

Abstract
To analyze the impact of various technical details on the results of QM/MM enzyme simulations, including the QM region size, catechol-O-methyltransferase (COMT) is studied as a model system using an approximate QM/MM method (DFTB3/CHARMM). The results show that key equilibrium and kinetic properties for methyl transfer in COMT exhibit limited variations with respect to the size of the QM region, which ranges from ~100 to ~500 atoms in this study. With extensive sampling, local and global structural characteristics of the enzyme are largely conserved across the studied QM regions, while the nature of the transition state (e.g., secondary kinetic isotope effect) and reaction exergonicity are largely maintained. Deviations in the free energy profile with different QM region sizes are similar in magnitude to those observed with changes in other simulation protocols, such as different initial enzyme conformations and boundary conditions. Electronic structural properties, such as the covariance matrix of residual charge fluctuations, appear to exhibit rather long-range correlations, especially when the peptide backbone is included in the QM region; this observation holds when a range-separated DFT approach is used as the QM region, suggesting that delocalization error is unlikely the origin. Overall, the analyses suggest that multiple simulation details determine the results of QM/MM enzyme simulations with comparable contributions.
Graphical Abstract:
1. Introduction
Combined quantum mechanical (QM)/ molecular mechanical (MM) simulations have been widely employed in the characterization and understanding of enzymatic processes.1–9 Systematically improving over the past few decades, QM/MM simulations have shown extensive success from drug discovery to enzyme engineering.4,9–11 However, QM/MM simulations still suffer some pressing challenges and limitations.12 For instance, in addition to the general concern in the accuracy of the QM method,13–15 the quality of the MM force field,16 the QM/MM boundary scheme17–19 and the QM/MM non-bonded parameters,20–22 a systematic method for the identification of the appropriate QM region has historically been elusive.23–26
The selection of a QM region is not a straightforward task and inappropriate choices, especially with inaccuracy in QM/MM interactions,22,27 can lead to computational artifacts and thus abjectly different predictions from experiment. Although large QM regions should be able to more realistically describe a system, enlarging a QM region may scale to prohibitively expensive levels. It has been shown that QM regions on the order of several hundreds to thousands of atoms are required for a variety of chemical properties such as energies,28–30 NMR shieldings,31,32 and other spectroscopic properties.33 This slowly converging nature of energy differences has prompted the analysis of QM region selection based on electronic structure properties such as the Fukui function25 and bond critical point.34 Some of these schemes suggest upwards of a couple of hundred atoms.25,35,36
While it is unequivocal that extensively large QM regions are necessary for convergent energies and many other electronic properties, it is unclear if large QM regions are most essential (among all possible methodological considerations) for a meaningful understanding of the catalytic mechanism of enzymes, such as the nature of the transition state and its stabilization relative to uncatalyzed reaction in solution. In particular, disagreement exists regarding how the QM region size impacts computed QM/MM free energy profiles,37,38 which require extensive sampling for convergent results.7,39–42 For instance, it was shown in catechol-O-methyltransferase (COMT), where 200–300 atoms are required for converged energies,35 that QM/MM free energy simulations using different QM choices can yield remarkably different if not opposite thermodynamic results while significantly affecting barrier heights.43 Yet, for the same system, PM6/MM calculations have shown that the free energy surface is fairly robust, especially with respect to the predicted energy barrier,44 although the reliability of the PM6 method used therein was questioned43,45 for properly capturing electronic structure over an extended region.
In this work, using COMT as a test enzyme system, we analyze the catalytic properties (Scheme 1) across previously studied QM partition schemes.43,45 Our work is complementary to previous studies35,43–45 because of several reasons. First, we use a different QM approach (DFTB346), thus comparison to previous studies helps better understand whether key observations depend on the QM level. Second, our free energy simulations explore much more of the protein’s conformational ensemble, in terms of not only the extensive length of individual metadynamics simulations but exploring different initial structures, which include both the crystal structure and snapshots taken from long (~400 ns) classical molecular dynamics simulations. Third, we also compare results from spherical boundary (GSBP47,48) and periodic boundary setups, for both the wild type enzyme and a more flexible variant (Y68A, see below) studied in previous experimental investigations,49 so as to evaluate the impact of fluctuations of remote residues. Finally, we also compute two kinetic properties of the system: secondary kinetic isotope effect (KIE) for the methyl transfer and barrier crossing transmission coefficient, κ,50 using path-integral51 and positive reaction flux52,53 simulations, respectively.
Scheme 1:
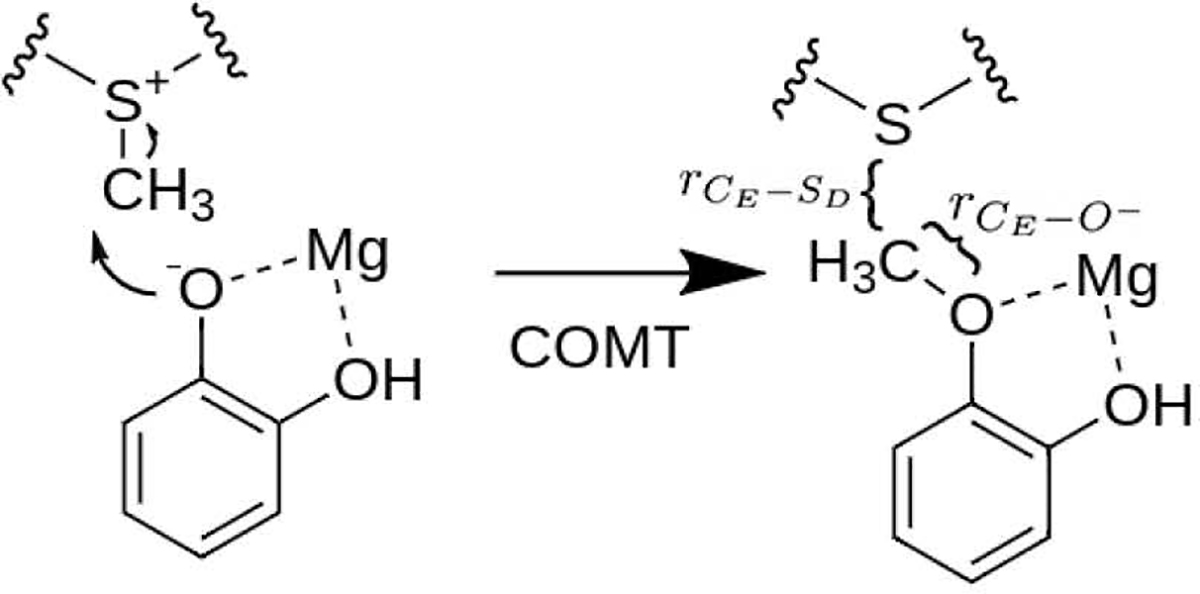
The methyl transfer reaction in catechol-O-methyltransferase (COMT).
The equilibrium and kinetic results make clear that the overall structural features, free energy landscape and nature of transition state exhibit limited variations across the different models with a QM region that ranges from ~100 atoms to more than 500 atoms, suggesting that the key catalytic features of the enzyme is largely conserved regardless of the extent of the QM region. Extensive sampling along with the addition of physical attributes such as dispersion interactions among the QM atoms, initial starting conformation, periodic vs. spherical boundary condition54 and the QM/MM partitioning scheme play at least equally recognizable differences in the computed free energy profile. On the other hand, covariance of residual charge fluctuations43 exhibits long-range correlation, especially when the QM region includes protein backbone atoms, suggesting that a large QM region is required to properly capture certain electronic structural properties; to probe whether charge delocalization error associated with popular GGAs55 (on which the DFTB3 model is based56,57), plays a major role in this observation, we have also conducted charge analysis using a range-separated Density Functional Theory (DFT) method enabled by recent developments of GPU based DFT approaches.58–60
2. Computational Methods
Human catechol-o-methyltransferase (COMT) with 3,5-dinitrocatechol and S-adenosyl methionine substrates (PDB ID: 3BWM)61 is used to set up the calculation (Fig. 1a), in which 3,5-dinitrocatecholate is modified to catecholate. All simulations are conducted using CHARMM62 and the CHARMM36 force field,63–65 along with the modified TIP3P model for water.66,67 Protonation state of titratable groups is determined via PropKa68,69 and hydrogen atoms are added with the HBUILD module of CHARMM. Four different QM partitions (M2–5, see illustration in Fig. 1b; also see Table S1 in the Supporting Information) previously studied43 are recreated by placing QM link atoms at the Cα-Cβ frontier while employing the Divided Frontier Charge (DIV) scheme.18 One partition (M5C) extends the QM region to include the backbone of most amino acids, leading to regions of contiguous peptide strands in line with the largest QM region in previous calculations.43 The smallest QM region in the previous work43 (M1) is not studied here as it does not include all the ligands of the Mg2+ in the QM region and therefore, in our opinion, does not represent a viable choice in realistic studies.
Figure 1:
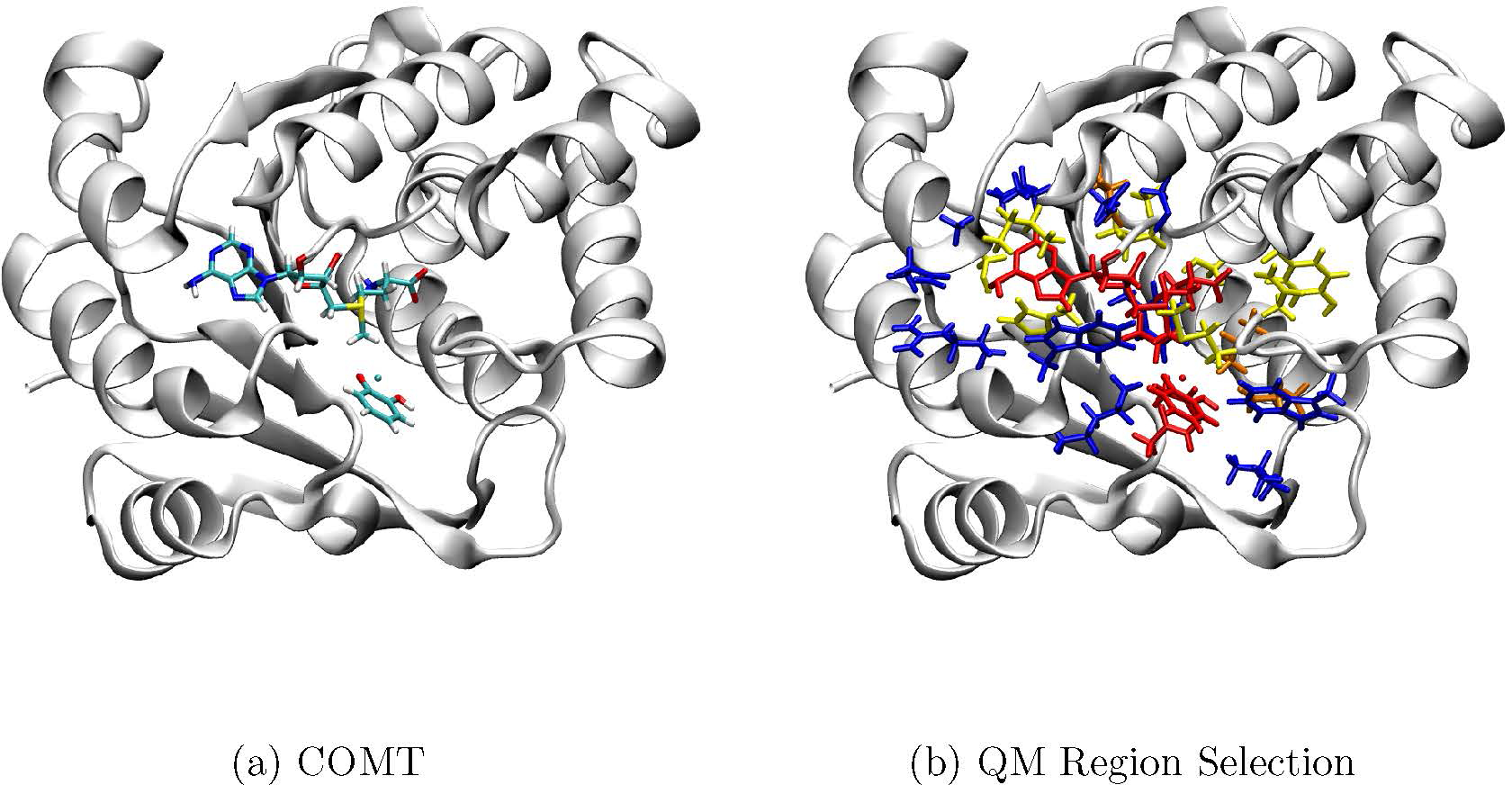
Illustration of the active site and QM region selections for the catechol-O-methyltransferase (COMT) system. (a) The active site that contains the co-factor, S-adenosyl methionine (SAM), the Mg2+ ion, the substrate catechol; (b) extending QM regions of M2 (red), M3 (+orange), M4 (+yellow), and M5 (+blue) where each region incorporates the previous colored regions. For amino acids, only the sidechains are included in the QM region in these partitioning scheme. In the M5C model, the QM region further includes the protein backbone atoms for residues in the M5 model, leading to a QM region consisting of 505 atoms.
The approximate Density Functional Tight Binding56 up to 3rd Order (DFTB3)46 with the 3OB parameterization,70,71 is used as the QM method. DFTB3 has been shown to provide fairly reliable structures and energies in biomolecules while providing 102–3 speedup in comparison to traditional DFT methods,57,72 making it particularly attractive for free energy simulations of biomolecules,73 although system-specific calibration is always essential.74 As shown in Fig. S1 in the Supporting Information, calculations for a gas-phase model system using trimethyl sulfide, catecholate, Mg2+ and coordinating waters find that DFTB3 underestimates the barrier while overestimating exergonicity for the methyl transfer reaction of interest as compared to two popular DFT methods; consistent with the larger exergonicity, the transition state is also earlier at the DFTB3 level compared to DFT results. For the model reaction in water (Fig. S2), similar to previous observations,75 DFTB3/MM free energy simulations underestimate the methyl transfer barrier compared to the experimental estimate (see discussion in76) by at least 4 kcal/mol. Nevertheless, the similarity in the general potential energy profiles in the condensed phase (Fig. S2b) supports the use of DFTB3 for the purpose of examining the impact of different methodological details, especially QM region size, on QM/MM results for COMT. Moreover, as noted below, DFTB3/MM simulations are able to capture the equilibrium and kinetic isotope effects for the methyl transfer in solution and COMT, further supporting the use of DFTB3 for describing the relevant transition state. To probe the effect of dispersion among DFTB3 atoms, each QM region is studied with and without Grimme’s D3 empirical dispersion model.77 As discussed in a recent review of semi-empirical QM methods for the treatment of non-covalent interactions in the context of biomolecular simulations,78 DFTB3, especially when supplemented with the D3 dispersion model, generally provides an adequate description of non-covalent interactions, although further improvements for highly charged interactions are warranted.
The primary set of systems is constructed based on the crystal structure (PDB code: 3BWM) using the Generalized Solvent Boundary Potential (GSBP) scheme, which has been shown to efficiently model long-range electrostatics in protein environments.47,48 System setups follow general procedures previously used in our group.22,74 In brief, the inner region has a radius of 27 Å and is explicitly solvated up to 25 Å about the center (chosen to be the active site Mg2+ ion); the inner-inner reaction field matrix, Mij, is described with spherical harmonics up to 20th order, and the outer region electrostatic potential is represented with a grid of size 0.5 Å using a focusing scheme.79 Non-bonded interactions are treated with extended electrostatics and a group-based cutoff scheme.80 Each system is allowed to equilibrate for no less than 200 ps using a 1 fs integration step at 300 K after minimization and heating simulations.
To test the sensitivity of QM/MM results to the GSBP partitioning, as well as the GSBP framework itself, classical MD simulations are performed for both wild type (WT) and the Y68A COMT with periodic boundary condition (PBC). Both systems are setup using CHARMM-GUI81 using a 76 Å cubic box in a 0.15 M KCl solution. MD simulations were run at 300 K using a 2 fs integration step and PME82 electrostatics in OpenMM83 for over 400 ns each, including an initial equilibration of 250 ps. Several snapshots from the classical simulations were then prepared for QM/MM PBC and GSBP simulations using the M2 QM selection based upon different protein configurations, leading to 8 additional systems (3 PBC and 5 GSBP, vide infra). Upon QM/MM conversion, each system is allowed to re-equilibriate for no less than 50 ps at 300 K using a 1 fs integration step. QM/MM PBC simulations employ PME electrostatics84,85 while GSBP simulations employ extended-electrostatics48 in the same fashion as described above.
Potentials of mean force (PMFs) for the methyl transfer reaction for the different QM/MM models are computed using well-tempered metadynamics with multiple walkers and the CHARMM-Plumed interface.86–89 The reaction coordinate ξ is chosen as the anti-symmetric stretch between the catecholate nucleophilic O− and the SAM sulfur/methyl group; i.e., (see Scheme 1). 28 walker seeds are generated along ξ from −1 to 2 Å for each system. Each walker is run for at least 200 ps each for a total of 5.6 ns of sampling for models M2–5(+D3), whereas 100 ps per walker is used for M5C. As shown in Fig. S3 in the Supporting Information, this level of sampling is found to be adequate for the convergence of PMF for the current system. Statistical analysis of the free energies were performed using block-analyzed reweighting of the resultant PMFs.90
Kinetic isotope effects and transmission coefficients are computed to analyze the nature of the transition state and the quality of the approximate reaction coordinate.91 The maxima of the computed PMFs are used to construct putative transition state ensembles using constrained MD simulations; since the experimental KIE was measured49 based on V/Km, the reactant state in the KIE calculations is taken to be SAM in a 25 Å water sphere. The putative transition state and reactant state ensembles are constructed based on 200 ps of sampling, leading to over 200,000 configurations for each. Secondary tritium KIEs on the SAM methyl are then computed using a path-integral free energy perturbation (PI-FEP) method.51 Convergent results require 32 beads representing hydrogen and at least 15,000 evenly spaced frames from the transition state/reactant ensemble with at least 100 Monte Carlo steps per classical frame. In total, calculation of the KIE via the PI-FEP approach requires the equivalence of around 50–100 ns of sampling, and thus is limited to models M2-M4. The transmission coefficient, κ, is obtained via the plateau of the normalized reactive flux correlation function,50,52,53,92 which is obtained from initiating 100 transition state frames, each with 100 randomized momenta along the reaction coordinate for M2–4.
3. Results and Discussion
3.1. Free Energy Profile
COMT has previously been extensively studied as a model system for methytransferases, which play a fundamental role in gene regulation93,94 and neurotransmitter metabolism.95 COMT inactivates catecholamines such as dopamine, norepinephrin and epinephrin through methylation donated by a SAM cofactor in a Mg2+-ligated active site.76,96,97 As methyltransferases play a key role in post-translational modification, especially in the area of neurobiology, COMT has been studied extensively, providing a crucial model in the understanding of methyltransfers.49 For instance, genetic polymorphism in COMT such as Val158Met has been shown to reduce COMT’s catabolic activity, leading to an apparent relationship to certain detrimental neuropsychiatric phenotypes.98
As COMT provides a rich base for theoretical models, energy and free energy surfaces have been computed using multiple levels of theory, ranging from full ab initio quantum mechanical active site models to QM/MM free energy simulations.35,43,44,76,99–101 However, seemingly disparate results were obtained between different studies, leading to discussions of error sources between methods. To date, there has been much disagreement in how large the QM region should be for reliable free energy results.
To evaluate the (in)sensitivity of the free energy profile to QM size, we show the COMT methylation transfer PMFs after extensive sampling based on increasing QM region sizes using DFTB3/MM multi-walker well-tempered metadynamics (Fig. 2), with a summary of critical energetics and reaction coordinate values with different QM regions summarized in Table 1. Overall, the computed free energy barriers are in the range of 10.1–13.0 kcal/mol (the correction due to transmission coefficient is generally modest, see Table 3), substantially lower than the experimental free energy barrier of 18.1–19.2 kcal/mol.102 This is expected based on the gas-phase and solution-phase model calculations shown in the Supporting Information; for example, as noted above, the DFTB3/MM simulations underestimate the reaction barrier in solution by at least 4 kcal/mol.75 It should be noted that although a recent DFTB3/MM study of COMT obtained a free energy barrier of 22.5 kcal/mol,100 the study was done starting with a mono-chelating catecholate motif that significantly differs from the initial structure in the current and other previous studies.35,43,44
Figure 2:
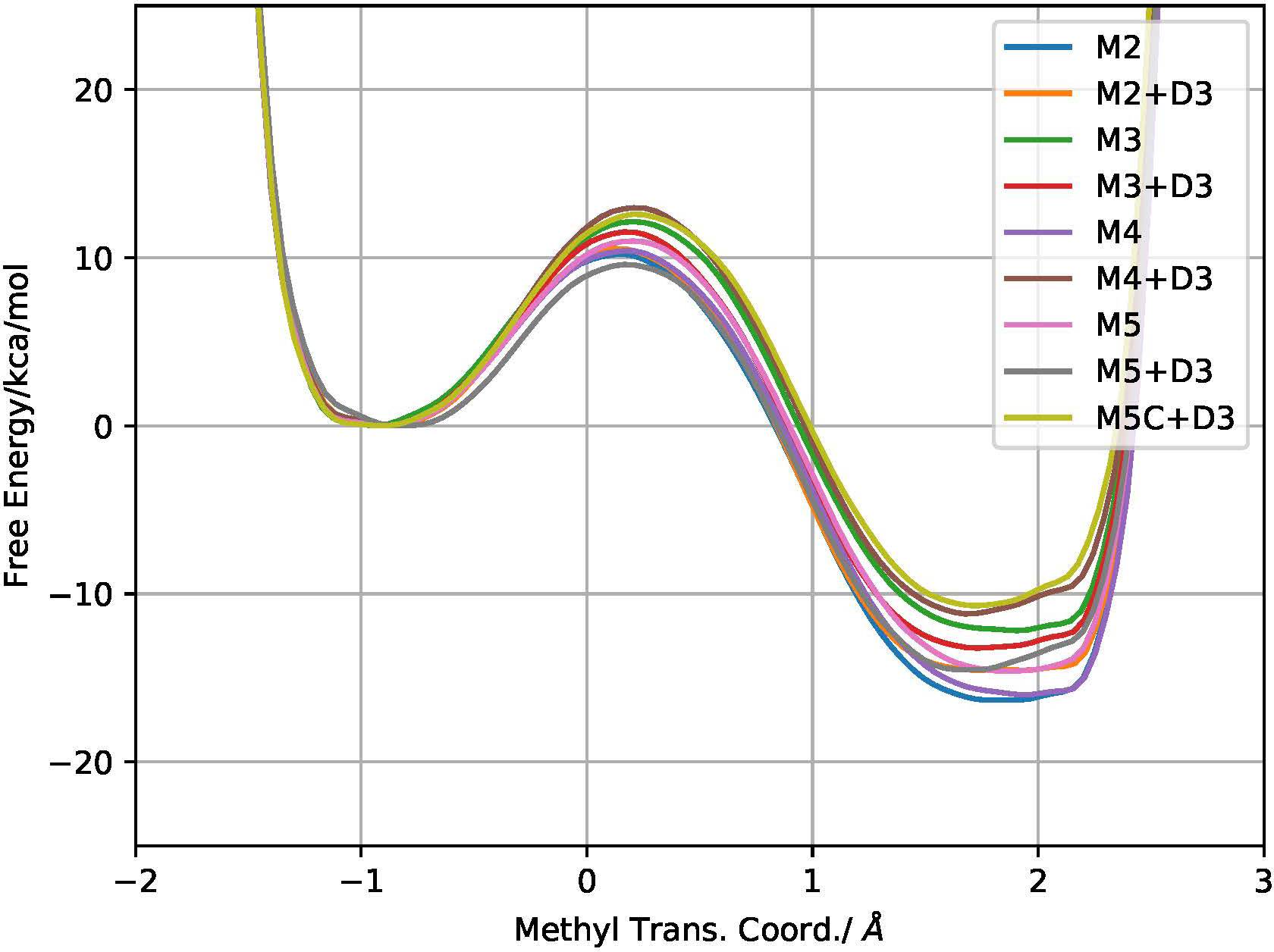
Free energy profiles from DFTB3/MM metadynamics simulations with different QM/MM models; ‘+D3’ results include the empirical dispersion contribution per respective model for the metadynamics simulations. Note that for M5C, due to the large QM region, only simulations including the empirical dispersion interactions among the QM atoms (M5C+D3) are conducted. Statistical errors along each surface are typically in the 0.1–0.3 kcal/mol range.
Table 1:
Key features for the computed free energy profiles shown in Fig. 2 of the main text; energetics (statistical uncertainty in parentheses) are in kcal/mol, and reaction coordinate values (RX: reactant; TS: transition state; PD: product) are in the unit of Angstrom.
| Models | ξRX | ξTS | ΔG‡ | ξPD | ΔG | |
|---|---|---|---|---|---|---|
|
| ||||||
| M2 | −0.88 | 0.16 | 10.2(0.2) | 46 | 1.77 | −16.3(0.1) |
| M2+D3 | −0.88 | 0.12 | 10.5(0.3) | 49 | 1.92 | −14.5(0.1) |
| M3 | −0.93 | 0.20 | 12.1(0.2) | 43 | 1.90 | −12.2(0.1) |
| M3+D3 | −0.88 | 0.16 | 11.5(0.2) | 48 | 1.73 | −13.2(0.1) |
| M4 | −0.93 | 0.17 | 10.4(0.2) | 47 | 1.90 | −16.0(0.1) |
| M4+D3 | −0.88 | 0.21 | 13.0(0.2) | 42 | 1.68 | −11.2(0.1) |
| M5 | −0.92 | 0.25 | 11.7(0.2) | 44 | 1.75 | −12.5(0.1) |
| M5+D3 | −0.81 | 0.12 | 10.1(0.2) | 59 | 1.76 | −13.4(0.1) |
| M5C+D3 | −0.92 | 0.20 | 12.6(0.2) | 42 | 1.71 | −10.7(0.1) |
The planarity of the methyl group is given as the average absolute value of the H-C-H-H improper dihedral in degrees from each transition state ensemble with the standard deviation given in parentheses, defined such that 0 is perfectly planar.
Table 3:
Computed secondary equilibrium and kinetic isotope effects for the methyl group in SAM, along with other key transition state properties and the transmission coeffcient, κ; energetics are in kcal/mol, and reaction coordinates are in the unit of Angstrom.
| Models | ξTS | ΔG‡ | a | 2o Isotope Effect b | κ | Effective ΔG‡ c |
|---|---|---|---|---|---|---|
|
| ||||||
| Equil. | 0.78 | |||||
| M2 | 0.16 | 10.2 | 46 (9.2) | 0.77 | 0.61 | 10.5 |
| M2+D3 | 0.12 | 10.5 | 49 (9.4) | 0.82 | 0.59 | 10.8 |
| M3 | 0.20 | 12.1 | 43 (9.0) | 0.81 | 0.36 | 12.7 |
| M3+D3 | 0.16 | 11.5 | 48 (9.4) | 0.78 | 0.25 | 12.3 |
| M4 | 0.17 | 10.4 | 47 (9.4) | 0.80 | 0.39 | 11.0 |
| M4+D3 | 0.21 | 13.0 | 42 (8.7) | 0.86 | 0.31 | 13.7 |
The planarity of the methyl group is given as the average absolute value of the H-C-H-H improper dihedral in degrees from each transition state ensemble with the standard deviation given in parentheses, defined such that 0 is perfectly planar.
The first row gives equilibrium isotope effect for the reaction in solution, and the rest are kinetic isotope effects for the enzymatic reaction. The experimental values are 0.83 and 0.79, respectively. 49,96,99,105
The effective ΔG‡ is the corresponding ΔG‡ value corrected by the transmission coefficient (i.e., increased by an amount of kBTln(κ−1).
The issue of primary interest here is not the absolute barrier itself, but its variations due to various methodological details; thus no attempt was made to adjust the reaction barrier by refitting the DFTB model, such as repulsive potentials.103 Along this line, we observe a modest level of sensitivity in both barrier and reaction exergonicity to the QM size with similar trends in barrier energy distributions (Fig. S4). With an overall spread of 2.9 kcal/mol in free energy barriers among the various models (as compared to ~8 kcal/mol for similar PM6/MM studies44 and ~20 kcal/mol for DFT/MM43), there is no apparent trend in the QM size dependence; the position of the barrier along the reaction coordinate is ~0.15 Å, with a minor variation of 0.13 Å among the different QM regions. Although the exergonicity is more liable to change (with a range of 5.6 kcal/mol), again the variations are much less severe than reported previously (~20 kcal/mol for similar PM6/MM studies44 but ~25 kcal/mol for DFT/MM43) while maintaining the qualitative exergonic nature of the reaction. Previous studies have noted that the sensitivity in exer/exogonicity can be attributed to the change from formally charged reactants to neutralized products.44 An interesting observation is that for larger QM models the effect of dispersion becomes increasingly important. For instance, inclusion of dispersion for M5 shifts the transition state earlier by 0.13 Å while decreasing the height by 1.6 kcal/mol. Meanwhile, inclusion of dispersion in M4 decreases the exergonicity by ~6 kcal/mol.
As discussed in previous work,17–19 partitioning schemes generally should avoid creation of the QM/MM boundary across polar covalent bonds to prevent over-polarization of the QM region. Even with this precaution, extra care must be taken to further prevent un-physical charge distribution through certain methods such as the DIV scheme used here.18 Nonetheless, in M5C+D3 we partition the QM region across peptide bonds to include the protein backbone so as to better compare to the recent study of Kulik and co-workers.35,43 While M5C+D3 is still within range compared to all other models, it is noted that M5C+D3 produces somewhat larger differences from M5+D3 (ΔΔG‡ = 2.5, ΔΔG = 2.7 kcal/mol).
All models discussed so far have been constructed in the GSBP framework based on the crystal structure, thus significant reorganization of the protein environment may not be captured due to the atoms fixed in the outer region of the GSBP setup.54 To evaluate contributions from slower fluctuations that involve distal regions, we have conducted a battery of free energy simulations with starting structures taken from longer classical molecular dynamics simulations of both WT- and Y68A-COMT using QM/MM GSBP and PBC simulations; the Y68A mutant is chosen so as to compare the magnitude of free energy variation due to different simulation protocols with the impact of mutation, which is 1.6 kcal/mol according to experiment.102
In classical molecular simulations, WT COMT stayed remarkably similar to the crystal structure except for a reorganization of Y38 and R201, while Y68A appeared to be more flexible, primarily due to a more mobile N-terminus with modest deviations in some key distances in the active site such as the methyl C-catecholate O distance, as well as rearrangements in the next shell (see Figs. S5–S8 in Supporting Information). This offers an excellent opportunity to verify the robustness of free energy results as a function of different structural models. For the WT enzyme, we initiate DFTB3/MM free energy simulations using GSBP with 190 ns (Y38-R201 elongation) and 250 ns (stabilized RMSD point for both WT & Y68A) snapshots. Similarly, we initiate GSBP simulations for Y68A starting with the 40 ns (SAM-Y143 elongation), 110 ns (N-Terminus leaves solution) and 250 ns snapshots from classical simulations. As further comparison, we perform PBC based DFTB3/MM free energy simulations for both WT and Y68A starting with the snapshot at 250 ns from the respective classical simulations.
Overall, as seen in Fig. 3 and Table S2, the PBC models do not differ substantially (sub kBT) from their most direct complementary GSBP models, due to the lack of any highly mobile regions in COMT. For WT simulations, the minor structural changes after longer MD runs can produce free energy barrier differences on the order of 1.5–1.8 kcal/mol. For Y68A, the significant deviations from WT crystal structure can produce free energy barriers upwards of 3.7 kcal/mol. However, these variations are on the order of the deviations observed due to different QM regions and within the observed experimental increase in free energy upon mutation, making it challenging to robustly capture the effect of the Y86A mutation.
Figure 3:
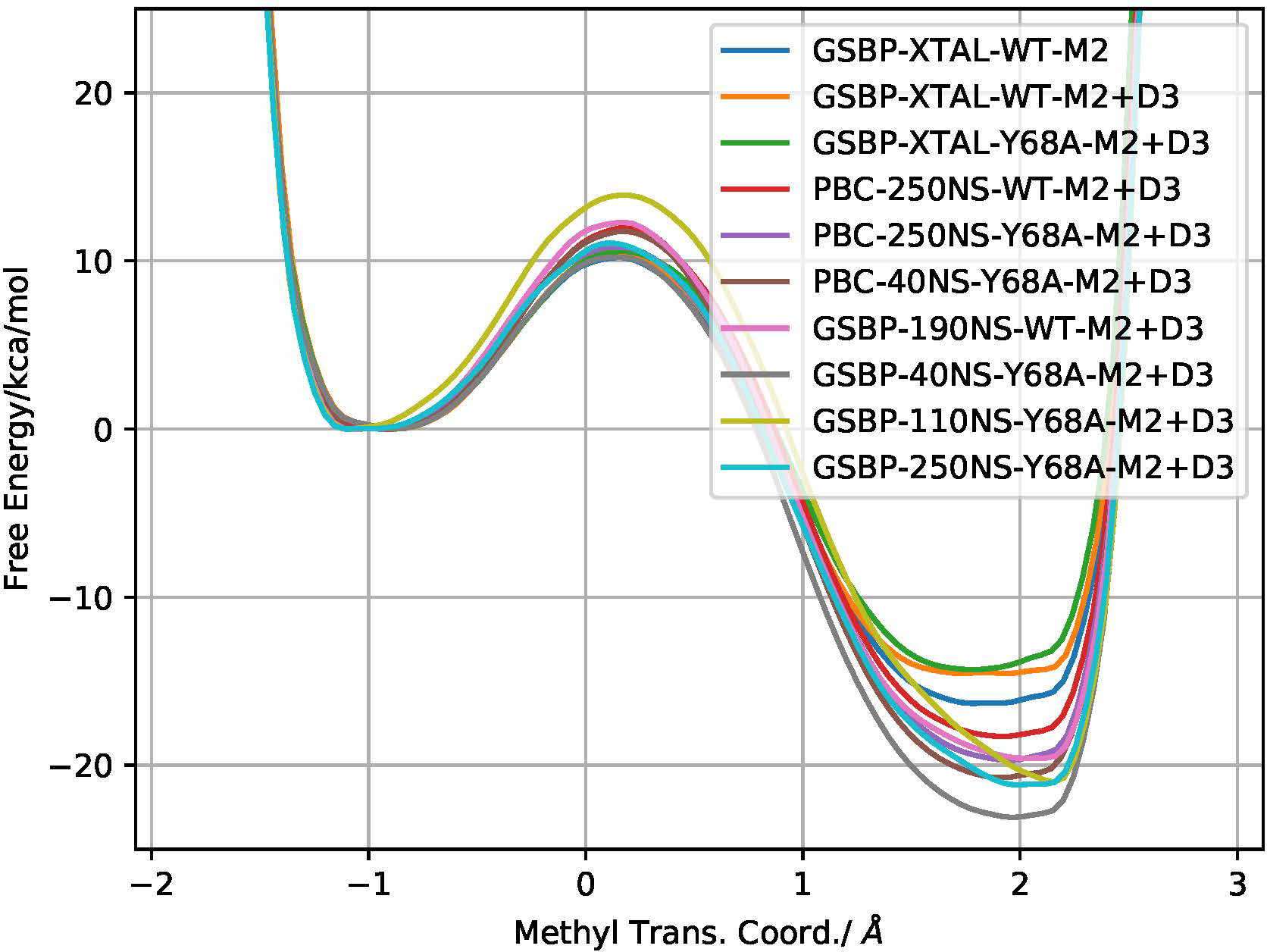
Free energy profiles from DFTB3/MM metadynamics simulations for different WT and Y86A models using the M2 QM region; ‘+D3’ results include the empirical dispersion contribution per respective model. Error along the PMF ranges typically from 0.1–0.3 kcal/mol. See Supporting Information Table S2 for tabulated form.
In short, compared to previous DFT/MM based umbrella sampling simulations,35,43 the current study suggests that once adequate sampling is conducted, the free energy profile exhibits modest sensitivity to the QM region size. Other methodological choices such as dispersion among the QM atoms, QM/MM partitioning scheme and the starting structure of the free energy simulations are at least as important to the free energy results. These variations make it challenging to consistently capture modest contributions due to the mutation of second-shell residues, such as Y68A.
3.2. Structural Features
To determine how each subsequent expansion of the QM region affects the free energy surfaces, we investigate key local and global structural features. One of the most important features is the retention of the core active site distances, particularly coordination to the Mg2+ ion. Monitoring all amino acid/substrate coordination distances across the reaction reveals that the active site is structurally impervious to QM region choice (Fig. 4). For example, as the reaction progresses, catecholate charge becomes neutralized via methyl transfer (see Fig. S10 in the Supporting Information), thus one may expect a slight loosening of the CAT-O−-Mg distance. Indeed, all models capture this trend of CAT-O−-Mg coordination elongation along with a slight tightening of D141/D169-Mg coordination as the reaction progresses, and extending the QM region does not noticeably affect these trends. However, for some simulations, without the inclusion of dispersion, catecholate coordination at the product side becomes destabilized (Fig. 4), an effect not observed in any simulations including dispersion.
Figure 4:
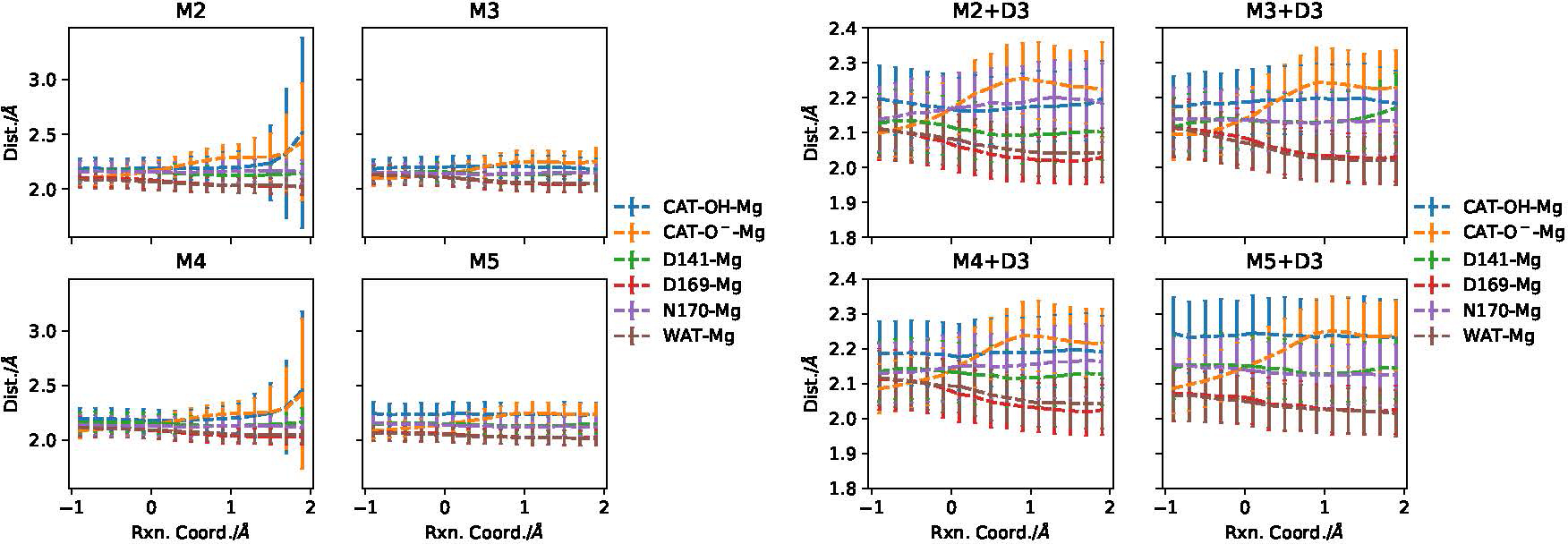
Evolution of O-Mg coordination distances in the active site across the reaction coordinate ξ for the QM/MM models with and without empirical dispersion (+D3). The main difference only lies in the destabilization of catecholate coordination at the product side in some of the simulations (M2 and M4) without the empirical dispersion.
With regards to other key active site structural characteristics, previously it was observed43 that CAT-OH participates in a low-barrier hydrogen bond with E199, a feature that model M2 does not capture as E199 is in the MM region. Even with larger QM regions, SHAKE was applied to all bonds involving hydrogen, barring proton transfer between catechol and E199. To make a fair comparison to previous simulations, PMF calculations are repeated without the SHAKE constraint in catecholate for the M3 model, which includes E199 in the QM region; the free energy profile for the proton transfer between catechol and E199 is also explicitly computed (Fig. 5). These observations confirm that the preferred product state does exhibit an unusually shared proton between CAT and E199; however, the exergonicity is only increased by ~1.8 kcal/mol, and the effect on the barrier height is even smaller (~0.5 kcal/mol lower if the proton is shared). Thus the allowance of this unusually short hydrogen bond/sharing is unlikely to change the free energy surface between M3 and larger models.
Figure 5:
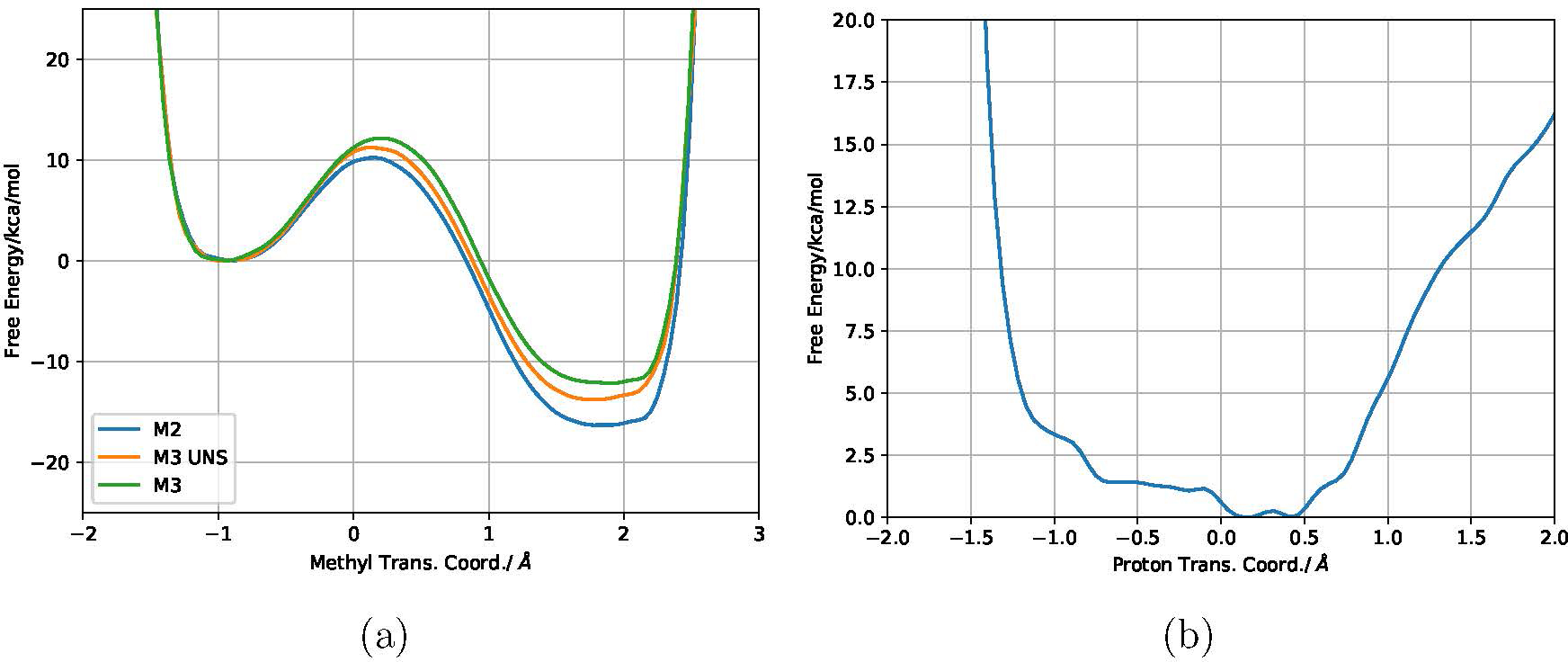
Effect of the strong hydrogen-bond between E199 and catecholate on the free energy surface (FES) in COMT. (a) FES of M2, M3 and M3 without the SHAKE protocol on the CAT-O-H bond (UNS). (b) FES of proton transfer given by the asymmetric stretch of the Catechol O, Catechol H, and an E199 acceptor oxygen.
Looking at more global structural characteristics, we determine the average number of inter-residue hydrogen bonds between different regions of COMT. As M2 is the minimalist model in this study, we next consider extending our structural analysis to the perseverance of hydrogen bonding networks within and outside the “core” M2 region. Using a distance and angular threshold of 3.0 Å and 20°, respectively, the number of core-core, core-outer, and outer-outer (not to confuse with the outer region in the GSBP setup) hydrogen bonds are computed for the reactant, transition state and product regions across the models; the M2 core selection includes only the M2 QM region, and the outer selection includes all protein atoms included in the M5 QM model. As shown in Table 1, hydrogen bonds are largely maintained throughout the reaction and between each model. Again, we see no significant structural change globally within variance in the protein regardless of the QM region size.
The lack of apparent structural changes between each model after extensive sampling reinforces the relative robustness of the free energy surfaces with regards to expanding QM regions. Similar analyses were done on select Y68A GSBP and PBC simulations due to Y68A’s relatively high flexibility (see Table S4 in Supporting Information). Compared to WT simulations, Y68A appears to have ~ one less hydrogen bond between the core and outer region, while the core-core/outer-outer regions are similar. Given that Y68, nominally in the region beyond the core, does not natively hydrogen bond with the core in WT, these results suggest that the Y68A mutation affects the active site in an indirect way, disrupting nearby hydrogen bonds instead. Further studies are necessary to explore these structural differences to firmly establish the mechanism for the reduced catalytic activity of Y68A.49
3.3. Kinetic Isotope Effect and Transmission Coefficient
To evaluate DFTB3 for characterizing the methyl transfer transition state, we compute the secondary proton/tritium kinetic isotope effect (KIE) for the main systems studied (i.e., the GSBP setups based on the crystal structure) using a path-integral free energy perturbation approach,51 which has been applied to not only H/D/T KIEs but also heavy-atom KIEs such as 18O KIEs for phosphoryl transfer reactions.79,104 As mentioned above, the cost of the path-integral simulations limited us to models M2-M4(+D3).
All models agree rather well with previous theoretically and experimentally observed inverse KIE (0.79) and equilibrium isotope effect (0.83).49,96,99,105 As these calculations depend on the local configuration and vibrational modes of the transition state, a minimal QM region (M2, M2+D3) leads to reasonable estimates of the KIE likely because DFTB3 gives fairly accurate vibrational frequencies for sulfur containing compounds.106 These results further support the use of DFTB3/MM for the characterization of methyl transfer transition state in COMT, and the conclusion that the key characteristics of the transition state does not change in any major way with respect to the QM size.
The transmission coefficients, κ, evaluated for different models (M2-M4, with and without D3) and the corresponding corrections to the effective activation free energy are also summarized in Table 3. For most models, the deviation of κ from unity is modest and in the range of 0.5–0.6, which is expected for a relatively simple methyl transfer reaction in the enzyme active site. With some models, however, the value of κ is smaller, suggesting perturbation in the active site structure, charge distribution and therefore the coupling of the transferring methyl group with the environment.53 For example, it appears that κ generally decreases as empirical dispersion is included to each QM region. Nevertheless, the contribution of κ to the effective activation free energy is overall on the order of kBT.
3.4. Electronic Structural Features: Charges and Charge Covariance
Finally, we discuss the dependence of electronic structural features of COMT on the QM region size. First, we note that the evolutions of the charges of Mg2+, the catechol and SAM during the reaction are generally insensitive to the QM region size (see Fig. S10 in the Supporting Information), again supporting that the overall feature of the catalyzed methyl transfer is preserved across different QM region selections. To probe the electronic structure at a more detailed level, we focus here on the covariance matrix of total charge amongst residues, as pioneered by Kulik and co-workers;43,107 similar to Ref.,43 for each amino acid residue (or co-factor), we compute the sum of Mulliken charges, and we monitor the charge covariance matrices in the reactant, transition state and product ensembles. The covariance matrices are organized based on the average distances to the Mg2+. Calculations for the M2 model suggest that the DFTB3/MM results based on the Mulliken analysis are generally consistent with higher level DFT/MM results based on the CM5 charge108 (see Figs. S11–S12 in the Supporting Information).
With a large QM region as in M5+D3, while there is non-negligible coupling between charge fluctuations between the core M2 region and residues further away (Fig. 6), we note that the magnitudes of such coupling are markedly reduced compared to those within the M2 core. Indeed, mapping of the covariance matrices onto the protein structure (Fig. 7) further highlights that only nearby residues are significantly involved. Focusing on couplings within the M2 core (Fig. 8), all QM/MM models display overall similar degrees of charge covariance. As M3 includes some second shell amino acids, particularly E199, that engage in strong hydrogen bonds to the substrates, there are large fluctuations attenuated to M3. Notably, even with a somewhat destabilized catecholate geometry in the product, the charge covariance in M2 remains remarkably similar to the other models. Thus, it appears that even minimalistic models such as M2 or M3 can adequately capture and screen the charge fluctuations during the course of the reaction in COMT.
Figure 6:
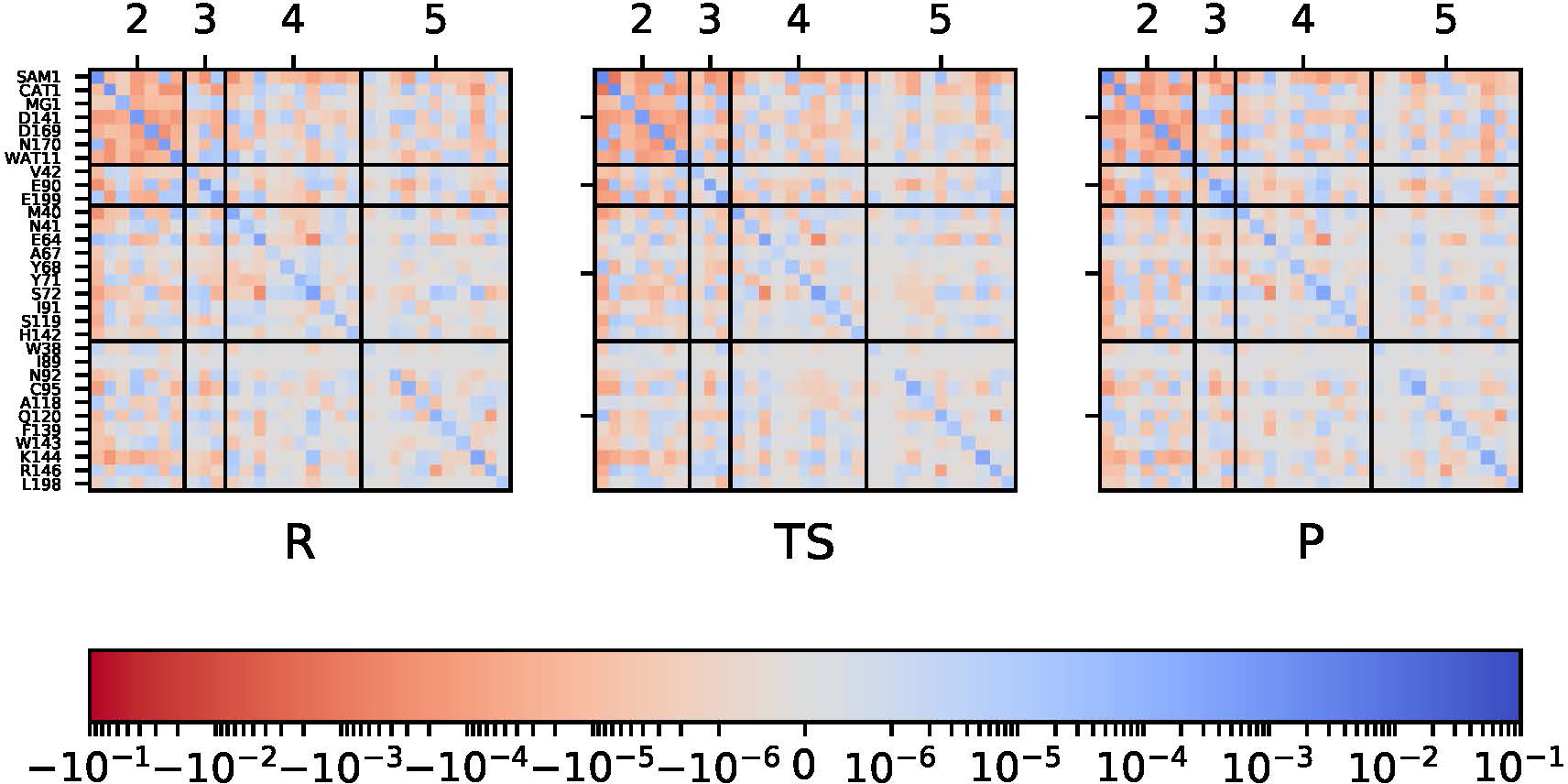
The charge covariance matrices at the DFTB3/MM level for the reactant, transition state and product bins (see Table 1) for the M5+D3 model. Grid spacing highlights the residues in different QM selection choices in the QM/MM models.
Figure 7:
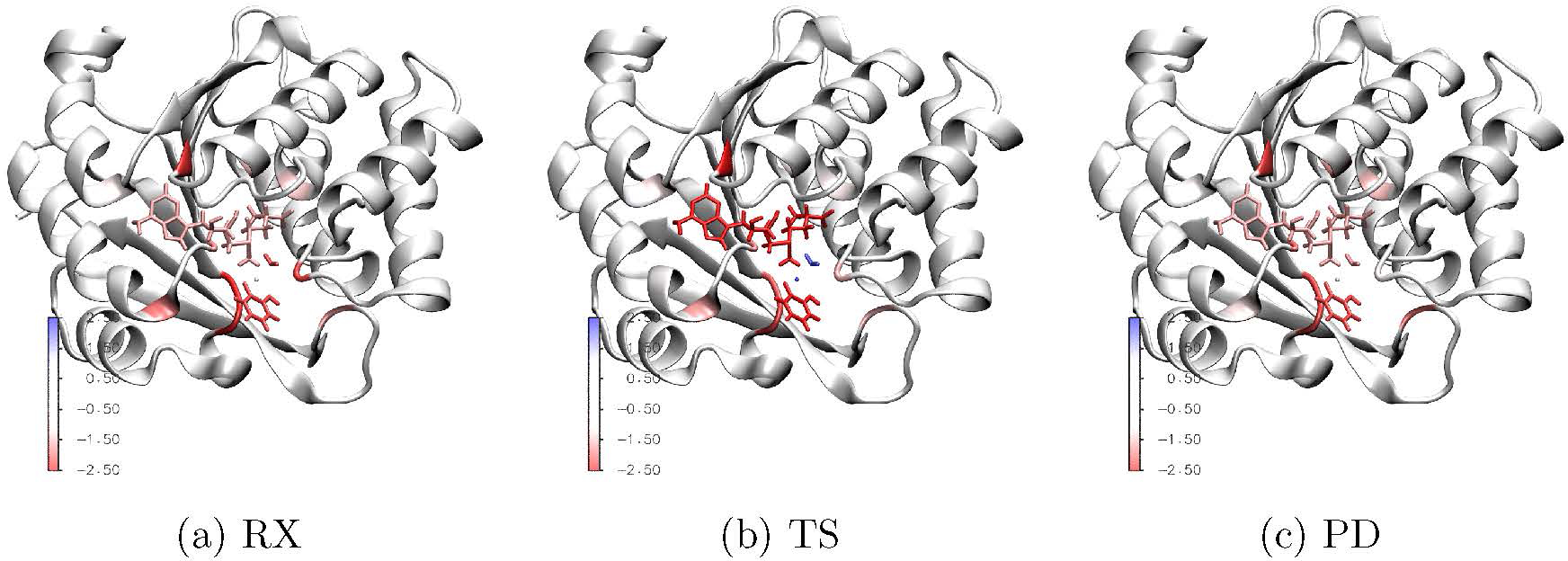
Max Covariance at the DFTB3/MM level (×105 for visual clarity) between any residue to those in the M2 region computed for M5+D3 mapped onto the protein structure; for results for the M5C+D3 model, see Fig. S20 in the Supporting Information.
Figure 8:
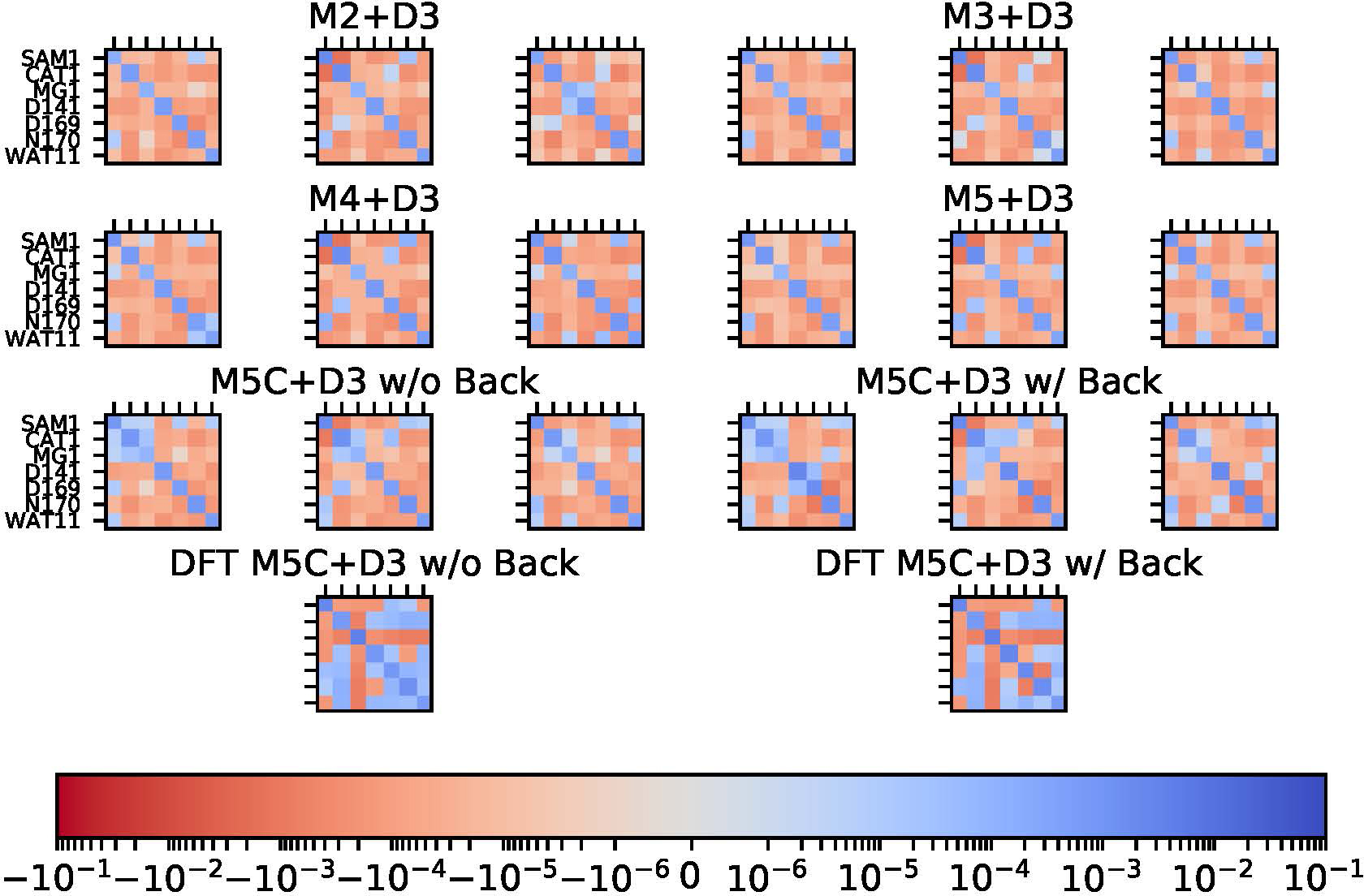
Charge covariance matrices at the DFTB3/MM level for the M2 region from different QM/MM simulations that include empirical dispersion; for each model, results for the reactant, transition state and product are shown separately. For the M5C+D3 model, charges are reported for each residue with those for the backbone atoms either included (‘w/Back’) or excluded (‘w/o Back’). Results for QM/MM simulations without the empirical dispersions are overall similar and thus not shown. For comparison, the last row shows the CM5 charges from DFT (ωPBEh/def2-SVP)/MM calculations using DFTB3/MM snapshots for the reactant state.
On the other hand, between M5C+D3 (Fig. 9, which does not include backbone charges) and M5+D3, we note several major differences in the charge covariance matrices. With M5C+D3, in the reactant state, there is a notable simultaneous charge movement amongst the charged center (SAM, CAT, MG). Moreover, the overall charge covariance seems shifted from the core M2/M3 regions and globally spread. For example, the ratio of extreme off-diagonal covariance values is 25 for M5+D3, while the ratios are generally much smaller and less than 5 with the M5C+D3 model (Fig. S13 in the Supporting Information), highlighting more uniform charge covariance across different regions. These observations are reinforced when including the backbone charges in the covariance calculations (Fig. S14 in the Supporting Information). Single-point charges using ωPBEh/def2-SVP and PBE/def2-SVP on the DFTB3 M5C+D3 structures reveal similar trends (e.g., Figs. S15 vs. S17, and Figs. S16 vs. S18 in the Supporting Information). While ωPBEh/def2-SVP modestly decreases the magnitude of charge covariances of residues in the outer compared to core residues, inclusion of the backbone leads to a more pronounced effect (Figs. S15 vs. S16, and Fig. S19 in the Supporting Information). Therefore, it appears that the electronic structure is perturbed substantially when the QM region includes protein backbone atoms to form contiguously bonded regions, and this trend is not due to the delocalization errors associated with typical GGA functionals.55 This is further illustrated by the ranges of total charge for each residue in M5+D3 and M5C+D3 calculations. As shown in Fig. 10, the range is significantly expanded with the M5C+D3 model; similar to previous discussion,43 the total charge of even non-polar residues such as Ile and Leu can fluctuate as much as 0.4 e once the protein backbone is included in the QM region.
Figure 9:
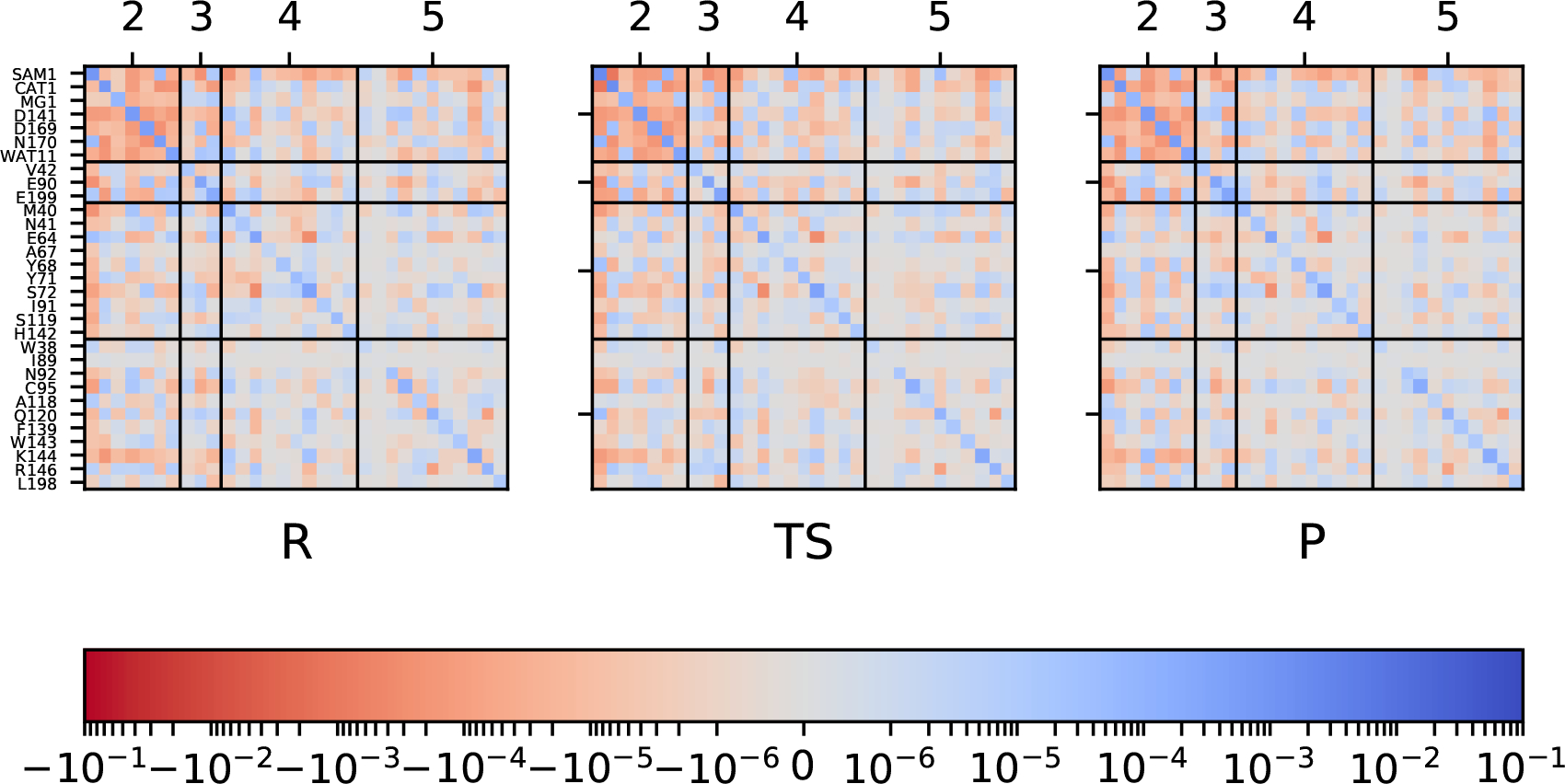
Similar to Fig. 6, but for the M5C+D3 model. To be consistent with Fig. 6, the results here only include charges for the sidechain atoms. Results include backbone charges for the M5C+D3 model are shown in Fig. S14 in the Supporting Information; results computed with DFT/MM single point calculations are also included in the Supporting Information (Figs. S15–S18).
Figure 10:
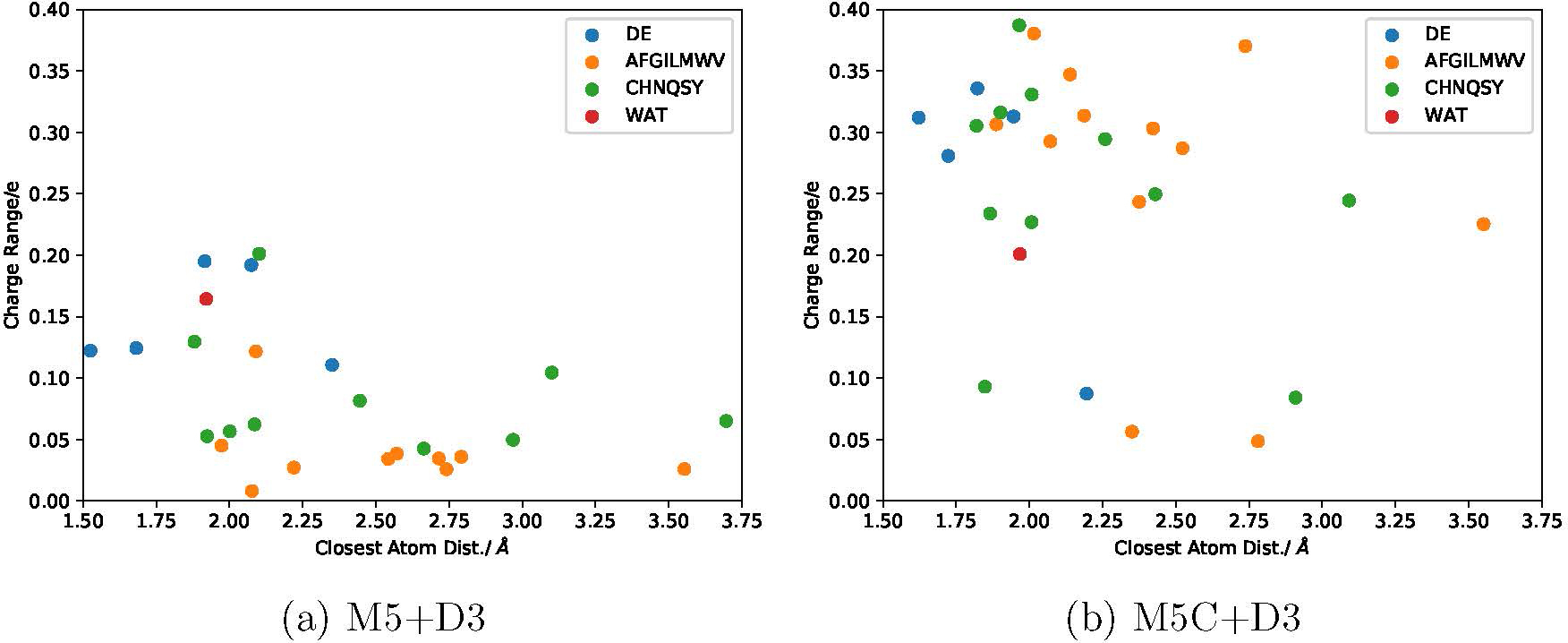
The range of total charge (i.e., maximum deviation from the integer reference) per residue from (a) M5+D3 and (b) M5C+D3 simulations; for the latter, the charges include backbone contributions. The charge range is plotted as a function of the closest distance of the residue to either SAM, CAT or MG. Charges are color coded into negatively charged (blue), non-polar (orange), polar (green), positively charged (red), and water (purple). All charges are DFTB3 Mulliken charges.
4. Concluding Remarks
Although the value of QM/MM simulations to mechanistic analysis of enzymes has been generally recognized, it remains non-trivial to balance computational accuracy and efficiency for arbitrary enzyme systems. One of the technical issues that has attracted much debate in recent years concerns the appropriate size of the QM region, with recommendations ranging from less than a hundred to several hundreds and approaching a thousand. It is an important issue since the proper QM size will also impact other methodological choices, such as multi-level free energy methods6,10,109,110 and machine learning approaches111,112 for improving the accuracy of low-level QM/MM energetics; if the proper QM region is routinely on the order of several hundreds, the applicability of these approaches will be substantially limited. Using COMT as a model system and an efficient but approximate DFT approach as the QM method, we aim to gain additional insights into this debate.
The most significant observation here is that the free energy profile, key structural/charge evolutions of the enzyme during the reaction as well as kinetic isotope effects show limited variations with respect to the QM region size, qualitatively similar to the observation from a previous semi-empirical QM/MM free energy study.44 For example, the position and height of the barrier change by 0.13 Å and 2.9 kcal/mol as the QM region is enlarged by more than 400 atoms. Similarly, even electronic structural properties such as residual charge covariance matrix for the core residues (M2 region) appear to be rather insensitive to the variation of the QM region. There are larger variations in the exergonicity, which is consistent with the consideration that the methyl transfer reaction leads to neutralization of a pair of formal charges, thus polarization of the enzyme environment is expected to make larger contributions; still, the magnitude of variation observed here is substantially smaller than those reported in previous work.43 Thus these results support the notion38,44 that the key features of enzyme catalysis can be captured with a modest QM region size. The significant variations in energies observed in previous studies appear to be largely quenched once extensive conformational sampling is included. Whether this is yet another manifestation of the enthalpy-entropy compensation phenomena often observed for biological systems113–115 deserves further analysis, although a preliminary analysis along the lines of previous studies116 (see Fig. S4 in the Supporting Information) does not indicate any such compensation effects. We note that without adequate sampling of dipolar relaxation in the enzyme environment, the effect of distant groups on the QM region can be overestimated. For example, when contributions from distant charged residues to the pKa of a titratable group were analyzed in a perturbative fashion,117,118 i.e., by computing the change of QM/MM interaction energies without re-sampling the MD trajectory after the MM partial charges on a charged residue are turned off, the effects were significantly overestimated (> 10 kcal/mol). For a similar reason, with limited sampling, the effect of altering the description (MM vs. QM) of distal residues can be exaggerated.
For large QM region calculations, in addition to the issue of dispersion, which evidently makes a notable contribution to the free energy profile in COMT, an important technical detail concerns the QM/MM partitioning scheme. While including only the sidechains in the QM region leads to rather short-ranged residual charge covariance, including the protein backbone into the QM region leads to significantly longer-ranged coupling in charge fluctuations. This observation can be explained by the fact that including protein backbone atoms leads to a larger set of contiguously bonded atoms. Therefore, it is plausible that the proper convergence of certain electronic structure properties indeed requires rather large QM regions, especially those that are highly sensitive to the charge density variation; indeed, according to the holographic electron density theorem,119 variation in the local charge density may impact charge density in remote regions since the electron density is analytic away from the nuclei. We note that the DFTB3 approach used here, due to the parameterization based on the PBE functional, suffers from delocalization error as other popular GGA functionals.120,121 Therefore, the long-range residual charge covariance observed in the M5C(+D3) model might be an exaggerated effect, as observed in the early development of the charge fluctuation model for proteins.122 However, we note that the trends in the charge fluctuations are largely conserved when a range-separated DFT method is used as the QM. Thus these results strongly suggest that different properties likely require different QM region sizes, a feature often observed in the analysis of complex systems. It is therefore encouraging to see extensive efforts in systematically determining QM region dependence with respect to different observables.15,26,28–31,35,123
While our computed KIEs for WT-COMT match experimental values rather well, investigation of the Y68A mutant offers a surprising challenge. On the one hand, although Y68A is more flexible, PBC and the more constrained GSBP boundary conditions lead to remarkably similar free energy results. On the other hand, the higher structural flexibility of Y68A offers different starting enzyme conformations that lead to deviations in the barrier height on the same order as experimentally measured effect of mutation (1.6 kcal/mol on the barrier height). More systematic analysis of the Y68A mutant is warranted to firmly establish the mechanism for the reduced activity induced by the second-shell mutation.
Finally, we recognize that the current study employs an approximate, semi-empirical approach (DFTB3) that has limitations in the description of both chemical and non-covalent interactions. For example, rather strong hydrogen bonds have been observed in recent analyses of high-resolution crystal structures,124,125 which might demand more accurate QM methods for proper description. Therefore, it remains important to cross-validate the current observations with more accurate QM methods. However, we emphasize that such analysis requires extensive sampling to properly converge the free energy profile, and short QM/MM simulations using reactant state conformations equilibrated with long classical MD simulations,36 while informative, are not replacement for proper free energy simulations. Moreover, as emphasized repeatedly in previous studies,7,74,126,127 it is essential (and often straightforward) to calibrate semi-empirical QM method for the specific system of interest (e.g., using proton affinities of key reactive groups) to avoid unrealistic errors in computed free energy profiles.128
Supplementary Material
Table 2:
The average number of hydrogen bonds (with standard deviations in parentheses) between atoms in different regions, where cc = core M2/core M2, co = core M2/M5 selection, oo = M5 selection/M5 selection.
| Ensemble a | QM/MM Model | cc | co | oo |
|---|---|---|---|---|
|
| ||||
| RX | M2+D3 | 0.43 (0.57) | 4.50 (1.36) | 3.28 (1.32) |
| M3+D3 | 0.60 (0.64) | 3.98 (1.41) | 2.98 (1.32) | |
| M4+D3 | 0.72 (0.65) | 3.92 (1.32) | 2.79 (1.26) | |
| M5+D3 | 0.52 (0.63) | 4.25 (1.42) | 3.38 (1.24) | |
| TS | M2+D3 | 0.64 (0.66) | 3.56 (1.33) | 3.16 (1.39) |
| M3+D3 | 0.51 (0.62) | 4.03 (1.45) | 3.56 (1.28) | |
| M4+D3 | 0.55 (0.63) | 3.71 (1.41) | 2.64 (1.24) | |
| M5+D3 | 0.47 (0.60) | 4.40 (1.44) | 3.21 (1.25) | |
| PD | M2+D3 | 0.55 (0.56) | 4.16 (1.33) | 3.23 (1.24) |
| M3+D3 | 0.43 (0.55) | 4.32 (1.45) | 3.51 (1.27) | |
| M4+D3 | 0.46 (0.57) | 3.80 (1.43) | 2.57 (1.17) | |
| M5+D3 | 0.52 (0.60) | 4.25 (1.40) | 3.22 (1.25) | |
A window size of 0.2 Å was used for the reactant (RX) and product (PD) while a window size of 0.1 Å was used for the transition state (TS).
Acknowledgement
This work was supported by grants from the NIH to Q.C. (R01 GM106443, R35 GM141930). Computational resources from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant number OCI-1053575, are greatly appreciated; part of the computational work was performed on the Shared Computing Cluster which is administered by Boston University’s Research Computing Services (URL: www.bu.edu/tech/support/research/). C.O. acknowledges financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via the SFB 1309–325871075. D.R. and Q.C. acknowledge discussions with Prof. Dan Major regarding convergence of path-integral based kinetic isotope effect calculations.
Footnotes
Supporting Information Available
Results for model methyl transfer reaction in the gas phase and solution; examples for the convergence behavior of metadynamics simulations; additional data for the QM/MM simulations using different QM models, boundary conditions (PBC vs. GSBP), starting conformations collected from long (~400 ns) classical MD simulations. Also included are charge covariance analyses with DFT/MM (PBE and ωPBEh) single point calculations on DFTB3/MM snapshots. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Warshel A; Levitt M Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [DOI] [PubMed] [Google Scholar]
- (2).Field MJ; Bash PA; Karplus M A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar]
- (3).Monard G; Merz KM Combined Quantum Mechanical/Molecular Mechanical Methodologies Applied to Biomolecular Systems. Acc. Chem. Res. 1999, 32, 904–911. [Google Scholar]
- (4).Gao J; Truhlar DG Quantum Mechanical Methods for Enzyme Kinetics. Ann. Rev. Phys. Chem. 2002, 53, 467–505. [DOI] [PubMed] [Google Scholar]
- (5).Friesner RA; Guallar V Ab intio QM and QM/MM methods for studying enzyme catalysis. Annu. Rev. Phys. Chem. 2005, 56, 389–427. [DOI] [PubMed] [Google Scholar]
- (6).Rosta E; Klähn M; Warshel A Towards Accurate Ab Initio QM/MM Calculations of Free-Energy Profiles of Enzymatic Reactions. J. Phys. Chem. B 2006, 110, 2934–2941. [DOI] [PubMed] [Google Scholar]
- (7).Riccardi D; Schaefer P; Yang Y; Yu H; Ghosh N; Prat-Resina X; Konig P; Li G; Xu D; Guo H; Elstner M; Cui Q Feature Article: Development of effective quantum mechanical/molecular mechanical (QM/MM) methods for complex biological processes. J. Phys. Chem. B 2006, 110, 6458–6469. [DOI] [PubMed] [Google Scholar]
- (8).Senn HM; Thiel W QM/MM studies of enzymes. Curr. Opin. Chem. Biol. 2007, 11, 182–187. [DOI] [PubMed] [Google Scholar]
- (9).Brunk E; Rothlisberger U Mixed Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States. Chem. Rev. 2015, 115, 6217–6263. [DOI] [PubMed] [Google Scholar]
- (10).Lu X; Fang D; Ito S; Okamoto Y; Ovchinnikov V; Cui Q QM/MM free energy simulations: recent progress and challenges. Mol. Simulat. 2016, 42, 1056–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lonsdale R; Mulholland A QM/MM Modelling of Drug-Metabolizing Enzymes. Curr. Top. Med. Chem. 2014, 14, 1339–1347. [DOI] [PubMed] [Google Scholar]
- (12).Cui Q; Pal T; Xie L Perspective: Biomolecular QM/MM Simulations: What are some of the “Burning Issues”? J. Phys. Chem. B 2021, 125, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Claeyssens F; Harvey JN; Manby FR; Mata RA; Mulholland AJ; Ranaghan KE; Schutz M; Thiel S; Thiel W; Werner HJ High-accuracy computation of reaction barriers in enzymes. Angew. Chem. Int. Ed. 2006, 45, 6856–6859. [DOI] [PubMed] [Google Scholar]
- (14).Cui Q Quantum Mechanical Methods in Biochemistry and Biophysics. J. Chem. Phys. 2016, 145, 140901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schulz CE; van Gastel M; Pantazis DA; Neese F Converged Structural and Spectroscopic Properties for Refined QM/MM Models of Azurin. Inorg. Chem. 2021, 60, 7399–7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Loco D; Lagardere L; Cisneros GA; Scalmani G; Frisch M; Lipparini F; Mennucci B; Piquemal JP Towards large scale hybrid QM/MM dynamics of complex systems with advanced point dipole polarizable embeddings. Chem. Sci. 2019, 10, 7200–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gao JL; Amara P; Alhambra C; Field MJ, A generalized hybrid orbital (GHO) method for the treatment of boundary atoms in combined QM/MM calculations. J. Phys. Chem. A 1998, 102, 4714–4721. [Google Scholar]
- (18).König PH; Hoffmann M; Frauenheim T; Cui Q A Critical Evaluation of Different QM/MM Frontier Treatments with SCC-DFTB as the QM Method. J. Phys. Chem. B 2005, 109, 9082–9095. [DOI] [PubMed] [Google Scholar]
- (19).Das D; Eurenius KP; Billings EM; Sherwood P; Chatfield DC; Hodoscek M; Brooks BR Optimization of quantum mechanical molecular mechanical partitioning schemes: Gaussian delocalization of molecular mechanical charges and the double link atom method. J. Chem. Phys. 2002, 117, 10534–10547. [Google Scholar]
- (20).Freindorf M; Gao JL Optimization of the Lennard-Jones parameters for a combined ab initio quantum mechanical and molecular mechanical potential using the 3–21G basis set. J. Comput. Chem. 1996, 17, 386–395. [Google Scholar]
- (21).Riccardi D; Li G; Cui Q The importance of van der Waals interactions in QM/MM simulations. J. Phys. Chem. B 2004, 108, 6467–6478. [DOI] [PubMed] [Google Scholar]
- (22).Roston D; Cui Q QM/MM Analysis of Transition States and Transition State Analogues in Metalloenzymes. Methods in Enzymol. 2016, 577, 213–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Meier K; Thiel W; van Gunsteren WF, On the effect of a variation of the force field, spatial boundary condition and size of the QM region in QM/MM MD simulations. J. Comp. Chem. 2012, 33, 363–378. [DOI] [PubMed] [Google Scholar]
- (24).Liao RZ; Thiel W Convergence in the QM-Only and QM/MM Modeling of Enzymatic Reactions: A Case Study for Acetylene Hydratase. J. Comp. Chem. 2013, 34, 2389–2397. [DOI] [PubMed] [Google Scholar]
- (25).Karelina M; Kulik HJ Systematic Quantum Mechanical Region Determination in QM/MM Simulation. J. Chem. Theory Comput 2017, 13, 563–576. [DOI] [PubMed] [Google Scholar]
- (26).Brunken C; Reiher M Automated Construction of Quantum–Classical Hybrid Models. J. Chem. Theory Comput. 2021, 17, 3797–3813. [DOI] [PubMed] [Google Scholar]
- (27).Watanabe HC; Cui Q Quantitative analysis and correction of QM/MM boundary artifacts in adaptive QM/MM methods. J. Chem. Theory Comput. 2019, 15, 3917–3928. [DOI] [PubMed] [Google Scholar]
- (28).Sumowski CV; Ochsenfeld C A Convergence Study of QM/MM Isomerization Energies with the Selected Size of the QM Region for Peptidic Systems. J. Phys. Chem. A 2009, 113, 11734–11741. [DOI] [PubMed] [Google Scholar]
- (29).Hu L; Söderhjelm P; Ryde U On the Convergence of QM/MM Energies. J. Chem. Theory Comput 2011, 7, 761–777. [DOI] [PubMed] [Google Scholar]
- (30).Roßbach S; Ochsenfeld C Influence of Coupling and Embedding Schemes on QM Size Convergence in QM/MM Approaches for the Example of a Proton Transfer in DNA. J. Chem. Theory Comput. 2017, 13, 1102–1107. [DOI] [PubMed] [Google Scholar]
- (31).Flaig D; Beer M; Ochsenfeld C Convergence of Electronic Structure with the Size of the QM Region: Example of QM/MM NMR Shieldings. J. Chem. Theory Comput. 2012, 8, 2260–2271. [DOI] [PubMed] [Google Scholar]
- (32).Hartman JD; Neubauer TJ; Caulkins BG; Mueller LJ; Beran GJO Converging nuclear magnetic shielding calculations with respect to basis and system size in protein systems. J. Biomol. NMR 2015, 62, 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Isborn CM; Götz AW; Clark MA; Walker RC; Martínez TJ Electronic Absorption Spectra from MM and ab Initio QM/MM Molecular Dynamics: Environmental Effects on the Absorption Spectrum of Photoactive Yellow Protein. J. Chem. Theory Comput. 2012, 8, 5092–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fuller III J; Wilson TR; Eberhart ME; Alexandrova AN Charge Density in Enzyme Active Site as a Descriptor of Electrostatic Preorganization. J. Chem. Info. Model. 2019, 59, 2367–2373. [DOI] [PubMed] [Google Scholar]
- (35).Kulik HJ; Zhang J; Klinman JP; Martínez TJ How Large Should the QM Region Be in QM/MM Calculations? The Case of Catechol O-Methyltransferase. J. Phys. Chem. B 2016, 120, 11381–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mehmood R; Kulik HJ Both Configuration and QM Region Size Matter: Zinc Stability in QM/MM Models of DNA Methyltransferase. J. Chem. Theory Comput. 2020, 16, 3121–3134. [DOI] [PubMed] [Google Scholar]
- (37).Ryde U How Many Conformations Need To Be Sampled To Obtain Converged QM/MM Energies? The Curse of Exponential Averaging. J. Chem. Theory Comput 2017, 13, 5745–5752. [DOI] [PubMed] [Google Scholar]
- (38).Das S; Nam K; Major DT Rapid Convergence of Energy and Free Energy Profiles with Quantum Mechanical Size in Quantum Mechanical–Molecular Mechanical Simulations of Proton Transfer in DNA. J. Chem. Theory Comput. 2018, 14, 1695–1705. [DOI] [PubMed] [Google Scholar]
- (39).Kästner J; Senn HM; Thiel S; Otte N; Thiel W QM/MM Free-Energy Perturbation Compared to Thermodynamic Integration and Umbrella Sampling: Application to an Enzymatic Reaction. J. Chem. Theo. Comput. 2006, 2, 452–461. [DOI] [PubMed] [Google Scholar]
- (40).Hudson PS; Woodcock HL; Boresch S Use of Interaction Energies in QM/MM Free Energy Simulations. J. Chem. Theo. Comput 2019, 15, 4632–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Giese TJ; York DM Development of a Robust Indirect Approach for MM → QM Free Energy Calculations That Combines Force-Matched Reference Potential and Bennett’s Acceptance Ratio Methods. J. Chem. Theo. Comput. 2019, 15, 5543–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Pohorille A; Jarzynski C; Chipot C Good Practices in Free-Energy Calculations. J. Phys. Chem. B 2010, 114, 10235–10253. [DOI] [PubMed] [Google Scholar]
- (43).Kulik HJ Large-scale QM/MM free energy simulations of enzyme catalysis reveal the influence of charge transfer. Phys. Chem. Chem. Phys. 2018, 20, 20650–20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jindal G; Warshel A Exploring the Dependence of QM/MM Calculations of Enzyme Catalysis on the Size of the QM Region. J. Phys. Chem. B 2016, 120, 9913–9921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Patra N; Ioannidis EI; Kulik HJ Computational Investigation of the Interplay of Substrate Positioning and Reactivity in Catechol O-Methyltransferase. PLOS ONE 2016, 11, e0161868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gaus M; Cui Q; Elstner M DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB). J. Chem. Theory Comput 2011, 7, 931–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Im W; Bernèche S; Roux B Generalized solvent boundary potential for computer simulations. J. Chem. Phys. 2001, 114, 2924–2937. [Google Scholar]
- (48).Schaefer P; Riccardi D; Cui Q Reliable treatment of electrostatics in combined QM/MM simulation of macromolecules. J. Chem. Phys. 2005, 123, 014905. [DOI] [PubMed] [Google Scholar]
- (49).Zhang J; Kulik HJ; Martinez TJ; Klinman JP Mediation of donor–acceptor distance in an enzymatic methyl transfer reaction. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 7954–7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chandler D Statistical-mechanics of isomerization dynamics in liquids and transition-state approximation. J. Chem. Phys. 1978, 68, 2959–2970. [Google Scholar]
- (51).Major DT; Gao J An Integrated Path Integral and Free-Energy Perturbation-Umbrella Sampling Method for Computing Kinetic Isotope Effects of Chemical Reactions in Solution and in Enzymes. J. Chem. Theo. Comput. 2007, 3, 949–960. [DOI] [PubMed] [Google Scholar]
- (52).Bergsma JP; Gertner BJ; Wilson KR; Hynes JT Molecular dynamics of a model SN2 reaction in water. J. Chem. Phys. 1987, 86, 1356. [Google Scholar]
- (53).Castillo R; Roca M; Soriano A; Moliner V; Tuñón I Using Grote-Hynes Theory To Quantify Dynamical Effects on the Reaction Rate of Enzymatic Processes. The Case of Methyltransferases. J. Phys. Chem. B 2008, 112, 529–534. [DOI] [PubMed] [Google Scholar]
- (54).Lu X; Cui Q Charging free energy calculations using the Generalized Solvent Boundary Potential (GSBP) and periodic boundary condition: a comparative analysis using ion solvation and reduction potential in proteins,. J. Phys. Chem. B 2013, 117, 2005–2018. [DOI] [PubMed] [Google Scholar]
- (55).Cohen AJ; Mori-Sanchez P; Yang WT Challenges for Density Functional Theory. Chem. Rev. 2012, 112, 289–320. [DOI] [PubMed] [Google Scholar]
- (56).Elstner M; Porezag D; Jungnickel G; Elsner J; Haugk M; Frauenheim T; Suhai S; Seifert G Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar]
- (57).Gaus M; Cui Q; Elstner M Density functional tight binding: application to organic and biological molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 49–61. [Google Scholar]
- (58).Kussmann J; Ochsenfeld C Pre-selective screening for matrix elements in linear-scaling exact exchange calculations. J. Chem. Phys. 2013, 138, 134114. [DOI] [PubMed] [Google Scholar]
- (59).Laqua H; Thompson TH; Kussmann J; Ochsenfeld C Highly Efficient, Linear-Scaling Seminumerical Exact-Exchange Method for Graphic Processing Units. J. Chem. Theory Comput. 2020, 16, 1456–1468. [DOI] [PubMed] [Google Scholar]
- (60).Kussmann J; Laqua H; Ochsenfeld C Highly Efficient Resolution-of-Identity Density Functional Theory Calculations on Central and Graphics Processing Units. J. Chem. Theory Comput. 2021, 17, 1512–1521. [DOI] [PubMed] [Google Scholar]
- (61).Rutherford K; Trong IL; Stenkamp RE; Parson WW Crystal structures of human 108V and 108M catechol O-methyltransferase. J. Mol. Biol. 2008, 380, 120–130. [DOI] [PubMed] [Google Scholar]
- (62).Brooks BR; III CLB; Mackerell AD; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).MacKerell AD; Bashford D; Bellott M; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiórkiewicz-Kuczera J; Yin D; Karplus M All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (64).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory. Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- (67).Neria E; Fischer S; Karplus M Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar]
- (68).Søndergaard CR; Olsson MHM; Rostkowski M; Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- (69).Olsson MHM; Søndergaard CR; Rostkowski M; Jensen JH PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [DOI] [PubMed] [Google Scholar]
- (70).Gaus M; Goez A; Elstner M, Parametrization and Benchmark of DFTB3 for Organic Molecules. J. Chem. Theory Comput. 2013, 9, 338–354. [DOI] [PubMed] [Google Scholar]
- (71).Lu X; Gaus M; Elstner M; Cui Q Parametrization of DFTB3/3OB for Magnesium and Zinc for Chemical and Biological Applications. J. Phys. Chem. B 2014, 119, 1062–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Cui Q; Elstner M Density Functional Tight Binding: Values of Semi-empirical Methods in an ab initio Era. Phys. Chem. Chem. Phys. 2014, 16, 14368–14377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Roston D; Demapan D; Cui Q Extensive Free Energy Simulations Identify Water as the Base in Nucleotide Addition by DNA Polymerase. Proc. Natl. Acad. Sci. USA 2019, 116, 25048–25056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Roston D; Lu X; Fang D; Demapan D; Cui Q Analysis of Phosphoryl Transfer Enzymes with QM/MM Free Energy Simulations. Methods in Enzymol. 2019, 607, 53–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Saez DA; Vogt-Geisse S; Inostroza-Rivera R; Kubař T; Elstner M; Toro-Labbé A; Vöhringer-Martinez E The effect of the environment on the methyl transfer reaction mechanism between trimethylsulfonium and phenolate. Phys. Chem. Chem. Phys. 2016, 18, 24033–24042. [DOI] [PubMed] [Google Scholar]
- (76).Roca M; Martí S; Andrés J; Moliner V; Tuñón I; Bertrán J; Williams IH Theoretical Modeling of Enzyme Catalytic Power: Analysis of “Cratic” and Electrostatic Factors in CatecholO-Methyltransferase. J. Am. Chem. Soc. 2003, 125, 7726–7737. [DOI] [PubMed] [Google Scholar]
- (77).Grimme S; Antony J; Ehrlich S; Krieg H A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- (78).Christensen AS; Kubar T; Cui Q; Elstner M, Semi-empirical Quantum Mechanical Methods for Non-covalent Interactions for Chemical and Biochemical Applications. Chem. Rev. 2016, 116, 5301–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Roston D; Demapan D; Cui Q Leaving Group Ability Observably Affects Transition State Structure in a Single Enzyme Active Site. J. Am. Chem. Soc. 2016, 138, 7386–7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Stote RH; States DJ; Karplus M On the treatment of electrostatic interactions in biololecular simulation. Journal of Chimie Physique 1991, 88, 2419–2433. [Google Scholar]
- (81).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- (82).Darden T; York D; Pedersen L Particle mesh Ewald: An N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- (83).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Riccardi D; Schaefer P; Cui Q pKa calculations in solution and proteins with QM/MM free energy perturbation simulations. J. Phys. Chem. B 2005, 109, 17715–17733. [DOI] [PubMed] [Google Scholar]
- (85).Nam K; Gao JL; York DM An efficient linear-scaling Ewald method for long-range electrostatic interactions in combined QM/MM calculations. J. Chem. Theory Comput. 2005, 1, 2–13. [DOI] [PubMed] [Google Scholar]
- (86).The PLUMED consortium, Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. [DOI] [PubMed] [Google Scholar]
- (87).Barducci A; Bussi G; Parrinello M Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [DOI] [PubMed] [Google Scholar]
- (88).Bonomi M; Branduardi D; Bussi G; Camilloni C; Provasi D; Raiteri P; Dona-dio D; Marinelli F; Pietrucci F; Broglia RA; Parrinello M PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar]
- (89).Raiteri P; Laio A; Gervasio FL; Micheletti C; Parrinello M Efficient Reconstruction of Complex Free Energy Landscapes by Multiple Walkers Metadynamics†. J. Phys. Chem. B 2006, 110, 3533–3539. [DOI] [PubMed] [Google Scholar]
- (90).Bussi G; Laio A Using metadynamics to explore complex free-energy landscapes. Nat. Rev. Phys. 2020, 2, 200–212. [Google Scholar]
- (91).Bolhuis PG; Chandler D; Dellago C; Geissler PL Transition path sampling: Throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002, 53, 291–318. [DOI] [PubMed] [Google Scholar]
- (92).Pan AC; Chandler D Dynamics of Nucleation in the Ising Model†. J. Phys. Chem. B 2004, 108, 19681–19686. [Google Scholar]
- (93).Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [DOI] [PubMed] [Google Scholar]
- (94).Bernstein BE; Mikkelsen TS; Xie X; Kamal M; Huebert DJ; Cuff J; Fry B; Meissner A; Wernig M; Plath K; Jaenisch R; Wagschal A; Feil R; Schreiber SL; Lander ES A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [DOI] [PubMed] [Google Scholar]
- (95).Lachman HM; Papolos DF; Saito T; Yu Y-M; Szumlanski CL; Weinshilboum RM Human catechol-O-methyltransferase pharmacogenetics: description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics 1996, 6, 243–250. [DOI] [PubMed] [Google Scholar]
- (96).Hegazi MF; Borchardt RT; Schowen RL. alpha.-Deuterium and carbon-13 isotope effects for methyl transfer catalyzed by catechol O-methyltransferase. SN2-like transition state. J. Am. Chem. Soc. 1979, 101, 4359–4365. [DOI] [PubMed] [Google Scholar]
- (97).Cao Y; Chen Z-J; Jiang H-D; Chen J-Z Computational Studies of the Regioselectivities of COMT-Catalyzed Meta-/Para-O Methylations of Luteolin and Quercetin. J. Phys. Chem. B 2014, 118, 470–481. [DOI] [PubMed] [Google Scholar]
- (98).Egan MF; Goldberg TE; Kolachana BS; Callicott JH; Mazzanti CM; Straub RE; Goldman D; Weinberger DR Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 6917–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Ruggiero GD; Williams IH; Roca M; Moliner V; Tuñón I QM/MM Determination of Kinetic Isotope Effects for COMT-Catalyzed Methyl Transfer Does Not Support Compression Hypothesis. J. Am. Chem. Soc. 2004, 126, 8634–8635. [DOI] [PubMed] [Google Scholar]
- (100).Saez DA; Zinovjev K; Tuñón I; Vöhringer-Martinez E Catalytic Reaction Mechanism in Native and Mutant Catechol-O-methyltransferase from the Adaptive String Method and Mean Reaction Force Analysis. J, Phys. Chem. B 2018, 122, 8861–8871. [DOI] [PubMed] [Google Scholar]
- (101).Roca M; Williams IH Transition-State Vibrational Analysis and Isotope Effects for COMT- Catalyzed Methyl Transfer. J. Am. Chem. Soc. 2020, 142, 15548–15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Zhang J; Klinman JP Enzymatic Methyl Transfer: Role of an Active Site Residue in Generating Active Site Compaction That Correlates with Catalytic Efficiency. J. Am. Chem. Soc. 2011, 133, 17134–17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Gaus M; Chou CP; Witek H; Elstner M Automatized Parametrization of SCC-DFTB Repulsive Potentials: Application to Hydrocarbons. J. Phys. Chem. A 2009, 113, 11866–11881. [DOI] [PubMed] [Google Scholar]
- (104).Roston D; Cui Q Substrate and Transition State Binding in Alkaline Phosphatase Analyzed by Computation of Oxygen Isotope Effects. J. Am. Chem. Soc. 2016, 138, 11946–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Kanaan N; Pernía JJR; Williams IH QM/MM simulations for methyl transfer in solution and catalysed by COMT: ensemble-averaging of kinetic isotope effects. ChemComm 2008, 6114. [DOI] [PubMed] [Google Scholar]
- (106).Gaus M; Lu X; Elstner M; Cui Q Parameterization of DFTB3/3OB for Sulfur and Phosphorus for Chemical and Biological Applications. J. Chem. Theo. Comput. 2014, 10, 1518–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Yang Z; Mehmood R; Wang M; Qi HW; Steeves AH; Kulik HJ Revealing quantum mechanical effects in enzyme catalysis with large-scale electronic structure simulation. React. Chem. Eng. 2019, 4, 298–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Marenich AV; Jerome SV; Cramer CJ; Truhlar DG Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory. Comput. 2012, 8, 527–541. [DOI] [PubMed] [Google Scholar]
- (109).König G; Hudson PS; Boresch S; Woodcock HL Multiscale free energy simulations: An efficient method for connecting classical MD simulations to QM or QM/MM free energies using Non-Boltzmann Bennett reweighting schemes. J. Chem. Theory Comput. 2014, 10, 1406–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Wang J; Liu W; Li P; Mo Y; Hu W; Zheng J; Pan X; Shao Y; Mei Y Accelerated Computation of Free Energy Profile at Ab Initio Quantum Mechanical/Molecular Mechanics Accuracy via a Semiempirical Reference Potential. 4. Adaptive QM/MM. J. Chem. Theory Comput 2021, 17, 1318–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Shen L; Yang WT Molecular Dynamics Simulations with Quantum Mechanics/Molecular Mechanics and Adaptive Neural Networks. J. Chem. Theory. Comput 2018, 14, 1442–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Pan X; Yang J; Van R; Epifanovsky E; Ho J; Huang J; Pu J; Mei Y; Nam K; Shao Y Machine-Learning-Assisted Free Energy Simulation of Solution-Phase and Enzyme Reactions. J. Chem. Theory Comput. 2021, 17, 5745–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Chodera JD; Mobley DL Entropy-Enthalpy Compensation: Role and Ramifications in Biomolecular Ligand Recognition and Design. Annu. Rev. Biophys. 2013, 42, 121–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Sharp K Entropy–enthalpy compensation: Fact or artifact? Protein Sci. 2001, 10, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Olsson TSG; Ladbury JE; Pitt WR; Williams MA Extent of enthalpy-entropy compensation in protein-ligand interactions. Protein Sci. 2011, 20, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Pinnick ER; Calderon CE; Rusnak AJ; Wang F Achieving fast convergence of ab initio free energy perturbationcalculations with the adaptive force-matching method. Theo. Chem. Acc. 2012, 131, 1146. [Google Scholar]
- (117).Riccardi D; Cui Q pKa analysis for the zinc-bound water in Human Carbonic Anhydrase II: benchmark for ”multi-scale” QM/MM simulations and mechanistic implications. J. Phys. Chem. A 2007, 111, 5703–5711. [DOI] [PubMed] [Google Scholar]
- (118).Ghosh N; Cui Q pKa of residue 66 in Staphylococal nuclease: insights from QM/MM simulations with conventional sampling. J. Phys. Chem. B 2008, 112, 8387–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Mezey PG The Holographic Electron Density Theorem and Quantum Similarity Measures. Mol. Phys. 1999, 96, 169–178. [Google Scholar]
- (120).Mori-Sánchez P; Cohen AJ; Yang W Many-electron self-interaction error in approximate density functionals. J. Chem. Phys. 2006, 125, 201102. [DOI] [PubMed] [Google Scholar]
- (121).Bao JL; Gagliardi L; Truhlar DG Self-Interaction Error in Density Functional Theory: An Appraisal. J. Phys. Chem. Lett 2018, 9, 2353–2358. [DOI] [PubMed] [Google Scholar]
- (122).Kaminski GA; Stern HA; Berne BJ; Friesner RA; Cao YXX; Murphy RB; Zhou RH; Halgren TA Development of a polarizable force field for proteins via ab initio quantum chemistry: First generation model and gas phase tests. J. Comp. Chem. 2002, 23, 1515–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Yang Z; Hajlasz N; Steeves AH; Kulik HJ Quantifying the Long-Range Coupling of Electronic Properties in Proteins with Ab Initio Molecular Dynamics. Chem. Methods 2021, 1, 362–373. [Google Scholar]
- (124).Qi HW; Kulik HJ Evaluating Unexpectedly Short Non-covalent Distances in X-ray Crystal Structures of Proteins with Electronic Structure Analysis. J. Chem. Info. Model. 2019, 59, 2199–2211. [DOI] [PubMed] [Google Scholar]
- (125).Zhou SM; Wang L Unraveling the structural and chemical features of biological short hydrogen bonds. Chem. Sci. 2019, 10, 7734–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Goyal P; Qian HJ; Irle S; Lu X; Roston D; Mori T; Elstner M; Cui Q Feature Article: Molecular Simulation of Water and Hydration Effects in Different Environments: Challenges and Developments for DFTB Based Models. J. Phys. Chem. B 2014, 118, 11007–11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Cui Q; Karplus M QM/MM Studies of the Triosephosphate Isomerase (TIM) Catalyzed Reactions: Verification of Methodology and Analysis of the Reaction Mechanisms. J. Phys. Chem. B 2002, 106, 1768–1798. [Google Scholar]
- (128).Yagi K; Ito S; Sugita Y Exploring the Minimum-Energy Pathways and Free-Energy Profiles of Enzymatic Reactions with QM/MM Calculations. J. Phys. Chem. B 2021, 125, 4701–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.