

Abstract
Background
The calcium/calmodulin‐dependent serine protein kinase gene (CASK) is an essential gene in mammals, critical for neurodevelopment. The purpose of this study is to expand the understanding of the diagnosis of CASK‐linked disorders.
Materials/Methods
From clinical and genetic mutational analyses, relevant data in 2 Han Chinese patients were collected and analyzed. Real‐time quantitative PCR (RT‐qPCR) was performed to investigate the CASK expression levels in the patients. The X‐chromosome inactivation (XCI) patterns of the patients and their nuclear families were tested by quantitation of methylation of the polymorphic human androgen receptor (HUMARA) locus.
Results
Two Han Chinese patients both presented with intellectual disability (ID), microcephaly with pontine and cerebellar hypoplasia (MICPCH). Two de novo mutations of c.82C>T (p.Arg28*) and c.846C>G (p.Tyr282*) in CASK have been investigated and predicted to be deleterious, which have produced truncated proteins. The functional protein association network of STRING (http://string‐db.org) generated three‐dimensional (3D) atomic models based on protein sequences in CASK and two Arg28 and Tyr282 residues were marked. RT‐qPCR showed lower copy numbers of CASK expression in the patients than in their parents, as well as the sex‐ and age‐ matched control groups. Patient 1 showed a skewed XCI pattern, while no related changes noted in patient 2.
Conclusions
Patients carrying different nonsense variants may have different degrees of different clinical phenotypes. This study expands the spectrum of genotype and phenotype correlations of CASK‐linked disorders in the Han Chinese ethnicity and provides new insights into the molecular mechanism.
Keywords: CASK, developmental disorder, Han Chinese, microcephaly, X‐chromosome inactivation
Patients carrying different nonsynonymous variants had different clinical phenotypes. This study expands the spectrum of genotype and phenotype correlations of CASK disorders in the Han Chinese ethnicity, provides new insights into the molecular mechanism.

1. BACKGROUND
The calcium/calmodulin‐dependent serine protein kinase gene (CASK) (NCBI Reference Sequence: NM_001126054.2) is an essential gene in mammals, critical for neurodevelopment. Mutations are associated with different neurological disorders, resulting in phenotypes that range from intellectual disability to lethality, manifested as intellectual disability (ID), microcephaly with pontine and cerebellar hypoplasia (MICPCH), epileptic encephalopathy, X‐linked ID (XLID) with or without nystagmus, and autistic spectrum disorder (ASD) (Lee et al., 2020; Popp et al., 2017; Seto et al., 2017). MICPCH and XLID are its main types of manifestation.
The CASK protein includes a Ca2+/calmodulin‐dependent kinase domain, two L27 domains (from Lin‐2, Lin‐7), a postsynaptic density‐9/discs large/zonula occludens‐1 (PDZ) domain, an src homology 3 (SH3) domain and a guanylate kinase domain (Hata et al., 1996). Through these protein domains, the CASK proteins are able to interact with other proteins.
In addition, they are mostly present in dendrites, axons, synapses, and the nuclei of neurons, wherein they control several cellular functions, including the formation of the dendritic spine, outgrowth and differentiation of axon, transcriptional control, and cellular metabolism (Kuo et al., 2010; Mukherjee et al., 2020; Pan et al., 2021; Patel et al., 2020; Srivastava et al., 2016). Expression is significantly high in the central nervous system (CNS), especially during the period of brain development. It can transmit to the nucleus and interact directly with transcriptional factors responsible for conventional brain structure formation (Murakami et al., 2019).
The diagnosis of a CASK disorder is based on a female who is heterozygous or on a male who is hemizygous for a CASK pathogenic variant (Moog & Kutsche, 2013). Many mutations in CASK were reported to lead to CASK‐linked disorders (Hayashi et al., 2017; Huang & Hsueh, 2009; Studtmann et al., 2019), while only few cases of CASK‐linked disorders have been reported in Han Chinese. We recently encountered two cases of females whose clinical characteristics were consistent with those of CASK‐linked disorders. By next‐generation sequencing (NGS), two de novo heterozygous mutations in CASK were detected. Herein, we present the clinical characteristics of these two patients and explore the possible underlying pathogenesis of the disease.
2. MATERIALS AND METHODS
2.1. Patients
Two Han Chinese patients were diagnosed with CASK‐linked disorders at the pediatric neurology clinic of the Shanghai Children's Hospital from 2019 to 2021. Both cases fulfilled the criteria for the diagnosis of CASK‐linked disorders. Family histories were negative for neurological disorders. Screening for inherited metabolic disorders were insignificant as well for both cases and their perinatal and neonatal courses were unremarkable.
2.2. Variation detection
After admission, whole blood samples were obtained from the patients and their parents. NGS was performed according to standard procedures (Yang, Cheng, et al., 2018; Yang, Yin, et al., 2018). Sequencing genomic DNA samples were sonicated, followed by hybridization with the NimbleGen 2.0 probe sequence capture array (Roche, Shanghai, China) to enrich the exon DNA (Joy Orient, China). By qPCR, libraries were first tested for enrichment and size distribution. The concentration was tested using the Agilent Bioanalyzer 2100. The samples were sequenced on the sequencer (Hiseq2500, Illumina, California, USA). Each sample was performed for two parallel reactions. Data filtering, mapping, and variant detection were applied. Exon‐enriched DNA was sequenced according to the manufacturer's instruction.
The raw image files were processed using the BclToFastq (Illumina, California, USA) for base calling and producing the raw data. Low‐quality variations were filtered out using the quality score of Q20. The sequencing reads were aligned to the NCBI human reference genome (hg19) using Burrows‐Wheeler Aligner (BWA). Samtools and Pindel were applied for analyzing the single‐nucleotide polymorphism (SNP) and sequence indexing.
Synonymous changes and SNPs with the minor allele frequency (MAF) higher than 5% were removed; nonsynonymous changes were filtered using SIFT software. The functional mutations and their relationship to CASK‐linked disorders were further analyzed. STRING server (http://string‐db.org) was used for functional enrichment analysis.
2.3. QPCR analysis for transcription levels of CASK
Total RNA from the patients and their parents were extracted by using a QIAGEN RNA Preparation Kit (QIAGEN Inc., CO, Germany) (Wang et al., 2020). The cDNA from these samples was used by reverse transcription and synthesized by PrimeScript™ Strand cDNA Synthesis Kit/RT Master Mix (Takara Shuzo Co., Ltd., Japan). The qPCR primers used to test the expression of CASK in the venous blood as follows: forward primer, 5′‐TGTAGCTGGAGGACGTGTTG‐3′ and reverse primer, 5′‐AGACGTCTACAGGCTTTCCG‐3′. Real‐time qPCR was done by using an SYBR Premix Ex Taq II in a LightCycler® 96 Instrument (Roche, AG Schweiz). The relative expressions of CASK in patients' nuclear family were investigated, as well as in sex‐ and age‐matched control groups. The data were expressed as mean ± SD of triplicate experiments (**p < .05, ***p < .01 vs. control group).
2.4. XCI analysis based on human androgen receptor ( AR ) gene polymorphism
Inactivation patterns of the X‐chromosome of the patients and their nuclear families were tested by a methylation‐based analysis on the human AR using a reported protocol (Cutler Allen et al., 1992; Kassim et al., 2004). Briefly, 200 ng of DNA from peripheral venous blood cells was digested with HpaII (New England Biolabs, USA) at 37°C overnight, followed by enzymatic inactivation. Both digested and nondigested DNA samples were used as a pattern to amplify the AR to control the quality. PCR amplification on both digested and undigested DNA was performed using primers specific to the methylation regions of the short tandem repeats of exon1 in AR. The fluorescence‐labeled PCR products were denatured at 95°C (5 min) and further analyzed by capillary gel electrophoresis using Image Lab software. Analysis was performed with Sub‐Cell GT Agarose Gel Electrophoresis Systems (Bio‐Rad Laboratories, Hercules, CA, USA).
3. RESULTS
3.1. Clinical manifestations of the patients
Patient 1 was examined in our clinic at the age of 6 months. She exhibited psychomotor delay with difficulty in holding head up, rolling over, sitting with support, reaching for toys, raking hands, and passing objects hand to hand. She did not turn to voice. Her head circumference was 36 cm which is on the below 3% percentile. On further examination, there was mild hypotonia. There was no nystagmus, special facial abnormalities, high arched‐palate, or hypohidrosis.
Brain magnetic resonance imaging (MRI) showed decreased volume of the bilateral cerebellar hemispheres and brain stem. Cistern was wide with bilaterally widened temporal intracranial space. On Gesell intelligence test (July 31, 2020): adaptability 45 points, big movement 42 points, fine movement 50 points, language 61 points, personal social 40 points; Peabody assessment: equivalent to the age of 1 month, GMQ 53, FMQ 52, TMP 48. BAEP: the differentiation of the left III/V wave was poor, the amplitude was flat, and the right side was normal. The electroencephalogram (EEG) was unremarkable. Her parents are unaffected.
Patient 2 was seen at the clinic at the age of 13 months. She was a product of in‐vitro fertilization. She presented with psychomotor developmental delay with difficulty in sitting independently, standing with support, crawling, pincer grasping, pointing, and looking for dropped or hidden objects. Her language development was also delayed. No words were formed even “Mama”, “Baba”, or “Dada.”. She also had feeding problems. On examination, there was mildly increased muscle tone with hyperreflexia. Microcephaly was noted. No nystagmus but with wide‐set eyes, wide nasal bridge, small nose, and high‐arched palate (Figure 1a). Hypohidrosis was observed.
FIGURE 1.
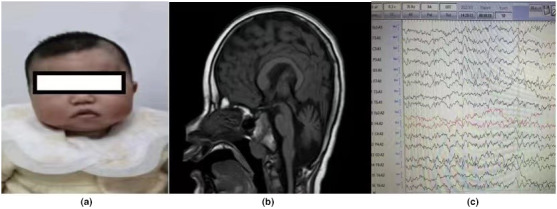
(a–c) The representive clinical characteristics of patient2: (a) Microcephaly with wide‐set eyes, wide nasal bridge, small nose andhigh‐arched palate. (b) Decreased volume of the bilateral cerebellarhemispheres and brain stem decreased on brain MRI. (c) EEG unremarkable.
Brain MRI showed that the volume of the bilateral cerebellar hemispheres and brain stem were decreased (Figure 1b). EEG was unremarkable (Figure 1c). Her parents are unaffected.
3.2. Gene sequencing analysis
Gene mutation analysis of patient 1 revealed a de novo nonsense mutation of c.82C>T (p.Arg28*) in CASK, resulting in a stop codon in a heterozygous‐like pattern. This mutation was not detected in the proband's parents (Figure S1). The locus 28 showed highly conserved by sequence conservation analysis (Figure S2). The other mutation carriers or variants with unknown pathogenic significance were detected (Table 1).
TABLE 1.
List of mutation carriers or variants with unknown pathogenic significance
| Gene | OMIM | Inheritance | HG19 position | Transcript | Nucleotide change | Zygous state | Source |
|---|---|---|---|---|---|---|---|
| AARS1 | 601065 | AD /AR | chr16: 70288599 | NM_001605 | c.2324_2325del (p.S775Cfs*22) | Heterozygous | Mother (heterozygous) |
| BCL11B | 606558 | AD | chr14: 99642394 | NM_138576 | c.779C>T (p.T260M) | Heterozygous | Father (heterozygous) |
| BRPF1 | 602410 | AD | chr3: 9785474 | NM_001003694 | c.2524C>T (p.R842W) | Heterozygous | Mother (heterozygous) |
| KCNB1 | 600397 | AD | chr20: 47990392 | NM_004975 | c.1705G>C (p.V569L) | Heterozygous | Father (heterozygous) |
| PHACTR1 | 608723 | AD | chr6: 13283783 | NM_030948 | c.1639G>T (p.A547S) | Heterozygous | Father (heterozygous) |
| PHF21A | 618725 | AD | chr11: 45967536 | NM_001101802 | c.1304A>G (p.N435S) | Heterozygous | Mother (heterozygous) |
| PRMT7 | 610087 | AR | chr16: 68380059 | NM_019023 | c.1067A>C (p.N356T) | Heterozygous | De novo |
| SLC36A2 | 608331 | AD /AR | chr5: 150726957 | NM_181776 | c.65C>A (p.S22*) | Heterozygous | Father (heterozygous) |
Patient 2 had a de novo heterozygous nonsense mutation of c.846C>G (p.Tyr282*) in CASK, resulting in a stop codon in a heterozygous‐like pattern. This mutation was not detected in the proband's parents (Figure S1).
3.3. STRING server for the functional enrichment analysis of nonsense mutations
STRING server‐generated 3D atomic models based on protein sequences in CASK. Two Arg28 and Tyr282 residues were marked (Figure 2). They were both in Ca2+/calmodulin‐dependent protein kinase II (CaMKII) domain and all the downstream domains (two L27 domains, a PDZ domain, an SH3 domain, and a guanylate kinase domain) can be affected.
FIGURE 2.
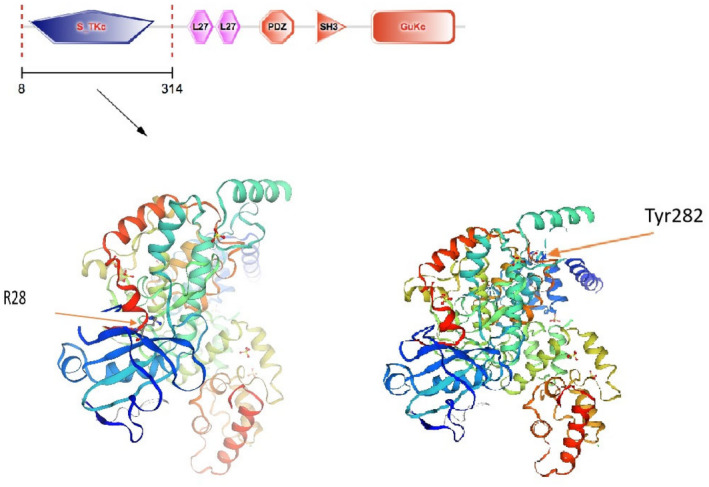
Predict 3D structure of CaMKII domain of CASK protein. Two Arg28 and Tyr282 residues were marked.
3.4. The transcription levels of CASK in the patients and in the control group
RT‐qPCR with the same primer sets confirmed decreased copy numbers of CASK expression in patients than their parents, as well as in sex‐ and age‐matched control groups (five males and five females). The discrepancy in the levels of CASK was not related to age or sex according to the results of the control groups (Figure 3).
FIGURE 3.
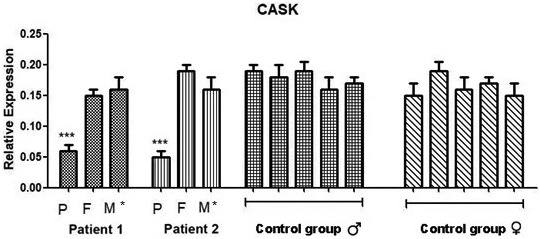
The relative expressions of CASK were investigated with RT‐qPCR in the patients' nuclear families, as well as in sex‐ and age‐matched control groups. The patients exhibited significantly lower expressions of the CASK compared to those of their parents (*P: Patient, F: Father, M: Mother). The discrepancy in the levels of CASK was not related to age or sex according to the results of the control groups. The data were expressed as mean ± SD of triplicate experiments (***p < .01 vs. control group).
3.5. Study skewed XCI patterns in the patients
Microsatellite PCR products of AR with or without digestion showed that the number of repeats within AR determines the size of the allele. In patient 1, PCR products derived from the undigested DNA yielded two peaks because of the different numbers of CAG repeats of the two alleles, 276 bp from the mother and 297 bp from the father (Figure 4). The X‐inactivation pattern was thought to be skewed if the proportion of the two alleles after digestion was at least 20:80 (Giorgio et al., 2017). The experiment showed that the patient had a skewed XCI pattern (13%:87%) with the paternal allele remarkably inactivated. In patient 2, PCR products derived from the undigested DNA yielded one peak of 291 bp, the same length of the chromosomes, respectively, inherited from parents (Figure 5). The AR polymorphism analysis did not show XCI pattern.
FIGURE 4.
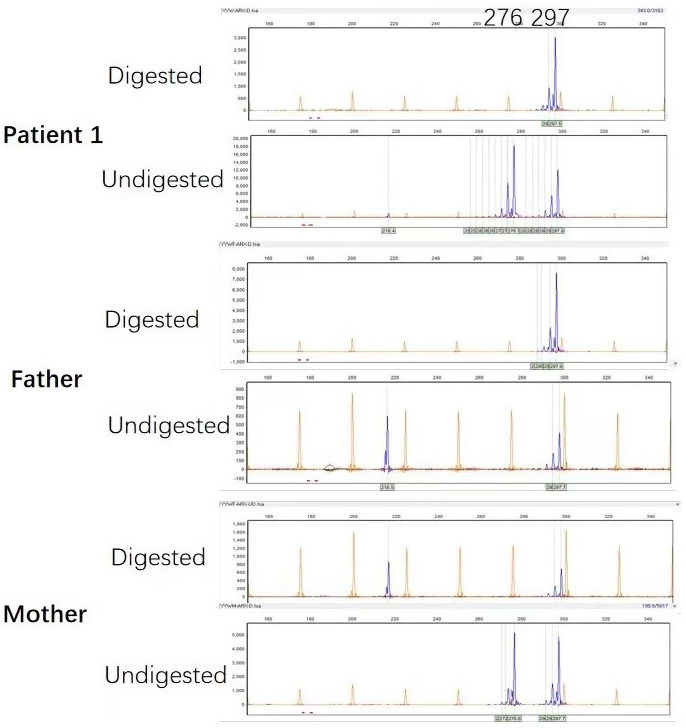
Patient 1's PCR products of the AR with and without HpaII digestion of the nuclear family. The peak represents the amplified AR allele. The PCR products of the patient derived from the undigested DNA yielded two peaks because of the different numbers of CAG repeats in the two alleles (276 bp from the mother and 297 bp from the father). However, after HpaII was digested, one peak (297 bp) appeared hypermethylated and the other peak (276 bp) was almost completely missing. The results revealed a skewed XCI pattern in the patient with the paternal allele highly inactivated.
FIGURE 5.
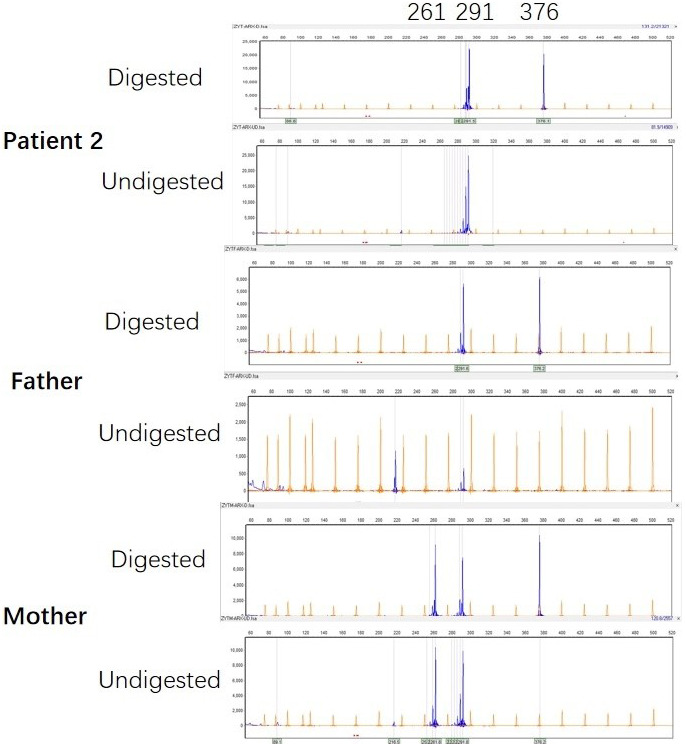
Patient 2's PCR products of the AR with and without HpaII digestion of the nuclear family. The peak represents the amplified AR allele. PCR products derived from the undigested DNA yielded one peak of 291 bp, the same length of the chromosomes, respectively, inherited from the parents. The AR polymorphism analysis did not show XCI pattern.
4. DISCUSSION
CASK domain arrangement is considered to be conserved in all metazoans, and individual domain structures present a high degree of evolutionary conservation (LaConte et al., 2014). Hence, mutations in CASK may cause pathogenesis via several mechanisms. There are several different computational programs available to predict or present the protein structures for mutated genes, STRING server was used in this study to present the affected protein domains based on 3D structural information. Identifying a point mutation interrupting a special protein interaction would be useful in dissecting its molecular function. In our study, two probands showed two nonsense mutations, resulting in two stop codons, which can affect all proteins in downstream domains (Figure 2). Although discovering mutations related to a particular disorder is informative, it is only the first step. Only after the characteristic of each mutation is identified, any potential therapeutic methods can be explored.
With the increase of family or sporadically affected females reported in variable clinical features, the clinical spectrum of CASK disorder is wider than previously reported. But the mechanism of pathogenesis in females has rarely been fully elucidated. Toshiyuki Seto et al. (2017) reported a novel CASK mutation identified in siblings, who exhibited different clinical manifestations due to different XCI patterns.
In our study, patient 1 had a de novo nonsense mutation of c.82C>T (p.Arg28*) in CASK. The codon coding arginine was substituted by a stop codon. The STRING server showed the mutated CASK protein was in CAMKII domain and all the downstream domains can be affected. RT‐ qPCR showed significantly lower levels of CASK expression than their parents. The discrepancy was not related to age or sex under the comparison with the sex‐ and age‐matched control groups, which indicated that the lower levels of CASK expression might be related to the nonsense mutation instead of other factors. Since CASK‐related syndromes exhibit X‐linked inheritance, female carriers may be unaffected or present with mild clinical manifestations. While patient 1 exhibited obvious clinical features of CASK disorder, we predicted that the possible hypothesis was related to the XCI pattern by methylation. By methylation‐sensitive PCR and AR CAG repeat polymorphism analysis, this patient showed a skewed XCI pattern. Hence we hypothesized that this patient's spontaneously mutated gene was on the activated X‐chromosome, which was unable to produce sufficient levels of CASK, resulting in a series of clinical manifestations. In a word, according to the clinical characteristics, patient 1 belonged to a syndrome of ID and MICPCH, with significantly lower levels of CASK expression, and XCI mechanism may exist.
Patient 2 had a de novo nonsense mutation of c.846C>G (p.Tyr282*) in CASK, which was not inherited from her parents. The codon coding tyrosine was substituted by a stop codon. STRING server showed the mutated CASK protein was in CAMKII domain and all the downstream domains can be affected. RT‐ qPCR showed significantly lower levels of CASK expression than their parents, as well as the sex‐ and age‐matched control groups. She did not show XCI pattern by AR polymorphism methylation‐based analysis. We hypothesized this patient may have other possible mechanisms like nonsense‐mediated mRNA decay (NMD) which can also lead to a reduction in CASK. NMD was a processing pathway in cells. Its mechanism prevents the production of potentially toxic truncated proteins by recognizing and degrading transcripts containing premature translation‐termination codon (PTC). Like a broom, it clears the wrong RNA to prevent its generative transformation into abnormal proteins, which leads to lower levels of CASK expression in this patient. The confirmed mechanism remains unclear. NMD mechanism or other further hypothesis need to be investigated. According to the clinical characteristics, patient 2 also belonged to a syndrome of ID and MICPCH.
MRI in patients with CASK mutations showed normal size of the corpus callosum, but with a reduced size of the cerebrum, midbrain, cerebellar vermis, or hemispheres (Takanashi et al., 2010). Both of our detected patients showed microcephaly with bilateral cerebellar hemispheres and brain stem decreased volume, which was considered to be relevant to the disruption of the CASK‐neurexin interaction (LaConte et al., 2018). The features of delayed development and brain images together with mild facial abnormality are strongly suggestive of this disorder. Prompt subsequent testing of CASK should be suggested (Moog et al., 2011).
CASK‐linked disorders are inherited in an X‐linked manner. Loss‐of‐function of CASK usually leads to the manifestation of CASK‐linked disorders in females. In both cases, the parents were examined and found to be without CASK mutation or neurological symptoms. Mutations in CASK are usually “de novo” and they affect more women than men (Rivas et al., 2017). As in our clinical cases, both are girls, in whom “de novo” mutation were confirmed while we could not see related changes in their parents.
5. CONCLUSIONS
In summary, patient 1, with a nonsense mutation of c.82C>T (p.Arg28*) in CASK, had early psychomotor delay, and MICPCH, without nystagmus, special facial abnormalities, epileptic encephalopathy, or ASD. Patient 2, with a nonsense mutation of c.846C>G (p.Tyr282*) in CASK, had early psychomotor developmental delay, mild facial abnormalities and MICPCH, without nystagmus, epileptic encephalopathy, or ASD. Owning to the identification of CASK mutations in patients with different clinical features, the clinical entity of CASK disorder is expanding. Our preliminary research put forward the possible mechanisms underlying the pathogenesis of our patients. The speculation of whether the patient 1's spontaneously mutated CASK was on the activated X‐chromosome and the possible NMD mechanism for patient 2's low levels of CASK expression remained to be further confirmed. The induction of patients' naturally formed somatic cells into stem cells would be a valuable way for further research.
In conclusion, two patients both presented with ID, microcephaly, and MICPCH, with mild clinical differences (with or without special facial abnormality), which expands the spectrum of genotype and phenotype correlations of CASK‐linked disorders in Han Chinese, providing new insights into the molecular mechanism of CASK‐linked disorders and helping to further explore the contribution of other protein–protein interactions. Due to the limited small number of patients, further large‐scale case studies are needed to investigate the genotype–phenotype correlations and specific treatment approaches of CASK‐linked disorders (Becker et al., 2020).
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTIONS
StudyDesign: Kunfang Yang, Fang Yuan, Zhiping Liu, Yun Ren and Yucai Chen. Data Collection: Kunfang Yang, Longlong Lin, Xiaoping Lan and Yun Ren. Statistical Analysis: Kunfang Yang, Longlong Lin, Fang Yuan, Xiaoping Lan. Data Interpretation: Longlong Lin and Jiaoyan Li. ManuscriptPreparation: Kunfang Yang, Xiaoguang Li, Zhiping Liu and Yilin Wang. Literature Search: Xiaoguang Li, Yilin Wang and Yucai Chen. Funds Collection: Yucai Chen.
ETHICAL COMPLIANCE
The parents of the probands were informed by the consultant in the Department of Pediatric Neurology about the purpose of the DNA analysis. The samples were investigated after obtaining informed consent from the parents, as well as the legal representatives of the sex‐ and age‐matched control groups. This work was approved by the Ethics Committee of Shanghai Children's Hospital.
Supporting information
Supinfo
ACKNOWLEDGMENTS
We would like to express our gratitude to the patients and their parents for their cooperation. All figures were permitted to be published by them. This work was supported by Shanghai key clinical specialty project (shslczdzk05705) and Key disciplines of top priority in Shanghai (2017ZZ02019).
Yang, K. , Lin, L. , Yuan, F. , Li, X. , Liu, Z. , Lan, X. , Wang, Y. , Ren, Y. , Li, J. , & Chen, Y. (2022). Two heterozygous mutations in the calcium/calmodulin‐dependent serine protein kinase gene (CASK) in cases with developmental disorders. Molecular Genetics & Genomic Medicine, 10, e2065. 10.1002/mgg3.2065
DATA AVAILABILITY STATEMENT
All datasets generated for this study are included in the Supplementary Materials.
REFERENCES
- Becker, M. , Mastropasqua, F. , Reising, J. P. , Maier, S. , Ho, M.‐L. , Rabkina, I. , Li, D. , Neufeld, J. , Ballenberger, L. , Myers, L. , Moritz, V. , Kele, M. , Wincent, J. , Willfors, C. , Sitnikov, R. , Herlenius, E. , Anderlid, B.‐M. , Falk, A. , Bölte, S. , & Tammimies, K. (2020). Presynaptic dysfunction in CASK‐related neurodevelopmental disorders. Translational Psychiatry, 10, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler Allen, R. , Zoghbi, H. Y. , Moseley, A. B. , Rosenblatt, H. M. , & Belmont, J. (1992). Methylation of Hpall and Hhal sites near the polymorphic CAG repeat in the human androgen‐receptor gene correlates with X chromosome inactivation. American Journal of Human Genetics, 51, 1229–1239. [PMC free article] [PubMed] [Google Scholar]
- Giorgio, E. , Brussino, A. , Biamino, E. , Belligni, E. F. , Bruselles, A. , Ciolfi, A. , Caputo, V. , Pizzi, S. , Calcia, A. , di Gregorio, E. , Cavalieri, S. , Mancini, C. , Pozzi, E. , Ferrero, M. , Riberi, E. , Borelli, I. , Amoroso, A. , Ferrero, G. B. , Tartaglia, M. , & Brusco, A. (2017). Exome sequencing in children of women with skewed X‐inactivation identifies atypical cases and complex phenotypes. European Journal of Paediatric Neurology, 21(3), 475–484. [DOI] [PubMed] [Google Scholar]
- Hata, Y. , Butz, S. , & Sudhof, T. C. (1996). CASK: A novel dlg/PSD95 homolog with an N‐terminal calmodulin‐dependent protein kinase domain identified by interaction with neurexins. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 16(8), 2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, S. , Uehara, D. T. , Tanimoto, K. , Mizuno, S. , Chinen, Y. , Fukumura, S. , Takanashi, J.‐i. , Osaka, H. , Okamoto, N. , & Inazawa, J. (2017). Comprehensive investigation of CASK mutations and other genetic etiologies in 41 patients with intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH). PLoS One, 12(8), e0181791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, T.‐N. , & Hsueh, Y.‐P. (2009). CASK point mutation regulates protein‐protein interactions and NR2b promoter activity. Biochemical and Biophysical Research Communications, 382(1), 219–222. [DOI] [PubMed] [Google Scholar]
- Kassim, S. , Zoheiry, N. M. , & Hamed, W. M. (2004). Androgen receptor gene methylation and exon one CAG repeat length in ovarian cancer: Differences from breast cancer. IUBMB Life, 56(7), 417–426. [DOI] [PubMed] [Google Scholar]
- Kuo, T.‐Y. , Hong, C.‐J. , Chien, H.‐L. , & Hsueh, Y. P. (2010). X‐linked mental retardation gene CASK interacts with Bcl11A/CTIP1 and regulates axon branching and outgrowth. Journal of Neuroscience Research, 88(11), 2364–2373. [DOI] [PubMed] [Google Scholar]
- LaConte, L. E. W. , Chavan, V. , Elias, A. F. , Hudson, C. , Schwanke, C. , Styren, K. , Shoof, J. , Kok, F. , Srivastava, S. , & Mukherjee, K. (2018). Two microcephaly‐associated novel missense mutations in CASK specifically disrupt the CASK‐neurexin interaction. Human Genetics, 137(3), 231–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaConte, L. E. W. , Chavan, V. , & Mukherjee, K. (2014). Identification and glycerol‐induced correction of misfolding mutations in the X‐linked mental retardation gene CASK. PLoS One, 9(2), e88276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , Kim, S. H. , Kim, B. , Lee, S. T. , Choi, J. R. , Kim, H. D. , Lee, J. S. , & Kang, H. C. (2020). Genetic diagnosis and clinical characteristics by etiological classification in early‐onset epileptic encephalopathy with burst suppression pattern. Epilepsy Research, 163, 106323. [DOI] [PubMed] [Google Scholar]
- Moog, U. , & Kutsche, K. (2013, November 26). CASK disorders. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., LJH B., Gripp K. W., & Amemiya A. (Eds.), GeneReviews® [internet]. University of Washington, Seattle; 1993–2021. Updated 2020 May 21. University of Washington. https://www.ncbi.nlm.nih.gov/books/ [PubMed] [Google Scholar]
- Moog, U. , Kutsche, K. , Kortüm, F. , Chilian, B. , Bierhals, T. , Apeshiotis, N. , Balg, S. , Chassaing, N. , Coubes, C. , das, S. , Engels, H. , van Esch, H. , Grasshoff, U. , Heise, M. , Isidor, B. , Jarvis, J. , Koehler, U. , Martin, T. , Oehl‐Jaschkowitz, B. , … Uyanik, G. (2011). Phenotypic spectrum associated with CASK loss‐of‐function mutations. Journal of Medical Genetics, 48(11), 741–751. [DOI] [PubMed] [Google Scholar]
- Mukherjee, K. , Patel, P. A. , & Rajan, D. S. (2020). Survival of a male patient harboring CASK Arg27Ter mutation to adolescence. Molecular Genetics & Genomic Medicine, 8, e1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, H. , Kimura, Y. , Enomoto, Y. , Tsurusaki, Y. , Akahira‐Azuma, M. , Kuroda, Y. , Tsuji, M. , Goto, T. , & Kurosawa, K. (2019). Discordant phenotype caused by CASK mutation in siblings with NF1. Human Genome Variation, 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Y. E. , Tibbe, D. , Harms, F. L. , Reißner, C. , Becker, K. , Dingmann, B. , Mirzaa, G. , Kattentidt‐Mouravieva, A. A. , Shoukier, M. , Aggarwal, S. , Missler, M. , Kutsche, K. , & Kreienkamp, H.‐J. (2021). Missense mutations in CASK, coding for the calcium−/calmodulin‐dependent serine protein kinase, interfere with neurexin binding and neurexin‐induced oligomerization. Journal of Neurochemistry, 157(4), 1331–1350. [DOI] [PubMed] [Google Scholar]
- Patel, P. A. , Liang, C. , Arora, A. , Vijayan, S. , Ahuja, S. , Wagley, P. K. , Settlage, R. , LaConte, L. E. W. , Goodkin, H. P. , Lazar, I. , Srivastava, S. , & Mukherjee, K. (2020). Haploinsufficiency of X‐linked intellectual disability gene CASK induces post‐ transcriptional changes in synaptic and cellular metabolic pathways. Experimental Neurology, 329, 113319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp, B. , Ekici, A. B. , Thiel, C. T. , Hoyer, J. , Wiesener, A. , Kraus, C. , Reis, A. , & Zweier, C. (2017). Exome Pool‐Seq in neurodevelopmental disorders. European Journal of Human Genetics, 25, 1364–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas, L. , Blanco, Ó. , Torreira, C. , Repáraz, A. , Melcón, C. , & Amado, A. (2017). Pontocerebellar hypoplasia secondary to CASK gene deletion. Case report. Revista Chilena de Pediatria, 88(4), 529–533. [DOI] [PubMed] [Google Scholar]
- Seto, T. , Hamazaki, T. , Nishigaki, S. , Kudo, S. , Shintaku, H. , Ondo, Y. , Shimojima, K. , & Yamamoto, T. (2017). A novel CASK mutation identified in siblings exhibiting developmental disorders with/without microcephaly. Intractable & Rare Diseases Research, 6(3), 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. , McMillan, R. , Willis, J. , Clark, H. , Chavan, V. , Liang, C. , Zhang, H. , Hulver, M. , & Mukherjee, K. (2016). X‐linked intellectual disability gene CASK regulates postnatal brain growth in a non‐cell autonomous manner. Acta Neuropathologica Communications, 4, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studtmann, C. , LaConte, L. E. W. , & Mukherjee, K. (2019). Comments on: A de novo in‐frame deletion of CASK gene causes early onset infantile spasms and supratentorial cerebral malformation in a female patient. American Journal of Medical Genetics. Part A, 179(12), 2514–2516. [DOI] [PubMed] [Google Scholar]
- Takanashi, J. , Arai, H. , Nabatame, S. , Hirai, S. , Hayashi, S. , Inazawa, J. , Okamoto, N. , & Barkovich, A. J. (2010). Neuroradiologic features of CASK mutations. AJNR. American Journal of Neuroradiology, 31, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Lin, L. , Xue, Y. , Wang, Y. , Liu, Z. , Ou, Z. , Wu, S. , Lan, X. , Zhang, Y. , Yuan, F. , Luo, X. , Wang, C. , Xi, J. , Sun, X. , & Chen, Y. (2020). MED12‐related disease in a Chinese girl: Clinical characteristics and underlying mechanism. Frontiers in Genetics, 11, 129. 10.3389/fgene.2020.00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, K. , Cheng, H. , Yuan, F. , Meng, L. , Yin, R. , Zhang, Y. , Wang, S. , Wang, C. , Lu, Y. , Xi, J. , Lu, Q. , & Chen, Y. (2018). CHRNE compound heterozygous mutations in congenital myasthenic syndrome. Medicine, 97(17), e0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, K. , Yin, R. , Lan, X. , Zhang, Y. , Cheng, H. , Wang, S. , Wang, C. , Lu, Y. , Xi, J. , Lu, Q. , Huang, J. , & Chen, Y. (2018). Dopa‐responsive dystonia in Han Chinese patients: One novel heterozygous mutation in GTP Cyclohydrolase 1 (GCH1) and three known mutations in TH . Medical Science Monitor, 24, 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo
Data Availability Statement
All datasets generated for this study are included in the Supplementary Materials.