

Abstract
This study aimed to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of vutiglabridin, a potential anti‐obesity treatment under development, for the first time in humans. A randomized, placebo‐controlled, single‐ and multiple‐ascending dose study (SAD and MAD, respectively) was performed in healthy Koreans and Whites. Subjects randomly received a single oral dose of 30–720 mg vutiglabridin or placebo at a ratio of 8:2 in the SAD study or 240–480 mg vutiglabridin or placebo once daily for 14 days in the MAD study. Food effect was also evaluated in 240 mg single dose group. Pharmacokinetics were evaluated through plasma concentrations, and pharmacodynamic biomarkers related to obesity or inflammation were analyzed. Safety and tolerability were assessed throughout the study. Single and multiple doses of vutiglabridin were generally well‐tolerated. The pharmacokinetic parameters show less than dose‐proportionality increase, and plasma concentrations increased more than two‐fold after multiple administrations. The mean half‐life of Koreans and Whites in the MAD study was 110 and 73 h, respectively. The systemic exposure of vutiglabridin was significantly increased when taken with a high‐fat meal, and the systemic exposure was lower in Whites than in Koreans. Vutiglabridin was well‐tolerated in healthy Koreans and Whites. The plasma concentration increased less than the dose‐proportionality manner. These results justify further investigation of vutiglabridin in patients with obesity.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
A treatment with multifunctional effects is needed for obesity. Because the chronic inflammation caused by obesity plays an essential role in the progression of metabolic disorders, suppressing inflammatory pathways may be another important treatment goal.
WHAT QUESTION DID THIS STUDY ADDRESS?
What is the safety, tolerability, pharmacokinetic, and pharmacodynamic characteristics of vutiglabridin, a novel anti‐obesity agent, in healthy Korean and White individuals?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Vutiglabridin was well‐tolerated in both Korean and White individuals. The pharmacokinetic parameters of vutiglabridin did not show dose‐proportionality, and after multiple administrations, vutiglabridin showed accumulation.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This study provides safety, pharmacokinetic, and pharmacodynamic information about vutiglabridin, which will be used for further trials for the treatment of obesity.
INTRODUCTION
Obesity is defined as the accumulation of excessive body fat that is caused by excessive energy intake that exceeds energy expenditure, which can lead to health risks. Obesity is a global public health concern and has been markedly increasing over the past 4 decades. 1 , 2 In 2016, ~ 11% of men and 15% of women aged 18 years and over in the global population were obese, and more than half a billion cases of obesity were reported. 3 Because obesity substantially increases the risk of many diseases, including fatty liver disease, type 2 diabetes mellitus, stroke, and myocardial infarction, it is a major health challenge that can lead to a decline in quality of life and life expectancy. 4
The management and treatment of obesity includes medical nutrition therapy, physical activity, psychological and behavioral intervention, pharmacotherapy, and bariatric surgery. 5 The fundamental treatments of obesity are lifestyle interventions, including physical activity, nutrition, and behavior therapy. However, lifestyle interventions do not always produce satisfactory results, and if the patient with obesity does not show adequate weight loss after 3–6 months of lifestyle intervention, pharmacotherapies are considered. 6 Obesity treatment drugs for weight control can be classified into two categories: (1) drugs that reduce food intake by acting on the central nervous system, or (2) drugs that reduce absorption of fat in the intestines. 7 Currently, there are five long‐term use drugs approved by the US Food and Drug Administration (FDA) for weight loss, and four of them control weight by controlling appetite (orlistat [Xenical, Alli], phentermine‐topiramate [Qsymia], naltrexone‐bupropion [Contrave], liraglutide [Saxenda], and semaglutide [Wegovy]). 8 Drugs that control appetite to reduce energy intake may induce the loss of lean body and muscle mass as well as total weight loss, which can increase the risk of sarcopenia and may disturb metabolic homeostasis. 9 , 10 , 11 Therefore, a treatment with multifunctional effects that enhances energy expenditure and reduces energy intake is needed. Moreover, because the chronic inflammation caused by obesity plays an essential role in the progression of metabolic disorders, suppressing inflammatory pathways may be another important treatment goal for anti‐obesity drugs. 12 , 13 , 14
Glabridin is a prenylated isoflavonoid of Glycyrrhiza glabra L. (licorice) roots. It has antioxidative and anti‐inflammatory effects and is therefore regarded as a potential treatment for various metabolic diseases. 15 , 16 Glabridin has a mode of action that inhibits the NOD‐like receptor family pyrin domain containing 3 (NLRP3) inflammasome, which plays a key role in obesity‐related inflammation. 17 In an obese mouse model, glabridin reduced body weight by ~ 25%, ameliorated fatty liver, and reduced plasma levels of cholesterol and triglycerides. 18 Moreover, glabridin enhanced the activation of AMP‐activated protein kinase and promoted fatty acid oxidation. 18 These findings suggest that glabridin could ameliorate obesity‐related metabolic disorders. Despite these properties, glabridin has not been developed as an anti‐obesity drug because of its low physicochemical stability, low bioavailability, and complicated extraction process from licorice. 19 , 20 Vutiglabridin (2‐(8,8‐dimethyl‐2,3,4,8,9,10‐hexahydropyrano[2,3‐f]chromen‐3‐yl)‐5‐ethoxyphenol) is a synthetic derivative of glabridin that has improved chemical stability compared to glabridin (Figure 1). Vutiglabridin has shown anti‐obesity effects, including weight reduction and increased energy expenditure, in a high‐fat diet (HFD)‐induced obese C57BL/6J mouse model. 21 Additionally, the expression of inflammation‐related genes (CCL2 and NFE2L2) in the liver and muscles were significantly altered in the vutiglabridin‐treated group compared to the non‐vutiglabridin‐treated group. Furthermore, the nonalcoholic fatty liver disease activity score showed that the inflammation score was lower in the vutiglabridin‐treated group than in the normal group in the HFD‐induced obese C57BL/6J mouse model. 21 Vutiglabridin is an isoflavone compound that is a racemic mixture containing a 1:1 ratio of the (R)‐ and (S)‐forms. It was discovered through a quantitative structure–activity relationship study. At equivalent dose levels, the body weight reduction effect of vutiglabridin (S)‐isomer surpassed that of (R)‐isomer in in the HFD‐induced obese mouse model. 21 Vutiglabridin had no effect on the central nervous system, respiratory system, or cardiovascular system in rats or beagle dogs. The no‐observed‐adverse‐effect level (NOAEL) values obtained from the rat, mouse, and beagle models were 30 mg/kg/day, 100 mg/kg/day, and 1000 mg/kg/day, respectively.
FIGURE 1.
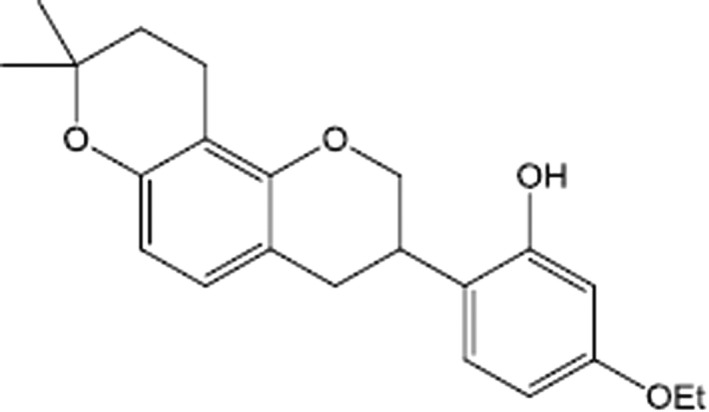
Chemical structure of vutiglabridin
Based on this understanding, this study aimed to evaluate the safety, tolerability, pharmacokinetic, and pharmacodynamic characteristics of vutiglabridin in healthy Korean and White individuals for the first time in humans.
METHODS
Subjects and study design
A randomized, double‐blinded, placebo‐controlled, single‐ and multiple‐ascending dose (SAD and MAD, respectively) study was performed in healthy Korean and White subjects. Male or female subjects aged between 19 and 50 years old with body mass index (BMI) ranging from 18 to 25 kg/m2 were recruited. Korean subjects were defined as being born in South Korea and having four ethnic Korean grandparents, and White subjects were defined as being European and having both parents and grandparents of European ancestry. Subjects’ health status were evaluated based on their medical history, physical examination, vital signs, 12‐lead electrocardiogram (ECG), and clinical laboratory test results. Subjects were excluded if they had a history of hypersensitivity to drugs, including licorice, or anticipated the use of any medication, herbal remedies, or vitamin supplements within 7 days prior to initial dosing. Additionally, because reproductive toxicity was observed in preclinical studies, male subjects with a testosterone level ≤2.49 ng/ml or ≥8.36 ng/ml were excluded. In the SAD study, there was a total of 60 healthy Korean subjects, and in the MAD study, there was a total of 20 healthy Korean subjects and 12 healthy White subjects. This study was reviewed and approved by the Ministry of Food and Drug Safety and the Institutional Review Board of Seoul National University Hospital (Seoul, Republic of Korea, approval number: 1806–098‐952). This study was performed in accordance with the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice and was registered with Clinical Research Information Service in Korea (KCT0003673) and in open registry (NCT04732988). An informed consent form was obtained from all study subjects before any study‐related procedures. The primary end point of this study was to evaluate safety, tolerability, and pharmacokinetic characteristics of vutiglabridin. In addition, the effect of food and ethnicity on characteristics of vutiglabridin and the effect of vutiglabridin on exploratory pharmacodynamic markers were evaluated as secondary end points.
According to FDA guidance, assuming a safety factor of 10 and a healthy male subject weight of 60 kg, the lowest dose of maximum recommended starting dose (MRSD) in humans was 30 mg based on the value of NOAEL of rats. 22 Additionally, in the efficacy test of vutiglabridin, the pharmacologically active dose (PAD) was expected to be 48.8 to 487.8 mg in the mouse model. Therefore, 30 mg, which is lower than PAD and close to MRSD, was determined as the first dose of the SAD study. In addition, the maximum tolerated dose of the preclinical study was found to be 1000 mg/kg in beagle dogs, and safety factors of 10 and 30 were applied, respectively. Assuming a healthy male subject of 60 kg, the doses of 3333 and 1111 mg were expected in humans. Accordingly, the doses in the SAD study were increased from 30 to 720 mg sequentially, and in the MAD study, the doses of 240 and 480 mg were set based on the PAD.
In the SAD study, six dose groups (30, 60, 120, 240, 480, and 720 mg) were evaluated, and the subjects in each dose group randomly received a single oral dose of vutiglabridin or matching placebo at a ratio of 8:2. The placebo formulation consisted of polyethylene glycol, microcrystalline cellulose, ethanol, and sterile water. The appearance of vutiglabridin and placebo was identical. Eligible subjects received a computer‐generated randomization number and the subjects’ randomization status were kept blinded to the subjects, investigators, and sponsor (i.e., double‐blind) throughout the study. To evaluate the effect of food, subjects received a single vutiglabridin 240 mg or matching placebo with and without a high‐fat meal (a fat content more than 30% and total calories more than 900 kcal) with 2 weeks of washout between each dose. In the MAD study, two dose groups (240 and 480 mg) were evaluated, and the subjects in each dose group randomly received a once‐daily oral dose of vutiglabridin or matching placebo at a ratio of 8:2 for 14 days. In the 480 mg group of the MAD study, vutiglabridin was administered to Korean and White individuals for ethnic comparison. Pharmacokinetic analysis was conducted with serial blood samples in the SAD and MAD studies, and the pharmacodynamic end points were only evaluated in the MAD study.
Safety and tolerability assessment
Safety was evaluated by monitoring adverse events, clinical laboratory tests, physical examinations, vital signs, and 12‐lead ECGs. In rat models, females that received multiple doses of vutiglabridin 1000 mg/kg/day showed an irregular menstrual cycle, and males that received multiple doses of vutiglabridin 300 or 1000 mg/kg/day showed decreased sperm motility and sperm count and an increased sperm malformation rate. Additionally, in the histopathological examination, several reproductive toxicities, including adrenal cortex hypertrophy, testicular seminiferous tubule degeneration, and uterine atrophy, were observed in both male and female rats receiving multiple doses of vutiglabridin 300 or 1000 mg/kg/day. Considering the reproductive toxicities observed in preclinical studies, several biomarkers related to reproduction were assessed through clinical laboratory tests, and semen tests were additionally performed in the MAD study. Testosterone, luteinizing hormone (LH), follicle‐stimulating hormone (FSH), and inhibin B tests were conducted at predose on day 1 and the poststudy visit (PSV). Semen tests were performed at screening and the PSV, in which semen volume, pH, number of white blood cells, sperm number, concentration, motility, and morphology were evaluated.
Determination of the plasma concentration
Serial plasma samples were collected for pharmacokinetic analysis. In the SAD study, blood samples were collected at the following time points: 0, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72, and 96 h postdose for the 30 and 60 mg dose groups and at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 72, 96, 144, and 192 h postdose for the 120 to 720 mg dose groups. In the MAD study, blood samples were collected at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 h on day 1 and at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 72, 96, 144, and 192 h on day 14.
The total plasma concentration of vutiglabridin was calculated by adding the concentration values of (R)‐vutiglabridin and (S)‐vutiglabridin. Plasma concentrations of the (S)‐isomer and (R)‐isomer were determined by liquid chromatography–tandem mass spectrometry using a Waters Xevo TQ‐S mass spectrometer (Waters). The assay ranges were 1 to 1500 ng/ml for the (R)‐isomer and 2 to 3000 ng/ml for the (S)‐isomer. Fifty microliters of calibration standard, quality control, or study sample was added to the tubes. Subsequently, 20 μl of internal standard ([±]‐ vutiglabridin‐d5) working solution and 500 μl of methanol‐acetonitrile‐formic acid (25:75:0.1) were added. The samples were then vortexed and centrifuged, and a portion of the supernatant was then transferred to an autosampler vial. The monitored mass transitions were 353.1 >147.0 m/z for vutiglabridin (both (R)‐ and (S)‐isomers) and 358.1 >147.0 m/z for vutiglabridin‐d5. The below the quantifiable limit was 1.0 ng/ml for (R)‐vutiglabridin and 2.0 ng/ml for (S)‐vutiglabridin.
Pharmacokinetic analysis
Pharmacokinetic parameters were calculated by noncompartmental methods using WinNonlin software version 8.1.1 (Certara). The maximum plasma concentration (Cmax) and time to reach Cmax (Tmax) were determined from the obtained concentration‐time data. The areas under the concentration‐time curves from time 0 to 24 h (AUC0‐24), to the last measurable timepoint (AUClast), and extrapolated to infinity (AUCinf) using the terminal elimination rate constant (λz) were calculated by the linear trapezoidal summation for the ascending concentrations and log trapezoidal summation for the descending concentrations. After multiple administrations, the area under the plasma concentration‐time curve over a dosing interval of 24 h at steady‐state (AUCtau,ss) was calculated. The terminal elimination half‐life (t 1/2) was calculated as the natural logarithm of two divided by λz. The apparent clearance (CL/F) was calculated as the amount of dose divided by AUCinf or AUCtau, and the apparent volume of distribution (Vd/F) was calculated as the amount of dose divided by AUCinf*λz or AUCtau*λz. The peak trough fluctuation (PTF) was calculated using the following formula: PTF (%) = 100*(Cmax‐minimum plasma concentration [Cmin]/average plasma concentration [Cavg]). The accumulation ratio was calculated as the ratio of AUCtau after the last administration of vutiglabridin in comparison with that after the administration of the first dose.
Pharmacodynamic analysis
In the MAD study, serum concentrations of leptin, adiponectin, insulin, C‐peptide, interleukin‐6 (IL‐6), tumor necrosis factor‐α (TNFα), and C‐C motif chemokine ligand 2 (CCL2) were evaluated as obesity‐related biomarkers. After treatment with the study drugs for 14 days, the change in markers from baseline (i.e., day 1 predose) was compared between the vutiglabridin‐ and placebo‐treated groups. To evaluate biomarkers related to obesity, blood samples were collected at the predose timepoints of day 1 and day 14 in the MAD study. The concentration of adiponectin was detected using EZHADP‐61 K Human Adiponectin enzyme‐linked immunosorbent assay (ELISA) kit (Millipore Corporation). 23 The concentrations for other biomarkers, except for adiponectin, were determined by the MILLIPLEX MAP Human Metabolic Hormone Magnetic Bead Panel, catalog number HMHEMAG‐34 K (Millipore Corporation). 24
Statistical analysis
SAS software version 9.4 (SAS Institute) was used for statistical analysis. With the data obtained in the SAD study, the dose‐proportionality of vutiglabridin was evaluated by the Kruskal–Wallis test and power model. The average of Cmax and AUC corrected by the dose (Cmax/dose and AUC/dose) was analyzed by using the Kruskal–Wallis test, and natural log‐transformed Cmax, AUClast, and AUCinf of vutiglabridin were analyzed by the power model of . 25 The equation of the power model was log‐transformed as follows: , where is the parameter, is the intercept, and is the dose‐proportionality coefficient. If the 90% confidence interval (90% CI) of the estimation included 1, dose‐proportionality was concluded. For food effect evaluation, the geometric mean ratio (GMR) and 90% CI of the fasted state to the fed state for AUClast and Cmax were calculated using analysis of variance (ANOVA). The food effect was considered a fixed effect, and the differences between subjects were considered a random effect. To evaluate the effect of ethnicity on the pharmacokinetic properties of vutiglabridin, the Cmax and AUClast calculated after a single dose of vutiglabridin 480 mg and the Cmax,ss and AUCtau,ss calculated after multiple doses were log‐transformed, and the GMR and 90% CI of the Korean subjects to White subjects were calculated using ANOVA. Correlation analysis was performed to evaluate the relationship between the AUCtau,ss and pharmacodynamic end points. A p value less than 0.05 was considered statistically significant.
RESULTS
Subject characteristics
In the SAD study, a total of 61 Korean subjects were enrolled and randomized, and 59 subjects completed the study. A total of 60 subjects who have received vutiglabridin (N = 48) or matching placebo (N = 12) more than once were included in the safety analysis set, and among 59 subjects who completed the study, 47 subjects received vutiglabridin and 12 subjects received matching placebo and were included in the pharmacokinetic analysis set. The mean age, height, weight, and BMI of subjects who completed the SAD study were 28.9 ± 6.8 years, 173.4 ± 5.9 cm, 68.9 ± 7.3 kg, and 22.9 ± 1.7 kg/m2, respectively. In the MAD study, a total of 32 subjects (20 Korean subjects and 12 White subjects) were enrolled and randomized, and 29 subjects (19 Korean subjects and 10 White subjects) completed the study. A total of 32 subjects who have received vutiglabridin (N = 25) or matching placebo (N = 7) more than once were included in the safety analysis set. Among a total of 29 subjects who completed the MAD study, 15 Korean subjects and eight White subjects received vutiglabridin, and four Korean subjects and two White subjects received placebo and were included in the pharmacokinetic and pharmacodynamic analysis sets. For Korean subjects who completed the MAD study, the mean age, height, weight, and BMI were 29.6 ± 5.1 years, 174.8 ± 4.7 cm, 70.5 ± 6.6 kg, and 23.0 ± 1.6 kg/m2, respectively, and for White subjects, the corresponding values were 28.5 ± 3.9 years, 175.8 ± 5.5 cm, 71.4 ± 5.8 kg, and 23.1 ± 1.7 kg/m2, respectively. The demographics were comparable between the two ethnicities. Although subjects regardless of sex were planned to be recruited, all subjects who were enrolled in the SAD and MAD studies were men.
Safety and tolerability
Single and multiple oral dosing of vutiglabridin were generally well‐tolerated by the subjects. In the SAD study, 13 treatment‐emergent adverse events (TEAEs) were reported by 10 subjects, and seven TEAEs were determined to be adverse drug reactions (ADRs; Table 1). Among the TEAEs, four serious adverse events (SAEs) occurred in one subject in the 720 mg dose group due to a car accident (neck sprain, lumbar sprain, contusion of the right knee, and sprain of the medial collateral ligament of the right knee). All TEAEs, except for those SAEs, were mild in their severity and spontaneously resolved. The incidence of TEAEs did not increase with the increase in the administered dose of vutiglabridin. In the MAD study, 17 TEAEs were reported in 10 subjects, and 16 TEAEs were determined to be ADRs (Table 2). Except for one TEAE that was moderate in severity (gastroesophageal reflux disease), all TEAEs were mild in severity and spontaneously resolved. There were no clinically significant changes observed in clinical laboratory tests, physical examinations, vital signs, 12‐lead ECG, or semen test results. Additionally, no clinically meaningful changes in the levels of testosterone, LH, FSH, and inhibin B were observed after administrations of investigational products.
TABLE 1.
Summary of TEAEs after a single administration of vutiglabridin
| System organ class, Preferred term | Vutiglabridin, 30 mg (N = 8) | Vutiglabridin, 60 mg (N = 8) | Vutiglabridin, 120 mg (N = 8) | Vutiglabridin, 240 mg (fasted) (N = 8) | Vutiglabridin, 240 mg (fed) (N = 8) | Vutiglabridin, 480 mg (N = 8) | Vutiglabridin, 720 mg (N = 8) | Placebo, (N = 12) |
|---|---|---|---|---|---|---|---|---|
| TEAEs | 1 (12.5) | 3 (37.5) | 2 (25.0) | 1 (12.5) | 0 (0.0) | 0 (0.0) | 1 (12.5) | 2 (16.7) |
| Gastrointestinal disorders | 1 (12.5) | 1 (12.5) | ||||||
| Gingival pain | 1 (12.5) | |||||||
| Stomatitis | 1 (12.5) | |||||||
| General disorders and administration site conditions | 1 (12.5) | |||||||
| Pyrexia | 1 (12.5) | |||||||
| Infections and infestations | 1 (12.5) | |||||||
| Upper respiratory tract infection | 1 (12.5) | |||||||
| Injury, poisoning and procedural complications | 1 (12.5) | |||||||
| Contusion | 1 (12.5) | |||||||
| Ligament sprain | 1 (12.5) | |||||||
| Musculoskeletal and connective tissue disorders | 1 (8.3) | |||||||
| Neck pain | 1 (8.3) | |||||||
| Nervous system disorders | 2 (25.0) | 1 (8.3) | ||||||
| Dizziness | 1 (12.5) | |||||||
| Dizziness postural | 1 (8.3) | |||||||
| Headache | 1 (12.5) | |||||||
| Respiratory, thoracic and mediastinal disorders | 1 (12.5) | |||||||
| Rhinorrhea | 1 (12.5) |
Note: Data are shown as the number of subjects (percentage of subjects).
Abbreviation: TEAE, treatment‐emergent adverse event.
TABLE 2.
Summary of TEAEs after multiple administrations of vutiglabridin
| System organ class, Preferred term | Korean subjects | White subjects | |||
|---|---|---|---|---|---|
| Vutiglabridin 240 mg (N = 8) | Vutiglabridin 480 mg (N = 8) | Placebo (N = 4) | Vutiglabridin 480 mg (N = 9) | Placebo (N = 3) | |
| TEAEs | 4 (50.0) | 3 (37.5) | 0 (0.0) | 2 (22.2) | 1 (33.3) |
| Gastrointestinal disorders | 1 (12.5) | 1 (12.5) | 1 (11.1) | 1 (33.3) | |
| Diarrhea | 1 (11.1) | 1 (33.3) | |||
| Dyspepsia | 1 (12.5) | ||||
| Gastroesophageal reflux disease | 1 (12.5) | ||||
| Investigations | 3 (37.5) | ||||
| Bacterial test positive | 3 (37.5) | ||||
| Blood cholesterol increased | 1 (12.5) | ||||
| Blood insulin increased | 2 (25.0) | ||||
| Blood lactate dehydrogenase increased | 1 (12.5) | ||||
| Metabolism and nutrition disorders | 1 (12.5) | ||||
| Decreased appetite | 1 (12.5) | ||||
| Nervous system disorders | 1 (11.1) | ||||
| Headache | 1 (11.1) | ||||
| Reproductive system and breast disorders | 1 (12.5) | ||||
| Hematospermia | 1 (12.5) | ||||
| Respiratory, thoracic and mediastinal disorders | 1 (12.5) | ||||
| Rhinorrhea | 1 (12.5) | ||||
| Skin and subcutaneous tissue disorders | 1 (12.5) | ||||
| Acne | 1 (12.5) | ||||
Note: Data are shown as the number of subjects (percentage of subjects).
Abbreviation: TEAE, treatment‐emergent adverse event.
Pharmacokinetics
After a single administration of vutiglabridin, the plasma concentrations of vutiglabridin rapidly increased (median Tmax: 1.5 to 3 h) and showed biphasic elimination profiles (Figure 2). The plasma concentration of total vutiglabridin increased less than the dose‐proportionality after a single oral administration of vutiglabridin (Table 3). The CL/F and Vd/F of the total vutiglabridin increased as the dose of vutiglabridin increased, and the t 1/2 became longer (Table 3). The 95% CIs of the slope of the log‐transformed Cmax and AUClast of total vutiglabridin were 0.60–0.79 and 0.71–0.85, respectively. For (S)‐vutiglabridin, the 95% CIs of the slope of the log‐transformed Cmax and AUClast were 0.60–0.77 and 0.74–0.85, respectively, and the corresponding values were 0.56–0.93 and 0.52–1.10 for (R)‐vutiglabridin. Further pharmacokinetic information about (S)‐vutiglabridin and (R)‐vutiglabridin are shown in Figure S1, and Tables S1 and S2.
FIGURE 2.
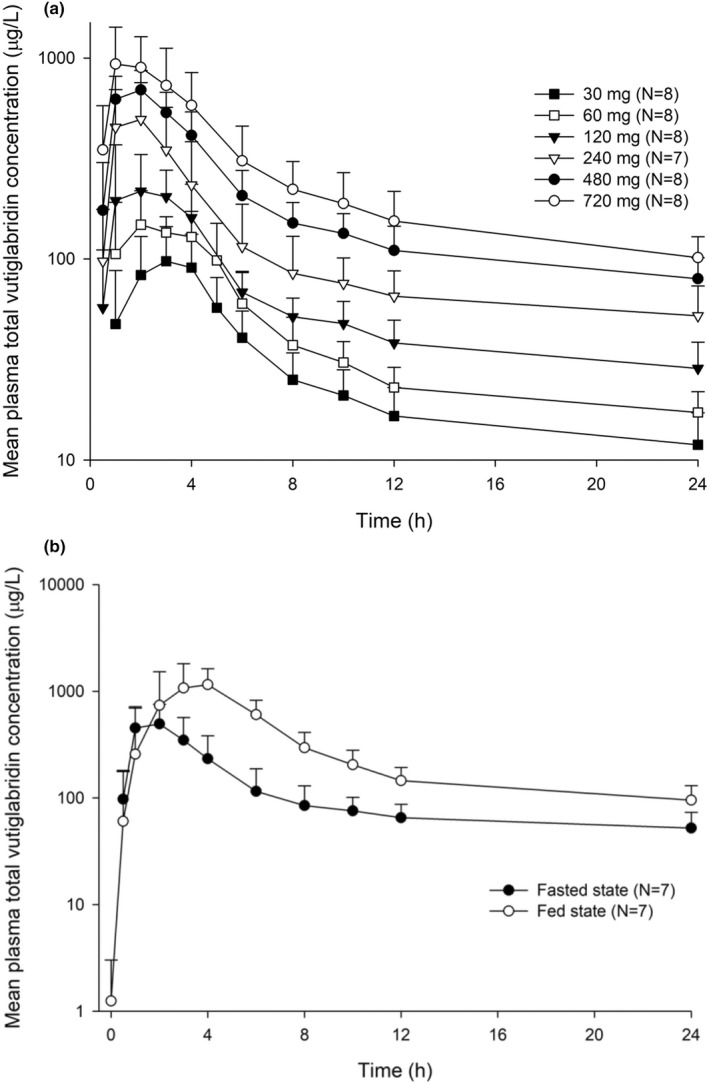
Mean plasma concentration‐time profiles of total vutiglabridin after a single oral administration of vutiglabridin. The error bars represent the standard deviations. (a) After single oral administration of vutiglabridin and (b) after a single oral administration of vutiglabridin 240 mg with or without a high‐fat meal
TABLE 3.
Pharmacokinetic parameters of total vutiglabridin after single and multiple administrations
| Dose groups | N | Tmax (Tmax,ss) (h) | Cmax (Cmax,ss) (μg/L) | AUClast (AUCtau) (h∙μg/L) | AUCinf (h∙μg/L) | t 1/2 (h) | CL/F (L/h) | Vd/F (L) | PTF (%) | Accumulation ratio |
|---|---|---|---|---|---|---|---|---|---|---|
| Single ascending dose study | ||||||||||
| 30 mg | 8 |
2.50 [2.00–4.02] |
119 ± 40.9 | 1220 ± 485 | 1710 ± 670 | 72.6 ± 9.33 | 20.1 ± 7.60 | 2100 ± 769 | ‐ | ‐ |
| 60 mg | 8 |
2.00 [1.00–5.00] |
181 ± 47.9 | 1680 ± 292 | 2280 ± 348 | 77.6 ± 40.0 | 26.8 ± 3.91 | 2880 ± 1130 | ‐ | ‐ |
| 120 mg | 8 |
1.50 [1.00–4.00] |
295 ± 98.0 | 3220 ± 723 | 3670 ± 965 | 77.7 ± 26.4 | 34.5 ± 8.18 | 3690 ± 1100 | ‐ | ‐ |
| 240 mg (fasted) | 7 |
2.00 [1.00–2.00] |
579 ± 263 | 5700 ± 2190 | 6320 ± 2490 | 71.5 ± 16.8 | 41.5 ± 10.5 | 4310 ± 1740 | ‐ | ‐ |
| 240 mg (fed) | 7 |
3.00 [2.00–4.00] |
1680 ± 650 | 13,900 ± 4090 | 15,900 ± 4810 | 87.6 ± 16.6 | 16.1 ± 4.01 | 2020 ± 559 | ‐ | ‐ |
| 480 mg | 8 |
2.00 [1.00–3.00] |
763 ± 142 | 9360 ± 1960 | 10,600 ± 2350 | 79.3 ± 23.4 | 47.2 ± 10.2 | 5370 ± 1840 | ‐ | ‐ |
| 720 mg | 8 |
2.00 [1.00–4.00] |
1030 ± 414 | 13,700 ± 4380 | 16,300 ± 6380 | 85.4 ± 25.7 | 49.1 ± 15.3 | 5730 ± 1730 | ‐ | ‐ |
| Multiple ascending dose study | ||||||||||
| Korean subjects 240 mg | 8 |
2.01 [1.00–4.00] |
624 ± 93.6 | 6540 ± 1460 | ‐ | 107 ± 37.6 | 38.7 ± 10.8 | 5840 ± 2020 | 169 ± 30.3 | 2.20 ± 0.40 |
| Korean subjects 480 mg | 7 |
2.00 [1.00–4.00] |
1110 ± 240 | 11,700 ± 2270 | ‐ | 113 ± 33.7 | 42.5 ± 8.64 | 7080 ± 2960 | 167 ± 61.8 | 2.70 ± 0.89 |
| White subjects 480 mg | 8 |
1.50 [1.00–4.00] |
923 ± 195 | 8320 ± 2610 | ‐ | 72.8 ± 22.9 | 61.8 ± 15.9 | 6120 ± 1010 | 213 ± 66.4 | 2.76 ± 0.85 |
Note: Data are shown as the mean ± standard deviation except for Tmax, which is shown as the median [minimum – maximum].
Abbreviations: AUCinf: area under the plasma concentration‐time curve from 0 h to infinity; AUClast, area under the plasma concentration‐time curve from 0 h to the last measurable time point; AUCtau, area under the plasma concentration‐time curve for a dosing interval at steady state; CL/F, apparent clearance; Cmax, maximal plasma concentration; Cmax,ss, maximal plasma concentration at steady state; PTF, peak trough fluctuation; t 1/2, elimination half‐life after a single dose; Tmax, time for the maximal plasma concentration; Tmax,ss, time for the maximal plasma concentration at steady state; Vd/F, apparent volume of distribution.
In the MAD study, vutiglabridin significantly accumulated in the plasma after once daily administrations for 14 days, and the mean accumulation ratio ranged from 2.20 to 2.76 (Table 3). The overall pharmacokinetic profiles were similar between single and multiple administrations in both ethnicities, and the plasma concentrations of vutiglabridin showed biphasic elimination profiles (Figure 3). After multiple administrations of vutiglabridin, the values of median Tmax, CL/F, and Vd/F of total vutiglabridin were similar to the values after a single administration. However, a longer t 1/2 was observed after multiple administrations (Table 3).
FIGURE 3.
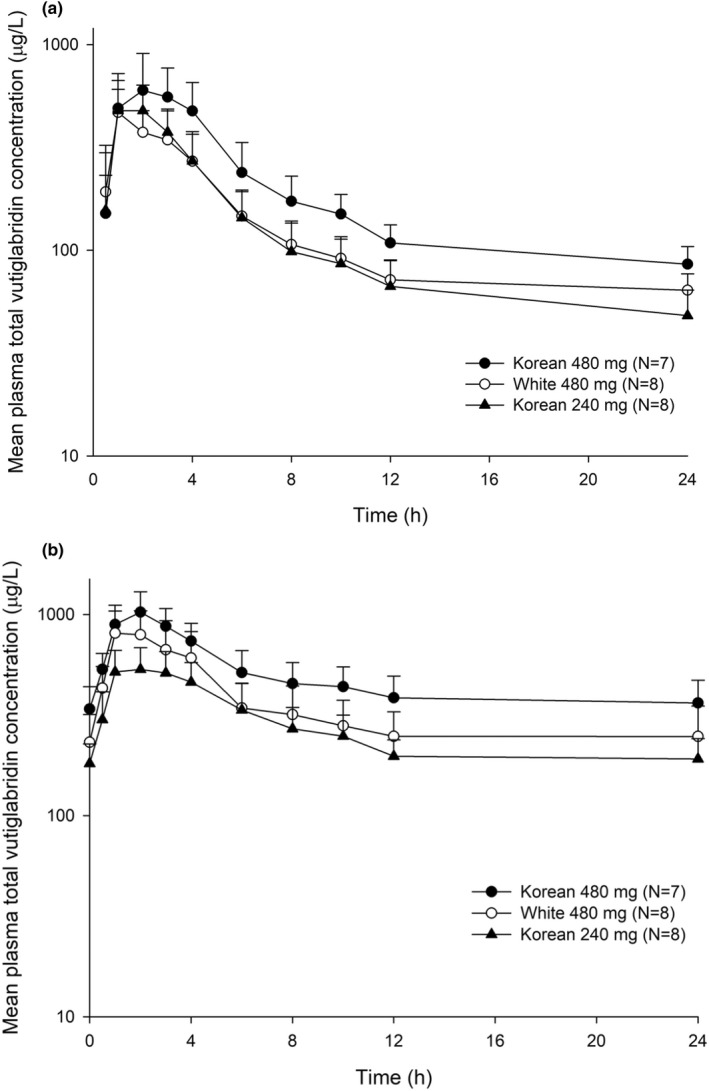
Mean plasma concentration‐time profiles of total vutiglabridin after multiple oral administrations of vutiglabridin. The error bars represent the SDs. (a) After the first dose during the multiple‐dose study, and (b) after the last dose during the multiple‐dose study
The systemic exposure of vutiglabridin significantly increased when 240 mg of vutiglabridin was taken with a high‐fat meal (Figure 2, Table 3). The median Tmax of vutiglabridin was delayed 1 h when taken with a high‐fat meal compared to the fasted state (Table 3). The GMRs (90% CIs) of the fed to fasted state for the Cmax and AUClast of total vutiglabridin were 2.95 (2.10–4.13) and 2.48 (2.10–2.93), respectively. The effect of food on the pharmacokinetics of (R)‐ and (S)‐vutiglabridin was similar to that of total vutiglabridin (Tables S1 and S2).
The systemic exposure of total vutiglabridin was higher in Korean subjects than in White subjects after single and multiple oral administrations of vutiglabridin 480 mg, and both (R)‐ and (S)‐vutiglabridin showed similarly higher exposure levels in Korean subjects than in White subjects (Figure 4, Tables S1 and S2). After a single administration of vutiglabridin 480 mg, the GMRs (90% CIs) of the White subjects to Korean subjects for Cmax and AUClast of the total vutiglabridin were 0.70 (0.53–0.93) and 0.68 (0.53–0.87), respectively, and those values after multiple administrations were 0.83 (0.67–1.02) and 0.70 (0.56–0.87) for Cmax,ss and AUCtau,ss, respectively.
FIGURE 4.
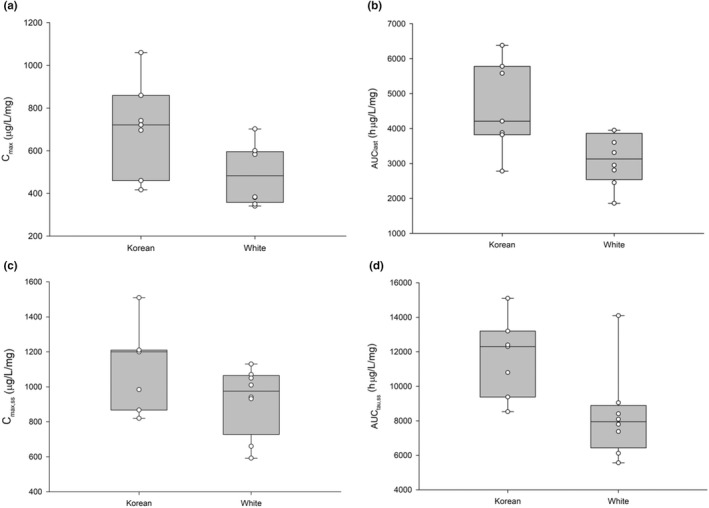
Comparison of the pharmacokinetic parameters of total vutiglabridin between Korean and White subjects. AUClast, area under the concentration‐time curves to the last measurable timepoint; AUCtau,ss, area under the plasma concentration‐time curve over a dosing interval at steady‐state; Cmax, maximum plasma concentration; Cmax,ss, maximum plasma concentration at steady‐state
Pharmacodynamics
After multiple vutiglabridin administrations, a trend toward increasing adiponectin levels and decreasing C‐peptide and CCL2 levels were observed compared with the placebo groups (Figure S2, Table S5). In Korean subjects, a tendency of exposure‐response correlation was observed in several pharmacodynamic markers (e.g., adiponectin, C‐peptide, CCL2, and insulin) after multiple administrations of vutiglabridin but the pharmacodynamic parameters showed considerable interindividual variability (Figure S2, Table S5). A significant positive correlation was observed between the AUCtau of total vutiglabridin and baseline corrected values of adiponectin (R = 0.4464, p < 0.05; Figure S2). For leptin, IL‐6, and TNFα, no specific changes were observed after vutiglabridin administration compared with baseline and placebo.
DISCUSSION
This study showed that vutiglabridin was well‐tolerated in healthy male Korean and White individuals. During the SAD and MAD studies, no subjects withdrew due to TEAEs, and the incidence of TEAEs did not increase as the dose of vutiglabridin increased (Tables 1 and 2). One subject in the SAD study had four SAEs due to a car accident 18 days after receiving 720 mg of vutiglabridin. Considering the onset time and the possible mechanism of action of vutiglabridin on exercise capacity or the central nervous system, those events were determined not to be related to the study drug. 21 Additionally, there were no vutiglabridin‐related or clinically significant changes in vital signs, 12‐lead ECGs, physical examinations, or semen tests.
The plasma concentration of vutiglabridin was increased less than dose‐proportionality (Figure 2, Table 3). Because the CL/F and Vd/F were simultaneously increased as the dose increased (Table 3), saturation of oral absorption process (i.e., decrease of oral bioavailability) is the likely reason for the observed nonlinear pharmacokinetics. The available pharmacokinetic data did not suggest a time‐dependent pharmacokinetic nonlinearity (e.g., auto‐induction or auto‐inhibition) after multiple administrations of vutiglabridin. In the 240 and 480 mg dose groups, the CL/F and Vd/F values were similar between the SAD and MAD studies. In addition, the AUCinf obtained from the SAD study were comparable with AUCtau obtained from the MAD study (Table 3).
The poor absorption of vutiglabridin was expected due to its hydrophobic property. It was also shown in the food effect study, showing increased systemic exposure of vutiglabridin ~2.3 to 4.0 times in the fed state compared to the fasted state. 21 , 26 Considering the results of the food effect study, to increase the absorption rate of vutiglabridin, it should be administered in the fed state. Because the effect of food can vary in different dose groups, a food effect study in a highest therapeutic dose needs to be evaluated in a further clinical study. Furthermore, other formulations that can increase the absorption should be considered. Additionally, considering that the half‐life of total vutiglabridin in the SAD study was longer than 70 h, once daily dosing or a longer dosing interval can be recommended to patients (Table 3).
The systemic exposure was lower by ~30% to 60% in White subjects after multiple administrations of vutiglabridin (Figure 3), although there were no significant differences in the demographic characteristics between Korean and White subjects. Asian individuals have a higher body fat percentage, intramyocellular lipid content, and liver fat content than White individuals, and this physiological difference may be one of the reasons for the pharmacokinetic differences between the White subjects and Korean subjects. 27 However, when considering that there was only one dose group of 480 mg that could compare these ethnic differences, further studies are needed to evaluate the exact factor affecting the different degrees of systemic exposure of vutiglabridin.
After multiple administrations of vutiglabridin, a trend toward increasing adiponectin levels compared to the placebo in Korean subjects was observed. Moreover, the C‐peptide levels and the CCL2 levels tended to decrease compared to the placebo in both ethnicities (Figure S2, Table S5). The baseline corrected values of adiponectin, C‐peptide, TNFα, and CCL2 increased as the dose of vutiglabridin increased, suggesting that the response increased as the exposure of vutiglabridin increased (Table S5). For adiponectin, a positive dose–response relationship and exposure‐response relationship were observed between the 240 and 480 mg dose groups and between the two ethnicities (Figure S2). Adiponectin plays an important role in energy metabolism and is known to be inversely correlated with BMI and fat accumulation. 28 , 29 The serum adiponectin level was decreased in the placebo groups after vutiglabridin administration (Figure S2, Table S5), and this phenomenon is probably due to the controlled diet and decreased physical activity during the study period. 30 , 31 Additionally, because adiponectin levels are known to show large daily variation and are affected by day/night rhythms, the controlled environment of the clinical study might have caused decreased adiponectin levels in the placebo groups. 32
After multiple administrations of vutiglabridin, a trend toward increasing adiponectin levels compared to the placebo in Korean subjects was observed. Moreover, the C‐peptide levels and the CCL2 levels tended to decrease compared to the placebo in both ethnicities (Figure S2, Table S5). The baseline corrected values of adiponectin, C‐peptide, TNFα, and CCL2 increased as the dose of vutiglabridin increased, suggesting that the response increased as the exposure of vutiglabridin increased (Table S5). For adiponectin, a positive dose–response relationship and exposure‐response relationship were observed between the 240 and 480 mg dose groups and between the two ethnicities (Figure S2). Adiponectin plays an important role in energy metabolism and is known to be inversely correlated with BMI and fat accumulation. 29 , 30 The serum adiponectin level was decreased in the placebo groups after vutiglabridin administration (Figure S2, Table S5), and this phenomenon is probably due to the controlled diet and decreased physical activity during the study period. 31 , 32 Additionally, because adiponectin levels are known to show large daily variation and are affected by day/night rhythms, the controlled environment of the clinical study might have caused decreased adiponectin levels in the placebo groups. 33
There are several limitations in this study. First, this study was conducted during relatively short study periods in healthy normal weight subjects who has no obesity‐related pathophysiologic markers. Although adiponectin has shown a positive dose–response relationship, the adiponectin levels at baseline and after multiple oral administrations of vutiglabridin in treatment groups varied largely (Figure S3). Further clinical studies in patients with obesity will be needed for a more accurate evaluation of the pharmacological effect of vutiglabridin. Second, only male subjects were enrolled in this study, although we recruited both male and female subjects. The pharmacokinetics and pharmacodynamics of vutiglabridin in women should be evaluated in further clinical studies.
CONCLUSION
Vutiglabridin was well‐tolerated in healthy Korean and White individuals at a single oral dose of up to 720 mg and at multiple oral doses of up to 480 mg. The plasma concentration of vutiglabridin increased less than the dose‐proportional manner. These results justify further investigation of vutiglabridin in patients with obesity.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. D.Y.Y., H.Y., S.L., K.S.Y., I.J.J., and J.O. designed the research. J.Y.N., D.Y.Y., S.L., K.S.Y., I.J.J., and J.O. performed the research. J.Y.N., D.Y.Y., S.L., and J.O. analyzed the data.
CONFLICT OF INTEREST
Sang‐Ku Yoo and Youngah Kim are current/former employees of Glaceum Inc. and hold stocks/shares. Glaceum Inc. provided funding for the research, and holds Patent US9783551B2, which grants intellectual properties (IPs) for the synthesis and use of the compounds in the article. All other authors declared no competing interests for this work.
Supporting information
Figure S1‐S3
Table S1‐S5
ACKNOWLEDGMENTS
This study was sponsored by Glaceum Incorporated. Joo Young Na received a scholarship from the BK21 FOUR education program.
Na JY, Yoon DY, Yoo H, et al. Safety, tolerability, pharmacokinetic, and pharmacodynamic characteristics of vutiglabridin: A first‐in‐class, first‐in‐human study. Clin Transl Sci. 2022;15:2744‐2757. doi: 10.1111/cts.13401
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available upon request from the corresponding author. The data are not publicly available due to confidentiality restrictions.
REFERENCES
- 1. Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nat Rev Genet. 2022;23(2):120‐133. doi: 10.1038/s41576-021-00414-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Williams EP, Mesidor M, Winters K, Dubbert PM, Wyatt SB. Overweight and obesity: prevalence, consequences, and causes of a growing public health problem. Curr Obes Rep. 2015;4(3):363‐370. doi: 10.1007/s13679-015-0169-4 [DOI] [PubMed] [Google Scholar]
- 3. OECD/WHO . Overweight and obesity. In: Health at a Glance: Asia/Pacific 2020: Measuring Progress Towards Universal Health Coverage. OECD Publishing; 2020. doi: 10.1787/a47d0cd2-en [DOI] [Google Scholar]
- 4. Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019;15(5):288‐298. doi: 10.1038/s41574-019-0176-8 [DOI] [PubMed] [Google Scholar]
- 5. Wharton S, Lau DCW, Vallis M, et al. Obesity in adults: a clinical practice guideline. CMAJ. 2020;192(31):E875‐E891. doi: 10.1503/cmaj.191707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Joo JK, Lee KS. Pharmacotherapy for obesity. J Menopausal Med. 2014;20(3):90‐96. doi: 10.6118/jmm.2014.20.3.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Erlanger SR, Henson EA. Classification and Pharmacological Management of Obesity. P T. 2008;33(12):724‐728. [PMC free article] [PubMed] [Google Scholar]
- 8. NIDDK. National Institute of Diabetes and Digestive and Kidney Diseases . Prescription medications to treat overweight & obesity. Accessed October 7, 2021. https://www.niddk.nih.gov/health‐information/weight‐management/prescription‐medications‐treat‐overweight‐obesity
- 9. Gill LE, Bartels SJ, Batsis JA. Weight Management in Older Adults. Curr Obes Rep. 2015;4(3):379‐388. doi: 10.1007/s13679-015-0161-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller SL, Wolfe RR. The danger of weight loss in the elderly. J Nutr Health Aging. 2008;12(7):487‐491. doi: 10.1007/BF02982710 [DOI] [PubMed] [Google Scholar]
- 11. Cava E, Yeat NC, Mittendorfer B. Preserving healthy muscle during weight loss. Adv Nutr. 2017;8(3):511‐519. doi: 10.3945/an.116.014506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Williams GM, Iatropoulos MJ. Alteration of liver cell function and proliferation: differentiation between adaptation and toxicity. Toxicol Pathol. 2002;30(1):41‐53. doi: 10.1080/01926230252824699 [DOI] [PubMed] [Google Scholar]
- 13. Hall AP, Elcombe CR, Foster JR, et al. Liver hypertrophy: a review of adaptive (adverse and non‐adverse) changes‐‐conclusions from the 3rd International ESTP Expert Workshop. Toxicol Pathol. 2012;40(7):971‐994. doi: 10.1177/0192623312448935 [DOI] [PubMed] [Google Scholar]
- 14. Rodríguez‐Hernández H, Simental‐Mendía LE, Rodríguez‐Ramírez G, Reyes‐Romero MA. Obesity and inflammation: epidemiology, risk factors, and markers of inflammation. Int J Endocrinol. 2013;2013:678159. doi: 10.1155/2013/678159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakagawa K, Kishida H, Arai N, Nishiyama T, Mae T. Licorice flavonoids suppress abdominal fat accumulation and increase in blood glucose level in obese diabetic KK‐A(y) mice. Biol Pharm Bull. 2004;27(11):1775‐1778. doi: 10.1248/bpb.27.1775 [DOI] [PubMed] [Google Scholar]
- 16. Simmler C, Pauli GF, Chen SN. Phytochemistry and biological properties of glabridin. Fitoterapia. 2013;90:160‐184. doi: 10.1016/j.fitote.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye Y, Liu K, Geng S, et al. A novel fluorescent recombinant cell‐based biosensor for screening NLRP3 inflammasome inhibitors. Sens Actuators B. 2019;301:126864. doi: 10.1016/j.snb.2019.126864 [DOI] [Google Scholar]
- 18. Lee JW, Choe SS, Jang H, et al. AMPK activation with glabridin ameliorates adiposity and lipid dysregulation in obesity. J Lipid Res. 2012;53(7):1277‐1286. doi: 10.1194/jlr.M022897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ito C, Oi N, Hashimoto T, et al. Absorption of dietary licorice isoflavan glabridin to blood circulation in rats. J Nutr Sci Vitaminol (Tokyo). 2007;53(4):358‐365. doi: 10.3177/jnsv.53.358 [DOI] [PubMed] [Google Scholar]
- 20. Ao M, Shi Y, Cui Y, Guo W, Wang J, Yu L. Factors influencing glabridin stability. Nat Prod Commun. 2010;5(12):1907‐1912. [PubMed] [Google Scholar]
- 21. Choi LS, Jo IG, Kang KS, et al. Discovery and preclinical efficacy of HSG4112, a synthetic structural analog of glabridin, for the treatment of obesity. Int J Obes (Lond). 2021;45(1):130‐142. doi: 10.1038/s41366-020-00686-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. FDA . Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers.
- 23. Cesur G, Ozguner F, Yilmaz N, Dundar B. The relationship between ghrelin and adiponectin levels in breast milk and infant serum and growth of infants during early postnatal life. J Physiol Sci. 2012;62(3):185‐190. doi: 10.1007/s12576-012-0193-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vass RA, Bell EF, Colaizy TT, et al. Hormone levels in preterm and donor human milk before and after Holder pasteurization. Pediatr Res. 2020;88(4):612‐617. doi: 10.1038/s41390-020-0789-6 [DOI] [PubMed] [Google Scholar]
- 25. Gough K, Hutchison M, Keene O, et al. Assessment of dose proportionality: report from the statisticians in the Pharmaceutical industry/Pharmacokinetics UK Joint Working Party. Drug Inf J. 1995;29(3):1039‐1048. doi: 10.1177/009286159502900324 [DOI] [Google Scholar]
- 26. Birkett DJ. Pharmacokinetics made easy 9: non‐linear pharmacokinetics. Aust Prescr. 1994;17(2):36‐38. doi: 10.18773/austprescr.1994.046 [DOI] [Google Scholar]
- 27. Ditzinger F, Price DJ, Ilie AR, et al. Lipophilicity and hydrophobicity considerations in bio‐enabling oral formulations approaches ‐ a PEARRL review. J Pharm Pharmacol. 2019;71(4):464‐482. doi: 10.1111/jphp.12984 [DOI] [PubMed] [Google Scholar]
- 28. Wulan SN, Westerterp KR, Plasqui G. Ethnic differences in body composition and the associated metabolic profile: a comparative study between Asians and Caucasians. Maturitas. 2010;65(4):315‐319. doi: 10.1016/j.maturitas.2009.12.012 [DOI] [PubMed] [Google Scholar]
- 29. De Rosa A, Monaco ML, Capasso M, et al. Adiponectin oligomers as potential indicators of adipose tissue improvement in obese subjects. Eur J Endocrinol. 2013;169(1):37‐43. doi: 10.1530/EJE-12-1039 [DOI] [PubMed] [Google Scholar]
- 30. Nigro E, Scudiero O, Monaco ML, et al. New insight into adiponectin role in obesity and obesity‐related diseases. Biomed Res Int. 2014;2014:658913. doi: 10.1155/2014/658913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Polito R, Monda V, Nigro E, et al. The important role of adiponectin and orexin‐A, two key proteins improving healthy status: focus on physical activity. Front Physiol. 2020;11:356. doi: 10.3389/fphys.2020.00356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reis CEG, Bressan J, Alfenas RCG. Effect of the diet components on adiponectin levels. Nutr Hosp. 2010;25(6):881‐888. [PubMed] [Google Scholar]
- 33. Scheer FAJL, Chan JL, Fargnoli J. Day/night variations of high‐molecular‐weight adiponectin and lipocalin‐2 in healthy men studied under fed and fasted conditions. Diabetologia. 2010;53(11):2401‐2405. doi: 10.1007/s00125-010-1869-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1‐S3
Table S1‐S5
Data Availability Statement
The data that support the findings of this study are available upon request from the corresponding author. The data are not publicly available due to confidentiality restrictions.