

Abstract
A phase I trial (NCT03447314; 204686) evaluated the safety and efficacy of GSK1795091, a Toll‐like receptor 4 (TLR4) agonist, in combination with immunotherapy (GSK3174998 [anti‐OX40 monoclonal antibody], GSK3359609 [anti‐ICOS monoclonal antibody], or pembrolizumab) in patients with solid tumors. The primary endpoint was safety; other endpoints included efficacy, pharmacokinetics, and pharmacodynamics (PD). Manufacturing of GSK1795091 formulation was modified during the trial to streamline production and administration, resulting in reduced PD (cytokine) activity. Fifty‐four patients received GSK1795091 with a combination partner; 32 received only the modified GSK1795091 formulation, 15 received only the original formulation, and seven switched mid‐study from the original to the modified formulation. Despite the modified formulation demonstrating higher systemic GSK1795091 exposure compared with the original formulation, the transient, dose‐dependent elevations in cytokine and chemokine concentrations were no longer observed (e.g., IP‐10, IL10, IL1‐RA). Most patients (51/54; 94%) experienced ≥1 treatment‐emergent adverse event (TEAE) during the study. Safety profiles were similar between formulations, but a higher incidence of TEAEs associated with immune responses (chills, fatigue, pyrexia, nausea, and vomiting) were observed with the original formulation. No conclusions can be made regarding GSK1795091 anti‐tumor activity due to the limited data collected. Manufacturing changes were hypothesized to have caused the change in biological activity in this study. Structural characterization revealed GSK1795091 aggregate size in the modified formulation to be twice that in the original formulation, suggesting a negative correlation between GSK1795091 aggregate size and PD activity. This may have important clinical implications for future development of structurally similar compounds.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Toll‐like receptor 4 (TLR4) agonists, such as lipopolysaccharide (LPS) and lipid A analogs, have demonstrated anti‐cancer effects in patients. Previous studies offer conflicting evidence on the active form (monomeric vs. aggregates) of LPS/lipid A analogs and suggest that different forms can stimulate different immune pathways.
WHAT QUESTION DID THIS STUDY ADDRESS?
This phase I study investigated the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of GSK1795091 with one of three immunotherapies in patients with solid tumors. In addition, the study addressed the effects of a manufacturing change on the biological activity of GSK1795091.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study enables a better understanding of the impact of the manufacturing process on drug aggregate morphology and activity. The formulation change led to the formation of particle aggregates with globular morphology, suggesting the initial dissolution of GSK1795091 in ethanol, instead of initial dissolution using sonication, was the likely contributor to the lowered biological activity of GSK1795091. This could have implications for the development of other lipid A analogs and TLR agonists.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Following manufacturing changes of LPS/other lipid A analogs and chemically manufactured active pharmaceutical ingredients that are prone to structural organization in solution, it is recommended to perform in vitro PD assessments to understand the impact on its biological activity prior to clinical assessment. PK and PD evaluations should be prioritized to ensure no clinically meaningful changes related to the manufacturing occur.
INTRODUCTION
Toll‐like receptors (TLRs) are membrane receptors that elicit innate and adaptive immune responses. 1 In humans, 10 TLRs have been identified and are functionally subdivided into intracellular TLRs that recognize viral and bacterial DNA and RNA, and cell surface TLRs that primarily recognize extracellular ligands, including microbial components, such as lipopolysaccharide (LPS). 1 , 2 LPS, a major component of the outer membrane of Gram‐negative bacteria, is the primary natural ligand of TLR4 3 and elicits an immune response through TLR4‐mediated signaling. 4 LPS has demonstrated an ability to induce cytokine production in vitro, anti‐cancer activity mediated by lipid A (a conserved portion of LPS) in vivo, and anti‐cancer activity in a phase II clinical trial. 5 , 6 , 7 Several lipid A analogs have been developed for the treatment of cancer. 8 , 9 , 10
The TLR4 agonist GSK1795091 is a synthetic lipid A analog that demonstrated immunomodulatory activity in preclinical in vivo studies, including an ability to induce production of proinflammatory cytokines, enhance antigen presentation, and regulate T‐cell responses, alone and in combination with a surrogate OX40 agonist. 11 In the first‐time‐in‐human (FTIH) study (204,685; NCT02798978), GSK1795091 demonstrated an acceptable safety profile, induced transient increases in cytokine levels, and led to changes in immune cell counts in a dose‐dependent manner. 12
Considerable evidence supports the use of TLR agonists as immune adjuvants in combination with other anti‐cancer treatments. The TLR4 agonists glucopyranosyl lipid A in a stable emulsion (GLASE) and OM‐174 are being investigated in combination with other therapies in several cancer types. 13 , 14 This phase I study evaluated the safety, tolerability, and efficacy of GSK1795091 in combination with several immunomodulatory agents with complementary mechanisms of action: GSK3174998 (anti‐OX40 monoclonal antibody), 15 GSK3359609 (anti‐inducible costimulatory [ICOS] monoclonal antibody), 16 or pembrolizumab (anti–programmed cell death protein 1 [PD‐1] monoclonal antibody), 17 under the hypothesis that combinations would lead to greater anti‐tumor immune response and clinical activity than with monotherapies alone. The study was designed in two parts: Part 1 (dose escalation) to evaluate the dose and safety of these combinations, and Part 2 (cohort expansion) to further evaluate the safety and efficacy of the selected dosing regimen. During Part 1, the manufacturing procedure for GSK1795091 was modified to reduce the manufacturing time and to optimize administration. Pharmacodynamic (PD) evaluation of GSK1795091 revealed that the modified formulation had significantly lower biological activity than the original formulation. The study was therefore terminated before the opening of Part 2, and both formulations underwent further investigation to understand the consequences of the manufacturing change.
Here we report data for primary (safety), secondary (pharmacokinetics [PK]), and exploratory (PK/PD response) endpoints from Part 1 of the NCT03447314 study, as well as data related to investigations of the cause of the change in biological activity. Findings that led to the discovery of an apparent manufacturing‐dependent correlation between the aggregate size and shape of GSK1795091 and its biological activity are highlighted, along with consequential learnings and recommendations for future development of TLR4 agonists and other structurally similar compounds.
MATERIALS AND METHODS
Formulation manufacturing process
The chemical structure of GSK1795091 is shown in Figure S1. The manufacturing procedure was adjusted to replace a sonication step with ethanol dissolution to dissolve the GSK1795091 active pharmaceutical ingredient (API). The final ethanol concentration in the modified formulation was low (0.05% v/v). The adjustment resulted in a reduction of manufacturing time from 2 days to 1 day and streamlined the analytical testing throughout the process. Additionally, the lower final GSK1795091 concentration in the preparation of the modified formulation allowed a simplified dosing procedure (with no dilution steps required at lower doses <200 ng) and reduced waste due to a longer in‐use stability period (24 vs. 8 h), which allows more time between dose preparation and administration. Full details of the manufacturing procedures and components of the original and modified GSK1795091 formulations are presented in Appendix S1 and in Table S1 and Table S2.
Full details for dynamic light scattering (DLS) and cryo‐transmission electron microscopy (TEM) analyses used to characterize the formulations are summarized in Appendix S1.
Study design and treatments
This was a phase I, open‐label, nonrandomized study (204686; NCT03447314) of the TLR4 agonist GSK1795091, administered with a combination partner (anti‐OX40 monoclonal antibody GSK3174998, anti‐ICOS monoclonal antibody GSK3359609, or the anti–PD‐1 monoclonal antibody, pembrolizumab) in patients with advanced solid tumor cancers. Part 1 utilized a Neuenschwander continual reassessment method design 18 to assess safety and tolerability of escalating doses of GSK1795091 with a fixed dose of the combination partner to identify doses for evaluation in Part 2 (Figure S2). Due to the ultra‐low drug concentrations of the GSK1795091 formulations, doses were administered by intravenous (IV) bolus injection. The administration kit consisted of a syringe and catheter (BD Insyte Autoguard catheter [no tubing] and BD Nexiva catheter [with tubing]). Dose and content recovery from both syringe−catheter configurations were determined to be 100% of label claim. However, due to the long length of tubing on the BD Nexiva catheter, a post‐administration flush volume with 3–5 ml of placebo diluent of was required to obtain 100% recovery. These data indicate that the formulation and administration kit were compatible and that there was no loss of GSK1795091 due to sticking on the inner surfaces of the administration kits. The starting dose of GSK1795091 was 50 ng; patients were allocated to one of three cohorts: Group A, receiving GSK1795091 + GSK3174998 (fixed dose of 24 mg IV); Group B, receiving GSK1795091 + GSK3359609 (fixed dose of 80 mg IV); or Group C, receiving GSK1795091 + pembrolizumab (fixed dose of 200 mg IV). GSK1795091 was administered once‐weekly by IV injection for 12 weeks, including an initial 2‐week safety run‐in period of GSK1795091 monotherapy. Combination treatment was initiated from Week 3, and the combination partner was administered by IV infusion once every 3 weeks. From Week 12, GSK1795091 was administered every 3 weeks to coincide with combination partner treatment. Patients received study treatment for 2 years unless one of the following occurred earlier: disease progression as determined by immune‐related Response Evaluation Criteria in Solid Tumors (RECIST) criteria, death, unacceptable toxicity, or protocol‐defined stopping criteria met. Following the discovery that the modified GSK1795091 formulation resulted in lower biological activity compared with the original formulation (measured as a reduction in cytokine production), enrollment into Part 1 was interrupted after considering the totality of data available, and screening of participants was paused to allow continued investigation into the cause (January 2020). The protocol was then amended to discontinue GSK1795091 and to allow patients to continue treatment with the combination partner as monotherapy (March 2020). Enrollment to the study was formally stopped before initiating Part 2 (September 2020).
The study was conducted in accordance with the principles of the Declaration of Helsinki and Council for International Organization of Medical Sciences International Ethical Guidelines and in accordance with the International Conference on Harmonisation Good Clinical Practice. Approval was obtained from institutional review boards and ethics committees before study initiation. Written informed consent was obtained from all patients before enrollment.
Patients
Adult patients with histological confirmation of an advanced solid tumor malignancy with available archival tumor tissue were included in this study. Included patients had disease progression after standard therapies (measurable disease per RECIST 1.1), an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, life expectancy of at least 12 weeks, and adequate organ function. Excluded patients had other malignancies, central nervous system metastases requiring steroids within 2 weeks of first study treatment, active autoimmune disease requiring systemic disease‐modifying or immunosuppressive treatment, a concurrent condition requiring immunosuppressive treatment, known human immunodeficiency virus infection, or current unstable liver or biliary disease. Patients were not permitted to have had prior treatment with OX40, ICOS, or TLR4 agonists, anti‐cancer therapy, or investigational therapy within 30 days or five half‐lives of the drug (whichever was shorter), radiation therapy (with exceptions), allogeneic or autologous bone marrow transplantation, or other solid organ transplantation. For complete inclusion and exclusion information see Appendix S1.
Study endpoints and assessments
The primary endpoints of this study were to evaluate the safety and tolerability of GSK1795091 in combination with GSK3174998, GSK3359609, or pembrolizumab. Secondary endpoints included evaluation of the anti‐tumor activity of GSK1795091 in combination (objective response rate [ORR] and disease control rate [DCR]) and PK of GSK1785091 (maximum observed concentration [Cmax], area under the curve over the dosing interval [AUC(0–τ)], terminal half‐life [t ½], clearance, and volume of distribution at steady state [Vss]). Accumulation ratios for trough concentrations (Ctrough) were not calculated as many pre‐dose concentrations were below the limit of quantification. Exploratory endpoints included evaluation of PK/PD relationships, such as between PK parameters and various blood‐derived PD markers of immunologic activity (e.g., granulocyte colony‐stimulating factor, interleukin [IL]‐6, IL‐8, IL‐10, IL‐1 receptor agonist [IL‐1Ra], interferon gamma‐induced protein [IP‐10], monocyte chemoattractant protein 1 [MCP‐1], and tumor necrosis factor α [TNFα]).
Patients were monitored for treatment‐emergent adverse events (TEAEs) from the start of study treatment until at least 30 days after the last dose of study treatment or the start of a new anti‐cancer therapy, whichever occurred first. Serious TEAEs and adverse events (AEs) of special interest were monitored until up to 90 days after the last dose of study treatment or until the start of a new anti‐cancer therapy, whichever occurred first. TEAEs were coded using the standard Medical Dictionary for Regulatory Activities, grouped by System Organ Class, and graded by the investigator according to the National Cancer Institute—Common Terminology Criteria for Adverse Events, version 4.0. Other safety assessments included physical examinations, assessment of vital signs, electrocardiograms, and clinical safety laboratory tests.
Anti‐cancer activity was evaluated according to RECIST 1.1 and immune‐related RECIST (irRECIST). Blood samples for PK and PD analysis of GSK1795091 were collected within 1 h before study drug administration and at additional timepoints following administration (see Appendix S1 for complete timings). Cytokine/chemokine levels in isolated plasma were analyzed using a custom multiplex immunoassay (Quanterix).
Statistical analyses
No specific statistical hypotheses were tested. The sample size for the study was planned to allow adequate characterization of safety, clinical activity, PK, and PD based on the study objectives. Safety and anti‐tumor activity were evaluated in patients who received ≥1 dose of any study treatment. In some analyses, results for combination partners were pooled due to limited sample sizes and to evaluate the effect of the formulation. PK was evaluated in patients who received ≥1 dose of GSK1795091 and from whom ≥1 post‐dose PK sample for GSK1795091 was measured. PK samples were analyzed using a previously validated method 19 and data were analyzed using standard noncompartmental methods and descriptive statistics. PD biomarkers were evaluated in patients who received ≥1 dose of any study drug, from whom ≥1 PD sample was obtained, analyzed, and measurable, and were summarized descriptively and/or graphically.
RESULTS
Physicochemical characterization of GSK1795091 original and modified formulations
Comparisons of the stability of the original and modified formulations are presented in Table S3; the chemical structure of GSK1795091 is shown in Figure S2. Both the original and modified GSK1795091 formulations met all the specification criteria during the testing timeframes (Table S3).
Characterization of the two formulations by cryo‐TEM revealed that the original GSK1795091 formulation gave rise to a heterogeneous particle population with no globular structures, while aggregated globular structures were mainly observed in samples of the modified formulation. The aggregated globular structures observed in the modified formulation were larger than the sheet‐like structures in the original formulation; this was consistent with DLS particle size analysis, which showed that the mean aggregate size of the modified formulation is approximately two times that of the original formulation (Figure 1). GSK1795091 in the modified formulation degraded faster than the original formulation. However, the modified formulation was within the specifications and suitable for dosing (Table S3).
FIGURE 1.
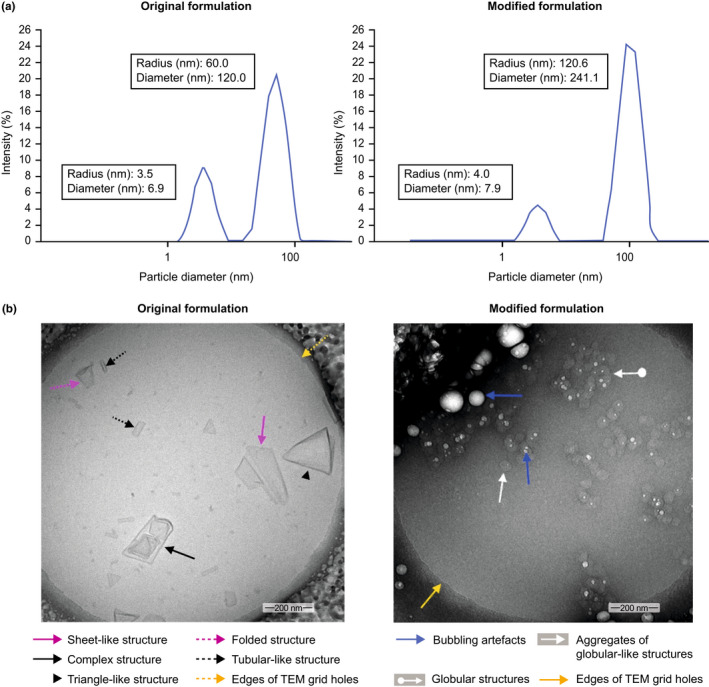
Structural characterization of the original and modified GSK1795091 formulations. Evaluation of the original and modified formulations using (a) dynamic light scattering, revealing that the diameter of GSK1795091 particles in the modified formulation was twice the diameter of the original formulation, and (b) cryo‐transmission electron microscopy (TEM) images, showing formation of large aggregates of globular‐like structures in the modified formulation while the original formulation has mostly sheet‐like morphologies with no globular structures. The globular‐like structures appeared to be sensitive to beam electron due to the presence of ethanol which evaporates under the effect of heat resulting in the dark shades in the image. Globular structures are small, round bodies or spherical objects formed by attractive forces between molecules or group of molecules. Complex structures are multidimensional arrangements of molecules or group of molecules.
Clinical study results
Patient disposition and baseline characteristics
The NCT03447314 study was carried out across eight sites in four countries (5 in the USA, 1 in Canada, 1 in The Netherlands, and 1 in Spain). In total, 54 patients were enrolled and assigned to receive GSK1795091 (TLR4 agonist) with an immunomodulatory combination partner (30 patients received GSK3174998 [anti‐OX40 monoclonal antibody], 11 received GSK3359609 [anti‐ICOS monoclonal antibody], and 13 received pembrolizumab; Figure 2). Most patients received the modified formulation of GSK1795091: 32 patients received only the modified formulation, 15 patients received only the original formulation, and seven patients received both formulations (switching from the original to the modified during the study). At the time of data cutoff (July 1, 2020), all patients had been discontinued from GSK1795091, 19 patients (35.2%) were ongoing in the study (off study treatment and in follow‐up [n = 15] or receiving combination partner monotherapy [n = 4] following GSK1795091 discontinuation), 8 (14.8%) withdrew from the study before completion, and 30 (55.6%) died (including 3 patients who withdrew from the study before completion). Among patients who died during the study, the primary cause of death was cancer. Baseline patient demographics and characteristics are summarized in Table 1. The baseline characteristics were overall similar between the patients who received only original or modified formulations.
FIGURE 2.
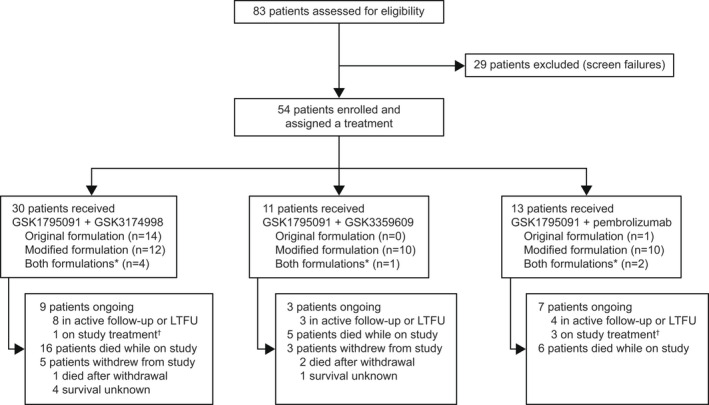
CONSORT flow diagram of study disposition. *Patients switched from original to modified GSK1795091 formulation during the study. †Patients continued combination partner monotherapy following GSK1795091 discontinuation. LTFU, long‐term follow‐up.
TABLE 1.
Summary of demographics and baseline characteristics (combination partners pooled) a
| Characteristic | Total (N = 54) | Original formulation b (N = 15) | Modified formulation b (N = 32) |
|---|---|---|---|
| Age (years), mean (SD) | 57.6 (12.7) | 60.2 (12.0) | 56.6 (12.7) |
| Female, n (%) | 28 (52.0) | 6 (40.0) | 18 (56.3) |
| BMI (kg/m2), mean (SD) | 26.9 (6.3) | 29.2 (5.1) | 25.7 (5.8) |
| Height (cm), mean (SD) | 168.4 (8.7) | 168.9 (9.9) | 168.1 (8.1) |
| Weight (kg), mean (SD) | 76.8 (19.9) | 83.5 (18.2) | 73.8 (19.3) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 8 (14.8) | 3 (20.0) | 5 (15.6) |
| Other | 45 (83.3) | 12 (80.0) | 26 (81.3) |
| Race, n (%) | |||
| Asian | 2 (3.7) | 1 (6.7) | 1 (3.1) |
| Caucasian | 47 (87.0) | 12 (80.0) | 28 (87.5) |
| Mixed race | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Missing data | 5 (9.3) | 2 (13.3) | 3 (9.4) |
Note: Data on tumor type, prior lines of therapy, and prior immunotherapy were not retrievable and could not be presented.
Abbreviations: BMI, body mass index; ICOS, inducible T‐cell costimulatory; PD‐1, programmed cell death protein 1; SD, standard deviation.
Data for combination partners were pooled for both formulations due to limited sample sizes. Combination partners included GSK3174998 (anti‐OX40 monoclonal antibody), GSK3359609 (anti‐ICOS monoclonal antibody), or pembrolizumab (anti–PD‐1 monoclonal antibody).
Data shown for patients who received either formulation alone. The seven patients who switched formulation during the study and received both formulations are included in the totals and are not shown separately by formulation.
Pharmacokinetics
Since the PK of GSK1795091 appeared to differ based on formulation, PK results are summarized separately for the two formulations. Due to limited sample sizes, PK results were pooled (regardless of treatment partner) and reported for each dose of GSK1795091. Key PK parameters are summarized in Table 2. Overall, geometric mean exposures (AUC[0–τ]) increased proportionally to dose for both formulations and were higher for the modified formulation compared with the original formulation at the equivalent dose levels. Terminal half‐life (t ½), geometric mean clearance, and geometric mean volume of distribution at steady state (Vss) were only estimable for the modified formulation. When estimable, t ½ ranged from 41 to 74 h, geometric mean clearance ranged from 0.053 to 0.084 L/h, and Vss ranged from 3.8 to 4.2 L across dose levels for the modified formulation. The Cmax for GSK1795091 with the different combination partners was similar between each formulation (Table S4), but no statistical test was conducted as this was not an objective of the study and sample sizes were limited. Cmax concentrations occurred shortly after the bolus injection; median tmax values ranged from 0.1 to 0.9 h after dosing.
TABLE 2.
Summary of Cmax and AUC(0–τ) for GSK1795091 on Day 1 for original and modified formulations (combination partners pooled) a
| Parameter (Day 1) | GSK1795091 dose | ||||||
|---|---|---|---|---|---|---|---|
| 50 ng | 100 ng | 150 ng | 200 ng | 250 ng | |||
| Cmax (pg/ml) | n | 7 | 4 | 2 | 0 | 0 | |
| Original | Geometric mean (%CV) | 4.7 (24.5) | 6.0 (51.7) | NC | NC | NC | |
| Median (range) | 4.7 (3.2–6.0) | 5.3 (4.0–11.9) | 11.0 (8.3–13.6) | NC | NC | ||
| n | 9 | 11 | 6 | 4 | 2 | ||
| Modified | Geometric mean (%CV) | 15.4 (24.5) | 31.0 (37.0) | 34.6 (26.4) | 57.0 (21.1) | NC | |
| Median (range) | 14.0 (13.1–28.8) | 32.3 (13.2–56.6) | 31.3 (26.8–53.4) | 60.0 (43.6–68.1) | 82.2 (65.0–99.4) | ||
| AUC(0–τ) (h × pg/ml) | n | 5 | 4 | 2 | 0 | 0 | |
| Original | Geometric mean (%CV) | 120.9 (131.0) | 122.4 (144.3) | NC | NC | NC | |
| Median (range) | 208.4 (35.0–276) | 146.8 (39.0–383) | 410.7 (310–512) | NC | NC | ||
| n | 7 | 9 | 5 | 3 | 1 | ||
| Modified | Geometric mean (%CV) | 846.5 (10.1) | 1521 (36.2) | 2117 (17.3) | 3336 (30.9) | NC | |
| Median (range) | 857.1 (721–990) | 1681 (645–2120) | 2116 (1670–2540) | 3615 (2390–4300) | 4166 (nc) | ||
Abbreviations: AUC, area under the curve; Cmax, concentration maximum; CV, coefficient of variation; ICOS, inducible T‐cell costimulatory; NC, noncalculable; PD‐1, programmed cell death protein 1.
Data for combination partners were pooled for both formulations due to limited sample sizes and to specifically evaluate the effect of the two formulations on the pharmacokinetic profile of GSK1795091. Combination partners included GSK3174998 (anti‐OX40 monoclonal antibody), GSK3359609 (anti‐ICOS monoclonal antibody), or pembrolizumab (anti–PD‐1 monoclonal antibody).
GSK1795091 concentration–time profiles for both formulations were not available at all doses for each combination partner, so data from different combination partners were pooled for each dose level and formulation of GSK1795091 (Figure 3). For both formulations, GSK1795091 concentration–time profiles for a full treatment cycle (168 h) were characterized by a biphasic elimination process, with a distribution phase occurring from 2 to 6 h post‐dose, followed by a more prolonged elimination phase. For both formulations, there were no signs of accumulation between cycles, and PK appeared to be time‐invariant. Median GSK1795091 concentrations were markedly higher (~3‐fold to ~16‐fold depending on dose and timepoint) with the modified formulation compared with the original formulation (Figure 3).
FIGURE 3.
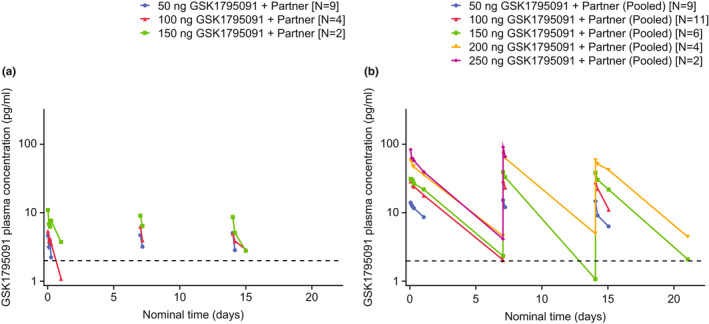
Median GSK1795091 pharmacokinetic concentration–time plots with (a) original formulation and (b) modified formulation (combination partners pooled)*. Horizontal dashed line represents the default lower limit of quantification for the assay (2.0 pg/ml). *Data for combination partners were pooled for both formulations due to limited sample sizes and to specifically evaluate the effect of the two formulations on the pharmacokinetic profile of GSK1795091. Combination partners included GSK3174998 (anti‐OX40 monoclonal antibody), GSK3359609 (anti‐ICOS monoclonal antibody), or pembrolizumab (anti–PD‐1 monoclonal antibody). ICOS, inducible T‐cell costimulatory; PD‐1, programmed cell death protein 1.
PK−PD response and analysis of biomarkers
A concentration‐dependent PD response was observed for most biomarkers evaluated (e.g., IP‐10, IL‐10, IL‐1Ra, IL‐6, and MCP‐1) in patients receiving the original GSK1795091 formulation, but not with the modified formulation (Figure 4). No PD response was observed for TNFα or safety markers (systolic blood pressure and pulse rate) with either of the formulations (Figure S3).
FIGURE 4.
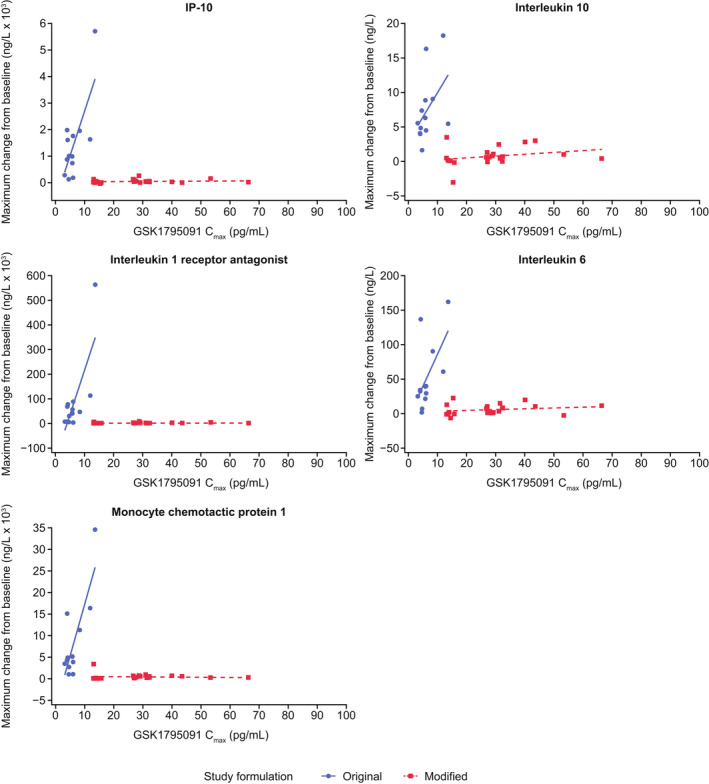
Maximum change from baseline in pharmacodynamic (PD) biomarkers versus GSK1795091 Cmax for original and modified formulations (combination partners pooled)*. *Data for combination partners were pooled for both formulations due to limited sample sizes and to specifically evaluate the effect of the two formulations on the PD profile of GSK1795091. Combination partners included GSK3174998 (anti‐OX40 monoclonal antibody), GSK3359609 (anti‐ICOS monoclonal antibody), or pembrolizumab (anti–PD‐1 monoclonal antibody). Cmax, concentration maximum; ICOS, inducible T‐cell costimulatory; IP‐10, interferon gamma‐induced protein 10; PD‐1, programmed cell death protein 1.
Transient increases in circulating plasma concentrations of IP‐10, IL‐6, MCP‐1, and IL‐1Ra peaked from 2 to 6 h and returned to baseline within 24 h of administration of the original GSK1795091 formulation only (Figure S4).
Safety
Dose‐limiting toxicities were observed in two patients in the study; both were assessed as related to GSK1795091 and led to discontinuation of treatment. One patient in Group A receiving 150 ng GSK1795091 (original formulation only) with GSK3174998 experienced Grade 3 blood alkaline phosphatase increase, Grade 3 alanine aminotransferase increase, Grade 3 aspartate aminotransferase increase, and Grade 3 hepatitis; and one patient in Group C receiving 100 ng GSK1795091 (original formulation only) with pembrolizumab experienced Grade 3 hypotension.
Overall, 51/54 patients (94.4%) who received GSK1795091 with a combination partner experienced ≥1 TEAE during the study, with 24/54 (44.4%) experiencing Grade ≥3 TEAEs (Table 3). The most common nonserious TEAEs (≥20% incidence) were chills (n = 22 [40.7%]), nausea (n = 20 [37.0%]), fatigue (n = 19 [35.2%]), anemia (n = 14 [25.9%]), vomiting (n = 12 [22.2%]), and decreased appetite (n = 11 [20.4%]). Two TEAEs were fatal (sepsis [n = 2; 50 ng GSK1795091 with GSK3359609 and pembrolizumab] and depressed level of consciousness [n = 1; 50 ng GSK1795091 with GSK3359609]) and were considered unrelated to treatment.
TABLE 3.
Summary of treatment‐emergent adverse events (occurring in ≥10% of patients) and grade ≥3 events for original and modified formulations (combination partners pooled) a
| n (%) | Any event | Grade ≥3 event | ||||
|---|---|---|---|---|---|---|
| Total (N = 54) | Original formulation b (N = 15) | Modified formulation b (N = 32) | Total (N = 54) | Original formulation b (N = 15) | Modified formulation b (N = 32) | |
| Any events | 51 (94.4) | 15 (100) | 29 (90.6) | 24 (44.4) | 4 (26.7) | 14 (43.8) |
| Blood and lymphatic system | ||||||
| Anemia | 14 (25.9) | 4 (26.7) | 8 (25.0) | 6 (11.1) | 1 (6.7) | 3 (9.3) |
| Gastrointestinal disorders | ||||||
| Nausea | 20 (37.0) | 8 (53.3) | 8 (25.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Vomiting | 12 (22.2) | 6 (40.0) | 4 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Constipation | 6 (11.1) | 1 (6.7) | 4 (12.5) | 1 (1.9) | 1 (6.7) | 0 (0.0) |
| Diarrhea | 6 (11.1) | 2 (13.3) | 4 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| General disorders and administration site conditions | ||||||
| Chills | 22 (40.7) | 10 (66.7) | 8 (25.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Fatigue | 19 (35.2) | 9 (60.0) | 8 (25.0) | 2 (3.7) | 0 (0.0) | 1 (3.1) |
| Pyrexia | 8 (14.8) | 3 (20.0) | 3 (9.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Investigations | ||||||
| Weight decreased | 7 (13.0) | 4 (26.7) | 2 (6.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Metabolism and nutrition disorders | ||||||
| Decreased appetite | 11 (20.4) | 2 (13.3) | 6 (18.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Musculoskeletal and connective tissue disorders | ||||||
| Arthralgia | 6 (11.1) | 2 (13.3) | 4 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Back pain | 6 (11.1) | 2 (13.3) | 2 (6.3) | 2 (3.7) | 1 (6.7) | 0 (0.0) |
| Nervous system disorders | ||||||
| Headache | 8 (14.8) | 4 (26.7) | 4 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Dizziness | 7 (13.0) | 2 (13.3) | 3 (9.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Vascular disorders | ||||||
| Hypertension | 7 (13.0) | 3 (20.0) | 3 (9.4) | 2 (3.7) | 0 (0.0) | 1 (3.1) |
Abbreviations: ICOS, inducible T‐cell costimulatory; PD‐1, programmed cell death protein 1.
Data for combination partners were pooled for both formulations due to limited sample sizes and to specifically evaluate the effect of the two formulations on the safety profile of GSK1795091. Combination partners included GSK3174998 (anti‐OX40 monoclonal antibody), GSK3359609 (anti‐ICOS monoclonal antibody), or pembrolizumab (anti–PD‐1 monoclonal antibody).
Data shown for patients who received either formulation alone. The seven patients who switched formulation during the study and received both formulations are included in the total and are not shown separately by formulation.
While the overall incidence of TEAEs was similar between patients receiving the original formulation only (15/15 [100%]) compared with the modified GSK1795091 formulation only (29/32 [90.6%]), the incidence of nausea, vomiting, chills, fatigue, pyrexia, weight reduction, headache, and hypertension was higher in patients receiving the original formulation only (Table 3). Many of these events are typically associated with active immune activation. While dose levels were pooled for safety analysis due to small sample sizes, a difference in the incidence of chills was observed between the two formulations at 50 ng GSK1795091 (4/9 patients [44.4%], original formulation only vs. 2/9 [22.2%], modified formulation only; data not shown). A dose–response trend was also observed for the next two dose levels with the original but not the modified formulation (original formulation only: 4/4 [100%] at 100 ng, 2/2 [100%] at 150 ng; modified formulation only: 4/11 [36.4%] at 100 ng, 0/6 [0%] at 150 ng; data not shown). Serious TEAEs were reported by 18 patients (33.3%), with a similar incidence between formulations (original formulation only: 4/15 [26.7%]; modified formulation only: 10/32 [31.3%]); three serious TEAEs experienced by two patients receiving the original formulation only were related to the study treatment (Table S5).
Overall, more than half of patients (36/54 [66.7%]) reported treatment‐related AEs. These were reported in 14/15 (93.3%) of those receiving original formulation only, and 16/32 (50.0%) of those receiving the modified formulation. Grade 3/4 treatment‐related AEs were reported in 7/54 patients (13.0%) and were similar between formulations (original formulation only: 2/15 [13.3%]; modified formulation only: 3/32 [9.4%]). No fatal treatment‐related AEs were observed.
Efficacy
There are limited data regarding the anti‐tumor activity of GSK1795091 in combination with either GSK3174998, GSK3359609, or pembrolizumab. This is due to the small sample sizes resulting from the change in formulation and discontinuation of the combination treatment (and subsequent continued treatment with monotherapy for some patients).
Three patients in the study achieved a partial response:
50 ng GSK1795091 and 24 mg GSK3174998 (n = 1/9 [11.1%]).
100 ng GSK1795091 and 200 mg pembrolizumab (n = 2/6 [33.3%]).
Thirteen patients in the study achieved stable disease:
50 ng GSK1795091 and 24 mg GSK3174998 (3/9 [33.3%]); 100 ng GSK1795091 and 24 mg GSK3174998 (3/6 [50.0%]); 150 ng GSK1795091 and 24 mg GSK3174998 (3/9 [33.3%]); 200 ng GSK1795091 and 24 mg GSK3174998 (1/4 [25.0%]).
50 ng GSK1795091 and 80 mg GSK3359609 (1/6 [16.7%]); 100 ng GSK1795091 and 80 mg GSK3359609 (1/5 [20.0%]).
50 ng GSK1795091 and 200 mg pembrolizumab (1/7 [14.3%]).
Across all cohorts, the ORR was 5.6% and the DCR was 27.8%.
Investigative follow‐up studies
In vivo and in vitro PD response assessment
The change in cytokine production observed between the two formulations was investigated in follow‐up nonclinical studies. Consistent with the findings in the clinical study, reduced production of IL‐6 was observed in naïve BALB/c mice following administration of the modified GSK1795091 formulation compared with the original (Figure S5A). Cytokine production was also lower in an in vitro human whole‐blood assay (healthy donors) with the modified formulation compared with the original formulation. However, no significant changes were observed between different API batches prepared using the same formulation method (Figure S5B).
In vitro evaluation of the impact of formulation manufacturing processes on PD response
The in vitro whole‐blood assay was repeated with several formulation preparation conditions, which were altered to determine which aspect may have resulted in the reduced PD response in patients receiving the modified formulation of GSK1795091 (Figure S6). No notable changes were seen between the original formulation that had been spiked with ethanol, that had been increased in ionic strength, or was prepared using a reduced sonication time, compared with the positive control (preclinical formulation and preclinical batch of GSK1795091). Initial dissolution of GSK1795091 in 100% ethanol before diluting to the final concentration in the modified formulation resulted in a reduced PD compared with the positive control. Sonication did not ameliorate the lowered PD response observed with the modified formulation.
DISCUSSION
This was a phase I study evaluating the safety, tolerability, and efficacy of GSK1795091 (TLR4 agonist) in combination with one of three immunomodulatory agents: GSK3174998 (anti‐OX40 monoclonal antibody), GSK3359609 (anti‐ICOS monoclonal antibody), or pembrolizumab (anti–PD‐1 monoclonal antibody). During the dose‐escalation part of the study (Part 1), the manufacturing procedure of GSK1795091 was modified to streamline the manufacturing process to make it more suitable for large‐scale manufacturing and simplify the clinical administration procedure. However, PD and PK analyses performed as the study was ongoing revealed that the modified formulation had substantially lowered biological activity, leading to discontinuation of GSK1795091 and early termination of the study. The further development of GSK1795091 has since been discontinued and no further studies are planned.
The PK of GSK1795091 was linear, dose‐proportional, and overall consistent with those of other synthetic lipid A analogs 8 , 9 and the FTIH study performed in healthy volunteers. 12 However, while the same original formulation was used in the FTIH study, GSK1795091 exposures for the original formulation were lower compared with those seen in the FTIH study at equivalent doses. The reason for the difference in GSK1795091 exposures between this and the FTIH study is unknown but may be related to differences in administration procedures between the trials or the different trial populations (healthy volunteers vs. patients with advanced solid tumors). After the formulation was modified and the administration procedure was changed to include an increased IV flush, GSK1795091 PK exposures increased.
While the modified GSK1795091 formulation/administration procedure resulted in increased GSK1795091 exposure compared with the original formulation, it was associated with a lack of PD response, in the form of diminished cytokine and chemokine response compared with the original formulation and the FTIH study. 12 Similar PD results were observed when comparing the modified versus original formulation in follow‐up in vivo and ex vivo experiments.
The overall incidence of TEAEs was similar between the two formulations. However, consistent with the diminished levels of cytokines following administration of the modified formulation, a lower incidence of certain TEAEs was observed, including ‘flu‐like’ events such as chills, fatigue, pyrexia, nausea, and vomiting. Due to limited data, no conclusions could be made regarding the anti‐tumor activity of GSK1795091 in combination with either GSK3174998, GSK3359609, or pembrolizumab.
The marked change in PD response between the modified and original formulations may be due to the differences in aggregate size and shape between GSK1795091 formulations. Physiochemical characterization of both formulations by cryo‐TEM revealed the aggregate size of the modified formulation to be twice that of the original formulation, with a globular morphology. This may have resulted in a configuration that impaired binding of GSK1795091 to the TLR4 receptor, leading to reduced downstream signaling. Lipid A aggregate morphology has also been linked to biological activity, which is dependent on the molecular shape of lipid A, 20 suggesting both size and shape implications of GSK1795091 aggregates. Previous studies offer conflicting evidence on whether LPS is active in monomer or aggregate form. Our data suggest that an aggregate form of GSK1795091 has lower biological activity, consistent with previous findings with LPS. 21 Conversely, aggregates can also be the active form of LPS and lipid A analogs. 22 It has been suggested that different forms of the same LPS can stimulate different immune pathways. 23 Together, the evidence suggests a complex interplay of monomeric/aggregate form, aggregate size, and morphology, and that other factors may modulate the activity of LPS and lipid A analogs.
Several follow‐up experiments were carried out to identify the cause for the lower biological activity of GSK1795091 following the formulation change. Follow‐up nonclinical data demonstrated similar levels of released cytokines between different API batches prepared using the same formulation method (original or modified), suggesting that the change in PD response stemmed from the adaptation of the formulation manufacturing procedure rather than the API itself. As the modified formulation included the addition of an ethanol dissolution step, it was hypothesized that ethanol in the preparation may have had an adverse effect on the activity of GSK1795091 as ethanol inactivates LPS in polysaccharide preparations. 24 Results from a whole‐blood assay follow‐up investigation indicated that the original formulation spiked with 0.05% ethanol did not negatively impact PD activity. However, the modified formulation included initial dissolution of GSK1795091 in 100% ethanol, suggesting this step is unfavorable to the bioactivity of GSK1795091 and in agreement with the aforementioned LPS inactivation procedure that used 60% ethanol. 24 These investigations concluded that the sonication step used for the original formulation appeared to be a critical step for producing a more biologically active form of GSK1795091. However, sonication of the modified formulation (i.e., after initial dissolution in 100% ethanol) did not enhance PD activity, suggesting that the biological activity of GSK1795091 is not recoverable by this technique. Sonication is the most extensively used method to create small unilamellar vesicles 25 and may have helped to prevent the formation of the larger, globular aggregates upon initial dissolution, in contrast to observations with the modified formulation. Other potential reasons for reduced PD activity such as increasing ionic strength (whole‐blood assay), compatibility with the injection equipment used, quality of the PK and PD assays, as well as drug preparation in the clinic (including dilution and administration) were also investigated and ruled out. A limitation of this study is that it was not originally designed to assess the effects of a formulation change on study drug efficacy, safety, or PK and PD parameters. Several other factors could also have contributed to the observed effects, such as dose, drug combinations, small sample sizes, etc. Our study was not designed to account for the potential impact of these factors post‐formulation‐change, so we cannot rule out some contribution to the resulting change in biological activity. Nonetheless, the nonclinical follow‐up studies support our clinical observations and provide confirmatory evidence that the formulation change is the main driver underlying the change in biological activity.
The quality testing and release specifications of the modified GSK1795091 formulation were in line with standard operating procedures and regulatory expectations for a small‐molecule injectable drug, meaning that the release specifications did not capture the differences that produced the change in biological activity. Thus, the change in activity was only discovered during the in‐stream clinical study assessments. Based on these findings, we would recommend in vitro PD assessment of the biological activity of TLR agonists and possibly other lipid A analogs, including chemically manufactured APIs that might be prone to structural organization in solution, following a significant manufacturing change. These findings may have important implications for the future development of other structurally similar compounds and suggest that considerations for release specifications should ensure similar activity of new formulations. For in‐study formulation changes, initial in vitro assessment to confirm comparable response is advised, particularly for drugs whose biological activity may be dependent on aggregate size and shape and those sensitive to manufacturing procedures, such as other lipid A and LPS analogs. 20 , 21 , 22 , 26 , 27
AUTHOR CONTRIBUTIONS
N.S., G.J.H., E.G., H.P., J.S., and S.A.P.P. performed the research. A.R.H. designed and performed the research. M.A., G.C., J.C., R.E., K.M., P.S., H.S., M.L.W., and C.M. designed the research and analyzed the data. All authors were involved in writing the manuscript.
FUNDING INFORMATION
This study was funded by GlaxoSmithKline (204686; NCT03447314).
CONFLICT OF INTEREST
A.R.H. received research funding (paid to institution) from GSK, Merck, Pfizer, MedImmune/Genentech, Roche, Janssen, BMS, AstraZeneca, Astellas, Boehringer‐Ingelheim, and Bayer; and provided consultation or attended advisory boards for GSK, Merck, Novartis, and Eisai. G.C., H.S., J.C., K.M., M.A., M.L.W., and R.E. are employed by GSK and hold GSK stock/shares. C.M. and P.S. are former employees of GSK. E.G. receives research funding from Novartis, Roche, Thermo Fisher, AstraZeneca, Taiho, and BeiGene; is a consultant/advisor for Roche/Genentech, F. Hoffmann/La Roche, Ellipses Pharma, Neomed Therapeutics 1 Inc., Boehringer Ingelheim, Janssen Global Service, SeaGen, Alkermes, Thermo Fisher, Bristol Myers Squibb, MabDiscovery, Anaveon, and F‐Star Therapeutics; has taken part in the speakers bureau for Merck Sharp & Dohme, Roche, Thermo Fisher, and Lilly; is a clinical trial principal investigator (PI) or co‐PI on studies carried out by Affimed Gmbh, Amgen SA, Anaveon AG, AstraZeneca AB, Biontech Gmbh, Catalym Gmbh, Cytomx, F. Hoffmann La Roche Ltd, F‐Star Beta Limited, Genentech Inc, Genmab B.V., Hutchison Medipharma Limited, Icon, Imcheck Therapeutics, Immunocore Ltd, Janssen‐Cilag SA, Medimmune Llc, Merck Kgga, Novartis Farmacéutica, S. A., Peptomyc, Ribon Therapeutics, Roche Farma S.A., Seattle Genetics Inc, Symphogen A/S, and Taiho Pharma USA Inc. N.S. provided consultation or attended advisory boards for AIMM Therapeutics, Boehringer Ingelheim, and Ellipses Pharma; received institutional research funding from AB Science, Abbvie, Actuate Therapeutics, Amgen, Array, AstraZeneca/MedImmune, Bayer, Blueprint Medicines, Boehringer Ingelheim, Bristol Myers Squibb, Cantargia, CellCentric, Cytovation, Deciphera, Genentech/Roche, GlaxoSmithKline, Incyte, Lilly, Merck Sharp & Dohme, Merus, Molecular Partners, Novartis, Pfizer, Pierre Fabre, Roche, Sanofi, Taiho, and Takeda (outside the submitted work). G.J.H. receives consulting fees and serves in an advisory role for Bicara, Boxer Capital, BMS, Exicure, General Catalyst, Kura, Maverick, Merck, Naveris, Prelude, Rain, Regeneron/Sanofi Genzyme, Remix, and SIRPant. He receives institutional grant support from Actuate, ASCO Conquer Cancer Foundation, Bicara, BMS, Elevar, Exicure, Gateway for Cancer Research, Genentech, GSK, Kartos, Kite, KSQ, NantKwest/Altor Bioscience, Regeneron, Repertoire, Sanofi Genzyme, Secura Bio, and V Foundation/ACCRF. Expert witness role: Aaronson Rappaport Feinstein & Deutsch, LLP; Ahmuty, Demers, & McManus, Esqs; and Wilson Elser Moskowitz Edelman & Dicker, LLP. H.P. received research/grant funding from the following sources: Adlai Nortye USA, Alpine Immune Sciences, Ambrx, Amgen, Aprea Therapeutics AB, Array BioPharma, Bayer, BeiGene, BJ Bioscience, Bristol Myers Squibb, Daiichi Pharmaceutical, Eli Lilly, Elicio Therapeutics, EMD Serono, Exelixis, Genentech, Gilead Sciences, GlaxoSmithKline, Gossamer Bio, Hoffman‐LaRoche, Hutchison MediPharma, ImmuneOncia Therapeutics, Incyte, Jounce Therapeutics, Mabspace Biosciences, MacroGenics, MedImmune, Medivation, Merck, Millennium, Mirati Therapeutics, Novartis Pharmaceuticals, Oncologie, Pfizer, PsiOxus Therapeutics, Puma Biotechnology, Regeneron, Pharmaceuticals, RePare Therapeutics, Seattle Genetics, Synermore Biologics, Taiho Pharmaceutical, TopAlliance Biosciences, Turning Point Therapeutics, Vedanta Biosciences, and Xencor Inc. J.S. has a leadership role at Dialectic Therapeutics and owns stocks and other ownership interests at Abbvie, Abbott Laboratories, Bristol Myers Squibb, Intuitive Surgical, Johnson & Johnson, Merck, and Regeneron; has consulting or advisory roles at Synlogic and Binhui Biopharmaceuticals, Ltd. S.A.P.P. receives research/grant funding through the institution from the following sources: AbbVie, Inc.; ABM Therapeutics, Inc.; Acepodia, Inc.; Alkermes; Aminex Therapeutics; Amphivena Therapeutics, Inc.; BioMarin Pharmaceutical, Inc.; Boehringer Ingelheim; Bristol Myers Squibb; Cerulean Pharma, Inc.; Chugai Pharmaceutical Co., Ltd; Curis, Inc.; Cyclacel Pharmaceuticals; Daiichi Sankyo; Eli Lilly; ENB Therapeutics; Five Prime Therapeutics; F‐Star Beta Limited; F‐Star Therapeutics; Gene Quantum; Genmab A/S; GlaxoSmithKline; Helix BioPharma Corp.; HiberCell, Inc.; Immunomedics, Inc.; Incyte Corp.; Jacobio Pharmaceuticals Co., Ltd; Lytix Biopharma AS; Medimmune, LLC; Medivation, Inc.; Merck Sharp and Dohme Corp.; Novartis Pharmaceuticals; Pieris Pharmaceuticals, Inc.; Pfizer; Principia Biopharma, Inc.; Puma Biotechnology, Inc.; Rapt Therapeutics, Inc.; Seattle Genetics; Silverback Therapeutics; Synlogic Therapeutics; Taiho Oncology; Tesaro, Inc.; TransThera Bio; NCI/NIH; P30CA016672 – Core Grant (CCSG Shared Resources). She has worked as a consultant for CRC Oncology.
Supporting information
Figure S1
Appendix S1
Table S1
ACKNOWLEDGMENTS
We thank all the patients for participating in the study, as well as the clinical site staff and site personnel for their involvement and supporting the trial. We thank Esther Hrabrick (member of the GSK study team) for support of the study, the Vaccines Belgium group for their early work on the asset and support of the oncology studies, and IQVIA as the contract research organization (CRO) partner. Editorial support (in the form of writing assistance, including development of the initial draft based on author direction, assembling tables and figures, collating authors' comments, grammatical editing, and referencing) was provided by Anna Polyakova, PhD, and Frankie Wignall, PhD, at Fishawack Indicia Ltd of Fishawack Health, UK.
Steeghs N, Hansen AR, Hanna GJ, et al. Manufacturing‐dependent change in biological activity of the TLR4 agonist GSK1795091 and implications for lipid A analog development. Clin Transl Sci. 2022;15:2625‐2639. doi: 10.1111/cts.13387
Peter Skrdla and Christopher Matheny: At the time of study design and initiation.
REFERENCES
- 1. Yu L, Wang L, Chen S. Dual character of Toll‐like receptor signaling: pro‐tumorigenic effects and anti‐tumor functions. Biochim Biophys Acta. 2013;1835(2):144‐154. doi: 10.1016/j.bbcan.2012.10.006 [DOI] [PubMed] [Google Scholar]
- 2. El‐Zayat SR, Sibaii H, Mannaa FA. Toll‐like receptors activation, signaling, and targeting: an overview. Bulletin of the National Research Centre. 2019;43(1):187. doi: 10.1186/s42269-019-0227-2 [DOI] [Google Scholar]
- 3. Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085‐2088. doi: 10.1126/science.282.5396.2085 [DOI] [PubMed] [Google Scholar]
- 4. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll‐like receptor‐4 mediates lipopolysaccharide‐induced signal transduction. J Biol Chem. 1999;274(16):10689‐10692. doi: 10.1074/jbc.274.16.10689 [DOI] [PubMed] [Google Scholar]
- 5. Otto F, Schmid P, Mackensen A, et al. Phase II trial of intravenous endotoxin in patients with colorectal and non‐small cell lung cancer. Eur J Cancer. 1996;32a(10):1712‐1718. doi: 10.1016/0959-8049(96)00186-4 [DOI] [PubMed] [Google Scholar]
- 6. Jeannin JF, Onier N, Lagadec P, von Jeney N, Stütz P, Liehl E. Antitumor effect of synthetic derivatives of lipid a in an experimental model of colon cancer in the rat. Gastroenterology. 1991;101(3):726‐733. doi: 10.1016/0016-5085(91)90532-p [DOI] [PubMed] [Google Scholar]
- 7. Parr I, Wheeler E, Alexander P. Similarities of the anti‐tumour actions of endotoxin, lipid A and double‐stranded RNA. Br J Cancer. 1973;27(5):370‐389. doi: 10.1038/bjc.1973.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Bono JS, Dalgleish AG, Carmichael J, et al. Phase I study of ONO‐4007, a synthetic analogue of the lipid a moiety of bacterial lipopolysaccharide. Clin Cancer Res. 2000;6(2):397‐405. [PubMed] [Google Scholar]
- 9. Isambert N, Fumoleau P, Paul C, et al. Phase I study of OM‐174, a lipid A analogue, with assessment of immunological response, in patients with refractory solid tumors. BMC Cancer. 2013;13:172. doi: 10.1186/1471-2407-13-172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vosika GJ, Barr C, Gilbertson D. Phase‐I study of intravenous modified lipid A. Cancer Immunol Immunother. 1984;18(2):107‐112. doi: 10.1007/BF00205743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao H‐X, Bhattacharya S, Matheny CJ, et al. Synergy of TLR4 agonist GSK1795091, an innate immune activator, with agonistic antibody against co‐stimulatory immune checkpoint molecule OX40 in cancer immunotherapy. J Clin Oncol. 2018;36(15_suppl):12055. doi: 10.1200/JCO.2018.36.15_suppl.12055 [DOI] [Google Scholar]
- 12. Hug BA, Matheny CJ, Burns O, Struemper H, Wang X, Washburn ML. Safety, pharmacokinetics, and pharmacodynamics of the TLR4 agonist GSK1795091 in healthy individuals: results from a randomized, double‐blind, placebo‐controlled, ascending dose study. Clin Ther. 2020;42(8):1519‐34 e33. doi: 10.1016/j.clinthera.2020.05.022 [DOI] [PubMed] [Google Scholar]
- 13. Cen X, Liu S, Cheng K. The role of toll‐like receptor in inflammation and tumor immunity. Front Pharmacol. 2018;9:878. doi: 10.3389/fphar.2018.00878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shetab Boushehri MA, Lamprecht A. TLR4‐based immunotherapeutics in cancer: a review of the achievements and shortcomings. Mol Pharm. 2018;15(11):4777‐4800. doi: 10.1021/acs.molpharmaceut.8b00691 [DOI] [PubMed] [Google Scholar]
- 15. Postel‐Vinay S, Lam VK, Ros W, et al. Abstract CT150: a first‐in‐human phase I study of the OX40 agonist GSK3174998 (GSK998) ± 00B1 pembrolizumab in patients (pts) with selected advanced solid tumors (ENGAGE‐1). Cancer Res. 2020;80:CT150‐CT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Angevin E, Barnette MS, Bauer TM, et al. INDUCE‐1: a phase I open‐label study of GSK3359609, an ICOS agonist antibody, administered alone and in combination with pembrolizumab in patients with advanced solid tumors. J Clin Oncol. 2017;35(15_suppl):TPS3113‐TPS. doi: 10.1200/JCO.2017.35.15_suppl.TPS3113 [DOI] [Google Scholar]
- 17. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med. 2015;372(21):2018‐2028. doi: 10.1056/NEJMoa1501824 [DOI] [PubMed] [Google Scholar]
- 18. Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med. 2008;27(13):2420‐2439. doi: 10.1002/sim.3230 [DOI] [PubMed] [Google Scholar]
- 19. Licea‐Perez H, Junnotula V, Bowen Chester L, et al. Development of an ultra‐sensitive assay for the determination of an aminoalkyl glucosaminide 4‐phosphate, GSK1795091, in plasma to support a first time in human study. Anal Methods. 2018;10(25):3074‐3080. doi: 10.1039/C8AY00571K [DOI] [Google Scholar]
- 20. Seydel U, Oikawa M, Fukase K, Kusumoto S, Brandenburg K. Intrinsic conformation of lipid A is responsible for agonistic and antagonistic activity. Eur J Biochem. 2000;267(10):3032‐3039. doi: 10.1046/j.1432-1033.2000.01326.x [DOI] [PubMed] [Google Scholar]
- 21. Takayama K, Mitchell DH, Din ZZ, Mukerjee P, Li C, Coleman DL. Monomeric re lipopolysaccharide from Escherichia coli is more active than the aggregated form in the limulus amebocyte lysate assay and in inducing Egr‐1 mRNA in murine peritoneal macrophages. J Biol Chem. 1994;269(3):2241‐2244. [PubMed] [Google Scholar]
- 22. Mueller M, Lindner B, Kusumoto S, Fukase K, Schromm AB, Seydel U. Aggregates are the biologically active units of endotoxin. J Biol Chem. 2004;279(25):26307‐26313. doi: 10.1074/jbc.M401231200 [DOI] [PubMed] [Google Scholar]
- 23. Sasaki H, White SH. Aggregation behavior of an ultra‐pure lipopolysaccharide that stimulates TLR‐4 receptors. Biophys J. 2008;95(2):986‐993. doi: 10.1529/biophysj.108.129197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Govers C, Tomassen MMM, Rieder A, Ballance S, Knutsen SH, Mes JJ. Lipopolysaccharide quantification and alkali‐based inactivation in polysaccharide preparations to enable in vitro immune modulatory studies. Bioact Carbohydr Diet Fibre. 2016;8(1):15‐25. doi: 10.1016/j.bcdf.2016.09.001 [DOI] [Google Scholar]
- 25. Akbarzadeh A, Rezaei‐Sadabady R, Davaran S, et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):102. doi: 10.1186/1556-276X-8-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brandenburg K, Hawkins L, Garidel P, et al. Structural polymorphism and endotoxic activity of synthetic phospholipid‐like amphiphiles. Biochemistry. 2004;43(13):4039‐4046. doi: 10.1021/bi0361158 [DOI] [PubMed] [Google Scholar]
- 27. Brandenburg K, Matsuura M, Heine H, et al. Biophysical characterization of triacyl monosaccharide lipid A partial structures in relation to bioactivity. Biophys J. 2002;83(1):322‐333. doi: 10.1016/S0006-3495(02)75172-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Appendix S1
Table S1