various metabolic pathways to address energy needs of the cell (46). However, further research revealed that AMPK activity is not only promoted by increases in the AMP/ATP ratio but also by several kinases, including serine/threonine kinase 11 (also known as liver kinase B1), TGF-β–activated kinase 1, and calcium/calmodulin-dependent kinase, which indicates that AMPK is a focal point for a variety of signaling pathways and an important regulator of physiology (47). AMPK activation has been shown to attenuate pathology in various disease models. The widely used oral antidiabetic drug metformin, which promotes AMPK activity, has been found to improve cardiac function and prevent cardiac remodeling in nondiabetic models of heart failure and models of myocardial infarction (48–50). In addition, AMPK has been a prominent target for reducing pathological tissue fibrosis and TGF-β signaling in many disease models, including in the lung (40, 51, 52), liver (53–55) and kidney (56, 57). Our study provides a proof-of-concept that the biased M3 mAChR ligand, PD 102807, can modulate AMPK activity. This finding has wide-ranging implications, given the prominent role of AMPK in regulating various organ systems.
Previous studies have focused on identifying M3 biased signaling. Studies by the Tobin lab have indicated that M3 engages in Gq-independent signaling, potentially by means of mechanisms that involve receptor phosphorylation. The authors generated a mutant, phosphorylation-negative M3 mAChR in which 15 residues were substituted to prevent phosphorylation of the receptor. Expression of these phosphorylation-negative receptors did not affect canonical M3-Gq signaling but did result in impaired insulin release from mouse pancreatic β-islet cells (28), impaired constriction of murine airways (27), and impaired learning in mice (29). These studies point to a substantial non–Gq-dependent input from M3 receptors in the regulation of various physiological processes. Other studies have shown that the M3 mAChR undergoes barcode phosphorylation when activated by full versus partial agonists (and varied by cell type), suggesting that barcode phosphorylation is a possible mechanism for determining biased M3 signaling as well as indicating that barcode phosphorylation may have wider implications for how canonical Gq signaling is transduced (58). Indeed, barcode phosphorylation has been demonstrated for several GPCRs, and it is possibly a key feature in GPCR signaling (8, 59).
The Roth lab created a designer receptor based on the M3 mAChR with two mutations in the orthosteric pocket that render it insensitive to acetylcholine but allow its activation by clozapine-N-oxide, an otherwise pharmacologically inert compound, to induce M3 signaling (60–62). An additional R165L mutation to this receptor renders it unable to activate Gq signaling but still able to recruit arrestins (63). This arrestin-biased receptor was found to promote insulin release from murine pancreatic β-islet cells, thereby further strengthening the case for M3 biased signaling as an important regulator of physiological processes. All these studies have focused on identifying biased signaling induced by mAChR agonists. However, no studies to date have identified a biased ligand that can selectively activate non-canonical pathways downstream of M3. Pronin and colleagues have suggested that the muscarinic ligand pilocarpine can act as a biased agonist for M3 (64). Pilocarpine is a partial agonist that induces M3-mediated Gq signaling, so the potential for pilocarpine to selectively induce non-Gq pathways may be dependent on receptor expression levels and cell type. This suggests that mAChR ligands may behave as either balanced or biased ligands depending on the cell system to which they are applied. In our study, pilocarpine behaves as a partial muscarinic agonist that induces both modest calcium mobilization and modest arrestin recruitment.
Although our studies demonstrate the capacity of PD 102807 to stimulate M3-/GRK-/arrestin-dependent signaling to regulate ASM function that is independent of Gq, we cannot definitively assert that such signaling and function is entirely G protein independent. Although M3 signaling stimulated by its cognate agonist acetylcholine (and the comparable MCh and carbachol) appears to be mediated exclusively through Gq (36), additional studies—along the lines of those performed in HEK293 cells to assess a role for multiple G protein subtypes in biased signaling by several GPCRs (43)—are required to eliminate any contribution of other G proteins. To date, no properties other than muscarinic receptor antagonism have been attributed to PD 102807 in published literature. Our data indicate a requirement for M3 mAChR in PD 102807–induced AMPK activation; additional/alternative mechanisms for the actions of PD 102807 are possible, pending further study.
One limitation of the present study is that we have primarily focused on studying PD 102807–induced signaling and effects in cell-based models. Isolated ASM cells heterologously expressing hTERT are our model of choice because some of these lines retain M3 mAChR expression; these cells are not primary cells per se, but they are derived from low-passage primary cells that have been transfected with hTERT, essentially making them senescence resistant. It has been established that primary cultures of human ASM cells rapidly lose M3 expression with progressive population doubling (typically between the second and fourth passages) and are, therefore, impractical as a model to study M3 signaling, particularly in experiments of long duration. In the past 15 years, hTERT ASM cells have been widely used and have been established as a useful model system that is representative of ASM physiology (65–67), specifically in analyses of M3 signaling or function (68–70).
Because more integrative models enabling further validation of the physiological and translational relevance of our findings are beyond the scope of this study, our future efforts will focus on evaluation of the impact of muscarinic biased signaling in the airways by using ex vivo precision-cut lung slices as well as in vivo models of lung disease. With more biased ligands and biased GPCR signaling pathways being identified, it is increasingly important to determine the impact of biased signaling on physiological processes. Although many studies are ongoing, biased signaling is already showing promise in pain management (oliceridine and TRV734) and in the treatment of diseases and conditions (in various stages of clinical or preclinical studies), including heart failure (carvedilol), and schizophrenia (UNC9994) (17). This study suggests that M3 mAChR-biased signaling in the airways may be targeted for therapeutic purposes.
In summary, in this study, we identify PD 102807 as a biased M3 mAChR ligand that promotes AMPK signaling in a GRK2/3-arrestin–dependent and Gq-independent manner in ASM cells. The ability of PD 102807 to inhibit pathogenic ASM hypercontractility is highly relevant to the treatment of obstructive airway diseases.
Acknowledgments
Acknowledgment
The authors thank Dr. Michel Bouvier for the BRET constructs pcDNA3-β2AR-RLuc and pcDNA3-GFP10-βarrestin2.
Footnotes
Supported by National Institutes of Health awards R01 HL140064 (T.P.) and P01 HL114471 (R.B.P. and J.L.B.).
Author Contributions: E.T., R.B.P., and T.P. designed research; E.T., B.M., K.C.-M., H.S., S.D.S., and T.P. performed research; D.A.D. and J.L.B. provided new reagents/analytic tools, expertise, and manuscript editing; E.T., B.M., and T.P. analyzed data; E.T., R.B.P., and T.P. wrote the paper.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0320OC on August 9, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. McDonald PH, Lefkowitz RJ. Beta-arrestins: new roles in regulating heptahelical receptors’ functions. Cell Signal . 2001;13:683–689. doi: 10.1016/s0898-6568(01)00203-0. [DOI] [PubMed] [Google Scholar]
- 2. Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. Beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem . 2008;283:5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- 3. Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci USA . 2008;105:9988–9993. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA . 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benovic JL, Kühn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proc Natl Acad Sci USA . 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. Beta-arrestin: a protein that regulates beta-adrenergic receptor function. Science . 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 7. Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature . 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 8. Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, et al. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal . 2011;4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, et al. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci USA . 2005;102:1442–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ren XR, Reiter E, Ahn S, Kim J, Chen W, Lefkowitz RJ. Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc Natl Acad Sci USA . 2005;102:1448–1453. doi: 10.1073/pnas.0409534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, et al. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA . 2008;105:14555–14560. doi: 10.1073/pnas.0804745105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patel PA, Tilley DG, Rockman HA. Beta-arrestin-mediated signaling in the heart. Circ J . 2008;72:1725–1729. doi: 10.1253/circj.cj-08-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Forkuo GS, Kim H, Thanawala VJ, Al-Sawalha N, Valdez D, Joshi R, et al. Phosphodiesterase 4 inhibitors attenuate the asthma phenotype produced by β2-adrenoceptor agonists in phenylethanolamine n-methyltransferase-knockout mice. Am J Respir Cell Mol Biol . 2016;55:234–242. doi: 10.1165/rcmb.2015-0373OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Penn RB, Bond RA, Walker JK. GPCRs and arrestins in airways: implications for asthma. Handb Exp Pharmacol . 2014;219:387–403. doi: 10.1007/978-3-642-41199-1_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walker JK, Penn RB, Hanania NA, Dickey BF, Bond RA. New perspectives regarding β(2)-adrenoceptor ligands in the treatment of asthma. Br J Pharmacol . 2011;163:18–28. doi: 10.1111/j.1476-5381.2010.01178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen LP, Al-Sawalha NA, Parra S, Pokkunuri I, Omoluabi O, Okulate AA, et al. β2-adrenoceptor signaling in airway epithelial cells promotes eosinophilic inflammation, mucous metaplasia, and airway contractility. Proc Natl Acad Sci USA . 2017;114:E9163–E9171. doi: 10.1073/pnas.1710196114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov . 2018;17:243–260. doi: 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. N Engl J Med . 1984;311:421–425. doi: 10.1056/NEJM198408163110701. [DOI] [PubMed] [Google Scholar]
- 19. Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H, et al. Inhibition of allergen-induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J . 2007;30:653–661. doi: 10.1183/09031936.00004907. [DOI] [PubMed] [Google Scholar]
- 20. Gosens R, Bos IS, Zaagsma J, Meurs H. Protective effects of tiotropium bromide in the progression of airway smooth muscle remodeling. Am J Respir Crit Care Med . 2005;171:1096–1102. doi: 10.1164/rccm.200409-1249OC. [DOI] [PubMed] [Google Scholar]
- 21. Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R, et al. Tiotropium inhibits pulmonary inflammation and remodelling in a guinea pig model of COPD. Eur Respir J . 2011;38:789–796. doi: 10.1183/09031936.00146610. [DOI] [PubMed] [Google Scholar]
- 22. Kistemaker LE, Bos IS, Hylkema MN, Nawijn MC, Hiemstra PS, Wess J, et al. Muscarinic receptor subtype-specific effects on cigarette smoke-induced inflammation in mice. Eur Respir J . 2013;42:1677–1688. doi: 10.1183/09031936.00112412. [DOI] [PubMed] [Google Scholar]
- 23. Oenema TA, Maarsingh H, Smit M, Groothuis GM, Meurs H, Gosens R. Bronchoconstriction induces TGF-β release and airway remodelling in guinea pig lung slices. PLoS One . 2013;8:e65580. doi: 10.1371/journal.pone.0065580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oenema TA, Smit M, Smedinga L, Racké K, Halayko AJ, Meurs H, et al. Muscarinic receptor stimulation augments TGF-β1-induced contractile protein expression by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol . 2012;303:L589–L597. doi: 10.1152/ajplung.00400.2011. [DOI] [PubMed] [Google Scholar]
- 25. Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, et al. UPLIFT Study Investigators A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med . 2008;359:1543–1554. doi: 10.1056/NEJMoa0805800. [DOI] [PubMed] [Google Scholar]
- 26. Kerstjens HA, Engel M, Dahl R, Paggiaro P, Beck E, Vandewalker M, et al. Tiotropium in asthma poorly controlled with standard combination therapy. N Engl J Med . 2012;367:1198–1207. doi: 10.1056/NEJMoa1208606. [DOI] [PubMed] [Google Scholar]
- 27. Bradley SJ, Wiegman CH, Iglesias MM, Kong KC, Butcher AJ, Plouffe B, et al. Mapping physiological G protein-coupled receptor signaling pathways reveals a role for receptor phosphorylation in airway contraction. Proc Natl Acad Sci USA . 2016;113:4524–4529. doi: 10.1073/pnas.1521706113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kong KC, Butcher AJ, McWilliams P, Jones D, Wess J, Hamdan FF, et al. M3-muscarinic receptor promotes insulin release via receptor phosphorylation/arrestin-dependent activation of protein kinase D1. Proc Natl Acad Sci USA . 2010;107:21181–21186. doi: 10.1073/pnas.1011651107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Poulin B, Butcher A, McWilliams P, Bourgognon JM, Pawlak R, Kong KC, et al. The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/arrestin-dependent manner. Proc Natl Acad Sci USA . 2010;107:9440–9445. doi: 10.1073/pnas.0914801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tompkins E, Mimic B, Sayed D, Penn RB, Pera T. Biased muscarinic ligand promotes m3-mediated grk2/3-dependent ampk signaling in airway smooth muscle cells. Am J Respir Crit Care Med . 2020;201:A4411. [Google Scholar]
- 31. Gosens R, Stelmack GL, Dueck G, McNeill KD, Yamasaki A, Gerthoffer WT, et al. Role of caveolin-1 in p42/p44 MAP kinase activation and proliferation of human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol . 2006;291:L523–L534. doi: 10.1152/ajplung.00013.2006. [DOI] [PubMed] [Google Scholar]
- 32. Carr R, III, Du Y, Quoyer J, Panettieri RA, Jr, Janz JM, Bouvier M, et al. Development and characterization of pepducins as Gs-biased allosteric agonists. J Biol Chem . 2014;289:35668–35684. doi: 10.1074/jbc.M114.618819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deshpande DA, Yan H, Kong KC, Tiegs BC, Morgan SJ, Pera T, et al. Exploiting functional domains of GRK2/3 to alter the competitive balance of pro- and anticontractile signaling in airway smooth muscle. FASEB J . 2014;28:956–965. doi: 10.1096/fj.13-240226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Olianas MC, Onali P. PD 102807, a novel muscarinic M4 receptor antagonist, discriminates between striatal and cortical muscarinic receptors coupled to cyclic AMP. Life Sci . 1999;65:2233–2240. doi: 10.1016/s0024-3205(99)00488-9. [DOI] [PubMed] [Google Scholar]
- 35. Augelli-Szafran CE, Jaen JC, Moreland DW, Nelson CB, Penvose-Yi JR, Schwarz RD. Identification and characterization of m4 selective muscarinic antagonists. Bioorg Med Chem Lett . 1998;8:1991–1996. doi: 10.1016/s0960-894x(98)00351-5. [DOI] [PubMed] [Google Scholar]
- 36. Wang YX, Kotlikoff MI. Muscarinic signaling pathway for calcium release and calcium-activated chloride current in smooth muscle. Am J Physiol . 1997;273:C509–C519. doi: 10.1152/ajpcell.1997.273.2.C509. [DOI] [PubMed] [Google Scholar]
- 37. Pera T, Deshpande DA, Ippolito M, Wang B, Gavrila A, Michael JV, et al. Biased signaling of the proton-sensing receptor OGR1 by benzodiazepines. FASEB J . 2018;32:862–874. doi: 10.1096/fj.201700555R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pera T, Hegde A, Deshpande DA, Morgan SJ, Tiegs BC, Theriot BS, et al. Specificity of arrestin subtypes in regulating airway smooth muscle G protein-coupled receptor signaling and function. FASEB J . 2015;29:4227–4235. doi: 10.1096/fj.15-273094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gao J, Ye J, Ying Y, Lin H, Luo Z. Negative regulation of TGF-β by AMPK and implications in the treatment of associated disorders. Acta Biochim Biophys Sin (Shanghai) . 2018;50:523–531. doi: 10.1093/abbs/gmy028. [DOI] [PubMed] [Google Scholar]
- 40. Kheirollahi V, Wasnick RM, Biasin V, Vazquez-Armendariz AI, Chu X, Moiseenko A, et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat Commun . 2019;10:2987. doi: 10.1038/s41467-019-10839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med . 2018;24:1121–1127. doi: 10.1038/s41591-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Merlin J, Evans BA, Csikasz RI, Bengtsson T, Summers RJ, Hutchinson DS. The M3-muscarinic acetylcholine receptor stimulates glucose uptake in L6 skeletal muscle cells by a CaMKK-AMPK-dependent mechanism. Cell Signal . 2010;22:1104–1113. doi: 10.1016/j.cellsig.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 43. Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun . 2018;9:341. doi: 10.1038/s41467-017-02661-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, et al. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med . 2011;364:2006–2015. doi: 10.1056/NEJMoa1014350. [DOI] [PubMed] [Google Scholar]
- 45. Morty RE, Königshoff M, Eickelberg O. Transforming growth factor-beta signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc Am Thorac Soc . 2009;6:607–613. doi: 10.1513/pats.200908-087RM. [DOI] [PubMed] [Google Scholar]
- 46. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol . 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med . 2016;48:e245. doi: 10.1038/emm.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loi H, Boal F, Tronchere H, Cinato M, Kramar S, Oleshchuk O, et al. Metformin protects the heart against hypertrophic and apoptotic remodeling after myocardial infarction. Front Pharmacol . 2019;10:154. doi: 10.3389/fphar.2019.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hermida N, Markl A, Hamelet J, Van Assche T, Vanderper A, Herijgers P, et al. HMGCoA reductase inhibition reverses myocardial fibrosis and diastolic dysfunction through AMP-activated protein kinase activation in a mouse model of metabolic syndrome. Cardiovasc Res . 2013;99:44–54. doi: 10.1093/cvr/cvt070. [DOI] [PubMed] [Google Scholar]
- 50. Nam DH, Kim E, Benham A, Park HK, Soibam B, Taffet GE, et al. Transient activation of AMPK preceding left ventricular pressure overload reduces adverse remodeling and preserves left ventricular function. FASEB J . 2019;33:711–721. doi: 10.1096/fj.201800602R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mansour HH, Omran MM, Hasan HF, El Kiki SM. Modulation of bleomycin-induced oxidative stress and pulmonary fibrosis by N-acetylcysteine in rats via AMPK/SIRT1/NF-κβ. Clin Exp Pharmacol Physiol . 2020;47:1943–1952. doi: 10.1111/1440-1681.13378. [DOI] [PubMed] [Google Scholar]
- 52. Chen X, Walther FJ, Sengers RM, Laghmani H, Salam A, Folkerts G, et al. Metformin attenuates hyperoxia-induced lung injury in neonatal rats by reducing the inflammatory response. Am J Physiol Lung Cell Mol Physiol . 2015;309:L262–L270. doi: 10.1152/ajplung.00389.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caligiuri A, Bertolani C, Guerra CT, Aleffi S, Galastri S, Trappoliere M, et al. Adenosine monophosphate-activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology . 2008;47:668–676. doi: 10.1002/hep.21995. [DOI] [PubMed] [Google Scholar]
- 54. Lin Q, Huang Z, Cai G, Fan X, Yan X, Liu Z, et al. Activating AMP-activated protein kinase mediates fibroblast growth factor 1 protection from nonalcoholic fatty liver disease in mice. Hepatology . 2021;73:2206–2222. doi: 10.1002/hep.31568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sato Y, Qiu J, Hirose T, Miura T, Sato Y, Kohzuki M, et al. Metformin slows liver cyst formation and fibrosis in experimental model of polycystic liver disease. Am J Physiol Gastrointest Liver Physiol . 2021;320:G464–G473. doi: 10.1152/ajpgi.00120.2020. [DOI] [PubMed] [Google Scholar]
- 56. Chen KH, Hsu HH, Lee CC, Yen TH, Ko YC, Yang CW, et al. The AMPK agonist AICAR inhibits TGF-β1 induced activation of kidney myofibroblasts. PLoS One . 2014;9:e106554. doi: 10.1371/journal.pone.0106554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thakur S, Viswanadhapalli S, Kopp JB, Shi Q, Barnes JL, Block K, et al. Activation of AMP-activated protein kinase prevents TGF-β1-induced epithelial-mesenchymal transition and myofibroblast activation. Am J Pathol . 2015;185:2168–2180. doi: 10.1016/j.ajpath.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Butcher AJ, Prihandoko R, Kong KC, McWilliams P, Edwards JM, Bottrill A, et al. Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J Biol Chem . 2011;286:11506–11518. doi: 10.1074/jbc.M110.154526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tobin AB, Butcher AJ, Kong KC. Location, location, location...site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci . 2008;29:413–420. doi: 10.1016/j.tips.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron . 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA . 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guettier JM, Gautam D, Scarselli M, Ruiz de Azua I, Li JH, Rosemond E, et al. A chemical-genetic approach to study G protein regulation of beta cell function in vivo. Proc Natl Acad Sci USA . 2009;106:19197–19202. doi: 10.1073/pnas.0906593106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nakajima K, Wess J. Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Mol Pharmacol . 2012;82:575–582. doi: 10.1124/mol.112.080358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pronin AN, Wang Q, Slepak VZ. Teaching an old drug new tricks: agonism, antagonism, and biased signaling of pilocarpine through m3 muscarinic acetylcholine receptor. Mol Pharmacol . 2017;92:601–612. doi: 10.1124/mol.117.109678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Blais-Lecours P, Laouafa S, Arias-Reyes C, Santos WL, Joseph V, Burgess JK, et al. Metabolic adaptation of airway smooth muscle cells to an sphk2 substrate precedes cytostasis. Am J Respir Cell Mol Biol . 2020;62:35–42. doi: 10.1165/rcmb.2018-0397OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Burgess JK, Ketheson A, Faiz A, Limbert Rempel KA, Oliver BG, Ward JPT, et al. Phenotype and functional features of human telomerase reverse transcriptase immortalized human airway smooth muscle cells from asthmatic and non-asthmatic donors. Sci Rep . 2018;8:805. doi: 10.1038/s41598-017-18429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oenema TA, Kolahian S, Nanninga JE, Rieks D, Hiemstra PS, Zuyderduyn S, et al. Pro-inflammatory mechanisms of muscarinic receptor stimulation in airway smooth muscle. Respir Res . 2010;11:130. doi: 10.1186/1465-9921-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gosens R, Dueck G, Rector E, Nunes RO, Gerthoffer WT, Unruh H, et al. Cooperative regulation of GSK-3 by muscarinic and PDGF receptors is associated with airway myocyte proliferation. Am J Physiol Lung Cell Mol Physiol . 2007;293:L1348–L1358. doi: 10.1152/ajplung.00346.2007. [DOI] [PubMed] [Google Scholar]
- 69. Gosens R, Rieks D, Meurs H, Ninaber DK, Rabe KF, Nanninga J, et al. Muscarinic M3 receptor stimulation increases cigarette smoke-induced IL-8 secretion by human airway smooth muscle cells. Eur Respir J . 2009;34:1436–1443. doi: 10.1183/09031936.00045209. [DOI] [PubMed] [Google Scholar]
- 70. Gosens R, Stelmack GL, Dueck G, Mutawe MM, Hinton M, McNeill KD, et al. Caveolae facilitate muscarinic receptor-mediated intracellular Ca2+ mobilization and contraction in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol . 2007;293:L1406–L1418. doi: 10.1152/ajplung.00312.2007. [DOI] [PubMed] [Google Scholar]