

Abstract
During tumorigenesis, DNA mutations in protein coding sequences can alter amino acid sequences which can change the structures of proteins. While the 3D structure of mutated proteins has been studied with atomic resolution, the precise impact of somatic mutations on the 3D proteome during malignant transformation remains unknown because methods to reveal in vivo protein structures in high through put are limited. Here, we measured the accessibility of the lysine ε-amine for chemical modification across proteomes using covalent protein painting (CPP) to indirectly determine alterations in the 3D proteome. CPP is a novel, high throughput quantitative mass spectrometric method that surveyed a total of 8,052 lysine sites across the 60 cell lines of the well-studied anti-cancer cell line panel (NCI60). Overall, 5.2 structural alterations differentiated any cancer cell line from the other 59. Structural aberrations in 98 effector proteins correlated with the select presence of 90 commonly mutated proteins in the NCI60 cell line panel, suggesting that different tumor genotypes reshape a limited set of effector proteins. We searched our dataset for druggable conformational aberrations and identified 49 changes in the cancer conformational landscape that correlated with the growth inhibition profiles of 300 drug candidates out of 50,000 small molecules. We found that alterations in heat shock proteins are key predictors of anti-cancer drug efficacy, which implies that the proteostasis network may have a general but hitherto unrecognized role in maintaining malignancy. Individual lysine sites may serve as biomarkers to guide drug selection or may be directly targeted for anti-cancer drug development.
Keywords: Structural proteomics, chemical footprinting, protein misfolding, conformational diagnostics, cancer diagnostics
Graphical Absract
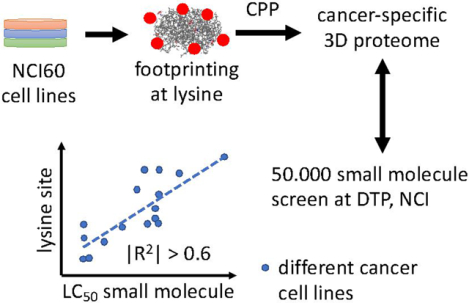
Introduction
Somatic mutations drive tumorigenesis by altering gene expression or protein function1, 2. Tumorigenic cells face the daunting challenge of propagating mutated proteins that are altered in 3D structure and that remodel protein-protein and protein-biomolecule interactions of non-mutated proteins. Today, cancer-specific abnormalities in protein abundance have been extensively studied with large scale proteomics3, 4 which revealed that heat shock factor 1 (HSF1) is often overexpressed in aggressive cancers5, 6. HSF1 is a master regulator of the proteostasis network that oversees protein folding and degradation. Its upregulation in aggressive cancers is likely due to the increasing number of proteins that might misfold as a result of the mutation burden. However, the extent of structural alteration in a proteome of transformed cells is not well understood. To gain a deeper understanding of the role of protein conformational changes in tumorigenesis, we surveyed the conformational landscape of the proteome in living cells7 (aka, the 3D proteome) with a novel high throughput chemical footprinting technique.
Protein structure determination is typically performed ex vivo and often requires highly purified proteins or protein complexes; consequently, these methods are not suitable for measuring conformational aberrations on a proteome scale. Recently, we developed Covalent Protein Painting (CPP), a method to measure conformational states of proteins in living cells8. CPP uses chemical methylation in living cells to quantify and compare the relative accessibility of the lysine ε-amine in proteins. Similar approaches have been used to measure protein unfolding9 or to determine lysine site accessibility of purified proteins and protein complexes in vitro10, 11. Unlike methods that rely on biophysical properties of proteins (like thermostability12), CPP uses a small covalent modification (dimethylation) to measure alterations in protein surface accessibility in the 3D proteome of living cells. Most lysine sites in a 3D proteome are accessible for chemical labeling. However, the chemical reactivity of the ε-amine of a lysine can be sterically limited by protein conformation or by the proximity of interacting proteins. While CPP quantifies the chemical reactivity of a lysine site and compares it between experimental conditions or cell lines, CPP remains agnostic about the molecular cause for the 3D rearrangements. Thus, CPP measures and quantifies a 3D proteome efficiently but does not reveal the reason for alterations in chemical accessibility.
In this study, we used CPP to quantify structural changes in the 3D proteome of different cancer cell lines. We measured the conformational landscapes of 60 different cancer cell lines and showed that mutations in cancer driver genes matched a limited number of conformational aberrations in the 3D proteome. In addition, conformational aberrations were correlated with the growth inhibitory effect of small molecules, demonstrating that the differential accessibility of distinct lysine sites for covalent modification may indicate potential target sites for anti-cancer drug development.
Materials and Methods
CPP labeling of cell lines
In the CPP strategy, solvent exposed ε-amines of lysine residues in proteins are dimethylated. Lysine residues that are sterically inaccessible due to protein-protein or biomolecule-protein interactions or altered protein conformation remain unmodified8. After denaturation and protein digestion, lysine residues that were initially inaccessible are exposed and are tagged with isotopically distinct dimethyl groups in a second labeling reaction. Isotope-tagged peptides are subsequently identified and quantified in an unbiased and high-throughput bottom-up proteomic experiment13. The intensity ratio of the isotope labeled lysine sites reports the relative proportion of molecules in which a lysine site was inaccessible for covalent modification, and the differences in relative inaccessibility of lysine sites in different tumors indicate structural changes in the 3D proteome that may be tumor specific. Cell culture dishes with adherent cells were placed on ice (3 °C) and cells were washed once in phosphate buffered saline (PBS, +Mg2+, +Ca2+) and then incubated in PBS with labeling reagents for 15 min at 3 °C as previously described8. Alternatively, in biological triplicate experiments, cryopreserved cell pellets of 107 cells (NCI60 cell line panel) were thawed on ice in phosphate buffered saline (PBS) in the presence of labeling reagents. Thawing cells were carefully resuspended in labeling buffer by repeated aspiration followed by periodic mixing during a 15 min incubation (3 °C). Samples were further processed as described in8. Surface accessible ε-amines of lysine sites were heavy-labeled with 13CD2O and NaBD3CN. Following digestion with Chymotrypsin, peptide N-termini and newly accessible lysine residues were dimethylated with CH2O and NaBD3CN. In the second reaction, the Schiff base that is formed between a lysine site and formaldehyde was reduced with deuteride anions (NaBD3CN) in order to differentiate chemically introduced dimethyl labels that include deuterium from natural dimethylation groups that predominantly contain hydrogen.
Liquid Chromatography and Mass Spectrometry
Peptide samples were loaded on EVPOSEP tips according to the manufacturer’s recommendations. Peptides were reversed-phase separated (Reprosil C18, 3 μm, 120 Å, 15 cm, i.d. 100 μm, PepSep) on an EVOSEP UHPLC system with a 45 min gradient (buffer B, 0 % to 50 % acetonitrile, 0.1 % formic acid) prior to electrospray ionization. Survey mass spectra and fragment ion spectra were acquired on an Orbitrap Lumos mass spectrometer (ThermoFisher Scientific) in data dependent mode and parallelizable time for ion injections. Survey mass spectra were recorded in the orbitrap with R = 120,000 at m/z = 200 with an automatic gain control (AGC) of 4*105 counts. Precursors were selected with a 10 ppm mass window (excluding isotopes) and a minimal abundance of 5*103 counts, and precursor ions were isolated with the quadrupole at a mass window of 1.6 m/z based on a charge +2 to +7 with an AGC of 2*104 ions. Isolated precursor ions were fragmented with collision-induced ionization at 35 % collisional energy, Q = 0.25, and 10 ms fragmentation time, and fragment ions were detected in the linear ion trap (LTQ). Mass spectrometric data was analyzed with IP2 (Integrated Proteomics). Fragment ion spectra were searched with the human curated Uniprot database (release 1.2018) assuming no enzyme specificity, and subsequently filtered for peptide identifications with at least one Chymotrypsin-specific endoproteolytic cleavage and one lysine in the peptide sequence. The ratio of light over heavy dimethylated peptide isomers was determined with Census (Integrated Proteomics) and the final surface accessibility for each lysine site was calculated in ProteinClusterQuant (PCQ)13.
Pearson correlation of protein sites within the NCI60 cell line panel
Surface accessibility ratios from 8,066 different protein sites acquired in 60 different cell lines were analyzed. When calculating Pearson correlations, ratio values of ±Infinity were converted to the maximum or minimum non-infinity values in the whole dataset and modified with a randomly selected offset value between 0 and 0.001 to avoid divisions by zero. Pairwise comparisons displayed in Figure 1 were assembled in a comparison matrix that included only protein sites that correlated with at least 4 other protein sites (Pearson’s correlation, |p| > 0.5), and the matrix of protein sites was hierarchically clustered using Euclidean distance and “complete linkage”. STRING functional proteome analysis of protein clusters in the comparison matrix was used to call significantly enriched GO terms.
Figure 1: Alterations in lysine site accessibility in different proteins that were observed simultaneously in the NCI60 cell line panel.
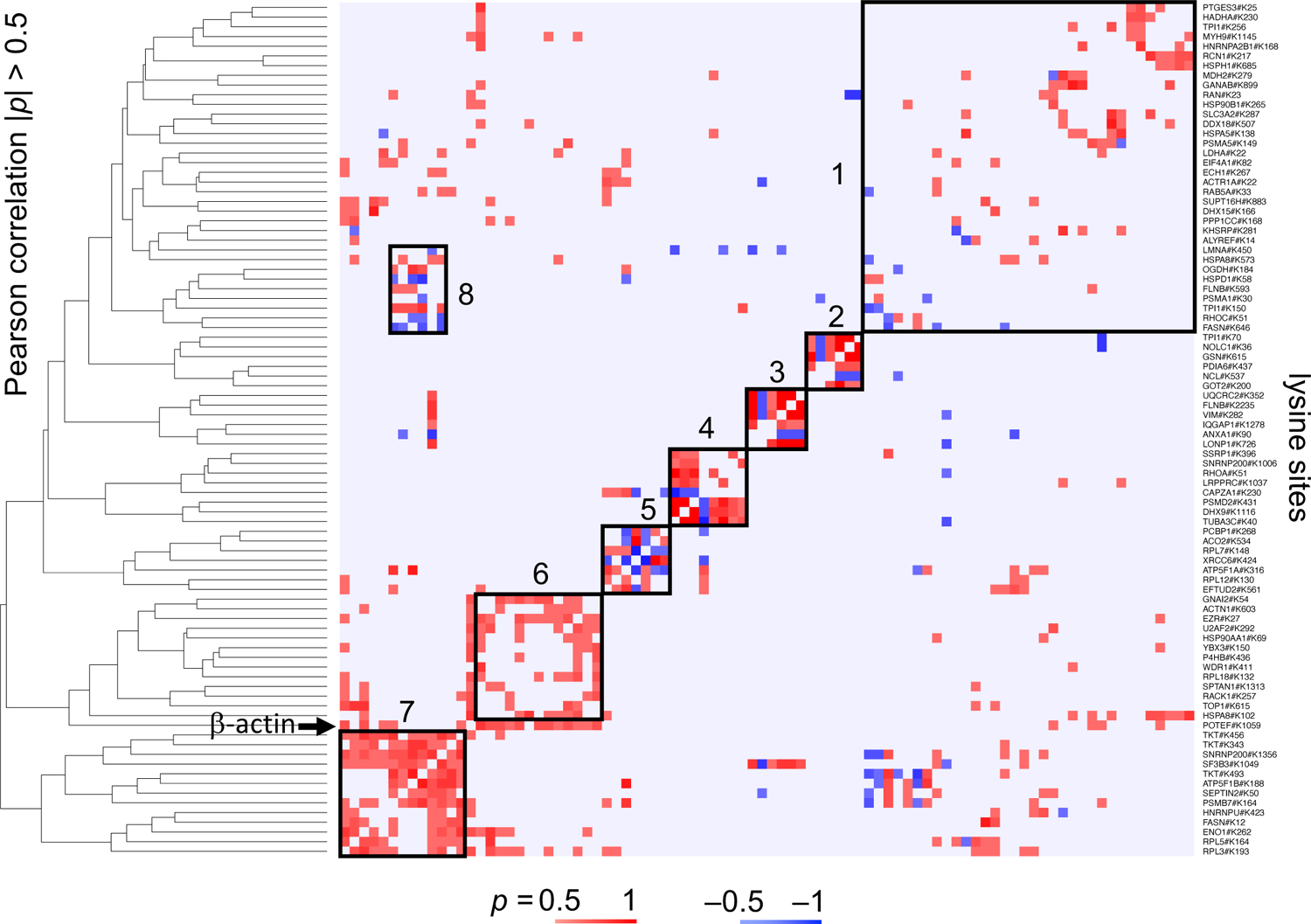
The heat map displays Pearson’s correlations between 88 lysine sites across all 60 cell lines. Sites were clustered by Euclidian distance based on the absolute value of the Pearson’s correlation. Red indicates positive and blue negative correlation. Protein names are listed on the right whereas the dendrogram based on the Euclidian distance is shown on the left and top of the diagram. GO terms for each group included protein folding and unfolded protein response (group 1, average node degree ⟨k⟩ = 6.4), oxidoreductase activity (group 2, ⟨k⟩ = 1.0), epithelial cell differentiation (group 3, ⟨k⟩ = 0.7), RNA helicase activity and ssDNA binding (group 4, ⟨k⟩ = 0.5) and mRNA metabolic processes (group 5, ⟨k⟩ = 1.2), cellular response to stress (group 6, ⟨k⟩ = 4.0), and mRNA metabolism (group 7, ⟨k⟩ = 2.8). Group 8 included a subset of proteins that were part of group 1 and 7 and matched GO terms localization to organelle, transmembrane transport, and chaperone-mediated protein folding (⟨k⟩ = 1.8).
Pearson correlation to drug growth inhibition
Using standard Pearson’s correlation, sites identified and quantified with at least 10 different measurements in at least 10 different cell lines were correlated with the LC50 values of small molecules reported in the chemical library screen of the DCTD at NCI (>50,000 compounds in total). The minimal variance of drug measurements was set to 0.5, and ±infinity values (fully accessible or inaccessible) were shifted to the minimal non-infinity value across all cell lines (Rmin = −0.5 or Rmax = +0.5). All drug-site correlations with |p| ≥ 0.6 were retained.
Results
Cancer Conformational Landscapes
We applied CPP to the 60 cell lines of the NCI60 cell line panel (NCI DCTD) to identify cancer specific alterations in the 3D proteome across a wide range of different types of cancers. A total of 275,536 peptide spectrum matches (PSM) were quantified, covering 8,052 lysine sites in 8,493 peptides and 3,308 protein groups with at least one peptide per site (Figure S1). When comparing individual cancer cell lines pairwise, an average of 5.2 significant changes in lysine site accessibility distinguished one cell line from any other single cell line (Student’s t-test, Benjamini-Hochberg (BH) corrected, q ≤ 0.05, Table S1). An average of 312 alterations differentiated one cell line from the other 59, and the average number of lysine sites that were identified in any two cell lines simultaneously was 162.5±14.8 lysine sites (average overlap). On average, 264.2 of 312 (85 %) lysine site alterations uniquely set any cancer cell line apart from all others. Overall, 15 % of the significantly altered lysine sites differed in more than one of the 59 pairwise comparisons per cell line. While the average overlap of detected lysine sites between cell lines was constant (162.5±14.8 overall), HCT-15 cells differed the most (847 lysine sites), followed by SK-MEL-5 melanoma (647) and HS-578T breast cancer cells (552) from the other 59 cell lines, respectively.
The 60 cancer cell lines of the NCI60 cell line panel originated from 9 different tissues and cell types (renal, non-small cells of the lung, breast, bone marrow, colon, ovary, melanocyte, prostate, and central nervous system (CNS)). When cell lines were pooled by tissue of origin and compared, conformational alterations were characteristic of the tissue of origin (Table S1). Non-adherent leukemia differed the most from any solid tissue tumor, with an average change in accessibility for covalent modification of 13.6 lysine sites. Breast, CNS, colon, melanoma, lung, ovarian, prostate, and renal tissue-derived tumors were distinguishable by alterations in accessibility of 4.25 lysine sites, on average. Tumor of prostate origin included the fewest lysine sites with differentially altered accessibility. Prostate, ovarian and lung tumors were all of epithelial cell origin and were indistinguishable by tissue origin.
Alterations in accessibility for covalent modification were covariant in a subset of lysine sites across several cancer cell lines. We found 88 lysine sites that differed in accessibility for covalent modification in ≥ 30 cell lines. When hierarchically clustered, these sites segregated into 8 groups with up to 13 lysine sites per group (Pearson’s correlation, |p| ≥ 0.5, Figure 1). Each group weakly associated with different Gene Ontology terms (GO, p < 0.05) which indicated that the functions of proteins within one group only partially overlapped. The lysine site ACTB#K359 in β-actin was an exception. ACTB#K359 was covariant with 23 of the 88 proteins that differed in ≥ 30 cell lines. The functional annotation of the 23 proteins was enriched for organonitrogen compound metabolism and regulation of apoptosis (average node degree ⟨k⟩ = 6.7). While the analysis might include pairs of lysine sites that occurred by chance, proteins with covariant alterations in lysine site accessibility clustered in small groups across all 60 cancer cell lines but displayed only limited functional connectivity.
Tumorigenesis Shapes the 3D Proteome
Conformational changes in the 3D proteome of cancer cells are a consequence of the pattern and time of acquisition of individual somatic mutations that do not result in lethal loss of function. Each of the NCI60 cancer cell lines evolved independently and comprises a distinct pattern of somatic mutations that alter the protein coding sequences in a total of 461 genes (COSMIC transcriptome analysis, excluding gene amplifications or deletions14). A proteogenomic study identified 4,771 protein sequence variants in the NCI60 cell lines, of which > 250 sequence variations were annotated in protein databases. This indicates a high level of proteoform variation in the cell lines that remains unexplained15. CPP data provides a measurement of altered protein conformation taking somatic mutations and other protein sequence variations indirectly into account. Out of 461 somatically mutated genes, only the protein product Calreticulin (of the gene CALR) was altered in lysine site accessibility and somatically mutated in the same cancer cell line. Lysine site CALR#K185 was significantly increased in accessibility in D244G-mutated Calreticulin (P27797) in HCT-116 cells. However, it remains to be shown that the altered lysine site accessibility is a caused by the point mutation D244G in Calreticulin, and simiar high accessibility of CALR#K185 was observed in additional cell lines in the absence of somatic mutations to the Calreticulin protein. We quantified lysine sites in an additional 132 protein products of the 461 somatically mutated genes, and 6 of the 132 proteins harbored at least one lysine site with an altered accessibility for covalent labeling in at least one of the 60 cell lines (Table S2). However, except for mutated Calreticulin, altered lysine site accessibility did not coincide with a somatic mutation. While the depth of 3D proteomic analysis with CPP is limited by incomplete coverage of the estimated 25,000 lysine sites (5,000 different proteins assumed) in the cellular proteome, only Calreticulin in one cell line was determined to have altered protein conformation and was also somatically mutated.
Next we explored the hypothesis that somatically mutated proteins might impinge on and alter protein conformations of non-mutated proteins to drive tumorigenesis. Somatically mutated proteins might harness non-mutated effector proteins with crucial biological functions during malignant transformation. Overall, lysine site coverage of common cellular proteins was better than the lysine site coverage of somatic mutated proteins. To find non-mutated proteins that harbor lysine sites that are altered in the presence of a mutated cancer driver gene, we searched our data for proteins with altered lysine site accessibility that coincided with a somatically mutated gene in ≥ 3 cancer cell lines. There are 103 out of 461 mutated genes that are mutated in ≥ 3 of the 60 cancer cell lines. A rank ordered list of the mutated genes is led by the tumor suppressor p53, which was mutated most frequently (31 cell lines), followed by PTEN (16 cell lines), FAT4, and KRAS (15 cell lines each). We found that 110 out of 8,052 (1.4 %) lysine sites in 98 non-mutated proteins correlated with the presence of somatic mutations in 90 different cancer driver genes (Figure 2). 40 out of the 110 lysine sites changed in accessibility for chemical modification by Δ > 20 %. A gene ontology analysis of all 98 proteins with altered lysine site accessibility indicated low interconnectivity (⟨k⟩ = 3.5) and a preference for effector protein functions in carbohydrate and RNA metabolism and in GTP-driven signal transduction. In contrast, the 103 somatically mutated proteins were highly interconnected (⟨k⟩ = 9.04) and were regulators rather than effectors of different cellular processes. Thus, somatic mutation of proteins with regulatory function induced distinct changes in lysine site accessibility in a small number of non-mutated, effector proteins.
Figure 2: CPP revealed correlations between 3D alterations and somatic mutations.
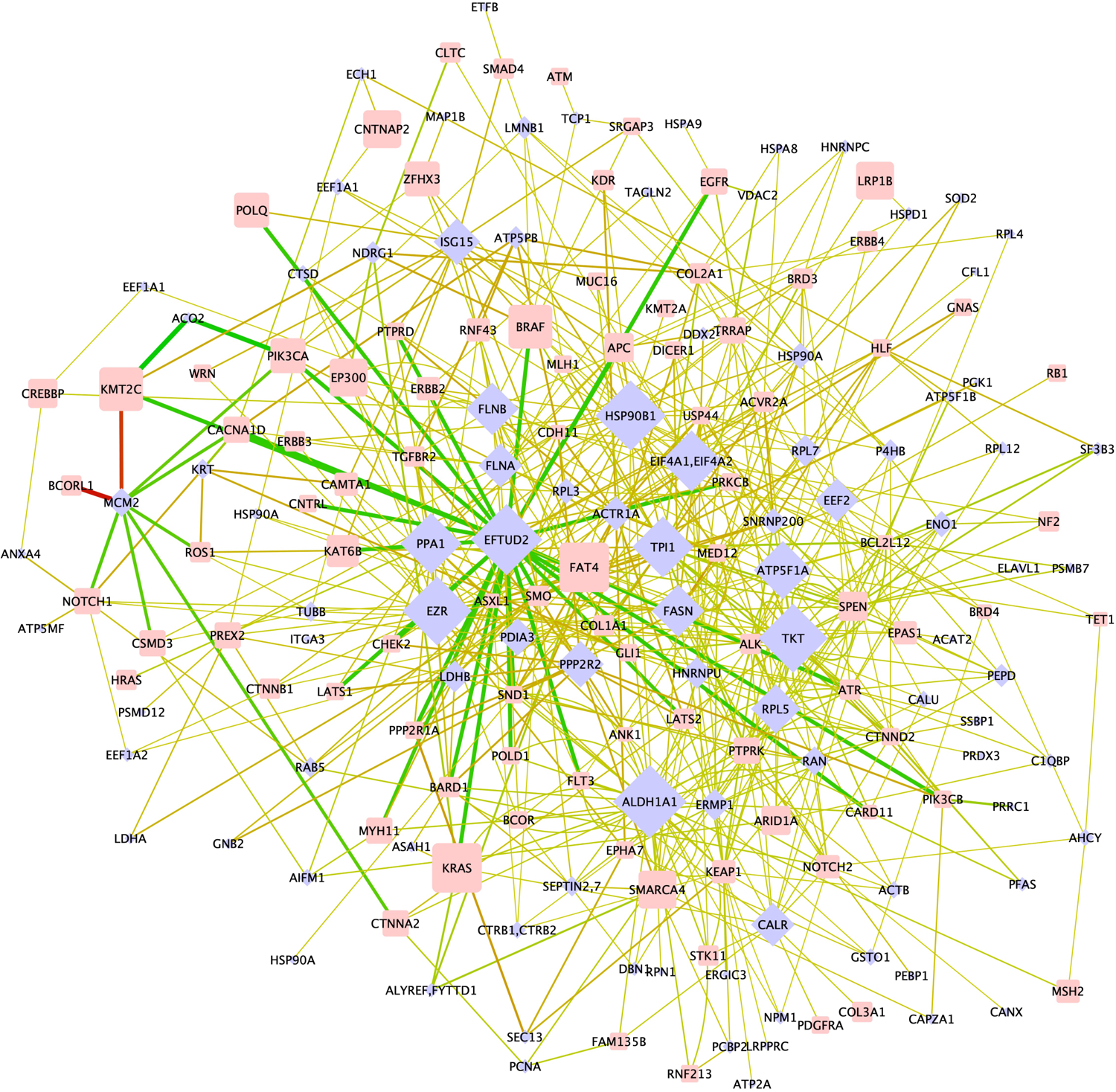
The bipartite network shows the correlation of somatic mutated proteins (oncogens and tumor suppressor genes, blue diamonds) to non-mutated effector proteins (rectangles) in which at least one lysine site is significantly altered in accessibility. The size of nodes scales with connectivity (effector proteins) or number of cell lines in which the protein is mutated (mutated proteins). Edges are colored green in case a decrease in surface accessibility was detected and red in case surface accessibility of the lysine site increased. The line thickness reflects the difference in lysine site accessibility (Δ) between cell lines with and without mutation in the somatically mutated protein.
The 98 effector proteins matched the 90 somatically mutated proteins 606 times in the bipartite network (⟨k⟩ = 6.2). On average, 7.7 different mutated proteins associated with a conformational change in the same effector protein, suggesting that mutations in different genes may cause the same structural changes in the 3D proteome and thus impinge on the same cellular pathways. However, mutated and structurally altered proteins were not part of the same cellular pathway. Only 22 of the 606 (3.8 %) edges in the network connected two proteins that are known to interact directly (Table S3). These proteins are members of protein folding and phosphorylation-mediated signal transduction pathways, which are often derailed upon malignant transformation (Material S1).
Protein nodes with a single edge were rare in the bipartite network. 19 of the 110 lysine sites associated with only one mutated protein (Material S1), and 8 of the 90 mutated proteins each correlated with a single lysine site that was altered in its accessibility (Table S4). Undiscovered lysine sites might correlate with additional somatic mutations which will increase the size and complexity of the bipartite network. Only 8 of the 110 lysine sites displayed a Δ > 50 % change in lysine site accessibility (Material S1, Figure S2, Table S5). These 8 lysine sites might serve as markers to differentiate tumors of colon, bone marrow, lung, breast, or ovary origin; however, the high redundancy in the bipartite network suggests that measuring alterations in a limited set of 110 lysine sites might be sufficient to show evidence of malignancy regardless of tumor origin.
Each cancer cell line displays a unique pattern of somatic mutations but somatic mutations in 90 proteins impacted the accessibility of lysine sites for covalent modification in 98 effector proteins. For example, 10 of the 60 cancer cell lines acquired oncogenic mutations in the catalytic subunit of the phosphatidyl inositol kinase p110α, PIK3CA, which is a known cancer driver gene16, 17. We found altered accessibility of 7 different lysine sites in 7 different proteins that correlated with the presence of oncogenic PIK3CA in the 10 cell lines (Figure 3A). Notably, a change in accessibility for covalent labeling in any of the 7 lysine sites was associated with at least one other somatically mutated protein besides mutated PIK3CA (p ≤ 0.05, Figure 3B).
Figure 3: Comparison of 3D proteome alterations that correlate with oncogenic PIK3CA mutation in the NCI60 cell line panel and that were identified upon de novo mutating PIK3CA in MCF10A cells.
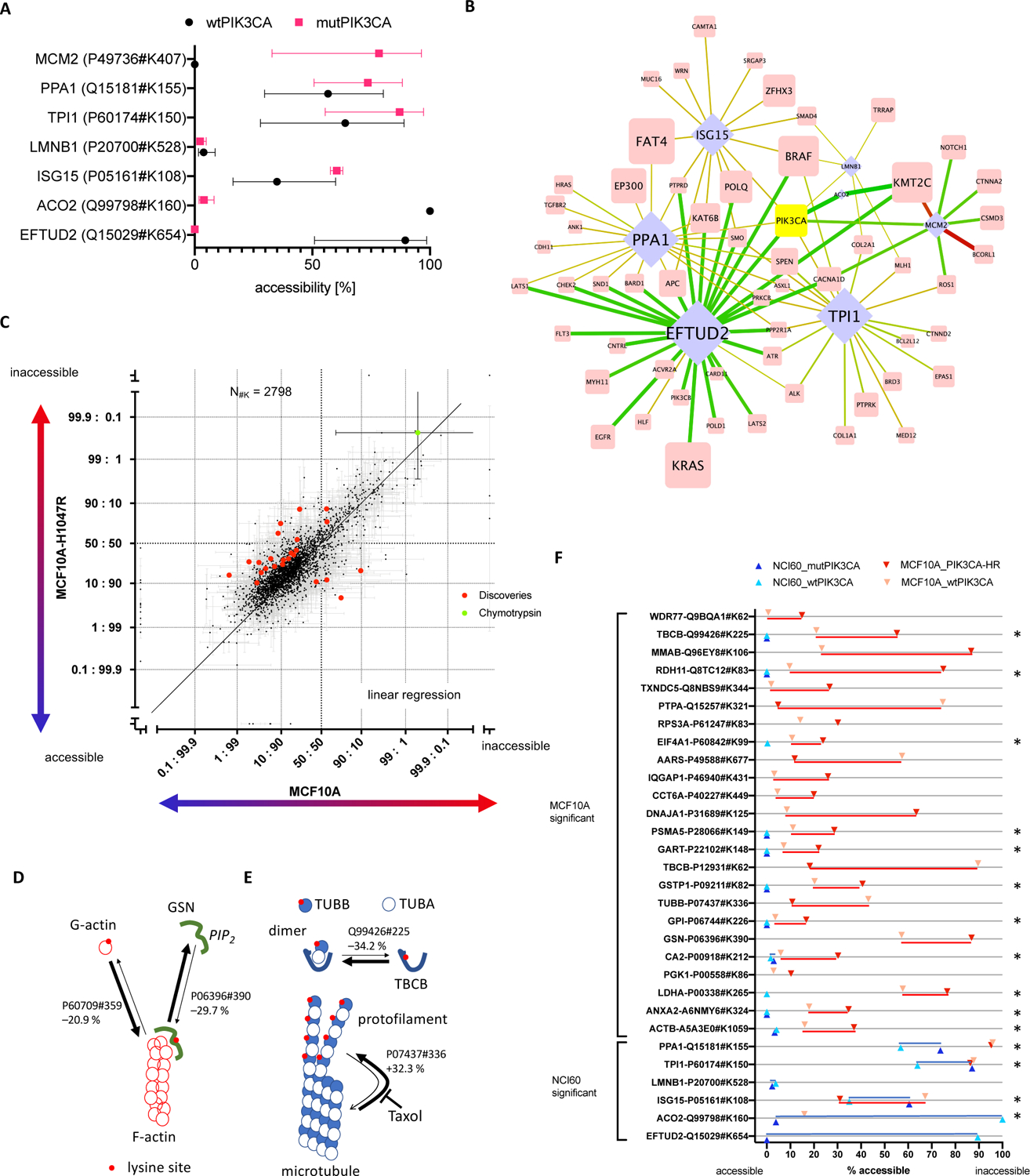
(A) Lysine site accessibilities for each effector protein that correlated with mutated and wt PIK3CA are indicated in red and black, respectively (error bars are standard deviation). (B) The bipartite network highlights interactions of PIK3CA in Figure 1C (identical figure annotation). Lysine site accessibility in six proteins differed significantly between 10 cell lines that harbored somatic mutations for PIK3CA (mutPIK3CA) and 50 cell lines with wtPIK3CA. (C) The scatterplot includes all lysine sites measured upon oncogenic activation of PIK3CA in MCF10A cells. Each dot represents an individual lysine site. Each lysine site was plotted with error of measurement (standard deviation) for its relative surface accessibility in MCF10A and MCF10A-H1047R cells (log2 converted value, inaccessible: accessible). The units “accessible” and “inaccessible” indicate that the lysine site was completely accessible or inaccessible. Lysine sites that were significantly altered in accessibility (q < 1%) are highlighted in red. Measurements obtained for Chymotrypsin site CRBT1#K54 (green dot) represent a positive control for “completely inaccessible” because Chymotrypsin was exogenously added following the initial labeling of the sample in CPP. The schematics (D) and (E) map changes in lysine site accessibility of proteins involved in actin and microtubule homeostasis. Protein-protein interactions and turnover of actin filaments and microtubules were altered upon oncogenic activation of PIK3CA in MCF10A cells. Red dots on protein surfaces pinpoint lysine sites accessible for CPP. Arrows indicate the homeostatic relationship between individual proteins and protein complexes. The thickness of the arrows show the relative change in the equilibrium upon mutation of PIK3CA according to the lysine site. (F) Comparison of significantly altered sites upon introduction of PIK3CA mutation in MCF10A cells (red) and presence of PIK3CA mutation in 10 out of 60 cancer cell lines (blue). Each measurement in lysine site accessibility is indicated and dark coloring refers to oncogenic PIK3CA, whereas light coloring represents the reference. Stars indicate lysine sites that were measured in both experiments.
In the absence of single, reciprocal associations between one mutated protein and one structural alteration in the 3D proteome of cancer cells, we decided to determine whether the complement of existing acquired somatic mutations determines which 3D proteome alterations arise from the introduction of a single de novo somatic mutation, H1047R in PIK3CA. Tumorigenic MCF10A-H1047R cells were clonally derived from parental, non-tumorigenic MCF10A cells18 and differ only by the oncogenic point mutation H1047R in PIK3CA, PIK3CAH1047R 19. We analyzed the 3D proteome of MCF10A breast epithelial cells with PIK3CAwt and MCF10A-H1047R cells with PIK3CAH1047R in biological triplicate experiments using multidimensional protein identification technology (MudPIT20), and identified 4,744 and 4,392 distinct lysine sites (14,401 and 18,377 peptide spectrum matches) in 2,391 and 2,284 proteins, respectively (Figure S3). 24 lysine sites in 24 proteins were altered in accessibility for covalent labeling upon oncogenic mutation of PIK3CA. Relative accessibility for chemical modification decreased in 20 and increased in 4 of the lysine sites (q ≤ 0.01, BH corrected, Figure 3C, Figure S4 and Table S6). Thus, introduction of the single oncogenic point mutation H1047R in PIK3CA altered the accessibility for covalent modification in only 0.8% of all lysine sites.
The molecular function of the 24 proteins with altered lysine accessibility revealed several distinct molecular pathways that might directly contribute to the malignant transformation of MCF10A cells upon H1047R mutation in PIK3CA. Specifically, lysine site GSN#K390 in gelsolin (P06396) shifted from predominantly accessible to partially inaccessible in the presence of elevated levels of the more highly phosphorylated phosphatidyl-inositol phosphates (PIP), PIP2 and PIP3 (Figure 3D). PIP3 suppresses gelsolin21, and PIP2 inhibits gelsolin’s ability to sever and cap actin filaments22. The lysine site ACTB#K359 in actin is accessible for covalent modification in soluble G-actin monomers, and we observed that the homeostatic equilibrium between free G-actin and fibrillar F-actin was shifted towards G-actin in the presence of PIK3CAH1047R. Most likely, constitutively active PIK3CAH1047R alters the equilibrium of gelsolin that severs actin filaments and thereby increases the relative amount of F-actin in MCF10A-H1047R cells.
Moreover, PIK3CAH1047R increased the accessibility of lysine site TUBB#K336 in 30 % of tubulin-β molecules (Figure 3E). In effect, it pushed the equilibrium of tubulin molecules polymerized in microtubules versus unbound tubulin-α and −β heterodimers towards an increased disassembly of microtubules and elevated levels of monomeric or heterodimeric tubulin-β23, 24. In addition, lysine site TBCB#K225 of the tubulin folding cofactor B protein (TBCB) displayed decreased accessibility in MCF10A-H1047R cells. TBCB binds and disassembles tubulin-α and −β heterodimers25 and altered accessibility of TBCB#K225 may reflect an altered homeostatic equilibrium in microtubule treadmilling. Microtubule treadmilling is directly targeted by the anti-cancer drug Taxol. Taxol prevents microtubule depolymerization but requires a narrow dosing regimen to control for its general toxicity26, 27. Taxol might critically interfere with an accelerated microtubule disassembly and thus preferentially inhibit cell growth and division of tumorigenic cells with malignant increased microtubule treadmilling.
Finally, oncogenic PIK3CAH1047R modulated the accessibility of a lysine residue in the proto-oncogene c-Src (P12931). Lysine site SRC#K62 increased in accessibility in 71 % of c-Src molecules, potentially activating its kinase. Activation of the c-Src kinase in the absence of a somatic mutation of the c-Src gene was previously observed28. C-Src influences survival, growth, and division of cells through its kinase activity at transmembrane protein complexes that assemble into cell-cell adhesion contacts29. Hart, et al.18 previously observed that mutating PIK3CAH1047R remodeled the proteome and transcriptome of the MCF10A cells. Here, we observed conformational alterations to the cytoskeleton that suggest specific alterations in the cytoskeletal homeostasis as a molecular mechanism by which malignant transformation is put into action in MCF10A-H1047R cells.
Mutating PIK3CAwt to PIK3CAH1047R in MCF10A cells enabled malignant transformation of MCF10A-H1047R cells through 3D changes in a small subset of proteins in the 3D proteome. However, the conformational changes and proteins differed from the 3D proteome alterations in the 10 NCI60 cell lines that already harbor mutated PIK3CA. 15 lysine sites were measured and quantified in both experiments but none of the conformational changes observed upon mutation of PIK3CAwt to PIK3CAH1047R in MCF10A cells were recapitulated in the 10 cancer cell lines of the NCI60 cell line panel that harbor mutated PIK3CA (Figure 3F). Therefore, we conclude that the tumor cell line specific, pre-existing somatic mutation patterns guide which conformational alterations in effector proteins accompany newly introduced somatic mutations during tumorigenesis.
Conformational Alterations Predict Anti-Cancer Drug Efficacy
Observing a common set of 98 conformational alterations in cancer cells suggests that 3D alterations might predict drug efficacy30. To test this hypothesis, we correlated the LC50 profiles of > 50,000 compounds with altered lysine site accessibility across all 60 cancer cell lines. LC50 profiles were previously reported by the Developmental Therapeutic Program at NCI31. Overall, the LC50 profiles of 300 small molecules correlated with the surface accessibility profiles of 49 lysine sites in 46 proteins 415 times (Pearson’s correlation |p| > 0.6, Figure 4A). 249 out of 415 correlations (edges) were positive, indicating that growth inhibition was more pronounced when lysine sites were less accessible for covalent modification. The remaining 166 correlations were negative, indicating that cell growth inhibition correlated with an increase in lysine site accessibility. 12 of 49 lysine sites (24.5 %) correlated with the LC50 profiles of a single drug, whereas 37 lysine sites (75.5 %) correlated with two or more small molecules. All edges were either positive or negative for 30 of the 37 lysine sites (82 %), whereas 7 lysine sites had both positive and negative edges. When the mechanism of action was known, small molecules did not directly bind to the proteins with altered lysine site accessibility (Material S1).
Figure 4: Correlation network between lysine site accessibility and small molecule-induced growth inhibition.
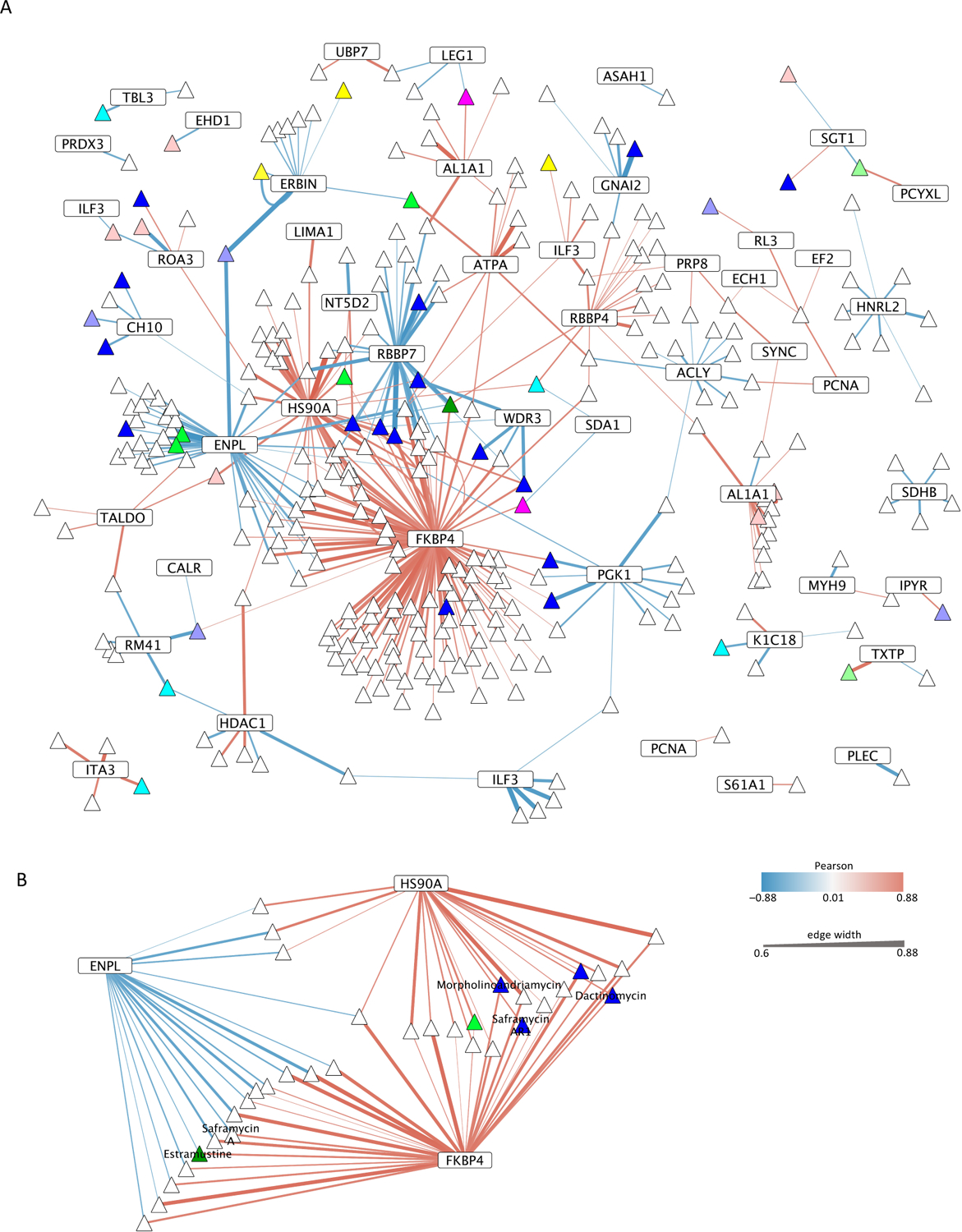
(A) The thickness of edges indicates the Pearson’s correlation of |p| > 0.6 in the bipartite network of lysine sites (rectangles) and small molecules (triangles). Proteins might harbor different lysine sites that correlated with small molecule activity, and thus the corresponding protein nodes are depicted several times in the network. Red edges refer to a positive correlation in which growth inhibition matched a reduced accessibility of a lysine site whereas blue edges denote a negative correlation in which growth inhibition matched increased accessibility of a lysine site. Node colors group small molecules with known mechanisms of action (lavender, antibiotic action; blue, DNA binding or intercalation or topoisomerase I or II inhibition; turquoise, V-ATPase inhibition; pink, Tubulin or Actin or Cofilin inhibition; dark green, hormone inhibition; light green, interaction with lipids, proteasome inhibition; yellow, HDAC, mitochondrion or proteasome inhibition; rosé, other mechanism of inhibition including amino acid biosynthesis, Akt, Stat3, Caspase 2,7,9, dinucleotide methyltransferase, inhibition of apoptosis (IAP), exportin). (B) The subnetwork depicts heat shock proteins HSP90A, HSP90B, and peptide isomerase FKBP4 which together correlated with the greatest number of small molecules. When known, non-scientific names of small molecules are indicated.
The 49 effector proteins were present in cellular pathways that were either directly or indirectly targeted by the growth inhibitory action of the small molecules. The majority of the 300 small molecules correlated with 3 lysine sites in 3 proteins that are part of the proteostasis network. Heat shock proteins HSP90AA1/HSP90AB1 (P07900, P08238) and HSP90B1 (P14625, Endoplasmin, GRP94) at lysine sites HSP90AA1#K327/HSP90AB1#K319 and HSP90B1#K493, respectively, and the peptidyl-prolyl cis-trans isomerase FKBP4 (FKBP4#K354, P02790) formed a large cluster with 156 different small molecules in the bipartite network (Figure 4B). HSP90 proteins and FKBP4 support folding or re-folding of nascent or misfolded proteins. The LC50 profile of 33 out of the 156 small molecules matched the relative accessibility of lysine sites in two of the three proteins, and one small molecule (NSC-673805) correlated with the relative accessibility of all three lysine sites. Increased inaccessibility of FKBP4#K354 and HSP90AA1#K327/HSP90AB1#K319 aligned positively with 118 and 38 growth inhibitors, respectively. In contrast, HSP90B1#K493 aligned negatively with the cytotoxicity of 41 small molecules. Thus, the relative accessibility of three distinct lysine sites in heat shock proteins of the HSP90 family and FKBP4 were sufficient to predict the growth inhibition efficacy of more than half of the 300 small molecules that were identified here. Overall, proteostasis may be more broadly derailed during tumorigenesis than previously recognized.
The mechanisms of action of the 156 small molecules are diverse and encompass DNA intercalation and translation inhibition, both of which broadly perturb proteome homeostasis. For example, the cytotoxicity patterns of Estramustine, Morpholinoadriamycin, Dactinomycin, Saframycin A, and Saframycin AR1 matched at least three of the four most connected lysine sites and proteins in the network including HSP90AA1/HSP90AB1, HSP90B1 and FKBP4. All three proteins participate in direct protein-protein interactions, and an altered lysine site accessibility might indicate a rise in client proteins that require re-folding through the proteostasis network in select tumor cell lines (Material S1). Thus, the results emphasize the importance of the proteostasis network for upholding malignant transformation. Most likely, the proteostasis network directly buffers the 3D proteome against an otherwise lethal increase in loss of function protein misfolding that might accompany tumorigenesis.
Furthermore, proteins RBBP4 and RBBP7 in the Nucleosome Remodeling Deacetylase (NuRD) protein complex showed overlapping associations with small molecules that correlated with heat shock proteins. RBBP4 and RBBP7 interchange in the NuRD complex and correlate positively and negatively with the growth inhibition of small molecules, respectively. Further, the histone deacetylase HDAC1 (Q13547) is part of the NuRD complex. Lysine site HDAC1#K123 correlated both positively and negatively with small molecules that inhibit cell growth (Material S1). Thus, lysine site accessibility and growth inhibition by small molecules interacting with the NuRD complex may indicate a hierarchy in conformational aberrations. However, the presence of both positive and negative drug correlations with a lysine site were rare (7 out of 49 lysine sites).
Decreased surface accessibility of phosphoglycerate kinase 1 site PGK1#K332 (P0058), of ATP-citrate synthase site ACLY#K944 (P53396), and of ATP5F1A#K194 in the mitochondrial ATP synthase-α (P25705) correlated with an increased cytotoxicity of several different small molecules. 3D alterations in these three proteins might contribute to the Warburg effect of tumor cells to produce adenosine triphosphate (ATP) through glycolysis rather than via aerobic respiration even though sufficient oxygen is available32, 33. Furthermore, lysine site-to-drug correlations highlighted hormone processing and signaling proteins. For example, Retinal dehydrogenase 1 sites ALDH1A1#K194 and ALDH1A1#K138 (P00352) matched the growth inhibitory effect of 13 and 7 different small molecules, respectively. The accessibility of Erbin#K63 (Q96RT1) in the adaptor protein for the tyrosine kinase receptor ERBB2 correlated with the growth inhibition exerted by 10 different small molecules.
Finally, infection of cells with retroviruses that integrate into the genome can drive malignant transformation. The interleukin enhancer-binding factor 3, ILF3 (Q12906), binds to viral dsRNA and inhibits translation of retroviral mRNAs34 as part of an innate anti-viral response. Lysine site ILF3#K419 correlated negatively with 6 different small molecules that have been described in part as DNA intercalators, whereas ILF3#K565 correlated positively with HDAC inhibitor analogues Romidepsin and NSC-709238, which reverse human immunodeficiency virus (HIV) latency in T cells35 (Figure S5). ILF3#K565 accessibility for chemical modification correlated negatively with HIV and SIV latency, reversing the effects of AZD5582 (NSC-787048) which binds and blocks the inhibitor of Apoptosis protein IAP36. Thus, differential lysine site accessibilities in the two distinct dsRNA binding domains of ILF3 indicated the sensitivity of cells to small molecules that interfere with the retrovirus activity and thus may block retrovirus-associated neoplastic transformation of infected cells.
Discussion
Here, we showed that tumorigenic mutations alter the surface accessibility of proteins that may shape the protein conformational landscape in cancer cells. In tumorigenesis, cells maximize their proliferation by defying extracellular clues to self-restrain. Even though tumorigenesis is stereotypic, each tumor exhibits a unique somatic mutation pattern. CPP analysis yielded a limited set of conformational alterations that correlated with the presence of select somatic mutated proteins. These conformational changes might be the result of altered protein-protein or protein-biomolecule interactions, post translational modifications, selective protein degradation, or other factors. Here, we showed that the individual patterns of somatic mutation impinge on a small number of effector proteins that are conformationally altered in tumorigenesis. Because conformational changes in effector proteins were stereotypic across the NCI60 cell line panel, it suggests that tumorigenesis relies on altering a limited set of effector proteins.
Furthermore, we looked for causative associations between somatic mutation and conformational change. De novo mutation of PIK3CA in MCF10A cells altered the accessibility of lysine sites for covalent modification in a few proteins. These alterations differed from those observed in the 10 cancer cell lines that had acquired oncogenic PIK3CA mutations during tumorigenesis. Thus, we suggest that the background of somatic mutations rather than the de novo acquired genetic aberration determines which specific conformational changes are induced in the 3D proteome, possibly because new genomic aberrations are tested against a unique genetic background for added growth advantage in tumor evolution.
Here, we used CPP to identify altered protein conformations and we correlated them with the efficacy of small molecules to inhibit cell growth. This proteomic approach to drug identification is fundamentally different than determining protein abundance in response to drug treatment37. By retrospectively correlating the growth inhibitory effect of small molecules with alterations in lysine site accessibility, we found that 49 conformational rearrangements in select effector proteins may predict the growth inhibition efficacy of 300 small molecules. The cancer conformational landscape suggests that tumorigenic mutations in regulatory proteins derail protein conformations, protein biomolecule associations or protein-protein interactions in non-mutated effector proteins and pathways. According to our results, at least 156 out of 300 cell growth-inhibiting small molecules are more effective in case the proteostasis network upholds malignant transformation. Further, we observed that oncogenic activation of PIK3CA alters the homeostatic equilibrium of microtubule treadmilling, likely increasing the load of the tubulin-β specific chaperone TBCB with tubulin-β molecules. Thus, our results imply that proteostasis might actively support malignant transformation.
By using CPP to quantify protein conformation in the proteostasis network, we might be able to target cancer more effectively during anti-cancer therapy. Non-tumorigenic cells are in a homeostatic optimum that evolved to minimize the overall load on the proteostasis network, but tumor cells gain somatic mutations that allow uncontrolled cell proliferation. However, this gain comes at the cost of a non-optimal activation or partial inactivation of cellular pathways. Because somatic mutations may cause cell physiological alterations that are suboptimal, the overall load on the proteostasis network will increase to compensate for a tumor-induced disequilibrium in the cellular proteome. In line with this interpretation, we found that alterations in the proteostasis network are almost independent of tumor type and mutational background. These alterations are therefore highly predictive for more than 150 small molecules that are known to limit cell proliferation. Notably, these small molecules are highly diversified in their mechanism of action.
The 49 protein rearrangements that indicate growth inhibition of small molecules may also be used as a diagnostic or sentinel to scout tumor cells for additional vulnerabilities. Indeed, most proteins with sentinel lysine sites are part of cellular networks that are pivotal for cell survival, such as proteostasis, chromatin remodeling, and metabolism. While our current knowledge of the 3D proteome is limited, it may soon be possible to design new drugs by selecting lysine sites. For example, a direct interruption of protein-protein interactions of FKBP4 or CHIP with HSP90 with novel small molecules might enhance the efficacy of existing anticancer therapies.
Supplementary Material
Material S1. Description of lysine sites directly involved in protein-protein interactions.
Figure S1. Summary of the NCI60 CPP results.
Figure S2. Subnetwork of protein sites that change by Δ > 50 %.
Figure S3. All lysine sites quantified in MCF10A and MCF10A-H1047R cells.
Figure S4. Volcano plot representation of lysine sites in MCF10A and MCF10A-H1047R cells.
Figure S5. Bipartite graphs of lysine sites to small molecule or somatic mutation correlations.
Figure S6. Additional information on protein to small molecule networks of select lysine sites.
Table S2. List of 6 somatic mutated genes that express a mutated protein with an altered lysine site in at least one of the 60 cancer cell lines.
Table S3. 22 pairs of somatic mutated proteins that correlate with altered lysine sites in proteins with direct functional relationship.
Table S4. All differentially accessible lysine sites that corelated with only one somatic mutated protein.
Table S5. Correlation between Δ ≥ 50 % in accessibility and somatically mutated proteins.
Table S6. Lysine sites with significantly changed accessibility in MCF10AH1047R cells.
Table S1. Pairwise comparison of the CPP results for all 60 cancer cell lines.
Acknowledgments:
We thank the National Cancer Institute Division of Cancer Treatment and Diagnosis (NCI DCTD) for providing us with the NCI60 cell line panel. We thank Claire Delahunty for reading the manuscript.
Funding:
Funding was provided by NIH grant R33CA212973-01 awarded to John R. Yates III.
Footnotes
Competing Interest Statement: The authors declare no competing interests.
Data Availability
Datasets are available from Massive and ProteomeXchange with the following accession numbers: MSV000085034 and PXD017794.
References
- 1.Li Y; Roberts ND; Wala JA; Shapira O; Schumacher SE; Kumar K; Khurana E; Waszak S; Korbel JO; Haber JE; Imielinski M; Akdemir KC; Alvarez EG; Baez-Ortega A; Beroukhim R; Boutros PC; Bowtell DDL; Brors B; Burns KH; Campbell PJ; Chan K; Chen K; Cortés-Ciriano I; Dueso-Barroso A; Dunford AJ; Edwards PA; Estivill X; Etemadmoghadam D; Feuerbach L; Fink JL; Frenkel-Morgenstern M; Garsed DW; Gerstein M; Gordenin DA; Haan D; Haber JE; Hess JM; Hutter B; Imielinski M; Jones DTW; Ju YS; Kazanov MD; Klimczak LJ; Koh Y; Korbel JO; Kumar K; Lee EA; Lee JJ-K; Li Y; Lynch AG; Macintyre G; Markowetz F; Martincorena I; Martinez-Fundichely A; Meyerson M; Miyano S; Nakagawa H; Navarro FCP; Ossowski S; Park PJ; Pearson JV; Puiggròs M; Rippe K; Roberts ND; Roberts SA; Rodriguez-Martin B; Schumacher SE; Scully R; Shackleton M; Sidiropoulos N; Sieverling L; Stewart C; Torrents D; Tubio JMC; Villasante I; Waddell N; Wala JA; Weischenfeldt J; Yang L; Yao X; Yoon S-S; Zamora J; Zhang C-Z; Weischenfeldt J; Beroukhim R; Campbell PJ; Group PSVW; Consortium P, Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578 (7793), 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B; Papadopoulos N; Velculescu VE; Zhou S; Diaz LA Jr.; Kinzler KW, Cancer genome landscapes. Science 2013, 339 (6127), 1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mertins P; Mani DR; Ruggles KV; Gillette MA; Clauser KR; Wang P; Wang X; Qiao JW; Cao S; Petralia F; Kawaler E; Mundt F; Krug K; Tu Z; Lei JT; Gatza ML; Wilkerson M; Perou CM; Yellapantula V; Huang KL; Lin C; McLellan MD; Yan P; Davies SR; Townsend RR; Skates SJ; Wang J; Zhang B; Kinsinger CR; Mesri M; Rodriguez H; Ding L; Paulovich AG; Fenyö D; Ellis MJ; Carr SA, Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534 (7605), 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo T; Luna A; Rajapakse VN; Koh CC; Wu Z; Liu W; Sun Y; Gao H; Menden MP; Xu C; Calzone L; Martignetti L; Auwerx C; Buljan M; Banaei-Esfahani A; Ori A; Iskar M; Gillet L; Bi R; Zhang J; Zhang H; Yu C; Zhong Q; Varma S; Schmitt U; Qiu P; Zhang Q; Zhu Y; Wild PJ; Garnett MJ; Bork P; Beck M; Liu K; Saez-Rodriguez J; Elloumi F; Reinhold WC; Sander C; Pommier Y; Aebersold R, Quantitative Proteome Landscape of the NCI-60 Cancer Cell Lines. iScience 2019, 21, 664–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scherz-Shouval R; Santagata S; Mendillo ML; Sholl LM; Ben-Aharon I; Beck AH; Dias-Santagata D; Koeva M; Stemmer SM; Whitesell L; Lindquist S, The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 2014, 158 (3), 564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter RL; Gökmen-Polar Y, HSF1 as a Cancer Biomarker and Therapeutic Target. Curr Cancer Drug Targets 2019, 19 (7), 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vasan N; Baselga J; Hyman DM, A view on drug resistance in cancer. Nature 2019, 575 (7782), 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bamberger C; Pankow S; Martínez-Bartolomé S; Ma M; Diedrich J; Rissman RA; Yates JR, Protein Footprinting via Covalent Protein Painting Reveals Structural Changes of the Proteome in Alzheimer’s Disease. Journal of proteome research 2021, 20 (5), 2762–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West GM; Tang L; Fitzgerald MC, Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass spectrometry-based strategy. Analytical chemistry 2008, 80 (11), 4175–85. [DOI] [PubMed] [Google Scholar]
- 10.Borotto NB; Zhou Y; Hollingsworth SR; Hale JE; Graban EM; Vaughan RC; Vachet RW, Investigating Therapeutic Protein Structure with Diethylpyrocarbonate Labeling and Mass Spectrometry. Analytical chemistry 2015, 87 (20), 10627–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y; Vachet RW, Covalent labeling with isotopically encoded reagents for faster structural analysis of proteins by mass spectrometry. Analytical chemistry 2013, 85 (20), 9664–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franken H; Mathieson T; Childs D; Sweetman GMA; Werner T; Tögel I; Doce C; Gade S; Bantscheff M; Drewes G; Reinhard FBM; Huber W; Savitski MM, Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nature protocols 2015, 10 (10), 1567–1593. [DOI] [PubMed] [Google Scholar]
- 13.Bamberger C; Martinez-Bartolome S; Montgomery M; Pankow S; Hulleman JD; Kelly JW; Yates JR 3rd, Deducing the presence of proteins and proteoforms in quantitative proteomics. Nat Commun 2018, 9 (1), 2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abaan OD; Polley EC; Davis SR; Zhu YJ; Bilke S; Walker RL; Pineda M; Gindin Y; Jiang Y; Reinhold WC; Holbeck SL; Simon RM; Doroshow JH; Pommier Y; Meltzer PS, The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res 2013, 73 (14), 4372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alfaro JA; Ignatchenko A; Ignatchenko V; Sinha A; Boutros PC; Kislinger T, Detecting protein variants by mass spectrometry: a comprehensive study in cancer cell-lines. Genome Med 2017, 9 (1), 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bailey MH; Tokheim C; Porta-Pardo E; Sengupta S; Bertrand D; Weerasinghe A; Colaprico A; Wendl MC; Kim J; Reardon B; Kwok-Shing Ng P; Jeong KJ; Cao S; Wang Z; Gao J; Gao Q; Wang F; Liu EM; Mularoni L; Rubio-Perez C; Nagarajan N; Cortes-Ciriano I; Zhou DC; Liang WW; Hess JM; Yellapantula VD; Tamborero D; Gonzalez-Perez A; Suphavilai C; Ko JY; Khurana E; Park PJ; Van Allen EM; Liang H; Group MCW; Cancer Genome Atlas Research, N.; Lawrence MS; Godzik A; Lopez-Bigas N; Stuart J; Wheeler D; Getz G; Chen K; Lazar AJ; Mills GB; Karchin R; Ding L, Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174 (4), 1034–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samuels Y; Wang Z; Bardelli A; Silliman N; Ptak J; Szabo S; Yan H; Gazdar A; Powell SM; Riggins GJ; Willson JK; Markowitz S; Kinzler KW; Vogelstein B; Velculescu VE, High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304 (5670), 554. [DOI] [PubMed] [Google Scholar]
- 18.Hart JR; Zhang Y; Liao L; Ueno L; Du L; Jonkers M; Yates JR 3rd; Vogt PK, The butterfly effect in cancer: a single base mutation can remodel the cell. Proc Natl Acad Sci U S A 2015, 112 (4), 1131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandelker D; Gabelli SB; Schmidt-Kittler O; Zhu J; Cheong I; Huang CH; Kinzler KW; Vogelstein B; Amzel LM, A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proceedings of the National Academy of Sciences of the United States of America 2009, 106 (40), 16996–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Washburn MP; Wolters D; Yates JR 3rd, Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature biotechnology 2001, 19 (3), 242–7. [DOI] [PubMed] [Google Scholar]
- 21.Patel VB; Zhabyeyev P; Chen X; Wang F; Paul M; Fan D; McLean BA; Basu R; Zhang P; Shah S; Dawson JF; Pyle WG; Hazra M; Kassiri Z; Hazra S; Vanhaesebroeck B; McCulloch CA; Oudit GY, PI3Kα-regulated gelsolin activity is a critical determinant of cardiac cytoskeletal remodeling and heart disease. Nature Communications 2018, 9 (1), 5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janmey PA; Stossel TP, Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature 1987, 325 (6102), 362–4. [DOI] [PubMed] [Google Scholar]
- 23.Nogales E; Wolf SG; Downing KH, Structure of the αβ tubulin dimer by electron crystallography. Nature 1998, 391 (6663), 199–203. [DOI] [PubMed] [Google Scholar]
- 24.Ichikawa M; Liu D; Kastritis PL; Basu K; Hsu TC; Yang S; Bui KH, Subnanometre-resolution structure of the doublet microtubule reveals new classes of microtubule-associated proteins. Nature Communications 2017, 8 (1), 15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serna M; Carranza G; Martín-Benito J; Janowski R; Canals A; Coll M; Zabala JC; Valpuesta JM, The structure of the complex between α‐tubulin, TBCE and TBCB reveals a tubulin dimer dissociation mechanism. Journal of Cell Science 2015, 128 (9), 1824. [DOI] [PubMed] [Google Scholar]
- 26.Weaver BA, How Taxol/paclitaxel kills cancer cells. Molecular biology of the cell 2014, 25 (18), 2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kellogg EH; Hejab NMA; Howes S; Northcote P; Miller JH; Díaz JF; Downing KH; Nogales E, Insights into the Distinct Mechanisms of Action of Taxane and Non-Taxane Microtubule Stabilizers from Cryo-EM Structures. Journal of Molecular Biology 2017, 429 (5), 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irby RB; Yeatman TJ, Role of Src expression and activation in human cancer. Oncogene 2000, 19 (49), 5636–5642. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y; Cao H; Chen J; McNiven MA, A direct interaction between the large GTPase dynamin-2 and FAK regulates focal adhesion dynamics in response to active Src. Molecular biology of the cell 2011, 22 (9), 1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franco M; Jeggari A; Peuget S; Böttger F; Selivanova G; Alexeyenko A, Prediction of response to anti-cancer drugs becomes robust via network integration of molecular data. Scientific Reports 2019, 9 (1), 2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoemaker RH, The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006, 6 (10), 813–23. [DOI] [PubMed] [Google Scholar]
- 32.DeBerardinis RJ; Chandel NS, We need to talk about the Warburg effect. Nature Metabolism 2020, 2 (2), 127–129. [DOI] [PubMed] [Google Scholar]
- 33.Warburg O, On the Origin of Cancer Cells. Science 1956, 123 (3191), 309. [DOI] [PubMed] [Google Scholar]
- 34.Harashima A; Guettouche T; Barber GN, Phosphorylation of the NFAR proteins by the dsRNA-dependent protein kinase PKR constitutes a novel mechanism of translational regulation and cellular defense. Genes Dev 2010, 24 (23), 2640–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Søgaard OS; Graversen ME; Leth S; Olesen R; Brinkmann CR; Nissen SK; Kjaer AS; Schleimann MH; Denton PW; Hey-Cunningham WJ; Koelsch KK; Pantaleo G; Krogsgaard K; Sommerfelt M; Fromentin R; Chomont N; Rasmussen TA; Østergaard L; Tolstrup M, The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS pathogens 2015, 11 (9), e1005142–e1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nixon CC; Mavigner M; Sampey GC; Brooks AD; Spagnuolo RA; Irlbeck DM; Mattingly C; Ho PT; Schoof N; Cammon CG; Tharp GK; Kanke M; Wang Z; Cleary RA; Upadhyay AA; De C; Wills SR; Falcinelli SD; Galardi C; Walum H; Schramm NJ; Deutsch J; Lifson JD; Fennessey CM; Keele BF; Jean S; Maguire S; Liao B; Browne EP; Ferris RG; Brehm JH; Favre D; Vanderford TH; Bosinger SE; Jones CD; Routy JP; Archin NM; Margolis DM; Wahl A; Dunham RM; Silvestri G; Chahroudi A; Garcia JV, Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578 (7793), 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saei AA; Beusch CM; Chernobrovkin A; Sabatier P; Zhang B; Tokat ÜG; Stergiou E; Gaetani M; Végvári Á; Zubarev RA, ProTargetMiner as a proteome signature library of anticancer molecules for functional discovery. Nature Communications 2019, 10 (1), 5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Material S1. Description of lysine sites directly involved in protein-protein interactions.
Figure S1. Summary of the NCI60 CPP results.
Figure S2. Subnetwork of protein sites that change by Δ > 50 %.
Figure S3. All lysine sites quantified in MCF10A and MCF10A-H1047R cells.
Figure S4. Volcano plot representation of lysine sites in MCF10A and MCF10A-H1047R cells.
Figure S5. Bipartite graphs of lysine sites to small molecule or somatic mutation correlations.
Figure S6. Additional information on protein to small molecule networks of select lysine sites.
Table S2. List of 6 somatic mutated genes that express a mutated protein with an altered lysine site in at least one of the 60 cancer cell lines.
Table S3. 22 pairs of somatic mutated proteins that correlate with altered lysine sites in proteins with direct functional relationship.
Table S4. All differentially accessible lysine sites that corelated with only one somatic mutated protein.
Table S5. Correlation between Δ ≥ 50 % in accessibility and somatically mutated proteins.
Table S6. Lysine sites with significantly changed accessibility in MCF10AH1047R cells.
Table S1. Pairwise comparison of the CPP results for all 60 cancer cell lines.
Data Availability Statement
Datasets are available from Massive and ProteomeXchange with the following accession numbers: MSV000085034 and PXD017794.