

Abstract
Background
Neurodevelopmental disorder with spastic diplegia and visual defects (NEDSDV) is a rare autosomal dominant syndrome, which is caused by the heterozygous germline loss‐of‐function variants in CTNNB1.
Methods
We evaluated the clinical and genetic findings of 24 previously undescribed Chinese patients affected by CTNNB1‐related disorders and explored the possible ethnicity‐related phenotypic variations.
Results
Twenty‐one loss‐of‐function variants were identified within these 24 NEDSDV patients, including 14 novel CTNNB1 variants and 7 recurrent ones. The prominent clinical manifestations in our cohort are developmental delay/intellectual disability (100%), motor delay (100%), speech impairment (100%), dystonia (87.5%) and microcephaly (69.6%). The common facial dysmorphisms were consistent with previous reports, including wide nasal bridge (58.3%), bulbous nose (45.8%), long philtrum (45.8%) and thin upper lip (45.8%). In addition, 19 patients (79.2%) in our cohort had mild visual defects, while one affected individual (4.2%) had familial exudative vitreoretinopathy. Notably, we discovered that 20 patients (83.3%) exhibited various behavioral abnormalities, which is described in Chinese patients for the first time.
Conclusion
We provided the largest known Chinese cohort with pathogenic CTNNB1 variants, which not only helps to expand the variant spectrum of CTNNB1 gene, but further delineates the typical phenotype of this disorder in Chinese population.
Keywords: CTNNB1 variants, genotype, Mainland China, neurodevelopmental disorder with spastic diplegia and visual defects (NEDSDV), phenotype
In this study, we delineated the genotypes and phenotypes of 24 Chinese NEDSDV patients, which broaden the variant spectrum of CTNNB1 gene and clinical phenotype spectrum of NEDSDV.

1. INTRODUCTION
CTNNB1 (MIM *116806) was first described to be related with intellectual disability by de Ligt et al. (2012). Since then, many individuals with CTNNB1 loss‐of‐function variants have been reported to suffer from a rare multisystem neurodevelopmental disorder, which was defined as neurodevelopmental disorder with spastic diplegia and visual defects (NEDSDV, MIM #615075). NEDSDV is an autosomal dominant disorder and its characteristic features include developmental delay/intellectual delay (DD/ID), speech impairment, microcephaly, motor delay, autistic spectrum disorder (ASD), hypotonia, progressive peripheral spasticity, dysmorphic craniofacial, and various degrees of visual abnormalities (Ho et al., 2021; Kharbanda et al., 2017; Kuechler et al., 2014; Rossetti et al., 2021; Tucci et al., 2014).
The CTNNB1 gene is located on chromosome 3p22.1 and encodes the ubiquitously expressed and evolutionarily conserved protein beta‐catenin, a 781‐amino acid protein with an N‐terminal domain (NTD), 12 central armadillo (ARM) repeat domains, and the C‐terminal domain (CTD) (Xu & Kimelman, 2007). As a multitasking protein, beta‐catenin is not only a core component of the cadherin complex but also a key factor in the canonical Wnt signaling, which plays an important role in stem cell renewal, as well as cell proliferation and differentiation during embryogenesis (Steinhart & Angers, 2018; Valenta et al., 2012). Many studies have identified that aberrant activation of beta‐catenin may promote various tumorigenesis, such as colorectal cancer and hepatocellular carcinoma (Bian et al., 2020; Zhang & Wang, 2020), while ablation of beta‐catenin would affect the nervous system development (Zechner et al., 2003).
To date, less than 100 different loss‐of‐function variants in CTNNB1 gene have been reported in the literatures, mostly from non‐Chinese populations. In this study, we reported the clinical characteristics and genetic findings of 24 mainland Chinese patients with pathogenic CTNNB1 variants. So far, this is the largest case series for NEDSDV patients of Chinese ethnicity. We also attempted to review the clinical features of our CTNNB1 variant patients together with 13 previously reported Chinese patients and 37 non‐Chinese patients with CTNNB1 variants (Ho et al., 2021; Ke & Chen, 2020; Li et al., 2017), and explore the possible ethnicity related phenotypic and genetic diversity in CTNNB1‐related NEDSDV.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
Written informed consent for publishing clinical information with photographs was obtained from the parents of patients. The study was conducted according to the principles of the Declaration of Helsinki and was approved by the Ethics Committee of Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University (XHEC‐D‐2022‐099).
2.2. Cohort
We gathered 24 patients with CTNNB1‐related neurodevelopmental disorder from 23 families in Mainland China: 14 males (58%) and 10 females (42%) were included in our cohort, with ages ranging from 0.6 to 11 years old. We abstracted the information about the developmental delay, autism diagnosis, and MRI or CT scans from the medical record. Besides, detailed information of patients was collected through the questionnaire completed by the parents, including the growth parameters, head circumference, development/neurological features, behavioral abnormalities, and other clinical features. Additionally, the pictures of the frontal and bilateral views of the face of the patients were provided by their parents. Two clinicians from Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University estimated the pictures of the patients to analyze their craniofacial features.
2.3. Genetic analysis
In our cohort, different sequencing methods and platforms were applied: exome sequencing (ES) were performed in most patients (23/24), while multigene panel testing were performed in 1 patient. The disease‐causing CTNNB1 variants identified from the genetic sequencing reports provided by the parents were rechecked and unified based on the human genome reference assembly GRCh37/hg19 and CTNNB1 transcript NM_001904.4.
2.4. Statistical analysis
We designed Chinese (our 24 patients and 13 Chinese patients previously reported) and non‐Chinese groups (37 non‐Chinese patients previously reported) with the same patients' number (n = 37) artificially to explore the similarities and differences between ethnicities. Relevant literatures were searched through PubMed database using the keywords “CTNNB1” and “Intellectual disability” or “Neurodevelopmental disorder”. We preferred to include the reports with series cases rather than single case reports, for more consistent evaluation criteria and description method. As reports about Chinese patients are still few, we included two single‐case reports in our comparison. Meanwhile, these cases should cover most of the phenotypes we concerned. Reports only focusing on ocular disease were excluded in our study. The statistical significance in phenotype and genotype differences of these two groups have been assessed by Fisher's exact test.
3. RESULTS
3.1. Cohort characteristics
A total of 24 molecularly diagnosed Chinese patients from 23 unrelated families were recruited into this study. All patients had no positive family history of CTNNB1‐related diseases except for patients 23 and 24, who are siblings. There were 10 females (42%) and 14 males (58%) with an average age of 3.07 years (range: 0.6 to 11y). The age of initial assessment was from 3 months to 2 years and the mean age of diagnosis was 1.94 years. None of these patients have been published in the literature. Their clinical findings were summarized in Tables 1 and 2 (details in Supplementary Table S1) and presented in Figure 1. For this study, eight previous publications reporting 13 Chinese patients and 37 patients from other countries were included for assessing the similarities and differences between Chinese NEDSDV patients and those of other ethnicities (Table 2) (Ho et al., 2021; Ke & Chen, 2020; Kharbanda et al., 2017; Kuechler et al., 2014; Li et al., 2017; Rossetti et al., 2021; Tucci et al., 2014; Wang, Zhao, et al., 2019).
TABLE 1.
Main clinical features of patients with CTNNB1 pathogenic variants in our cohort
| Feature | N | Total | % |
|---|---|---|---|
| Dysmorphisms | |||
| Primary microcephaly (<3rd percentile) | 3 | 20 | 15.0 |
| Microcephaly (<3rd percentile) | 16 | 23 | 69.6 |
| Narrow forehead | 1 | 24 | 4.2 |
| Fair skin | 2 | 24 | 8.3 |
| Sparse hair | 3 | 24 | 12.5 |
| Sparse eyebrows | 9 | 24 | 37.5 |
| Long eyelashes | 10 | 24 | 41.7 |
| Wide nasal bridge | 14 | 24 | 58.3 |
| Bulbous nose | 11 | 24 | 45.8 |
| Long philtrum | 11 | 24 | 45.8 |
| Thin upper lip | 11 | 24 | 45.8 |
| Ear anomalies | 11 | 24 | 45.8 |
| Short stature/delayed growth (<3rd percentile) | 3 | 24 | 12.5 |
| Visual defects | |||
| Astigmatism | 3 | 24 | 12.5 |
| Shortsighted | 1 | 24 | 4.2 |
| Hyperopia | 11 | 24 | 45.8 |
| Strabismus | 15 | 24 | 62.5 |
| Amblyopia | 1 | 24 | 4.2 |
| Retinopathy/Vitreous opacities/FEVR | 2 | 24 | 8.3 |
| Behavioral features | |||
| Autism spectrum disorder | 3 | 24 | 12.5 |
| Impulsiveness | 7 | 24 | 29.2 |
| Anxiety | 8 | 24 | 33.3 |
| Hyperactivity | 5 | 24 | 20.8 |
| Self‐injury | 2 | 24 | 8.3 |
| Repetitive behaviors | 8 | 24 | 33.3 |
| Sleep disturbance | 17 | 24 | 70.8 |
| Other clinical features | |||
| Gastroesophageal reflux | 1 | 24 | 4.2 |
| Diarrhea/constipation | 6 | 24 | 25.0 |
| Recurring upper respiratory tract infections | 1 | 24 | 4.2 |
| Congenital heart defect | 1 | 24 | 4.2 |
| Allergies | 1 | 24 | 4.2 |
Abbreviation: FEVR, familial exudative vitreoretinopathy.
TABLE 2.
Incidences and percentages of phenotypes in Chinese CTNNB1 variant patients and comparison with the literatures
| Clinical features (Not every characteristic is recorded for all patients) | 24 Novel Chinese patients | 13 Previously published Chinese patients (Ho et al., Ke et al., Li et al., Wang et al.) | Total 37 Chinese patients | 37 Previously published non‐Chinese patients (Kharbanda et al., Kuechler et al., Rossetti et al., Tucci et al) | p‐value (by Fisher exact test) |
|---|---|---|---|---|---|
| Gender (M:F) | 14:10 | 6:7 | 20:17 | 17:20 | .6423 |
| Developmental/neurological | |||||
| Microcephaly | 16/23 (69.6%) | 8/11 (72.7%) | 24/34 (70.6%) | 32/37 (86.5%) | .1464 |
| Dystonia | 21/24 (87.5%) | 11/12 (91.7%) | 32/36 (88.9%) | 36/37 (97.3%) | .1992 |
| Motor delay | 24/24 (100%) | 12/12 (100%) | 36/36 (100%) | 37/37 (100%) | 1.0000 |
| Speech impairment (age ≥3) | 5/5 (100%) | 3/4 (75.0%) | 8/9 (88.9%) | 35/36 (97.2%) | .3636 |
| DD/ID | 18/18 (100%) | 13/13 (100%) | 31/31 (100%) | 35/36 (97.2%) | 1.0000 |
| Brain imaging anomalies | 7/13 (53.8%) | 0/10 (0%) | 7/23 (30.4%) | 5/32 (15.6%) | .2078 |
| Craniofacial dysmorphism | 22/24 (91.7%) | 12/12 (100%) | 34/36 (94.4%) | 35/36 (97.2%) | 1.0000 |
| Behavioral anomalies | 20/24 (83.3%) | NA | 20/24 (83.3%) | 25/37 (67.6%) | .2373 |
| Visual defects | 20/24 (83.3%) | 9/13 (69.2%) | 29/37 (78.4%) | 25/36 (69.4%) | .4328 |
| Variant type | |||||
| Nonsense | 10/24 (41.7%) | 5/13 (38.5%) | 15/37 (40.5%) | 18/37 (48.6%) | .6403 |
| Frameshift | 11/24 (45.8%) | 5/13 (38.5%) | 16/37 (43.2%) | 13/37 (35.1%) | .6343 |
| Splice site | 3/24 (12.5%) | 3/13 (38.5%) | 6/37 (16.2%) | 2/37 (5.4%) | .2611 |
| Missense | 0/24 (0%) | 0/13 (0%) | 0/37 (0%) | 2/37 (5.4%) | .4932 |
| Inframe mutation | 0/24 (0%) | 0/13 (0%) | 0/37 (0%) | 1/37 (2.7%) | 1.0000 |
| Whole gene | 0/24 (0%) | 0/13 (0%) | 0/37 (0%) | 1/37 (2.7%) | 1.0000 |
Abbreviations: DD, developmental delay; F, female; ID, intellectual disability; M, male; NA, not applicable.
FIGURE 1.
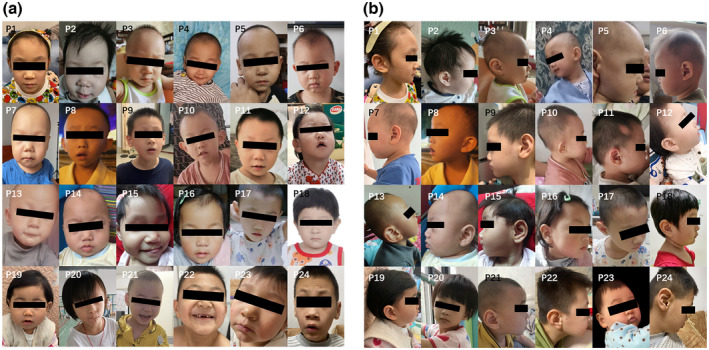
Frontal (a) and lateral (b) facial photos of 24 Chinese individuals with CTNNB1 loss‐of‐function variants
3.2. Dysmorphic features
Twenty‐two patients in our cohort presented with similar but not prominent facial features to what had been previously reported in the literature (Table 1 and Supplementary Table S2). Characteristic facial phenotype included long eyelashes (10/24), wide nasal bridge (14/24), bulbous nasal tip (11/24), long philtrum (11/24), and thin upper lip (11/24). One affected individual was found to have narrowed forehead and nine with sparse eyebrows. Intriguingly, we also noticed 11 of these patients showed large or protruding ears, which had been first reported in Ho et al's case (Ho et al., 2021), hence we proposed this may be a notable feature of Chinese. Besides, one patient had aural deformities and received auricular repairment. The detailed dysmorphic features of individuals were summarized in Supplementary Table S2, and clinical photos of affected individuals were shown in Figure 1.
3.3. Development and growth
The growth details for our cohort were summarized in Supplementary Tables S1 and S3. All 24 patients in our cohort were described with motor delay involving fine and gross motor areas, while 5 patients showed speech impairment, including four could only speak single word or phases over the age of 3 years, and one only speaks short sentences at 6 years. Sixteen patients were diagnosed with microcephaly (occipitofrontal head circumference [OFC] below 3rd percentile for sex, age, and ethnicity), including two had primary microcephaly (at birth). Meanwhile, most patients had age‐appropriate weight and height except for seven of them presenting short stature (height below 3rd percentile) or low weight (body weight below 3rd percentile).
3.4. Neurological features
DD/ID was described in the medical records of 18 patients. Among them, 12 cases were professionally assessed and diagnosed with mild‐to‐severe DD/ID. However, due to the differences in evaluation institutions and reporting forms, only seven of them had a full‐scale score, while the remaining five did not. As a common syndrome of CTNNB1‐related disorder, dystonia was also presented in 21 patients of our cohort. Meanwhile, abnormal gait was noted in all 21 patients who were able to walk with or without help. Thirteen patients had received neurological examination, and seven of them showed abnormal brain magnetic resonance imaging (MRI) or computed tomography (CT) findings, including widened extracerebral spaces (n = 2), white matter abnormalities (n = 2), enlargement of ventricles (n = 2), and delayed myelination (n = 1). Besides, 2/6 patients had abnormal electroencephalography (EGG), with one of them developing seizures. Their neurological features were summarized in Table 2 and Supplementary Table S1.
3.5. Behavioral features
As reported, the majority of our patients exhibited different behavioral abnormalities similar to those observed in Western patients (Table 1 and Supplementary Table S1). Among them, 3 patients were noticed to suffer from ASD, and two of them were finally diagnosed by formal autism assessment. Furthermore, 11 patients were more anxious or hyperactive in daily life, 8 patients exhibited repetitive behaviors, 7 patients were reported as impulsiveness, and 2 patients showed overly sensitivity to touch or voice. Even more, two patients had once self‐injury behaviors. Sleep disturbances were also found in 18 out of 24 patients, mainly including difficulties to fall asleep and night terror.
3.6. Ocular findings
Consistent with previous reports, our patients also presented a series of visual defects (Table 1 and Supplementary Table S2), including astigmatism, shortsighted, hyperopia, strabismus, amblyopia, and even familial exudative vitreoretinopathy (FEVR). Among these, strabismus is the most common one, with 15 patients presenting this symptom. One patient was noticed unable to chase objects with her eyes after birth, and then was diagnosed with binocular cataract and bilateral FEVR. At the age of 9 months, she was treated with vitrectomy combined with a lens and retinal resection.
3.7. Bone and joint deformity
Nine patients showed flatfoot, three cases exhibited strephexopodia, while four cases showed limited finger/wrist movement, and four individuals had spinal deformity. In addition, one patient had polydactyly, and had been treated by surgery.
3.8. Other clinical features
Six patients were found to have diarrhea or constipation, and the gastroesophageal reflux was also noticed in one patient. One patient had atrial septal defect and recurring upper respiratory tract infections.
3.9. Genotype
We identified 21 different variants in 24 patients, all are loss‐of‐function variants, including 11 frameshift (45.8%), 10 nonsense (41.7%), and 3 splice site variants (12.5%). Fourteen out of these variants were novel, which were not described in the literature, ClinVar, Genome aggregation database (gnomAD) and Human Gene Mutation Database (HGMD), including c.495 + 2dup, c.1857_1858del, c.1154dup, c.735‐1G > T, c.299_300del, c.987dup, c.1843dup, c.2010del, c.931del, c.1760_1761del, c.1853dup, c.950del, c.1689_1690dup, and c.2098dup. All variants were de novo, except for one variant that had not been verified in father and another variant that had not ruled out low‐grade parental mosaicism. All of the variants were classified as pathogenic or likely pathogenic variants based on the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). These variants were predicted to result in various amino acid changes, while amino acids 90 and 587 were the most common sites of variation, each accounting for 12.5% (3/24). The genotypic information in our cohort and the pathogenic variants previously reported in the literature are showed in Figure 2 and summarized in Table 3.
FIGURE 2.
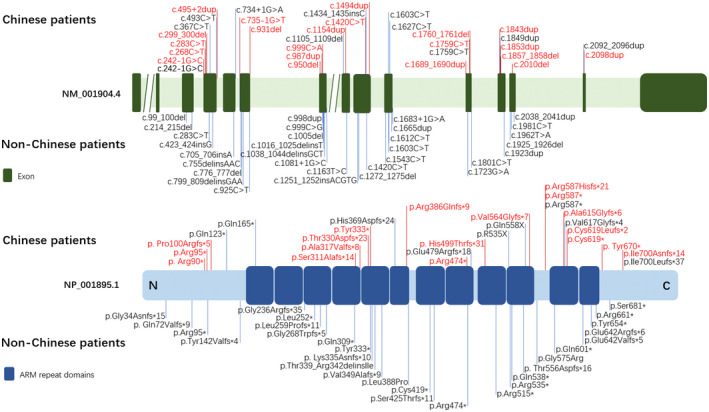
Summary of CTNNB1 pathogenic variants in 37 Chinese patients and 37 non‐Chinese patients. The variants in our study are marked in red
TABLE 3.
Overview of all CTNNB1 pathogenic variants identified in our cohort
| Patient | Genomic position | cDNA position | Protein position | Exon | Variant type | Inheritance | ACMG classification | Reported variant |
|---|---|---|---|---|---|---|---|---|
| P1 | chr3:41266700 | c.495 + 2dup | splice variant | Intron 4 | Splicing | De novo | Likely pathogenic | Novel |
| P2 | chr3:41277893–41,277,894 | c.1857_1858del | p.(Cys619*) | 12 | Nonsense | De novo | Pathogenic | Novel |
| P3 | chr3:41274904 | c.1154dup | p.(Arg386Glnfs*9) | 8 | Frameshift | De novo | Pathogenic | Novel |
| P4 | chr3:41277290 | c.1759C > T | p.(Arg587*) | 11 | Nonsense | De novo | Pathogenic | Reported |
| P5 | chr3:41277290 | c.1759C > T | p.(Arg587*) | 11 | Nonsense | De novo | Pathogenic | Reported |
| P6 | chr3:41275328 | c.1494dup | p.(His499Thrfs*31) | 9 | Frameshift | De novo | Pathogenic | Reported |
| P7 | chr3:41267150 | c.735‐1G > T | Splice variant | Intron 5 | Splicing | De novo | Pathogenic | Novel |
| P8 | chr3:41266471 | c.268C > T | p.(Arg90*) | 4 | Nonsense | De novo | Pathogenic | Reported |
| P9 | chr3:41266502–41,266,503 | c.299_300del | p.(Pro100Argfs*5) | 4 | Frameshift | De novo | Pathogenic | Novel |
| P10 | chr3:41268761 | c.999C > A | p.(Tyr333*) | 7 | Nonsense | De novo | Pathogenic | Reported |
| P11 | chr3:41268749 | c.987dup | p.(Thr330Aspfs*23) | 7 | Frameshift | De novo | Pathogenic | Novel |
| P12 | chr3:41266486 | c.283C > T | p.(Arg95*) | 4 | Nonsense | De novo | Pathogenic | Reported |
| P13 | chr3:41277879 | c.1843dup | p.(Ala615Glyfs*6) | 12 | Frameshift | De novo | Pathogenic | Novel |
| P14 | chr3:41278134 | c.2010del | p.(Tyr670*) | 13 | Nonsense | De novo | Pathogenic | Novel |
| P15 | chr3:41267347 | c.931del | p.(Ser311Alafs*14) | 6 | Frameshift | De novo | Pathogenic | Novel |
| P16 | chr3:41277291–41,277,292 | c.1760_1761del | p.(Arg587Hisfs*21) | 11 | Frameshift | De novo | Pathogenic | Novel |
| P17 | chr3:41277889 | c.1853dup | p.(Cys619Leufs*2) | 12 | Frameshift | De novo | Pathogenic | Novel |
| P18 | chr3:41268712 | c.950del | p.(Ala317Valfs*8) | 7 | Frameshift | De novo | Pathogenic | Novel |
| P19 | chr3:41277220–41,277,221 | c.1689_1690dup | p.(Val564Glyfs*7) | 11 | Frameshift | De novo | Pathogenic | Novel |
| P20 | chr3:41266444 | c.242‐1G > C | Splice variant | Intron 3 | Splicing | NK | Likely pathogenic | Reported |
| P21 | chr3:41279528 | c.2098dup | p.(Ile700Asnfs*14) | 14 | Frameshift | De novo | Pathogenic | Novel |
| P22 | chr3:41275254 | c.1420C > T | p.(Arg474*) | 9 | Nonsense | De novo | Pathogenic | Reported |
| P23 | chr3:41266471 | c.268C > T | p.(Arg90*) | 4 | Nonsense | NK | Pathogenic | Reported |
| P24 | chr3:41266471 | c.268C > T | p.(Arg90*) | 4 | Nonsense | NK | Pathogenic | Reported |
Note: Genomic, cDNA, and protein position are given as well as affected exons. All genomic positions are based on CRCh37/hg19; CTNNB1 reference sequence is NM_001904.4; NK, not known; *, the stop codon; ACMG, American College of Medical Genetics and Genomic.
4. DISCUSSION
Few Chinese NEDSDV patients had been reported up till now. Herein, we described the clinical and molecular genetic characterization of 24 mainland Chinese NEDSDV individuals to expand the genotype and phenotype spectrum of NEDSDV. We also compared the major clinical presentations of our 24 patients and 13 previous reported Chinese NEDSDV individuals with 37 patients of other ethnic backgrounds, attempting to elucidate the ethnicity‐related features of this disorder (Ho et al., 2021; Ke & Chen, 2020; Kharbanda et al., 2017; Kuechler et al., 2014; Li et al., 2017; Rossetti et al., 2021; Tucci et al., 2014; Wang, Zhao, et al., 2019).
This study discovered 21 different CTNNB1 variants, including 14 novel pathogenic variants, which broadened CTNNB1 variant spectrum. Most of the variants reported in our cohort are localized in ARM repeat domains and no particular mutational hotspots identified. So far, all of the reported NEDSDV‐related CTNNB1 variants are heterozygous variants, except that recently Taylor et al reported a patient with bi‐allelic variant which was inherited from her unaffected parents (Taylor et al., 2022). The majority of reported pathogenic variants in CTNNB1 are loss‐of‐function variants such as frameshift, nonsense, or splicing variants, except for a few cases with missense variants (Kuechler et al., 2014; Rossetti et al., 2021). Similar to previous reports, all of our patients had heterozygous loss‐of‐function variants. As for the type of variants, there is no significant difference between the Chinese group and the non‐Chinese group, with the nonsense (40.5% vs 48.6%, p = .6403) and frameshift (43.2% vs 35.1%, p = .6343) variants being the most frequent. In addition, most of the reported variants arise de novo, although some familial cases have been described (Ho et al., 2021; Kuechler et al., 2014; Wang, Zhao, et al., 2019). Consistent with these results, the great majority of our variants are de novo. However, we noticed that both siblings in our cohort carried a heterozygous CTNNB1 nonsense variant c.268C > T, p.(Arg90*). Neither parent was confirmed to carry this variant by means of Sanger sequencing of whole blood DNA, indicating the possibility of parental germline mosaicism.
DD/ID is the cardinal clinical feature of CTNNB1‐related disorders. In vivo experiments with beta‐catenin knockout mice also demonstrated that haploinsufficiency of this gene leads to dysregulation of synaptic plasticity, neuronal network connectivity, and synaptic adhesion, providing the underlying pathogenic mechanism for neurodevelopmental disorder (Tucci et al., 2014; Wickham et al., 2019). Consistent with previous reports, 18 patients in our cohort were professionally assessed and all diagnosed as DD/ID. We further compared the frequencies of DD/ID in Chinese patients (31/31) and non‐Chinese patients (35/36), and there was no statistically significant difference (100% vs 97.2%, p = 1.0000). We also revealed that there was no statistically significant difference between two groups in other neurological features including microcephaly (70.6% vs 86.5%, p = .1464), motor delay (100% vs 100%, p = 1.0000), dystonia (88.9% vs 97.3%, p = .1992), speech impairment (88.9% vs 97.2%, p = .3636), and brain imaging anomalies (30.4% vs 15.6%, p = .2078).
There was no significant difference concerning the craniofacial dysmorphism between Chinese cohort (34/36) and non‐Chinese cohort (35/36). The main common facial features in our cohort are bulbous nasal tip, wide nasal bridge, long philtrum, and thin upper lip. Noteworthy, one of the most common dysmorphism features in our patients is relatively large ears, which has only been reported in Chinese up to now. Nearly half of the individuals in our study presented it, and in Ho′s cohort, its incidence was higher (8/9). It indicates the large ears may be an ethnic‐specific phenotype, but we still cannot draw a definitive conclusion as this diagnosis is very subjective to clinicians.
Meanwhile, the visual defect is also a common symptom with an incidence of 78.4% in Chinese and 69.4% in non‐Chinese. And among the specific manifestations of visual defect, the strabismus is the most frequent one. A previous study had suggested that the Wnt/beta‐catenin signaling pathway plays a pivotal role in the proper formation of both the anterior segment of the eye and neurovascular retina (Wang, Liu, et al., 2019), which may help to explain why many reported cases with CTNNB1 variants have mild‐to‐severe visual symptoms. Most notably, patient 15, who had a truncating variant (p.Ser311Alafs*14) in exon 6, was diagnosed with binocular cataract, FEVR, and retinal detachment, showing the most serious ocular phenotype. Since the relationship between CTNNB1 haploinsufficiency and FEVR was first reported in 2016 (Dixon et al., 2016), a few cases had been reported and most of the patients were of Asian ethnicity (Coussa et al., 2020; Ke & Chen, 2020; Li et al., 2017; Panagiotou et al., 2017; Sun et al., 2019; Wang, Zhao, et al., 2019). However, our results showed that there was no statistically significant difference between Chinese patients (7/37) and non‐Chinese patients (2/36) in the frequency of FEVR. Since reports focusing on eye diseases were not included in our study, further research with larger sample size is needed to confirm whether ethnicity plays a role in phenotypic variations of NEDSDV. In addition, patient 15 also had an atrial septal defect. Congenital heart disease was also observed in previously published patients, but it was not specifically mentioned (Ke & Chen, 2020; Kuechler et al., 2014; Rossetti et al., 2021). Considering that canonical Wnt signaling and beta‐catenin play a critically important role in proper cardiogenesis (Piven & Winata, 2017), the occurrence of this phenotype may be related to the disruption of beta‐catenin transcriptional activity.
In this study, we described the behavioral features in Chinese patients for the first time. Twenty out of our 24 patients (83.3%) showed behavioral abnormalities, with anxious (33.3%), hyperactivity (20.8%), repetitive (33.3%) or impulsive behaviors (29.2%), and sleep disturbances (70.8%) were the most frequent findings. There was no significant difference in behavioral abnormalities between Chinese cohort (20/24) and non‐Chinese cohort (25/37). As reported in Western patients, our study also recorded three patients with ASD phenotype. Previous study had identified that CTNNB1 directly interacts with multiple top ASD risk genes (O'Roak et al., 2012). Likewise, autism‐associated behavioral defects had been noticed in the Ctnnb1 conditional knockout mice (Dong et al., 2016). Notably, we found that one of these three patients presented polydactyly, a phenotype that was first reported by Ke et al. in a Chinese patient (Ke & Chen, 2020). To our knowledge, this is the second Chinese case. Since the same symptom was not detected in patients from outside China, we considered polydactyly may be a unique phenotype in Chinese patients. However, further studies are needed to confirm our observation.
This study had several limitations. First of all, clinical information obtained from medical records and questionnaires may be limited due to varying medical criteria between hospitals and recall bias. Secondly, we focused only on studies with multiple non‐Chinese patients, single‐case studies, and reports focusing on eye disease were not included in this study. Given the limitations of our study, further studies with larger sample size are needed to draw more definitive conclusions.
5. CONCLUSION
In summary, we delineated the genotypes and phenotypes of 24 Chinese NEDSDV patients, which broaden the CTNNB1 variant spectrum and clinical phenotype spectrum of NEDSDV. We further explored the possible ethnicity‐related phenotypic variations in CTNNB1‐related NEDSDV. However, no ethnic‐specific feature had been noticed except relatively large ears.
AUTHOR CONTRIBUTIONS
Dan Yan and Yongkun Zhan made acquisition of patients' information, assembly and analysis of data, and manuscript writing. Yu Sun reviewed and edited the manuscript; Na Xu helped in data analysis. Yongguo Yu designed the study and did administration, financial support, and final approval of the manuscript. All authors have read and approved the final manuscript.
FUNDING INFORMATION
This work was supported by the National Key R&D Program of China (No. 2019YFC1005100, to YGY); the National Natural Science Foundation of China (No. 81873671 and No. 82070914, to YGY); the Shanghai Science and Technology Commission (No. 19140904500, to YGY); Shanghai Municipal Education Commission‐Gaofeng Clinical Medicine Grant Support (No. 20191908, to YGY); Shanghai Municipal Commission of Health and Family Planning (No. 20204Y0450, to YKZ).
CONFLICT OF INTEREST
The authors have no relevant financial or non‐financial interests to disclose.
Supporting information
Table S1 Main clinical features of individuals with CTNNB1 pathogenic variants
TABLE S2 Dysmorphic features in patients with CTNNB1 pathogenic variants
TABLE S3 Growth parameters of individuals with CTNNB1 pathogenic variants
ACKNOWLEDGMENTS
We would like to acknowledge the affected patients and their families for the participation in the study.
Yan, D. , Sun, Y. , Xu, N. , Yu, Y. , Zhan, Y. , & the Mainland Chinese League of NEDSDV Rare Disease (2022). Genetic and clinical characteristics of 24 mainland Chinese patients with CTNNB1 loss‐of‐function variants. Molecular Genetics & Genomic Medicine, 10, e2067. 10.1002/mgg3.2067
Yongguo Yu and Yongkun Zhan equal contribution as co‐corresponding author.
Contributor Information
Yongguo Yu, Email: yuyongguo@shsmu.edu.cn.
Yongkun Zhan, Email: zhanyongkun@xinhuamed.com.cn.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- Bian, J. , Dannappel, M. , Wan, C. , & Firestein, R. (2020). Transcriptional regulation of Wnt/beta‐catenin pathway in colorectal cancer. Cells, 9(9), 2125. 10.3390/cells9092125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussa, R. G. , Zhao, Y. , DeBenedictis, M. J. , Babiuch, A. , Sears, J. , & Traboulsi, E. I. (2020). Novel mutation in CTNNB1 causes familial exudative vitreoretinopathy (FEVR) and microcephaly: Case report and review of the literature. Ophthalmic Genetics, 41(1), 63–68. 10.1080/13816810.2020.1723118 [DOI] [PubMed] [Google Scholar]
- de Ligt, J. , Willemsen, M. H. , van Bon, B. W. , Kleefstra, T. , Yntema, H. G. , Kroes, T. , Vulto‐van Silfhout, A. T. , Koolen, D. A. , de Vries, P. , Gilissen, C. , del Rosario, M. , Hoischen, A. , Scheffer, H. , de Vries, B. B. A. , Brunner, H. G. , Veltman, J. A. , & Vissers, L. E. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. The New England Journal of Medicine, 367(20), 1921–1929. 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- Dixon, M. W. , Stem, M. S. , Schuette, J. L. , Keegan, C. E. , & Besirli, C. G. (2016). CTNNB1 mutation associated with familial exudative vitreoretinopathy (FEVR) phenotype. Ophthalmic Genetics, 37(4), 468–470. 10.3109/13816810.2015.1120318 [DOI] [PubMed] [Google Scholar]
- Dong, F. , Jiang, J. , McSweeney, C. , Zou, D. , Liu, L. , & Mao, Y. (2016). Deletion of CTNNB1 in inhibitory circuitry contributes to autism‐associated behavioral defects. Human Molecular Genetics, 25(13), 2738–2751. 10.1093/hmg/ddw131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, S. , Tsang, M. H. , Fung, J. L. , Huang, H. , Chow, C. B. , Cheng, S. S. , Luk, H. M. , Chung, B. H. Y. , & Lo, I. F. (2021). CTNNB1‐related neurodevelopmental disorder in a Chinese population: A case series. American Journal of Medical Genetics. Part A, 188(1), 130–137. 10.1002/ajmg.a.62504 [DOI] [PubMed] [Google Scholar]
- Ke, Z. , & Chen, Y. (2020). Case report: A de novo CTNNB1 nonsense mutation associated with neurodevelopmental disorder, retinal detachment, polydactyly. Frontiers in Pediatrics, 8, 575673. 10.3389/fped.2020.575673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda, M. , Pilz, D. T. , Tomkins, S. , Chandler, K. , Saggar, A. , Fryer, A. , McKay, V. , Louro, P. , Smith, J. C. , Burn, J. , Kini, U. , De Burca, A. , FitzPatrick, D. R. , Kinning, E. , & Study, D. D. D. (2017). Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. European Journal of Medical Genetics, 60(2), 130–135. 10.1016/j.ejmg.2016.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuechler, A. , Willemsen, M. H. , Albrecht, B. , Bacino, C. A. , Bartholomew, D. W. , van Bokhoven, H. , van den Boogaard, M. J. H. , Bramswig, N. , Büttner, C. , Cremer, K. , Czeschik, J. C. , Engels, H. , van Gassen, K. , Graf, E. , van Haelst, M. , He, W. , Hogue, J. S. , Kempers, M. , Koolen, D. , … Wieczorek, D. (2014). De novo mutations in beta‐catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Human Genetics, 134(1), 97–109. 10.1007/s00439-014-1498-1 [DOI] [PubMed] [Google Scholar]
- Li, N. , Xu, Y. , Li, G. , Yu, T. , Yao, R. E. , Wang, X. , & Wang, J. (2017). Exome sequencing identifies a de novo mutation of CTNNB1 gene in a patient mainly presented with retinal detachment, lens and vitreous opacities, microcephaly, and developmental delay: Case report and literature review. Medicine (Baltimore), 96(20), e6914. 10.1097/MD.0000000000006914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak, B. J. , Vives, L. , Girirajan, S. , Karakoc, E. , Krumm, N. , Coe, B. P. , Levy, R. , Ko, A. , Lee, C. , Smith, J. D. , Turner, E. H. , Stanaway, I. B. , Vernot, B. , Malig, M. , Baker, C. , Reilly, B. , Akey, J. M. , Borenstein, E. , Rieder, M. J. , … Eichler, E. E. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485(7397), 246–250. 10.1038/nature10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagiotou, E. S. , Sanjurjo Soriano, C. , Poulter, J. A. , Lord, E. C. , Dzulova, D. , Kondo, H. , Hiyoshi, A. , Chung, B. H. , Chu, Y. W. , Lai, C. H. Y. , Tafoya, M. E. , Karjosukarso, D. , Collin, R. W. J. , Topping, J. , Downey, L. M. , Ali, M. , Inglehearn, C. F. , & Toomes, C. (2017). Defects in the cell signaling mediator beta‐catenin cause the retinal vascular condition FEVR. American Journal of Human Genetics, 100(6), 960–968. 10.1016/j.ajhg.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piven, O. O. , & Winata, C. L. (2017). The canonical way to make a heart: Beta‐catenin and plakoglobin in heart development and remodeling. Experimental Biology and Medicine (Maywood, N.J.), 242(18), 1735–1745. 10.1177/1535370217732737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti, L. Z. , Bekheirnia, M. R. , Lewis, A. M. , Mefford, H. C. , Golden‐Grant, K. , Tarczy‐Hornoch, K. , Briere, L. C. , Sweetser, D. A. , Walker, M. A. , Kravets, E. , Stevenson, D. A. , Bruenner, G. , Sebastian, J. , Knapo, J. , Rosenfeld, J. A. , Marcogliese, P. C. , Undiagnosed Diseases Network , & Wangler, M. F. (2021). Missense variants in CTNNB1 can be associated with vitreoretinopathy‐Seven new cases of CTNNB1‐associated neurodevelopmental disorder including a previously unreported retinal phenotype. Molecular Genetics & Genomic Medicine, 9(1), e1542. 10.1002/mgg3.1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhart, Z. , & Angers, S. (2018). Wnt signaling in development and tissue homeostasis. Development, 145(11), dev146589. 10.1242/dev.146589 [DOI] [PubMed] [Google Scholar]
- Sun, W. , Xiao, X. , Li, S. , Jia, X. , Wang, P. , & Zhang, Q. (2019). Germline mutations in CTNNB1 associated with syndromic FEVR or norrie disease. Investigative Ophthalmology & Visual Science, 60(1), 93–97. 10.1167/iovs.18-25142 [DOI] [PubMed] [Google Scholar]
- Taylor, R. L. , Soriano, C. S. , Williams, S. , Dzulova, D. , Ashworth, J. , Hall, G. , Gale, T. , Lloyd, I. C. , Inglehearn, C. F. , Toomes, C. , Douzgou, S. , & Black, G. C. (2022). Bi‐allelic mutation of CTNNB1 causes a severe form of syndromic microphthalmia, persistent foetal vasculature and vitreoretinal dysplasia. Orphanet Journal of Rare Diseases, 17(1), 110. 10.1186/s13023-022-02239-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucci, V. , Kleefstra, T. , Hardy, A. , Heise, I. , Maggi, S. , Willemsen, M. H. , Hilton, H. , Esapa, C. , Simon, M. , Buenavista, M. T. , McGuffin, L. , Vizor, L. , Dodero, L. , Tsaftaris, S. , Romero, R. , Nillesen, W. N. , Vissers, L. E. , Kempers, M. J. , Vulto‐van Silfhout, A. , … Nolan, P. M. (2014). Dominant beta‐catenin mutations cause intellectual disability with recognizable syndromic features. The Journal of Clinical Investigation, 124(4), 1468–1482. 10.1172/JCI70372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta, T. , Hausmann, G. , & Basler, K. (2012). The many faces and functions of beta‐catenin. The EMBO Journal, 31(12), 2714–2736. 10.1038/emboj.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Zhao, Y. , Yang, L. , Han, S. , & Qi, M. (2019). Identification of a novel splice mutation in CTNNB1 gene in a Chinese family with both severe intellectual disability and serious visual defects. Neurological Sciences, 40(8), 1701–1704. 10.1007/s10072-019-03823-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Liu, C. H. , Huang, S. , & Chen, J. (2019). Wnt Signaling in vascular eye diseases. Progress in Retinal and Eye Research, 70, 110–133. 10.1016/j.preteyeres.2018.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, R. J. , Alexander, J. M. , Eden, L. W. , Valencia‐Yang, M. , Llamas, J. , Aubrey, J. R. , & Jacob, M. H. (2019). Learning impairments and molecular changes in the brain caused by beta‐catenin loss. Human Molecular Genetics, 28(17), 2965–2975. 10.1093/hmg/ddz115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , & Kimelman, D. (2007). Mechanistic insights from structural studies of beta‐catenin and its binding partners. Journal of Cell Science, 120(Pt 19), 3337–3344. 10.1242/jcs.013771 [DOI] [PubMed] [Google Scholar]
- Zechner, D. , Fujita, Y. , Hülsken, J. , Müller, T. , Walther, I. , Taketo, M. M. , Crenshaw, E. B. , Birchmeier, W. , & Birchmeier, C. (2003). β‐Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Developmental Biology, 258(2), 406–418. 10.1016/s0012-1606(03)00123-4 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , & Wang, X. (2020). Targeting the Wnt/beta‐catenin signaling pathway in cancer. Journal of Hematology & Oncology, 13(1), 165. 10.1186/s13045-020-00990-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Main clinical features of individuals with CTNNB1 pathogenic variants
TABLE S2 Dysmorphic features in patients with CTNNB1 pathogenic variants
TABLE S3 Growth parameters of individuals with CTNNB1 pathogenic variants
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.