

Abstract
We analyzed the P2X4 receptor structure–activity relationship of a known antagonist 5, a 1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione. Following extensive modification of the reported synthetic route, 4-pyridyl 21u (MRS4719) and 6-methyl 22c (MRS4596) analogues were most potent at human (h) P2X4R (IC50 0.503 and 1.38 μM, respectively, and selective versus hP2X1R, hP2X2/3R, hP2X3R). Thus, the naphthalene 6-, but not 7-position was amenable to substitution, and an N-phenyl ring aza-scan identified 21u with 3-fold higher activity than 5. Compounds 21u and 22c showed neuroprotective and learning- and memory-enhancing activities in a mouse middle cerebral artery occlusion (MCAO) model of ischemic stroke, with potency of 21u > 22c. 21u dose-dependently reduced infarct volume and reduced brain atrophy at 3 and 35 days post-stroke, respectively. Relevant to clinical implication, 21u also reduced ATP-induced [Ca2+]i influx in primary human monocyte-derived macrophages. This study indicates the translational potential of P2X4R antagonists for treating ischemic stroke, including in aging populations.
Graphical Abstract

INTRODUCTION
The P2X4 receptor (P2X4R), one of seven homotrimeric ATP-gated P2X cation channels, is involved in immune, musculoskeletal, endocrine, and renal/urinary functions,1–4 while three identical P2X subunits can form functional cation channels, heterotrimeric channels, such as combinations of P2X4 with P2X1, P2X2, or P2X7 subunits. Heteromeric P2X channels often differ in ligand activity and other pharmacological properties from homotrimeric channels. In the central nervous system, P2X4R action is usually mediated by brain macrophages such as microglial cells and by infiltrating monocytes.2 However, a presynaptic P2X4R facilitates GABA release in the arcuate nucleus, a neural circuit that controls feeding.5 The P2X4R is largely internalized within acidic compartments but can be cycled to the cell surface upon resensitization.6,7 There is a known co-expression in immune cells, such as microglial cells,8 of P2X4R with P2X7R, both of which are targets for the development of antagonists for the treatment of pain and inflammation. The P2X4R and P2X7R interact physically in the cell membrane and can form heterotrimers, but a distinct phenotype was not observed.9 Furthermore, antagonizing the microglial P2X4R is considered a promising target for multiple sclerosis.10,11 P2X4R antagonists are efficacious in reducing chronic neuropathic pain in vivo, including herpetic pain and in stroke protection.12,13 ATP released from the dorsal horn neurons through the vesicular nucleotide transporter (VNUT) activates P2X4R to regulate the release of pain-inducing mediators interleukin 1-beta (IL-1β) and brain-derived neurotrophic factor (BDNF).14–16 Therefore, P2X4R antagonists prevent the release of inflammatory mediators and reduce pain. However, in stroke models such as the middle cerebral artery occlusion (MCAO) model in mouse, the protective window for P2X4R antagonists is limited to the initial phase when activated myeloid cells exacerbate the ischemic damage.13,17 The P2X4R is also found on alveolar macrophages and in bronchial smooth muscle cells.18,19 P2X4R activation facilitates mast cell degranulation and allergic responses.20 In bone-marrow-derived mast cells, co-stimulation of P2X4R and prostaglandin EP3 receptors leads to degranulation.21 Although the anti-inflammatory activity of P2X4R antagonists is of interest in drug discovery, P2X4R agonists might also have translational potential. In cardiomyocytes, stimulation of a P2X4R protects against heart failure through nitric oxide synthase (NOS).22,23 The macrophage P2X4R is involved in the response to infection, suggesting another potential therapeutic application of selective agonists.1,24
Representative antagonists for this receptor are shown in Chart 1. Methods for measuring P2X4R inhibitory activity and molecular modeling of the receptor are reported.26 The antidepressant drug paroxetine (structure not shown) was the first antagonist of the P2X4R receptor to be discovered from the screening of chemical libraries,28 and another antidepressant duloxetine was later found to antagonize the P2X4R equipotently but with greater P2X4/P2X7 selectivity than paroxetine.29 Phenoxazine derivative 1 was found to antagonize the P2X4R with IC50 values of 0.19 μM (h, human) and 2.10 μM (r, rat), and its sulfonamide derivative 2 was less potent,25 while a carbamazepine derivative (structure not shown) was found to antagonize the P2X4R but with major species differences.30 Compound 3 is often used as a selective, competitive P2X4R antagonist, with an IC50 of 0.75 μM at rP2X4R.31 Compound 4 displayed an IC50 of 0.54 μM at the hP2X4R.32 However, compounds 1–4 have limited aqueous solubility. 1H-Naphtho[1,2-b][1,4]diazepine-2,4-(3H,5H)-dione-5-[3-(2,5-dihydro-5-thioxo-1,2,4-oxadiazol-3-yl)phenyl] sodium salt (5, NP-1815-PX) was shown to be P2X4R-selective and to have antiallodynic properties.12 We have chosen 5 as our lead compound here due to its demonstrated in vitro activity at rat, mouse, and human P2X4R, in vivo activity, and water solubility. Compound 5 was also shown to be inactive at the P2X7R.12 Compound 6 was reported as a selective pharmacological tool for the P2X4R with 0.21 μM activity, but its preclinical development ended due to a risk of CYP3A4 induction.27 Other diverse compounds have been reported to interact with the receptor, including natural products ivermectin and ginsenosides,33,34 both of which act as positive allosteric modulators (PAMs). Ethanol and zinc ions also act as allosteric modulators of the P2X4R.33,35 Plant alkaloid dehydrocorybulbine induces a nociceptive effect by blocking the P2X4R.36
Chart 1.

Commonly Used Non-Nucleotide P2X4R Antagonists, Showing IC50 Values at the hP2X4Ra2,25–27
aQpatch is an automated patch clamp system for whole cells (https://sophion.com/).
Various zebrafish P2X4R X-ray structures with both agonist-bound and apo-states have suggested a major conformational reorganization in the active state.37,38 The ligand binding sites have been characterized from the X-ray structures, and in conjunction with site-directed mutagenesis and molecular modeling. Most of the known P2XR antagonist classes are considered negative allosteric modulators (NAMs). The P2X4R subunit is dolphin-shaped, on which the antagonist binding sites are widely distributed, and the agonist binding site is located between two subunits.
Due to the lack of binding site information, we studied the structure–activity relationship (SAR) of the 5-aryl-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione series related to 5 in traditional ways. Analogues were synthesized and characterized in channel assays, comparing activity at hP2X1R and hP2X3R (luminescence assays using a luminescent Ca2+-sensitive photoprotein) and at hP2X2/3R and hP2X4R (fluorescence assays using a fluorescent Ca2+-sensitive dye). We characterized two most potent antagonists 21u and 22c for neuroprotective and neuro-rehabilitative activity in an in vivo ischemic stroke model.
RESULTS
Chemical Synthesis.
The syntheses of 1,5-dihydro-2H-naphtho[1, 2-b][1,4]diazepine-2,4(3H,5H)-dione derivative 512 and its analogues, in particular compounds 5, 22b, and 22c, were performed largely by following the route reported by Sakuma et al.39 However, to accomplish the envisaged SAR analysis, it was necessary to adapt other synthetic protocols in the schemes. For nitration of 2-naphthols (7a–c, Scheme 1), a quick literature search returned many results, and most high-yielding reactions used exotic reagents/conditions. Initially, a mild and high-yielding protocol that employed readily available chemicals was tried (Table S1; Supporting Information; entry 1) but resulted in only trace amounts of the desired product.40 Since nitronaphthols are acidic, an acidic workup did not affect the yield. Then, the traditional nitration using nitric acid with acetic acid or dichloromethane as solvent at low temperatures resulted in moderate yields. However, a similar condition at rt (CH2Cl2, 1.1 equiv fuming HNO3, 5 min) gave the desired nitronaphthol in a good, isolated yield. Under mild phase-transfer conditions, the yield was reduced.41
Scheme 1.

Synthesis of Substituted Naphthalene Core Intermediates 13a-da
aReagents and conditions: (a) CH2Cl2, fuming HNO3, rt, 5 min, 51–62%; (b) CH2Cl2, triethylamine (TEA), Tf2O, 0 °C → rt, 18 h, 66–85%; (c) toluene, 3-cyanoaniline, PPh3, Pd(PPh3)4, K2CO3, 110 °C, 16 h, 66–85%; (d) tetrahydrofuran (THF)-MeOH (2:1), Pd–C, H2, rt, 4 h, 62–93%; (e) acetic acid, bromine, rt, 1 h, 77%; (f) THF, ethyl chloromalonate, TEA, 0 °C → rt, 3 h, 70% (12a); (g) toluene-N,N-dimethylformamide (DMF) (9:1), monomethyl malonate, N,N′-dicyclohexylcarbodiimide (DCC), rt, 3 h, KO-t-Bu, rt, 3 h, 52% (11a → 13a); (h) toluene, malonyl chloride, 0 °C → rt, 80 °C, 20 min, 110 °C, 10 min, 40–68%; (i) THF, KO-t-Bu, rt, 3 h, quant. yield (12a → 13a); (j) acetic acid, bromine, 50 °C, 7 h, 81%; (k) DMF, N-bromosuccinimide (NBS), 50 °C, 5 h, 81%.
Nitronaphthols 8a–c (Scheme 1) were sulfonylated, and the products were subjected to Buchwald–Hartwig amination to give 10a–c. The nitro groups in these compounds were reduced to amines by catalytic hydrogenation to give 1,2-diaminonaphthalene derivatives 11a–c. With an intention to characterize the naphthalene ring SAR, 11a was subjected to bromination, which gave 4-bromonaphthalene derivative 11d, and the structure was confirmed by one-dimensional (1D) and two-dimensional (2D) NMR experiments. The 1,2-diamino compounds were converted to the corresponding diazepinodiones 13a–d in moderate yields by reacting with malonyl chloride in refluxing toluene. Compounds 11d and 13d were prepared via bromination of 11a and 13a at slightly elevated temperatures in comparable yields. To increase the formation of 13a, a sequential addition of monomethyl malonate, DCC, potassium t-butoxide gave a cyclized moiety in a slightly improved conversion (51%) compared to the malonyl chloride procedure (40%).42 A similar one-pot procedure using ethyl chloromalonate gave only low product amounts. However, a two-step protocol improved the yield to 70% overall, in which ethyl chloromalonate was coupled to the α-amino group, and the intermediate was purified and reacted with potassium t-butoxide to form diazepine analogue 13a.
To add functionality to the naphthalene core, the 6/7-methyl groups in 13b and 13c were modified (Scheme 2). Various benzylic bromination methods were tried, involving NBS and Br2 as reactants, AIBN and benzoyl peroxide as radical initiators, and benzene, 1,2-dichloroethane, CCl4, and CH2Cl2 as solvents. Among these combinations, NBS, (BzO)2, CH2Cl2/benzene gave the best conversion and isolated yields ranging from 40–65%. However, this process also gave varying amounts of 4-bromo derivatives (13e, 14b, and 16b), which were inseparable using normal silica gel chromatography. The benzyl bromides were reacted with nucleophiles to give the methylthio analogue 13f, and azide analogues 13g, h, j, and k. The azide group in 7-azidomethylene analogue 13g was converted to an amine by a Staudinger reduction, and the amine was acetylated to give 13i.
Scheme 2.

Synthesis of Intermediates from Aromatic Methyl Functionalizationa
aReagents and conditions: (a) benzene, NBS, (BzO)2, 85 °C, 18 h, 44–65%; (b) DMF, sodium thiomethoxide, rt, 1 h, quant. conv.; (c) DMF, NaN3, 50 °C, 18 h, 44–82% (overall yield); (d) THF-H2O (9:1), PMe3, rt, 5 h; (e) THF, acetic anhydride, TEA, rt, 18 h, 89% over two-steps; (f) CH2Cl2, NBS, (BzO)2, 85 °C, 3 h, 41%.
The bromine at the 4-position of naphthalene was used as a handle to install aryl, alkane, alkyne, and arylalkyne/heteroarylalkyne groups (Scheme 3). A palladium-catalyzed Suzuki coupling gave 13l, while a Sonogashira coupling gave 13m–o and 17. A direct Pd–C hydrogenation of 17 gave trimethylsilylethylene derivative 18, while tetra-n-butylammonium fluoride (TBAF)-mediated deprotection of tetramethylsilane (TMS) followed by catalytic saturation gave 4-ethylnaphthalene derivative 13p.
Scheme 3.

Synthesis of Intermediates Derived from Pd-Coupling Chemistry on 4-Bromonaphthalene 13da
aReagents and conditions: (a) 1,2-dimethoxyethane, NEt3, PPh3, Pd(PPh3)2Cl2, acetylene, 55 °C, 6 h, 50–80%; (b) MeOH, 10% Pd–C, H2 (bubble), rt, 5 h, (c) 1,2-DME, H2O, PhB(OH)2, Pd(PPh3)4, Na2CO3, 90 °C, 5 h, 77%; (d) THF, TBAF, rt, 18 h, 63%; (e) EtOAc, 10% Pd–C, H2 (bubble), rt, 5 h, 61%.
The nitrile groups in compounds 13a–p were converted to N-hydroxyimidamides by reaction with hydroxylamine hydrochloride in the presence of triethylamine as a base (Scheme 4). Reacting these compounds with thiocarbonyldiimidazole (thio-CDI) in the presence of DBU gave the desired 1,5-dihydro-2H-naphtho[1, 2-b][1,4]diazepine-2,4(3H,5H)-dione analogues 21a–p. The amino-analogue 21q was made from the corresponding azido-analogue 21g by a trimethylphosphine-mediated Staudinger reduction. For better solubility, the compounds may be converted to the sodium salt (5, 22b,c) by reacting with an equimolar amount of dilute sodium hydroxide followed by lyophilization. All of the final products tested biologically, except 21a–c and 21r, were purified by HPLC (Figure S1, Supporting Information).
Scheme 4.

Synthesis of Analogues of 5 with Substitutions at the 4/6/7-Positions of the Naphthalene Ringa
aReagents and conditions: (a) THF-MeOH (1:2), TEA, NH2OH·HCl, 70 °C, 2 h, 59%—quantitative; (b) anhyd. CH3CN, DBU, thio-CDI, 0 °C → rt, 2 h, 2–80% (c) CH3CN-H2O (1:1), 1 equiv NaOH, quantitative; (d) THF-H2O (9:1), PMe3, rt, 5 h, 82%.
Synthesis of toluidine analogue 21r proceeded with ease (Scheme 5), but the synthesis of the aza-analogues proved difficult. Initial efforts to couple isonicotinonitrile with nitronaphthol triflate 9a failed, which led to screening of various higher-generation Pd catalysts and ligands, bases, and solvents for the Buchwald–Hartwig reaction (Table S2). The use of 1,4-dioxane as solvent dramatically increased yield. The solvent effect of dioxane43,44 was such that any combination of catalyst, ligand, and base gave product 10s in good yield. The method of choice for the synthesis of 10s–v was entry 8 in Table S2, although it gave a relatively slightly lower yield due to a more challenging purification. After nitro reduction to an amine, we faced difficulty in generating 13s, possibly because of low solubility in toluene and low reactivity of the cyanopyridine-linked amine, or both. The procedure via intermediate like 12a (characterized as 12as) did not give the desired product, either by use of bases sodium t-butoxide or NaH or by heating in toluene with/without triethylamine. Hence, various solvent systems were compared for the malonyl chloride reaction. Pyridine as a solvent degraded the starting material, while in 3:1 toluene-dioxane the reaction proceeded with a slightly higher yield compared to toluene/1,2-DCE (2:1, by volume). For the preparation of 13u, an excess of malonyl chloride (5 equiv) and lower concentrations of reactants were exploited to avoid the formation of a dimer from two molecules of 11u bridged by one malonyl moiety. Disappointingly, 13s treated with 10 equiv of hydroxylamine gave a diazepinedione ring-opened product both at 70 °C and at rt. However, with 2 equiv hydroxylamine at rt for 2 h, the desired 20s was the major product. This hydroxyimidamide reacted with thio-CDI to produce 21s in moderate yields. The similar reaction sequence to generate 21s from 9a led to the synthesis of other aza-analogues 21t–v in acceptable yields.
Scheme 5.

Synthesis of Magic-Methyl/Aza-Scan Compounds 21r–va
aReagents and conditions: (a) toluene, Ar-NH2, PPh3, Pd(PPh3)4, K2CO3, 110 °C, 16 h, 95% (for 10r); hetAr-NH2, PPh3, Pd(PPh3)4, Cs2CO3, 110 °C, 16 h, 55–77% (for 10s–v); (b) THF-MeOH (2:1), Pd–C, H2, rt, 4 h, 62–93%; (c) toluene, malonyl chloride, 0 °C → rt, 80 °C, 20 min, 110 °C, 10 min, 54% (for 13r); 3:1 toluene-dioxane, malonyl chloride, rt → 110 °C, 30 min, 24–53% (for 13s–v); (d) THF-MeOH (1:2), 10 equiv TEA, 10 equiv NH2OH·HCl, 70 °C, 2 h, quantitative (for 20r); THF-MeOH (1:2), 2 equiv TEA, 2 equiv NH2OH·HCl, rt, 2 h, 75–91% (for 20s–v); (e) anhyd. CH3CN, DBU, thio-CDI, 0 °C → rt, 2 h, 45–75%.
Pharmacological Evaluation.
In Vitro Characterization.
Twenty compounds were tested in ion channel assays for inhibition of human (h) P2XRs (Table 1, Figure S2, Supporting Information), including at homotrimeric P2X1R, P2X3R, and P2X4R and at a heteromeric P2X2/3R. The assays used HEK-293 cells stably transfected with the P2X4R and CHO-K1 cells stably transfected with either P2X1R, P2X2/3R, and P2X3R in 96- and 386-well plates. Detection of each well utilized either a luminescence assay using a luminescent Ca2+-sensitive photoprotein for hP2X1R and hP2X3R or a fluorescent Ca2+-sensitive dye for hP2X2/3R and hP2X4R. Full dose–response curves were determined for both reference agonist and antagonist at each receptor (not shown). The IC50 values reported for the test compounds represent the inhibition of an ~EC80 concentration of the reference agonist (Table 1, footnote a). A robust Z′ (RZ′) parameter was determined for each assay plate and ranged from 0.683 to 0.903, which indicated the robust quality of the assay. The antagonists were not evaluated at the mouse (m) P2X4R, but we note that the lead compound 5 was as potent in blocking mP2X4R effects in vitro as at hP2X4R, and it also showed considerable in vivo efficacy in the mouse.12
Table 1.

| |||||
|---|---|---|---|---|---|
| compound | structure | hP2X1R (IC50 or % inhib.)c |
hP2X2/3R (IC50 or % inhib.)c |
hP2X3R (IC50 or % inhib.)c |
hP2X4R (IC50 or % inhib.)c |
| TNP-ATP d | 0.120 | 0.064 | 0.018 | 2.77 | |
| 5 | R1 = R2 = R3 = H | −6% | 10% | 3% | 1.35 |
| 22b | R1 = CH3, R2 = R3 = H | 6% | 9% | 13% | 17.9 |
| 22c e | R1 = H, R2 = CH3, R3 = H | −4% | 11% | 6% | 1.38 |
| 21d | R1 = R2 = H, R3 = Br | 5% | 11% | 7% | 9.51 |
| 21e | R1 = CH3, R2 = H, R3 = Br | −2% | −5% | 17% | 11.8 |
| 21f | R1 = CH2SCH3, R2 = R3 = H | ND | ND | ND | ND |
| 21g | R1 = CH2N3, R2 = R3 = H | 8% | 4% | 13% | 6% |
| 21h | R1 = CH2N3, R2 = H, R3 = Br | −13% | −4% | 15% | 8% |
| 21i | R1 = CH2NHAc, R2 = R3 = H | 26% | −5% | 9% | 16% |
| 21j | R1 = H, R2 = CH2N3, R3 = H | −5% | 2% | 0% | 21.3 |
| 21k | R1 = H, R2 = CH2N3, R3 = Br | ND | ND | ND | ND |
| 21l | R1 = R2 = H, R3 = Ph | 14% | 10% | 11% | 20.7 |
| 21m | R1 = R2 = H, R3 = C≡C−Ph | 14% | 5% | 7% | −4% |
| 21n | R1 = R2 = H, R3 = C≡C−(3-Py) | 9% | 13% | 9% | 34% |
| 21o | R1 = R2 = H, R3 = C≡C−n-Bu | 16% | 1% | 18% | 15% |
| 21p | R1 = R2 = H, R3 = Et | 6% | 10% | 6% | 5.13 |
| 21q | R1 = CH2NH2, R2 = R3 = H | 8% | 0% | 9% | 9% |
| 21r | X = CCH3, W = Y = Z = ch | 16% | 5% | 12% | 14.0 |
| 21s | W = N, X = Y = Z = CH | 18% | −6% | 18% | 13.5 |
| 21t | X = N, W = Y = Z = CH | 15% | 25% | 6% | 14.6 |
| 21u e | Y = N, W = X = Z = CH | 15% | 0% | 11% | 0.503 |
| 21v | Z = N, W = X = Y = CH | 31% | 15% | 17% | 6.48 |
ND, not determined.
Measured at hP2X4R (fluorescence), at hP2X1R and hP2X3R (luminescence), and at hP2X2/3R (fluorescence). Fixed agonist concentration roughly corresponds to the predetermined EC80 value of each: CTP, 10 μM at P2X4R; α,β-meATP: 1.0 μM at P2X1R, 5.0 μM at P2X2/3R, 1.0 μM at P2X3R. None of the analogues of 5 tested here had activity as P2XR agonists.
IC50 in μM, or percent inhibition at 30 μM concentration in italics. Each data point was determined in quadruplicate.
2′,3′-O-Trinitrophenyl-adenosine-5′-triphosphate (TNP-ATP) was used as a reference antagonist. Reference agonists displayed the following EC50 values in these assays: CTP, 5.28 μM at P2X4R; α,β-meATP, 0.658 μM at P2X1R, 3.17 μM at P2X2/3R, 1.10 μM at P2X3R.
Compounds evaluated in vivo.
The 6-methyl substitution of the naphthalene core (22c) did not lower the inhibitory activity of known antagonist 5. However, a 7-methyl substitution alone in 22b decreased activity by 13-fold, and larger/polar substitution with azidomethyl 22h or acetamidomethyl 22i at this position rendered the molecule inactive. A large azidomethyl group at the 6-position (21j) was destabilizing, although not to the same extent as 7-substituted analogues. However, a 4-bromo substitution in the absence (21d) or presence of 6-methyl (21e) only moderately reduced activity.
The results of the aza-scan showed the substantial effect of a ring nitrogen, either to enhance (4-pyridyl, 21u) or to reduce (21s, 21t, 21v) inhibitory activity with respect to reference antagonist 5. The active compounds were selective for the P2X4R; the most inhibition observed at any other receptor was at the P2X1R, which was ≤31% at 30 μM for compounds 21v and 21i.
In addition to showing that the P2X4R antagonists were selective for that receptor subtype within the purinergic family, we carried out broader screening at other cell surface signaling proteins. Off-target binding activity of 5, 22c, 21d, and 21u was determined at 45 receptors, channels, and transporters by the NIMH Psychoactive Drug Screening Program (PDSP).45 Significant binding of 21u (Ki, nM, mean ± standard error of the mean (SEM)) occurred only at D3 dopamine (0.77 ± 0.37) and 5-HT2C serotonin (6.0 ± 0.1) receptors (see Supporting Information Tables S3 and S4 for details). There were no significant interactions at 10 μM for 5, 22c, and 21d.
In Vivo Characterization of Neuroprotective Activity of Lead Compounds 21u and 22c.
Based on IC50 values, we chose the two highest activity P2X4R antagonists for an in vivo activity assay. We previously showed that the commercially available P2X4R inhibitor 5-(3-bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e][1,4]diazepin-2-one (3, 5-BDBD, 1 mg/kg/day × 3 days) reduced stroke infarct size in mice in the MCAO model when administered 4 h after the onset of acute stroke for a total of 3 days.13 Using the same pharmacological paradigm, we infused (by subcutaneous implantation of Alzet minipump 1003D) the 3-pyridyl analogue 21u at 0.5–3.0 mg/kg/day after ischemic stroke for a total of 3 days. We found that doses 1.5 and 3.0 mg/kg caused significant neuroprotection using total hemispheric infarct volume size as an outcome. The optimal dose of 1.5 mg/kg showed a greater level of reduction in cortical infarct volume compared to a high dose. This observation implied a ceiling effect at the latter dose (Figure 1). Interestingly, 21u (1.5 mg/kg × 3 day) also showed a neuroprotective effect by reducing infarct damage (after 3 days of stroke) and tissue atrophy (after 35 days of stroke) in middle-aged mice (Figure 2), suggesting antistroke efficacy across age and the translational relevance of 21u in the treatment of stroke. In studying the less potent 6-methyl analogue 22c, which has a 3-fold higher IC50 value than 21u, we tested the higher dose of 5 mg/kg/day. We found a moderate but significant decrease in total hemispheric infarct volume while effects were not significant on cortical or striatal infarct volume alone (Figure 3).
Figure 1.

Dose-dependent effect of 21u on infarct volume after ischemic stroke. Representative triphenyltetrazolium chloride- (TTC) stained sections showing infarct area (dotted line) in the top panel. Three-day treatment with 21u (0.5–3 mg/kg/day × 3 days continuous infusion with an Alzet minipump) in young adult male mice (8–12 weeks old) significantly reduced total hemispheric and cortical infarct volume at doses of 1.5 and 3.0 mg/kg (n = 5–6 mice/dose; total 21 male mice) measured at 3 days after stroke. **p < 0.01; vs Veh *p < 0.05; vs Veh in respective brain regions (one-way analysis of variance (ANOVA) followed Tukey’s post hoc test). Data are mean ± standard deviation (S.D.).
Figure 2.

Effect of 21u on infarct volume and brain atrophy in middle-aged mice. (A) Representative TTC-stained sections showing infarct area (dotted line) in the top panel. Three-day treatment with 21u (1.5 mg/kg/day × 3 days continuous infusion with an Alzet minipump) in 12- to 13-month-old mice significantly reduced total hemispheric and cortical infarct volume (n = 5/group = 10 mice total; 6 male and 4 female) measured at 3 days after stroke. (*p < 0.05; vs Veh in respective brain regions, unpaired t-test). Data are mean ± S.D. (B) Representative CV-stained sections showing % reduction in brain tissue. Consistent with decrease in infarct size, 3-day treatment with 21u (1.5 mg/kg/day × 3 days continuous infusion with an Alzet minipump) significantly prevented tissue loss when measured 35 days after stroke (n = 7–9/group = 16 mice total; 8 male and 8 female; *p < 0.05; vs Veh unpaired T test). Data are mean ± S.D.
Figure 3.

Effect of treatment with compound 22c on infarct volume after ischemic stroke (MCAO). Representative TTC-stained sections showing infarct area (dotted line) in the top panel. Three-day treatment with 22c (5.0 mg/kg/day × 3 days continuous infusion with an Alzet minipump) significantly (*p < 0.05; vs Veh, unpaired t-test) reduced total hemispheric infarct volume in male young adult mice at 3 days after stroke (8–12 weeks old) (n = 5/treatment group = 10 mice; all male). Data are expressed as mean ± S.D.
Overall, our in vivo data are consistent with our prior finding that short-term blockade of P2X4R confers sustained neuroprotection after stroke,13 as the reduced atrophy suggests a prolonged benefit.
Effect of 21u on Post-Stroke Sensory Motor Functions.
Following ischemic stroke (MCAO), vehicle-and 21u-treated groups did not show any differences in the total exploratory behavior as measured by total beam breaks in open-field task at any time point, suggesting no effect of this antagonist on gross locomotion activity (Figure 4A). Contrary to our expectations, treatment with 21u did not improve motor coordination and balance as assessed using rotarod (Figure 4B). These data suggest that 21u treatment may not affect sensory motor function during the recovery phase of stroke. Male and female separate analyses did not show any sex-specific effect (not shown here).
Figure 4.
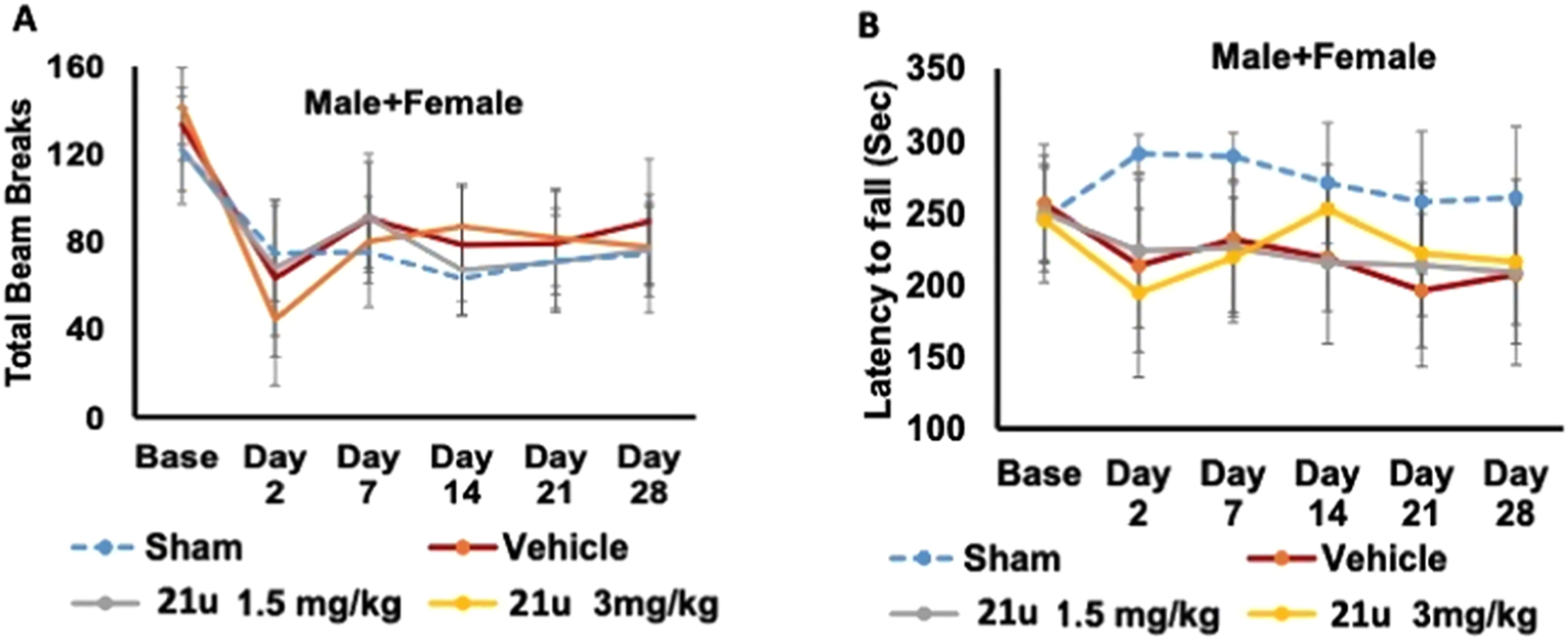
Effect of 21u on post-stroke sensorimotor deficits in middle-aged mice (11–12 months old). (A) Treatment with 21u neither caused any change in total locomotor activity (B) nor affected motor balance coordination in the rotarod task (rotating speed; 4–40 rpm, 5 min) over 4 weeks after stroke. Data are mean ± S.D. (n = 8–15/treatment group = 45 mice total; 25 males and 20 females).
Effect of Treatment with 21u on Post-Stroke Learning and Memory Deficit.
Both 21u- and vehicle-treated mice were assessed on the Novel Object Recognition Task (NORT)49 4 weeks after MCAO in middle-aged mice (Figure 5). Mice showed a dose-dependent improvement in learning and memory function after stroke, and a dose of 3 mg/kg showed significant retention of memory (p < 0.05 vs Veh.) after stroke suggesting neuro-rehabilitative effects after short-term treatment with 21u.
Figure 5.

Treatment with 21u improved post-stroke learning and memory in middle-aged mice. 21u-treated middle-aged mice showed dose-dependent improvement in learning and memory after stroke and reached statistical significance at a dose of 3 mg/kg (*p < 0.05 vs Veh, one-way ANOVA followed by Tukey’s multiple comparison test), measured in the Novel Object Recognition Test (n = 5–11/treatment group = 32 mice total; 19 males and 13 females). Data are mean ± S.D.
Effects of 21u on Calcium Influx in Human Monocyte-Derived Macrophages.
Primary human monocytes were isolated and matured into macrophages as described in the Experimental Methods section and plated on an Ibidi cell imaging dish overnight before the assay. Human cells responded to ATP (50 μM) with an increase in intracellular Ca2+ response. Compound 21u caused a dose-dependent (0–3 μM) inhibition of the ATP-induced (50 μM) increase in calcium influx (Figure 6A), resulting in a significant decrease of the peak Ca2+ altitude at doses of 0.6 μM or higher (Figure 6B). The data suggest that 21u can inhibit P2X4R-mediated Ca2+ influx in human subjects, with implication for clinical translation.
Figure 6.

(A) Calcium transients in response to ATP (50 μM) in primary human monocyte-derived macrophages (HMDM) of healthy subjects (N = 21–43 total cells from three separate subjects with two technical replicate). HMDMs were pretreated with 0, 0.1, 0.3, 0.6, 1, and 3 μM 21u for 15 min prior to the exposure of ATP; time-lapse images were acquired up to 4 min (repeating every 6 s) using a Zeiss LSM880 laser scanning confocal microscope. (B) Each bar shows the quantification of cytosolic Ca2+ peak amplitude without (0) or with 21u. Data are mean ± SEM. *P < 0.05, **P < 0.01 vs ATP only (21u at 0); one-way ANOVA with Tukey’s multiple comparisons test. F = Average fluorescence intensity after ATP exposure, F0 = fluorescence intensity prior to ATP exposure.
DISCUSSION
To achieve the desired aromatic functionalization and aza-scan modifications, we explored extensive modification of the reported synthetic route to a series of P2X4R antagonists derived from compound 5. We identified optimal multistep reaction conditions, including reagents, catalysts, solvent, and time/temperature of reaction needed to perform the appropriate synthetic transformations. Among the most challenging steps was the introduction of a malonyl group, which required a two-step process for several of the analogues. In addition, difficulties with nitration, Buchwald–Hartwig amination, and hydroxyimidamide formation were tackled to access the target molecules.
SAR experiments were conducted on novel compounds for their in vitro potency and selectivity at P2X4R. Although only ~μM activity at the target P2X4R was reached, this compound set expands the number of useful antagonists and provides a guide for future derivatization. There was no off-target inhibition of three other P2XR channels and few interactions with other receptors. Hence, based on the limited SAR, it may be safely assumed that large/polar variations at the 7-position of the naphthalene ring is not preferred, while the 6-position is amenable to substitution in 22c. This is consistent with sparse biological data disclosed in the patent by Sakuma et al.39 Also, an aza-scan of the N-phenyl ring indicated a strong preference for the 4-pyridyl group (21u) compared to other positions of nitrogen incorporation in the ring. Among the selective P2X4R blockers, the two most potent compounds, 21u and 22c, were further characterized for in vivo neuroprotective efficacy in a murine stroke model.
Thrombolytic alteplase (t-PA) and endovascular thrombectomy are the existing therapies to treat ischemic stroke. While they have increased reperfusion and improved clinical outcomes, nearly half of ischemic stroke patients do not recover to achieve an independent lifestyle. Mortality and morbidity remain high after ischemic stroke, indicating a high unmet medical gap necessitating novel approaches. A principal objective is to develop a new antistroke medical therapy by blocking the pro-inflammatory myeloid P2X4R, which contributes to inflammation-mediated injury during stroke.46
To study whether in vitro P2X4R inhibition activity of 21u and 22c can be translated into in vivo efficacy, we studied their neuroprotective and neuro-rehabilitative activity in a mouse model of ischemic stroke produced by transient occlusion of middle cerebral artery and followed by reperfusion. Three-day continuous administration of either P2X4R antagonist after stroke reduced infarct volume in young adult mice, although the efficacy and dose varied. Consistent with in vitro data, 21u was more efficacious and showed more potent neuroprotective effects at a dose of 1.5 mg/kg. The efficacy of 21u in reducing infarct size and tissue atrophy in middle-aged mice after stroke suggest its therapeutic potential in the vulnerable aging population. Adding to the potential for clinical translation, 21u was able to inhibit ATP-induced calcium influx in primary human monocyte-derived macrophages, suggesting that 21u could be effective in humans. Memory loss is the most common post-stroke sequel. The effectiveness of 21u in improving learning and memory tasks further reinforces its translationally relevant therapeutic potential even during the long-term functional recovery period after stroke. The latter finding is an extension of our previous observation13 where we showed the short-term blockade of P2X4R improves during long-term recovery, which is mediated by neuroprotective factors such as BDNF. It is well established that BDNF is involved in maintaining synaptic plasticity, neuronal survival, learning and memory, and neuroplasticity after stroke.47 Therefore, it is plausible that neuroprotective and neuro-rehabilitative effects of 21u are achieved via increased neuroprotective factors during recovery phase after stroke. These novel P2X4R antagonists with established potency and selectivity at both murine and human P2X4Rs, coupled with in vivo efficacy at neuroprotection in ischemic stroke, provide preclinical proof of concept for translation into new therapeutic small molecule drugs for this critical unmet medical condition.
CONCLUSIONS
A focused but broad-based SAR study was performed on compound 5, which led to 21u, 3-fold more potent for P2X4R than the reference compound 5 and comparably selective. Synthesis was achieved after optimization of some key steps: viz., nitration, diazepinedione formation, Buchwald–Hartwig amination, and hydroxyimidamide formation reactions. The narrow SAR revealed the 6-position of naphthalene as a potential modification site along with N-(4-pyridyl) moiety, paving the way for further improvements. Compound 21u displayed neuroprotective and neuro-rehabilitative properties in a murine middle cerebral artery ischemic stroke model with an effective dose of 1.5 mg/kg. This study indicates the translational potential of P2X4R antagonists for treating ischemic stroke, including in aging populations.
EXPERIMENTAL METHODS
Chemical Synthesis.
Reagents and Instrumentation.
6/7-Methyl-naphthalene-2-ol derivatives and aminopyridine nitriles were purchased from Enamine (New Jersey). All other chemicals and solvents were from Sigma-Aldrich (St. Louis, MO). Anhydrous solvents were obtained directly from commercial sources. All reactions were carried out under argon using anhydrous solvents. Rt or rt refers to 25 ± 2 °C. NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts are given in ppm (δ), calibrated to the residual solvent or TMS signals for hydrogen, carbon, and internally calibrated by solvent frequency for other nuclei (MestReNova 10.0.2). Exact mass measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) mass spectrometer equipped with a standard electrospray ionization (ESI) and modular LockSpray interface. Reversed-phase high-performance liquid chromatography (RP-HPLC) was performed using a Phenomenex Luna 5 μm C18(2)100A, AXIA, 21.2 mm × 250 mm column. Purity was determined using Agilent C18-XDB, 5 μm, 4.6 mm × 250 mm column, and a 0 → 100% linear gradient of acetonitrile/10 mM triethylammonium acetate as mobile phase at 1 mL/min flow rate for 20 min. Purity of all of the tested compounds was >95% at 254 nm and/or respective absorption wavelengths are in nm, unless noted otherwise.
1-Nitronaphthalen-2-ol (8a).
To a solution of 2-naphthol (100 mg, 0.69 mmol) in dichloromethane (2 mL) at rt was added fuming nitric acid (>99.5% HNO3, 32 μL, 0.08 mmol) and the mixture was stirred for additional 5 min. Silica gel (0.5 g) was added, and volatiles were evaporated to adsorb the product on the silica gel. Purification by silica gel chromatography afforded 8a as yellow crystals (80 mg, 61%, Rf = 0.3, thin-layer chromatography (TLC) eluent = 10% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 12.19 (s, 1H), 8.93 (d, J = 8.8 Hz, 1H), 8.01 (d, J = 9.0 Hz, 1H), 7.83 (dd, J = 8.1, 1.3 Hz, 1H), 7.74 (ddd, J = 8.6, 7.0, 1.4 Hz, 1H), 7.51 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 7.27 (d, J = 9.0 Hz, 1H). HRMS m/z [M − H]− for C10H7O3N calculated 188.0348, found 188.0346.
7-Methyl-1-nitronaphthalen-2-ol (8b).
Following the procedure described for the synthesis of 8a, compound 7b (100 mg, 0.63 mmol) gave 8b as yellow crystals (65 mg, 51%, Rf = 0.3, TLC eluent = 10% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 12.21 (s, 1H), 8.74 (s, 1H), 7.95 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.34 (dd, J = 8.3, 1.5 Hz, 1H), 7.18 (d, J = 9.0 Hz, 1H), 2.59 (s, 3H). HRMS m/z [M − H]− for C11H9O3N calculated 202.0504, found 202.0504.
6-Methyl-1-nitronaphthalen-2-ol (8c).
Following the procedure described for the synthesis of 8a, compound 7c (150 mg, 0.95 mmol) gave 8c as yellow crystals (101 mg, 52%, Rf = 0.3, TLC eluent = 10% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 12.21 (s, 1H), 8.83 (d, J = 8.9 Hz, 1H), 7.93 (d, J = 9.1 Hz, 1H), 7.60 (s, 1H), 7.57 (d, J = 9.2 Hz, 1H), 7.23 (d, J = 9.0 Hz, 1H), 2.51 (s, 3H). HRMS m/z [M − H]− for C11H9O3N calculated 202.0504, found 202.0507.
1-Nitronaphthalen-2-yl trifluoromethanesulfonate (9a).
To a solution of 8a (190 mg, 1.0 mmol) in dichloromethane (5 mL) at 0 °C was added triethylamine (0.35 mL, 2.5 mmol), followed by trifluoromethanesulfonic anhydride (0.2 mL, 1.2 mmol), and the mixture was stirred at rt for 2 h. The reaction mixture was partitioned between water-dichloromethane. The organic layer was separated, dried over anhydrous Na2SO4, and evaporated. The residue was purified by silica gel chromatography to afford 9a as a light yellow solid (275 mg, 85%, Rf = 0.25, TLC eluent = 10% ethyl acetate in hexanes; Note: if the reaction is incomplete, NaHCO3 workup would leave starting material in aqueous medium, and 9a was extracted into the organic phase, while the starting material 8a was recovered after acidification, followed by extraction). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 9.2 Hz, 1H), 7.99 (d, J = 8.2 Hz, 1H), 7.95−7.89 (m, 1H), 7.77 (ddd, J = 8.5, 6.9, 1.3 Hz, 1H), 7.71 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.55 (d, J = 9.2 Hz, 1H). 19F NMR (376 MHz, CDCl3) δ −73.04. HRMS m/z [M − H]− for C11H5NO5F3S calculated 319.9841, found 319.9842.
7-Methyl-1-nitronaphthalen-2-yl trifluoromethanesulfonate (9b).
Following the procedure described for the synthesis of 9a, compound 8b (65 mg, 0.32 mmol) gave 9b as yellow crystals (85 mg, 80%, Rf = 0.3, TLC eluent = 10% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.05 (d, J = 9.1 Hz, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.66 (s, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.46 (d, J = 9.1 Hz, 1H), 2.58 (s, 3H). 19F NMR (376 MHz, chloroform-d) δ −73.09.
6-Methyl-1-nitronaphthalen-2-yl trifluoromethanesulfonate (9c).
Following the procedure described for the synthesis of 9a, compound 8c (180 mg, 0.886 mmol) gave 9c as yellow crystals (195 mg, 66%, Rf = 0.3, TLC eluent = 10% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.01 (d, J = 9.1 Hz, 1H), 7.82 (d, J = 8.8 Hz, 1H), 7.75 (s, 1H), 7.59 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 9.1 Hz, 1H), 2.58 (s, 3H). 19F NMR (376 MHz, chloroform-d) δ −73.09.
3-((1-Nitronaphthalen-2-yl)amino)benzonitrile (10a).
To a round-bottom (RB) flask with a stir bar were added 9a (275 mg, 0.856 mmol), triphenylphosphine (PPh3, 225 mg, 0.856 mmol), tetrakis(triphenylphosphine)palladium (Pd(PPh3)4, 99 mg, 0.086 mmol), potassium carbonate (K2CO3, 119 mg, 0.856 mmol), and 3-cyanoaniline (152 mg, 1.284 mmol). The flask was evacuated and filled with nitrogen, followed by toluene (10 mL), and the mixture was heated to 110 °C for 16 h. The solvent was evaporated, and the residue was partitioned between dichloromethane and water. The organic layer was separated, washed with 0.2 M hydrochloric acid and brine, and dried over anhydrous Na2SO4. The solvent was evaporated and the residue was purified by silica gel chromatography to afford 10a as brown-orange crystals (210 mg, 85%, Rf = 0.25, TLC eluent = 50% CH2Cl2 in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.00 (s, 1H), 8.38 (d, J = 8.7 Hz, 1H), 7.87 (d, J = 9.2 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.66 (ddd, J = 8.6, 7.0, 1.4 Hz, 1H), 7.57−7.43 (m, 5H), 7.37 (d, J = 9.1 Hz, 1H). HRMS m/z [M + H]+ for C17H12N3O2 calculated 290.0930, found 290.0929.
3-((7-Methyl-1-nitronaphthalen-2-yl)amino)benzonitrile (10b).
Following the procedure described for the synthesis of 10a, compound 9b (80 mg, 0.24 mmol) gave 10b as a brown-orange crystal (45 mg, 62%, Rf = 0.25, TLC eluent = 50% CH2Cl2 in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.91 (s, 1H), 8.15 (s, 1H), 7.81 (d, J = 9.1 Hz, 1H), 7.67 (d, J = 8.2 Hz, 1H), 7.56−7.41 (m, 4H), 7.34−7.28 (m, 2H), 2.55 (s, 3H). HRMS m/z [M + H]+ for C18H13N3O2 calculated 304.1086, found 304.1083.
3-((6-Methyl-1-nitronaphthalen-2-yl)amino)benzonitrile (10c).
Following the procedure described for the synthesis of 10a, compound 9c (195 mg, 0.582 mmol) gave 10c as a brown-orange crystal (120 mg, 68%, Rf = 0.25, TLC eluent = 50% CH2Cl2 in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.98 (s, 1H), 8.29 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 9.1 Hz, 1H), 7.56 (s, 1H), 7.54−7.41 (m, 4H), 7.35 (d, J = 9.1 Hz, 1H), 2.50 (s, 3H).
3-Methyl-5-((1-nitronaphthalen-2-yl)amino)benzonitrile (10r).
Following the procedure described for the synthesis of 10a, compound 9a (100 mg, 0.311 mmol) gave 10r as an orange crystal (90 mg, 95%, Rf = 0.25, TLC eluent = 50% CH2Cl2 in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.04 (s, 1H), 8.42 (d, J = 8.8 Hz, 1H), 7.87 (d, J = 9.1 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.67 (dd, J = 8.6, 7.0 Hz, 1H), 7.47 (t, J = 7.5 Hz, 1H), 7.41−7.34 (m, 2H), 7.29 (s, 2H), 2.43 (s, 3H).
2-((1-Nitronaphthalen-2-yl)amino)isonicotinonitrile (10s).
Compound 9a (50 mg, 0.156 mmol), 2-amino-4-cyanopyridine (28 mg, 0.233 mmol), Pd(PPh3)4 (18 mg, 0.016 mmol), triphenylphosphine (41 mg, 0.156 mmol), and Cs2CO3 (76 mg, 0.233 mmol) where charged to a round-bottom flask and deoxygenated with vacuum-argon cycles. Dioxane (2 mL) was added and the mixture was stirred at 110 °C for 18 h. The solvent was evaporated, and the residue was partitioned between dichloromethane and water. The organic layer was separated, washed with brine, and dried over anhydrous Na2SO4. The solvent was evaporated, and the residue was purified by silica gel chromatography to afford 10s as brown-orange crystals (30 mg, 66%, Rf = 0.25, TLC eluent = CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 8.88 (s, 1H), 8.49−8.42 (m, 1H), 8.17 (dd, J = 9.0, 6.6 Hz, 2H), 8.01 (d, J = 9.1 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.68 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 7.54 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 7.17 (t, J = 1.1 Hz, 1H), 7.14 (dd, J = 5.1, 1.3 Hz, 1H). HRMS m/z [M + H]+ for C16H10N4O2 calculated 291.0882, found 291.0881.
5-((1-Nitronaphthalen-2-yl)amino)nicotinonitrile (10t).
Following the procedure described for the synthesis of 10s, compound 9a (100 mg, 0.311 mmol) gave 10t as an orange crystal (50 mg, 55%, Rf = 0.60, TLC eluent = 2% MeOH in CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 8.76 (d, J = 2.7 Hz, 1H), 8.72 (s, 1H), 8.64 (d, J = 1.8 Hz, 1H), 8.32 (d, J = 8.7 Hz, 1H), 7.96 (d, J = 9.1 Hz, 1H), 7.89−7.77 (m, 2H), 7.71 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.58−7.49 (m, 1H), 7.40 (d, J = 9.1 Hz, 1H).
4-((1-Nitronaphthalen-2-yl)amino)picolinonitrile (10u).
Following the procedure described for the synthesis of 10s, compound 9a (200 mg, 0.63 mmol) gave 10u as a yellow solid (140 mg, 77%, Rf = 0.30, TLC eluent = 100% CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 8.51 (d, J = 5.6 Hz, 1H), 8.18−8.10 (m, 2H), 8.06 (d, J = 9.0 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.73 (ddd, J = 8.6, 7.0, 1.4 Hz, 1H), 7.64−7.56 (m, 2H), 7.40 (d, J = 2.3 Hz, 1H), 7.16 (dd, J = 5.7, 2.4 Hz, 1H). HRMS m/z [M + H]+ for C16H10N4O2 calculated 291.0882, found 291.0883.
4-((1-Nitronaphthalen-2-yl)amino)picolinonitrile (10v).
Following the procedure described for the synthesis of 10s, compound 9a (200 mg, 0.63 mmol) gave 10v as a yellow solid (140 mg, 77%, Rf = 0.20, TLC eluent = 50% CH2Cl2 in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.04 (s, 1H), 8.40 (d, J = 9.2 Hz, 1H), 8.17 (d, J = 8.7 Hz, 1H), 8.02 (d, J = 9.2 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.74 (dd, J = 8.5, 7.4 Hz, 1H), 7.67 (ddd, J = 8.6, 6.9, 1.3 Hz, 1H), 7.53 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 7.35 (d, J = 7.4 Hz, 1H), 7.11 (d, J = 8.4 Hz, 1H). HRMS m/z [M + H]+ for C16H10N4O2 calculated 291.0882, found 291.0880.
3-((1-Aminonaphthalen-2-yl)amino)benzonitrile (11a).
To a solution of compound 10a (210 mg, 0.725 mmol) in 1:2 methanol-THF (12 mL) was added 10% Pd–C (100 mg) and the mixture was subjected to a stream of bubbled hydrogen for 4 h at rt. The catalyst was filtered off, and the filtrate was concentrated under vacuum. The residue was purified by silica gel chromatography to afford 5a as a yellowish solid (166 mg, 88%, Rf = 0.50, TLC eluent = 100% CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 8.33−8.12 (m, 3H), 7.98−7.80 (m, 2H), 7.72 (d, J = 8.6 Hz, 1H), 7.67−7.57 (m, 3H), 7.45 (d, J = 7.5 Hz, 1H), 7.27 (d, J = 6.7 Hz, 1H), 5.87 (s, 2H). HRMS m/z [M + H]+ for C17H14N3 calculated 260.1188, found 260.1184.
3-((1-Amino-7-methylnaphthalen-2-yl)amino)benzonitrile (11b).
Following the procedure described for the synthesis of 11a, compound 10b (45 mg, 0.148 mmol) gave 11b as a yellowish solid (25 mg, 62%, Rf = 0.5, TLC eluent = 100% CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 8.13 (dt, J = 8.4, 2.1 Hz, 1H), 8.03 (s, 1H), 7.78−7.58 (m, 3H), 7.54 (dt, J = 8.5, 2.2 Hz, 1H), 7.49−7.40 (m, 1H), 7.27 (d, J = 9.2 Hz, 2H), 5.86 (s, 1H), 4.73 (s, 2H), 2.97 (d, J = 2.4 Hz, 3H). HRMS m/z [M]+ for C18H15N3 calculated 273.1266, found 273.1266.
3-((1-Amino-6-methylnaphthalen-2-yl)amino)benzonitrile (11c).
Following the procedure described for the synthesis of 11a, compound 10c (120 mg, 0.396 mmol) gave 11c as a yellowish solid (100 mg, 93%, Rf = 0.5, TLC eluent = 100% CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 7.74 (d, J = 8.6 Hz, 1H), 7.59 (s, 1H), 7.38−7.32 (m, 1H), 7.23 (d, J = 8.4 Hz, 3H), 7.17 (d, J = 8.6 Hz, 1H), 7.05 (d, J = 7.6 Hz, 1H), 6.87 (d, J = 6.9 Hz, 2H), 5.43 (s, 1H), 4.36 (s, 2H), 2.52 (s, 3H). MS m/z [M + H]+ for C18H15N3 calculated 274.1339, found 274.1.
3-((1-Amino-4-bromonaphthalen-2-yl)amino)benzonitrile (11d).
Compound 11a (13 mg, 0.05 mmol) was dissolved in glacial acetic acid (0.3 mL) and was added bromine (3 μL, 0.06 mmol). The reaction mixture was stirred for 0.5–1 h at rt, and the solvent was evaporated under reduced pressure. The residue was partitioned between sat. NaHCO3 and ethyl acetate. The organic layer was separated, dried over anhydr. Na2SO4, and evaporated. The residue was purified by silica gel column chromatography to afford 11d as a dark green solid (13 mg, 77%, Rf = 0.5, TLC eluent = 100% CH2Cl2). 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.92 (s, 1H), 7.59 (t, J = 7.4 Hz, 1H), 7.56−7.45 (m, 2H), 7.31 (t, J = 8.0 Hz, 1H), 7.07 (d, J = 7.5 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.85 (s, 1H). HRMS m/z [M + H]+ for C17H12BrN3 calculated 338.0199, found 338.0202.
3-((1-Aminonaphthalen-2-yl)amino)-5-methylbenzonitrile (11r).
Following the procedure described for the synthesis of 11a, compound 10r (90 mg, 0.297 mmol) gave 11r as a greenish solid (51 mg, 63%, Rf = 0.3, TLC eluent = 100% CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 7.92−7.80 (m, 2H), 7.52 (tt, J = 7.4, 3.5 Hz, 2H), 7.34 (d, J = 8.6 Hz, 1H), 7.22 (d, J = 8.6 Hz, 1H), 6.90 (s, 1H), 6.70 (dt, J = 10.0, 1.8 Hz, 2H), 5.40 (s, 1H), 4.40 (s, 2H), 2.28 (s, 3H). HRMS m/z [M + H]+ for C18H16N3 calculated 274.1344, found 274.1342.
2-((1-Aminonaphthalen-2-yl)amino)isonicotinonitrile (11s).
Following the procedure described for the synthesis of 11a, compound 10s (90 mg, 0.310 mmol) gave 11s as a greenish-yellow solid (50 mg, 62%, Rf = 0.4, TLC eluent = 5% methanol in CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 8.32 (dd, J = 5.0, 0.9 Hz, 1H), 7.86 (tt, J = 8.2, 3.3 Hz, 3H), 7.60−7.50 (m, 2H), 7.36 (d, J = 8.6 Hz, 1H), 7.26 (d, J = 8.6 Hz, 1H), 6.90 (dd, J = 5.1, 1.4 Hz, 1H), 6.55 (s, 1H), 6.51 (d, J = 1.1 Hz, 1H), 4.45 (s, 2H). HRMS m/z [M + H]+ for C16H12N4 calculated 261.1140, found 261.1140.
5-((1-Aminonaphthalen-2-yl)amino)nicotinonitrile (11t).
Following the procedure described for the synthesis of 11a, compound 10t (100 mg, 0.344 mmol) gave 11t as a greenish-yellow solid (50 mg, 56%, Rf = 0.6, TLC eluent = 5% methanol in CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 8.38 (d, J = 2.9 Hz, 1H), 8.31 (d, J = 1.8 Hz, 1H), 7.91−7.80 (m, 2H), 7.60−7.50 (m, 2H), 7.36 (d, J = 8.6 Hz, 1H), 7.21 (d, J = 8.6 Hz, 1H), 7.02 (dd, J = 2.8, 1.8 Hz, 1H), 5.62 (s, 1H), 4.40 (s, 2H). HRMS m/z [M + H]+ for C16H12N4 calculated 261.1140, found 261.1143.
4-((1-Aminonaphthalen-2-yl)amino)picolinonitrile (11u).
Following the procedure described for the synthesis of 11a, compound 10u (150 mg, 0.52 mmol) gave 11u as a yellowish solid (100 mg, 74%, Rf = 0.3, TLC eluent = 5% methanol in CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 8.31 (d, J = 5.7 Hz, 1H), 7.86 (dt, J = 6.6, 3.6 Hz, 3H), 7.56 (dt, J = 6.3, 3.3 Hz, 2H), 7.36 (d, J = 8.6 Hz, 1H), 7.20 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 2.4 Hz, 1H), 6.70 (dd, J = 5.8, 2.4 Hz, 1H), 5.97 (s, 1H), 4.38 (s, 2H). HRMS m/z [M + H]+ for C16H12N4 calculated 261.1140, found 261.1135.
6-((1-Aminonaphthalen-2-yl)amino)picolinonitrile (11v).
Following the procedure described for the synthesis of 11a, compound 10v (140 mg, 0.48 mmol) gave 11v as a yellowish solid (60 mg, 48%, Rf = 0.6, TLC eluent = 100% CH2Cl2). 1H NMR (400 MHz, chloroform-d) δ 7.85 (ddt, J = 7.9, 5.0, 3.1 Hz, 3H), 7.58−7.42 (m, 4H), 7.34 (d, J = 8.6 Hz, 1H), 7.26 (d, J = 9.4 Hz, 1H), 7.12 (d, J = 7.2 Hz, 1H), 6.52 (d, J = 8.6 Hz, 1H), 6.44 (s, 1H), 4.45 (s, 2H). HRMS m/z [M + H]+ for C16H12N4 calculated 261.1140, found 261.1140.
Ethyl 3-((2-((3-Cyanophenyl)amino)naphthalen-1-yl)amino)-3-oxopropanoate (12a).
To an ice-cold solution of compound 11a (100 mg, 0.386 mmol) in THF (2 mL) was added triethylamine (81 μL, 0.578 mmol), followed by ethyl malonyl chloride (55 μL, 0.424 mmol), and the mixture was stirred at rt for 3 h. The reaction was quenched by adding sat. NaHCO3, and the product was extracted in ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, evaporated, and the residue was purified by silica gel column chromatography to afford 12a as a white solid (100 mg, 70%, Rf = 0.6, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.61 (s, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 8.9 Hz, 1H), 7.63−7.50 (m, 2H), 7.46 (ddd, J = 8.1, 6.8, 1.1 Hz, 1H), 7.30 (t, J = 7.9 Hz, 1H), 7.21 (t, J = 1.9 Hz, 1H), 7.18−7.09 (m, 1H), 6.94 (s, 1H), 4.34 (q, J = 7.1 Hz, 2H), 3.66 (s, 2H), 1.37 (t, J = 7.1 Hz, 2H).
Ethyl 3-((2-((4-Cyanopyridin-2-yl)amino)naphthalen-1-yl)-amino)-3-oxopropanoate (12sa).
Following the procedure described for the synthesis of 12a, compound 10s (50 mg, 0.192 mmol) gave 12sa as a white solid (55 mg, 76%, Rf = 0.4, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.54 (s, 1H), 8.29 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.90−7.74 (m, 4H), 7.58 (t, J = 7.7 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1H), 6.91 (s, 1H), 6.88 (d, J = 5.3 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 3.68 (s, 2H), 1.38 (t, J = 7.1 Hz, 3H). HRMS m/z [M + H]+ for C21H18N4O3 calculated 375.1457, found 375.1451.
3-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)benzonitrile (13a).
Method A.
To a suspension of compound 11a (35 mg, 0.135 mmol) in anhydrous toluene (2 mL) at 0 °C was added malonyl chloride dropwise (15 μL, 0.162 mmol) with vigorous stirring. The reaction mixture was heated to 80 °C for 20 min and then at 110 °C for 10 min. The reaction mixture was cooled, and saturated NaHCO3 solution and 5% iPrOH-CH2Cl2 were added. The organic layer was separated, dried over anhydrous Na2SO4, and evaporated under reduced pressure. The residue was purified by silica gel chromatography to afford 13a as a white solid (18 mg, 40%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.61 (s, 1H), 8.10 (d, J = 8.5 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.68−7.57 (m, 4H), 7.53 (d, J = 5.1 Hz, 2H), 6.93 (dd, J = 8.9, 1.7 Hz, 1H), 3.63 (s, 2H). HRMS m/z [M + H]+ for C20H14N3O2 calculated 328.1086, found 328.1081.
Method B.
To a solution of 12a (100 mg, 0.268 mmol) in THF (3 mL) was added a solution of potassium t-butoxide (1 M in THF, 0.3 mL, 0.295 mmol) and the mixture was stirred at rt for 3 h. 1 M aq. HCl was added, and the product was extracted in ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel chromatography to afford 13a as a light yellow solid (90 mg, quantitative yield).
Method C.
Compound 11a (100 mg, 0.386 mmol) was dissolved in a mixture of toluene (2 mL) and DMF (0.2 mL). Monomethyl malonate (45 μL, 0.424 mmol) and DCC (88 mg, 0.424 mmol) were added sequentially, and the mixture was stirred at rt for 3 h (conversion to 12b was confirmed by TLC, Rf = 0.6, eluent = 50% ethyl acetate in hexanes). Potassium t-butoxide (1 M in THF, 0.46 mL, 0.46 mmol) was added and stirred for an additional 3 h at rt. 1 M aq. HCl was added, and the product was extracted in ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel chromatography to afford 13a (65 mg, 52%).
3-(10-Methyl-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b]-[1,4]diazepin-5-yl)benzonitrile (13b).
Following the procedure described for the synthesis of 13a (Method A), compound 11b (420 mg, 1.53 mmol) gave 13b as a white solid (270 mg, 51%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.60 (s, 1H), 7.88 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 9.4 Hz, 3H), 7.53 (d, J = 5.1 Hz, 2H), 7.47 (d, J = 8.3 Hz, 1H), 6.85 (d, J = 8.9 Hz, 1H), 3.62 (s, 2H), 2.61 (s, 3H). HRMS m/z [M + H]+ for C21H15N3O2 calculated 342.1243, found 342.1247.
3-(9-Methyl-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b]-[1,4]diazepin-5-yl)benzonitrile (13c).
Following the procedure described for the synthesis of 13a (Method A), compound 11c (100 mg, 0.366 mmol) gave 13c as a white solid (85 mg, 68%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.34 (s, 1H), 8.06 (d, J = 8.6 Hz, 1H), 7.69−7.45 (m, 6H), 6.88 (d, J = 8.9 Hz, 1H), 3.63 (d, J = 2.6 Hz, 2H), 2.55 (s, 3H). HRMS m/z [M + H]+ for C21H15N3O2 calculated 342.1243, found 342.1241.
3-(7-Bromo-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b]-[1,4]diazepin-5-yl)benzonitrile (13d).
Method A.
To a solution of 13a (10 mg, 0.031 mmol) in acetic acid (0.3 mL) was added bromine (8.0 μL, 0.155 mol) and the mixture was stirred at 50 °C for 7 h. Volatile materials were evaporated under reduced pressure, and the residue was treated with saturated NaHCO3 and dichloromethane. The organic layer was separated and dried over anhydrous Na2SO4. The solvent was evaporated, and the residue was purified by silica gel column chromatography to afford 13d as a white solid (10 mg, 81%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.72 (s, 1H), 8.26 (dd, J = 15.8, 8.0 Hz, 2H), 7.77 (p, J = 6.9 Hz, 2H), 7.71−7.61 (m, 2H), 7.57 (dd, J = 13.8, 6.3 Hz, 2H), 7.28 (s, 1H), 3.65 (s, 2H). HRMS m/z [M + H]+ for C20H12N3O2Br calculated 406.0191, found 406.0186.
Method B.
A solution of compound 7a (8 mg, 0.025 mmol) and NBS (22 mg, 0.125 mmol) in DMF was heated to 50 °C for 5 h. DMF was removed by rotary evaporation under high vacuum, and the residue was portioned between saturated NaHCO3 and dichloromethane. The organic layer was separated and dried over anhydrous Na2SO4. The solvent was evaporated, and the residue was purified by silica gel column chromatography to afford 13d as a white solid (8 mg, 81%).
3-(10-((Methylthio)methyl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzonitrile (13f).
Compound 14a (8.0 mg, 0.019 mmol) was dissolved in DMF (0.5 mL), and sodium thiomethoxide (4.0 mg, 0.057 mmol) was added. After stirring the mixture at rt for 1 h, 1 mL of each 1 M HCl and water was added, and the product was extracted in ethyl acetate. The organic layer was separated, dried over sodium sulfate, evaporated, and the residue was dried under high vacuum. MS analysis showed only the desired product (13f) mass, which was carried forward to the next step without purification (8.0 mg, quantitative, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). HRMS m/z [M + H]+ for C22H17N3O2S calculated 388.1120 found 388.1120.
Compounds 13e,g,h: 3-(10-(Azidomethyl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzonitrile (13g).
Compound 14a (along with 14b and 13e, 40 mg, 0.095 mmol) was dissolved in DMF and added NaN3 (62 mg, 0.95 mmol). The reaction mixture was heated to 50 °C for 18 h. The solvent was rotary evaporated under high vacuum, and the residue was partitioned between water and ethyl acetate. The organic layer was separated, dried, and evaporated. The residue was purified by silica gel flash chromatography to afford 13g (30 mg, 82%, Rf = 0.25, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.95 (s, 1H), 8.09 (s, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.70−7.48 (m, 5H), 6.95 (d, J = 9.0 Hz, 1H), 4.62 (s, 2H), 3.64 (s, 2H). MS m/z [M + H]+ for C21H14N6O2 calculated 383.1251 found 383.1. Compound 13e+13h could be separated in this step (Rf = 0.30, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.17 (s, 1H), 8.70 (s, 1H), 8.29 (d, J = 8.6 Hz, 0H), 8.21−8.11 (m, 1H), 7.91 (s, 1H), 7.73−7.37 (m, 7H), 7.31−7.27 (m, 1H), 7.16 (s, 1H), 4.64 (s, 1H, naphthalene-7-CH2N3 of 13h), 3.64 (s, 1H, 1,4-diazepine-CH2 of 13h), 3.62 (s, 2H, 1,4-diazepine-CH2 of 13e), 2.63 (s, 3H, naphthalene-7-Me of 13e). MS for 13e; m/z [M + H]+ for C21H14N3O2Br calculated 420.0342; 422.0322 found 420.0; 422.0. MS for 13h; m/z [M + H]+ for C21H13N6O2Br calculated 461.0356; 463.0336 found 461.0; 463.0.
N-((5-(3-Cyanophenyl)-2,4-dioxo-2,3,4,5-tetrahydro-1H-naphtho[1,2-b][1,4]diazepin-10-yl)methyl)acetamide (13i).
The azido intermediate 13g (25 mg, 0.065 mmol) was dissolved in 9:1 THF-H2O (1 mL), and trimethylphosphine (PMe3, 20 μL, 0.196 mmol) was added. After stirring the reaction mixture at rt for 5 h, volatiles were rotary evaporated. The residue 15 was dried under high vacuum (25 mg, Rf = 0.35, TLC eluent = 20% methanol in dichloromethane). To the intermediate 15 (0.065 mmol) in THF (2 mL) were added triethylamine (18 μL, 0.130 mmol) and acetic anhydride (13 μL, 0.130 mmol), and the mixture was stirred at rt for 18 h. Water was added to the reaction mixture, and the product was extracted several times in ethyl acetate. The combined organic layers were dried over sodium sulfate, filtered, the filtrate evaporated to afford 13i, and used as such in the next step without purification/characterization (25 mg, 89%, Rf = 0.35, TLC eluent = 5% methanol in dichloromethane).
Compounds 13j and 13k: 3-(9-(Azidomethyl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzonitrile (13j).
Following the method described for the synthesis of 13g, compound 16a,b (25 mg, 0.060 mmol) gave 13j as a major product (10 mg, 44%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.22 (s, 1H), 8.19 (d, J = 8.6 Hz, 1H), 7.82 (s, 1H), 7.73−7.40 (m, 5H), 6.96 (d, J = 8.9 Hz, 1H), 4.57 (s, 2H), 4.62 (s, 0.3H, naphthalene-6-CH2N3 of 13k), 3.64 (s, 2H). HRMS m/z [M + H]+ for C21H14N6O2 calculated 383.1256 found 383.1255.
3-(2,4-Dioxo-7-phenyl-1,2,3,4-tetrahydro-5H-naphtho[1,2-b]-[1,4]diazepin-5-yl)benzonitrile (13l).
To a round-bottom flask equipped with a stir bar were added 13d (30 mg, 0.074 mmol), phenylboronic acid (14 mg, 0.111 mmol), Pd(PPh3)4 (9 mg, 0.008 mmol), and sodium carbonate (23 mg, 0.222 mmol), and the mixture was subjected to two to three vacuum-argon degassing cycles. To the above mixture were added 1,2-dimethoxyethane (2 mL) and water (0.2 mL) and heated to 90 °C for 5 h. The solvent was evaporated, and the residue was partitioned between water and dichloromethane. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and the filtrate evaporated. The residue was purified by silica gel flash chromatography to obtain 13l as a white solid (23 mg, 77%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.97 (s, 1H), 8.22 (d, J = 8.4 Hz, 1H), 7.95−7.88 (m, 1H), 7.76 (ddd, J = 8.4, 6.9, 1.3 Hz, 1H), 7.67−7.40 (m, 4H), 7.31 (dd, J = 7.2, 2.2 Hz, 2H), 6.88 (s, 1H), 3.82−3.56 (m, 2H). HRMS m/z [M + H]+ for C26H17N3O2 calculated 404.1399, found 404.1399.
3-(2,4-Dioxo-7-(phenylethynyl)-1,2,3,4-tetrahydro-5H-naphtho-[1,2-b][1,4]diazepin-5-yl)benzonitrile (13m).
Following the protocol described for the synthesis of 17, 13d (25 mg, 0.062 mmol) gave 13m as a light brown solid (17 mg, 65%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.35 (d, J = 4.3 Hz, 1H), 8.29 (dd, J = 8.3, 1.5 Hz, 1H), 8.21 (dd, J = 8.0, 1.6 Hz, 2H), 7.79 (dddd, J = 16.5, 12.2, 6.7, 2.3 Hz, 3H), 7.71−7.50 (m, 4H), 7.45−7.35 (m, 2H), 7.26 (s, 1H), 3.65 (s, 2H). HRMS m/z [M + H]+ for C28H17N3O2 calculated 428.1399 found 428.1405.
3-(2,4-Dioxo-7-(pyridin-3-ylethynyl)-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzonitrile (13n).
Following the protocol described for the synthesis of 17, 13d (20 mg, 0.049 mmol) gave 13n as a light brown solid (17 mg, 80%, Rf = 0.5, TLC eluent = ethyl acetate). 1H NMR (400 MHz, chloroform-d) δ 9.43 (s, 1H), 8.88−8.80 (m, 1H), 8.61 (dd, J = 5.0, 1.7 Hz, 1H), 8.47−8.39 (m, 1H), 8.30−8.20 (m, 1H), 7.89 (dt, J = 7.9, 1.9 Hz, 1H), 7.82−7.73 (m, 2H), 7.60 (d, J = 1.9 Hz, 1H), 7.35 (dd, J = 7.9, 4.9 Hz, 1H), 7.21 (s, 1H), 3.65 (s, 2H). HRMS m/z [M + H]+ for C27H16N4O2 calculated 429.1372 found 429.1351.
3-(7-(Hex-1-yn-1-yl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho-[1,2-b][1,4]diazepin-5-yl)benzonitrile (13o).
Following the protocol described for the synthesis of 17, 13d (20 mg, 0.05 mmol) gave 13o as a light brown solid (10 mg, 50%, Rf = 0.4, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.55 (s, 1H), 8.37 (dd, J = 8.0, 1.5 Hz, 1H), 8.24−8.15 (m, 1H), 7.73 (dddd, J = 19.1, 8.1, 6.9, 1.4 Hz, 2H), 7.64 (dq, J = 4.5, 1.6 Hz, 1H), 7.61−7.48 (m, 2H), 7.05 (s, 1H), 3.64 (s, 2H), 2.52 (td, J = 7.2, 2.5 Hz, 2H), 1.73−1.60 (m, 2H), 1.59−1.42 (m, 2H), 0.97 (t, J = 7.3 Hz, 3H). HRMS m/z [M + H]+ for C26H21N3O2 calculated 408.1712 found 408.1707.
3-(2,4-Dioxo-7-((trimethylsilyl)ethynyl)-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzonitrile (17).
A round-bottom flask equipped with a stir bar was charged with 13d (30 mg, 0.074 mmol), triphenylphosphine (20 mg, 0.074 mmol), Pd(PPh3)2Cl2 (10 mg, 0.016 mmol), and copper(I) iodide (CuI, 6 mg, 0.030 mmol) and subjected to two to three vacuum-argon degassing cycles. To the above mixture were added 1,2-dimethoxyethane (1 mL), triethylamine (0.25 mL), and trimethylsilylacetylene (21 μL, 0.148 mmol) sequentially, and the mixture was heated to 55 °C for 6 h [Note: 5–6 equiv of volatile alkynes and 6–18 h heating may be needed]. To the reaction mixture was added silica gel, and the solvent was evaporated to load it on a flash chromatography column to purify 17 as a white solid (25 mg, 80%, Rf = 0.4, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.80 (s, 1H), 8.42−8.30 (m, 1H), 8.22 (dd, J = 7.8, 1.5 Hz, 1H), 7.74 (dddd, J = 16.1, 8.2, 7.0, 1.4 Hz, 2H), 7.64 (dt, J = 8.7, 1.5 Hz, 2H), 7.59−7.48 (m, 2H), 7.12 (s, 1H), 3.62 (d, J = 1.4 Hz, 2H), 0.30 (s, 9H). HRMS m/z [M + H]+ for C25H21N3O2Si calculated 424.1481 found 424.1479.
Compounds 18, 19: 3-(7-Ethynyl-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzonitrile (19).
To a solution of compound 17 (with traces of 18, 25 mg, 0.059 mmol) was added TBAF (1 M in THF, 90 μL, 0.089 mmol), and the mixture was stirred at rt for 18 h. Silica gel was added, and the mixture was subjected to rotary evaporation to load it on a flash chromatography column to purify 18 and 19 as white solids.
Data for 19 (13 mg, 63%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes).
1H NMR (400 MHz, chloroform-d) δ 9.20 (s, 1H), 8.46−8.37 (m, 1H), 8.19 (dd, J = 7.8, 1.4 Hz, 1H), 7.77 (dddd, J = 16.6, 8.2, 6.9, 1.4 Hz, 2H), 7.70−7.49 (m, 3H), 7.19 (s, 1H), 3.65 (d, J = 1.4 Hz, 2H), 3.49 (s, 1H). HRMS m/z [M + H]+ for C22H13N3O2 calculated 352.1086 found 352.1087.
Data for 18 (3 mg, 12%, Rf = 0.4, TLC eluent = 50% ethyl acetate in hexanes).
1H NMR (400 MHz, chloroform-d) δ 8.80 (s, 1H), 8.23−8.11 (m, 1H), 8.08−7.94 (m, 1H), 7.84−7.47 (m, 4H), 6.76 (s, 1H), 3.63 (t, J = 2.4 Hz, 2H), 3.03−2.82 (m, 2H), 0.94−0.70 (m, 2H), 0.02 (s, 9H). HRMS m/z [M + H]+ for C25H25N3O2Si calculated 428.1794 found 428.1792.
3-(7-Ethyl-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)benzonitrile (13p).
Compound 19 (13 mg, 0.037 mmol) was dissolved in ethyl acetate (1.5 mL), and 10%Pd–C (15 mg) was added. A stream of hydrogen gas was bubbled through the reaction mixture for 5 h. The palladium catalyst was filtered, the filtrate was concentrated, and the residue was purified by silica gel flash column chromatography to afford 13p as a white solid (8 mg, 61%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 9.31 (s, 1H), 8.26−8.15 (m, 1H), 8.08 (dd, J = 8.3, 1.4 Hz, 1H), 7.77−7.58 (m, 3H), 7.58−7.49 (m, 2H), 6.75 (s, 1H), 3.72−3.56 (m, 2H), 2.98 (dp, J = 22.4, 7.4 Hz, 2H), 1.38−1.13 (m, 3H). HRMS m/z [M + H]+ for C22H17N3O2 calculated 356.1399 found 356.1396.
3-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)-5-methylbenzonitrile (13r).
Following the procedure described for the synthesis of 13a (Method A), compound 11r (51 mg, 0.187 mmol) gave 13r as a yellowish solid (34 mg, 54%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.71 (s, 1H), 8.13 (d, J = 8.5 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.74 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 7.71−7.60 (m, 2H), 7.46−7.39 (m, 1H), 7.35 (s, 1H), 6.96 (d, J = 9.0 Hz, 1H), 3.64 (s, 2H), 2.40 (s, 3H). HRMS m/z [M + H]+ for C21H16N3O2 calculated 342.1243, found 342.1246.
2-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)isonicotinonitrile (13s).
Following the procedure described for the synthesis of 13a (Method A), compound 11s (30 mg, 0.103 mmol) gave 13s as a light brown solid (12 mg, 35%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.77 (s, 1H), 8.64−8.56 (m, 1H), 8.17−8.08 (m, 2H), 7.95−7.86 (m, 1H), 7.77−7.61 (m, 3H), 7.47 (dd, J = 5.0, 1.4 Hz, 1H), 6.90 (d, J = 9.0 Hz, 1H), 3.72 (d, J = 12.0 Hz, 1H), 3.64 (dd, J = 11.9, 1.7 Hz, 1H). HRMS m/z [M + H]+ for C19H13N4O2 calculated 329.1039, found 329.1035.
5-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)nicotinonitrile (13t).
Compound 11t (50 mg, 0.172 mmol) was dissolved in a mixture of anhydrous toluene-dioxane (2:1, 5 mL), and at rt, the mixture was treated with malonyl chloride (25 μL, 0.26 mmol). The reaction mixture was heated to 110 °C for 30 min in a reflux condenser with vigorous stirring. The solvents were evaporated under reduced pressure, and the residue was partitioned between dilute aq. NaHCO3 solution and ethyl acetate. The product was extracted with ethyl acetate repeatedly until the organic layer showed no product in TLC. The organic layers were combined, dried, and evaporated to give 13t as a light brown solid (30 mg, 53%, Rf = 0.1, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.81 (d, J = 1.9 Hz, 0H), 8.69 (d, J = 2.5 Hz, 0H), 8.34 (s, 1H), 8.10 (d, J = 8.4 Hz, 1H), 8.03 (t, J = 2.1 Hz, 0H), 7.94 (d, J = 8.0 Hz, 1H), 7.77 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.74−7.65 (m, 2H), 6.93 (d, J = 8.9 Hz, 1H), 3.68 (s, 2H). HRMS m/z [M + H]+ for C19H13N4O2 calculated 329.1039, found 329.1040.
4-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)picolinonitrile (13u).
Malonyl chloride (48.6 μL, 70.48 mg, 0.5 mmol) was added into a solution of 11u (26 mg, 0.1 mmol) in anhydrous toluene-dioxane (3:1, 10 mL) at 0 °C. The resulting solution was stirred vigorously at rt for 5 min, then at 110 °C for 30 min in a reflux condenser. The volatiles were evaporated under reduced pressure, and the residue was purified by silica gel flash column chromatography to give a light yellow solid (13.2 mg, 40%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.94 (s, 1H), 8.72 (d, J = 5.4 Hz, 1H), 8.16 (d, J = 8.4 Hz, 1H), 7.95 (dd, J = 8.1, 1.3 Hz, 1H), 7.83−7.63 (m, 4H), 7.52 (dd, J = 5.5, 2.2 Hz, 1H), 6.97 (d, J = 8.9 Hz, 1H), 3.76−3.60 (m, 2H). HRMS m/z [M + H]+ for C19H13N4O2 calculated 329.1039, found 329.1038.
6-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)picolinonitrile (13v).
Following the procedure described for the synthesis of 13t, compound 11v (60 mg, 0.23 mmol) gave 13v as a light yellow solid (25 mg, 33%, Rf = 0.1, TLC eluent = 50% ethyl acetate in hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.28 (s, 1H), 8.04 (ddd, J = 21.6, 15.4, 8.3 Hz, 3H), 7.92 (d, J = 8.1 Hz, 1H), 7.76−7.62 (m, 4H), 6.91 (d, J = 8.9 Hz, 1H), 3.71 (d, J = 12.0 Hz, 1H), 3.62 (dd, J = 11.9, 1.8 Hz, 1H). HRMS m/z [M + H]+ for C19H13N4O2 calculated 329.1039, found 329.1034.
3-(10-(Bromomethyl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho-[1,2-b][1,4]diazepin-5-yl)benzonitrile (14a).
A mixture of compound 13b (50 mg, 0.146 mmol), N-bromosuccinimide (NBS, 32 mg, 0.176 mmol), and benzoyl peroxide (36 mg, 0.148 mmol) in benzene (2 mL) was heated to 85 °C for 18 h. To the reaction mixture were added dichloromethane, water, and a pinch of sodium bisulfite. The organic layer was separated, and the aqueous layer was extracted several times with dichloromethane. Combined organic layer was dried over anhydrous sodium sulfate and rotary evaporated. The residue was purified by silica gel column chromatography to afford mixture of products 14a,b, and 7e (40 mg, 65%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). NMR analysis showed 14a as a major product. 1H NMR (400 MHz, chloroform-d) δ 9.08 (s, 1H), 8.18 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.71−7.51 (m, 6H), 6.95 (d, J = 9.0 Hz, 1H), 4.73 (d, J = 3.1 Hz, 2H), 3.64 (d, J = 5.3 Hz, 3H). MS m/z [M + H]+ for C21H14N3O2Br calculated 420.0342, 422.0322, found 420.0, 422.0.
Mass analysis of the mixture showed the presence of 14b. MS m/z [M + H]+ for C21H13N3O2Br2 calculated 499.9427, found 499.9.
3-(9-(Bromomethyl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho-[1,2-b][1,4]diazepin-5-yl)benzonitrile (16a).
Following the procedure described for the synthesis of 14a, using 13c (50 mg, 0.146 mmol) and dichloroethane as a solvent gave the desired product mixtures 16a,b as a light brown solid (25 mg, 41%, Rf = 0.3, TLC eluent = 50% ethyl acetate in hexanes). NMR and mass analyses showed 16a as a major product. 1H NMR (400 MHz, chloroform-d) δ 10.01 (s, 1H), 8.23 (d, J = 8.6 Hz, 1H), 7.85 (s, 1H), 7.75−7.43 (m, 6H), 6.94 (d, J = 9.0 Hz, 1H), 4.65 (s, 2H), 3.61 (s, 2H). HRMS m/z [M + H]+ for C21H14N3O2Br calculated 420.0348, found 420.0349.
(Z)-3-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)-N′-hydroxybenzimidamide (20a).
To compound 13a (75 mg, 0.229 mmol) in THF-MeOH (1:2) was added hydroxylamine hydrochloride (160 mg, 2.29 mmol) followed by triethylamine (0.32 mL, 2.29 mmol) and heated to 70 °C for 2 h in a reflux condenser. The solvents were evaporated, and the residue was triturated with water. The precipitates were collected by filtration and dried to afford 20a as a yellowish solid and used as such in the next step without further purification (75 mg, 91%, Rf = 0.3, TLC eluent = 100% ethyl acetate). HRMS m/z [M + H]+ for C20H16N4O3 calculated 361.1301, found 361.1296.
(Z)-N′-Hydroxy-3-(10-methyl-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzimidamide (20b).
Following the procedure described for the synthesis of 20a, compound 13b (14 mg, 0.041 mmol) gave 20b as a whitish solid (14 mg, 91%, Rf = 0.3, TLC eluent = 100% ethyl acetate). HRMS m/z [M + H]+ for C21H18N4O3 calculated 375.1457, found 375.1451.
(Z)-N′-Hydroxy-3-(9-methyl-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzimidamide (20c).
Following the procedure described for the synthesis of 20a, compound 13c (21 mg, 0.062 mmol) gave 20c as a whitish solid (17 mg, 74%, Rf = 0.3, TLC eluent = 100% ethyl acetate). HRMS m/z [M + H]+ for C21H18N4O3 calculated 375.1457, found 375.1463.
(Z)-3-(7-Bromo-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b]-[1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20d).
Following the procedure described for the synthesis of 20a, compound 13d (28 mg, 0.069 mmol) gave 20d as a whitish solid (18 mg, 59%, Rf = 0.3, TLC eluent = 100% ethyl acetate). HRMS m/z [M + H]+ for C20H15N4O3Br calculated 439.0406, found 439.0399.
(Z)-3-(7-Bromo-10-methyl-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20e) and (Z)-N′-Hydroxy-3-(10-((methylthio)methyl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)benzimidamide (20f).
To compound 13f (8 mg, 0.021 mmol) in THF-MeOH (1:1) was added hydroxylamine hydrochloride (15 mg, 0.21 mmol) followed by triethylamine (30 μL, 0.21 mmol) and heated to 70 °C for 2 h under a reflux condenser. Water was added, and the product was extracted into ethyl acetate several times (until UV spots were absent on TLC). The combined organic layers were dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated. The residue was dried under high vacuum to afford 20f as a brownish solid and used as such in the next step without further purification/characterization (7 mg, 81%, Rf = 0.3, TLC eluent = 100% ethyl acetate).
(Z)-3-(10-(Azidomethyl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20g).
Following the procedure described for the synthesis of 20f, compound 13g (as a mixture of 13e and 13h, 30 mg, 0.078 mmol) gave 20g as a brown solid (35 mg, quantitative, Rf = 0.3, TLC eluent = 100% ethyl acetate). Data for 20g: HRMS m/z [M + H]+ for C21H17N7O3 calculated 416.1471, found 416.1474.
(Z)-N-((5-(3-(N′-Hydroxycarbamimidoyl)phenyl)-2,4-dioxo-2,3,4,5-tetrahydro-1H-naphtho[1,2-b][1,4]diazepin-10-yl)methyl)-acetamide (20i).
Following the procedure described for the synthesis of 20f, compound 13i (25 mg, 0.065 mmol) gave 20i as a brown solid (25 mg, quantitative, Rf = 0.3, TLC eluent = 10% methanol in dichloromethane). HRMS m/z [M + H]+ for C23H21N5O4 calculated 432.1672, found 432.1679.
(Z)-3-(9-(Azidomethyl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20j).
Following the procedure described for the synthesis of 20f, compound 13j (as a mixture with 13k, 13 mg, 0.034 mmol) gave 20j as a brown solid (15 mg, quantitative, Rf = 0.4, TLC eluent = 100% ethyl acetate). HRMS m/z [M + H]+ for C21H17N7O3 calculated 416.1471, found 416.1468. Data for 20k: MS m/z [M + H]+ for C21H16N7O3Br calculated 494.0571, found 494.1, 496.1.
(Z)-3-(2,4-Dioxo-7-phenyl-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20l).
Following the procedure described for the synthesis of 20f, compound 13l (23 mg, 0.057 mmol) gave 20l as a brown solid (17 mg, 68%, Rf = 0.3, TLC eluent = 100% ethyl acetate). HRMS m/z [M + H]+ for C26H20N4O3 calculated 437.1614, found 437.1615.
(Z)-3-(2,4-Dioxo-7-(phenylethynyl)-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20m).
Following the procedure described for the synthesis of 20f, compound 13m (18 mg, 0.04 mmol) gave 20m as a brown solid (17 mg, 93%, Rf = 0.3, TLC eluent = ethyl acetate). HRMS m/z [M + H]+ for C28H20N4O3 calculated 461.1614, found 461.1614.
(Z)-3-(2,4-Dioxo-7-(pyridin-3-ylethynyl)-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20n).
Following the procedure described for the synthesis of 20f, compound 13n (16 mg, 0.037 mmol) gave 20n as a brown solid (15 mg, 93%, Rf = 0.1, TLC eluent = ethyl acetate). HRMS m/z [M + H]+ for C27H19N5O3 calculated 462.1566, found 462.1560.
(Z)-3-(7-(Hex-1-yn-1-yl)-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20o).
Following the procedure described for the synthesis of 20f, compound 13o (10 mg, 0.023 mmol) gave 20o as a brown solid (12 mg, quantitative, Rf = 0.2, TLC eluent = ethyl acetate). HRMS m/z [M + H]+ for C26H24N4O3 calculated 441.1927, found 441.1923.
(Z)-3-(7-Ethyl-2,4-dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b]-[1,4]diazepin-5-yl)-N′-hydroxybenzimidamide (20p).
Following the procedure described for the synthesis of 20f, compound 13p (8 mg, 0.023 mmol) gave 20p as a brown solid (10 mg, quantitative, Rf = 0.3, TLC eluent = 100% ethyl acetate). HRMS m/z [M + H]+ for C22H20N4O3 calculated 389.1614, found 389.1617.
(Z)-3-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)-N′-hydroxy-5-methylbenzimidamide (20r).
Following the procedure described for the synthesis of 20a, compound 13r (30 mg, 0.088 mmol) gave 20r as a light brown solid (34 mg, quantitative, Rf = 0.3, TLC eluent = ethyl acetate). HRMS m/z [M + H]+ for C21H18N4O3 calculated 375.1457, found 375.1459.
(Z)-2-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)-N′-hydroxyisonicotinimidamide (20s).
Compound 13s (30 mg, 0.092 mmol) was dissolved in THF-MeOH (1:1 v/v, 3 mL) and hydroxylamine hydrochloride (13 mg, 0.184 mmol) was added followed by triethylamine (26 μL, 0.184 mmol), and the mixture was stirred at rt for 1–2 h (or reaction completion as indicated by TLC). The volatiles were evaporated under reduced pressure, and the residue was partitioned between water-ethyl acetate. The product was extracted repeatedly with ethyl acetate (4–5 times, or as indicated by TLC), combined, and the solvent was evaporated and dried under high vacuum to afford 20s as a light brown solid (25 mg, 75%, Rf = 0.2, TLC eluent = ethyl acetate). HRMS m/z [M + H]+ for C19H15N5O3 calculated 362.1253, found 362.1255.
(Z)-5-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)-N′-hydroxynicotin-imidamide (20t).
Following the procedure described for the synthesis of 20s, compound 13t (30 mg, 0.092 mmol) gave 20t as a light brown solid (25 mg, 75%, Rf = 0.3, TLC eluent = ethyl acetate).
(Z)-4-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)-N′-hydroxypicolin-imidamide (20u).
Following the procedure described for the synthesis of 20s, compound 13u (13.2 mg, 0.04 mmol) gave 20u as a light brown solid (11.52 mg, 80%, Rf = 0.3, TLC eluent = ethyl acetate). 1H NMR (400 MHz, DMSO) δ 10.98 (s, 1H), 9.84 (s, 1H), 8.60 (d, J = 5.5 Hz, 1H), 8.26 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 9.0 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.63 (t, J = 7.4 Hz, 1H), 7.55 (d, J = 2.1 Hz, 1H), 7.40 (dd, J = 5.5, 2.1 Hz, 1H), 7.02 (d, J = 8.9 Hz, 1H), 5.85 (s, 2H), 4.01 (q, J = 7.1 Hz, 2H). HRMS m/z [M + H]+ for C19H15N5O3 calculated 362.1253, found 362.1255.
(Z)-6-(2,4-Dioxo-1,2,3,4-tetrahydro-5H-naphtho[1,2-b][1,4]-diazepin-5-yl)-N′-hydroxypicolin-imidamide (20v).
Following the procedure described for the synthesis of 20s, compound 13v (25 mg, 0.076 mmol) gave 20v as a light brown solid (25 mg, 91%, Rf = 0.5, TLC eluent = ethyl acetate). HRMS m/z [M + H]+ for C19H15N5O3 calculated 362.1253, found 362.1256.
5-(3-(5-Thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione (NP-1815-PX, Nonsalt Form; 21a).
Compound 20a (75 mg, 0.21 mmol) was suspended in anhydrous acetonitrile (3 mL), DBU (125 μL, 0.84 mmol) was added at 0 °C, and the mixture was stirred for 15 min. Thiocarbonyldiimidazole (56 mg, 0.32 mmol) was added and the mixture was stirred at rt for 1.5 h. Dichloromethane and 1 M HCl were added, the layers were separated, and the aqueous layer was extracted several times with 5% isopropanol in dichloromethane. The combined organic layer was dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated, and the residue was purified by silica gel flash column chromatography to afford 21a as white/yellowish solid. The compound was purified further by preparative-TLC (35 mg, 42%, Rf = 0.35, TLC eluent = 15% methanol in dichloromethane). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 8.24 (d, J = 8.5 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 7.7 Hz, 1H), 7.74−7.64 (m, 2H), 7.64−7.57 (m, 1H), 7.54 (t, J = 7.9 Hz, 1H), 7.41−7.33 (m, 1H), 7.02 (d, J = 9.0 Hz, 1H), 3.75 (d, J = 11.9 Hz, 1H), 3.17 (d, J = 11.9 Hz, 1H). HRMS m/z [M + H]+ for C21H14N4O3S calculated 403.0865, found 403.0866.
10-Methyl-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)-phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione (21b).
Following the procedure described for the synthesis of 21a, compound 20b (14 mg, 0.037 mmol) gave 21b as a white/yellowish solid (7 mg, 45%, Rf = 0.35, TLC eluent = 15% methanol in dichloromethane). 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 8.05 (s, 1H), 7.83 (dd, J = 8.0, 5.6 Hz, 2H), 7.65 (d, J = 8.7 Hz, 2H), 7.58 (t, J = 7.9 Hz, 1H), 7.43 (dd, J = 14.3, 7.6 Hz, 2H), 6.93 (d, J = 9.0 Hz, 1H), 3.73 (d, J = 11.8 Hz, 1H), 3.16 (d, J = 11.8 Hz, 1H), 2.54 (s, 3H). HRMS m/z [M + H]+ for C22H16N4O3S calculated 417.1021, found 417.1025.
9-Methyl-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)-phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione (21c).
Following the procedure described for the synthesis of 21a, compound 20c (17 mg, 0.045 mmol) gave 22c as a white/yellowish solid (10 mg, 53%, Rf = 0.35, TLC eluent = 15% methanol in dichloromethane). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 8.17 (d, J = 8.7 Hz, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.69 (d, J = 11.3 Hz, 2H), 7.65−7.58 (m, 2H), 7.50 (dd, J = 16.0, 8.4 Hz, 2H), 6.96 (d, J = 9.0 Hz, 1H), 3.74 (d, J = 11.8 Hz, 1H), 3.17 (d, J = 11.8 Hz, 1H), 2.48 (s, 3H). HRMS m/z [M + H]+ for C22H16N4O3S calculated 417.1021, found 417.1025.
7-Bromo-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)-phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21d).
Following the procedure described for the synthesis of 21a, compound 20d (18 mg, 0.041 mmol) gave 21d as a white/yellowish solid (12 mg, 50%; Rf = 0.3, TLC eluent = 15% methanol in dichloromethane; purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 30/70 → 50/50 for 40 min, flow rate = 5.0 mL/min, Rt = 26.8 min). 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 8.36−8.28 (m, 1H), 8.16−8.08 (m, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.79 (qd, J = 7.1, 3.5 Hz, 2H), 7.67 (d, J = 2.2 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.39 (dd, J = 7.9, 2.1 Hz, 1H), 7.32 (s, 1H), 3.83 (d, J = 11.9 Hz, 1H), 3.17 (d, J = 11.9 Hz, 1H). HRMS m/z [M + H]+ for C21H13N4O3SBr calculated 480.9970, found 480.9976.
10-((Methylthio)methyl)-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21f).
Following the procedure described for the synthesis of 21a, compound 20f (7.0 mg, 0.017 mmol) gave 21f as a white/yellowish solid (2.07 mg, 22%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 35/65 → 50/50 for 40 min, flow rate = 5.0 mL/min, Rt = 22.8 min). 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 8.11 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 7.7 Hz, 1H), 7.68 (d, J = 9.0 Hz, 1H), 7.64−7.51 (m, 2H), 7.42−7.32 (m, 1H), 6.98 (d, J = 8.9 Hz, 1H), 3.90 (s, 2H), 3.76 (d, J = 11.8 Hz, 1H), 3.16 (d, J = 11.8 Hz, 1H), 2.01 (s, 2H). HRMS m/z [M + H]+ for C23H18N4O3S2 calculated 463.0899, found 463.0901.
7-Bromo-10-methyl-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21e).
Following the procedure described for the synthesis of 21a, mixture of compounds 21a,e,g,h (35 mg, 0.084 mmol) gave 21a,e,g,h as a white/yellowish solid (Rf = 0.5, TLC eluent = 15% methanol in dichloromethane and 0.1% acetic acid). The products were separated by RP-HPLC using linear gradient of CH3CN-10 mM TEAA in H2O 30/70 → 45/55 for 40 min, flow rate = 5.0 mL/min. This reaction also gave triethylamine salt of 21a (2.26 mg, 6%, Rt = 24.5 min). Data for 21e: 1.79 mg, 4.5%, Rt = 33.6 min. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.12 (s, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.62 (dd, J = 8.6, 1.5 Hz, 1H), 7.56 (t, J = 7.9 Hz, 1H), 7.37 (dd, J = 7.9, 2.2 Hz, 1H), 7.23 (s, 1H), 6.54 (s, 1H), 3.80 (d, J = 11.9 Hz, 1H), 3.17 (d, J = 11.9 Hz, 1H), 2.58 (s, 3H). HRMS m/z [M + H]+ for C22H15N4O3SBr calculated 495.0126, found 495.0126.
Data for 10-(Azidomethyl)-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]-diazepine-2,4(3H)-dione Triethylamine Salt (21g).
15 mg, 32%, Rt = 26.3 min. 1H NMR (400 MHz, DMSO-d6) δ 10.92 (s, 1H), 8.25 (s, 1H), 7.96 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 7.7 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 7.64 (d, J = 1.9 Hz, 1H), 7.59 (dd, J = 8.4, 1.6 Hz, 1H), 7.55 (t, J = 7.9 Hz, 1H), 7.37 (dd, J = 7.8, 2.2 Hz, 1H), 7.04 (d, J = 9.0 Hz, 1H), 6.54 (s, 1H), 4.71 (s, 2H), 3.77 (d, J = 11.8 Hz, 1H), 3.17 (d, J = 11.8 Hz, 1H). MS m/z [M + H]+ for C22H15N7O3S calculated 458.1030, found 458.1.
Data for 10-(Azidomethyl)-7-bromo-5-(3-(5-thioxo-4,5-dihydro1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]-diazepine-2,4(3H)-dione Triethylamine Salt (21h).
1.04 mg, 2%, Rt = 35.7 min. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 8.33 (s, 1H), 8.14 (d, J = 8.6 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.76 (d, J = 8.6 Hz, 1H), 7.68 (s, 1H), 7.57 (t, J = 7.8 Hz, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.32 (s, 1H), 6.54 (s, 1H), 4.78 (s, 2H), 3.85 (d, J = 12.0 Hz, 1H), 3.18 (d, J = 11.9 Hz, 1H). MS m/z [M + H]+ for C22H14N7O3SBr calculated 536.0135, found 536.1. Purity − 88.0% at 254 nm.
N-((2,4-Dioxo-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)-phenyl)-2,3,4,5-tetrahydro-1H-naphtho[1,2-b][1,4]diazepin-10-yl)-methyl)acetamide Triethylamine Salt (21i).
Following the procedure described for the synthesis of 21a, compound 20i (25.0 mg, 0.058 mmol) gave 21i as a white/yellowish solid (7.07 mg, 19%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 20/80 → 40/60 for 40 min, flow rate = 5.0 mL/min, Rt = 24.4 min). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 8.45 (t, J = 5.9 Hz, 1H), 8.13 (s, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.82 (dt, J = 7.8, 1.4 Hz, 1H), 7.68 (d, J = 9.0 Hz, 1H), 7.60−7.48 (m, 3H), 7.38 (dd, J = 8.1, 2.1 Hz, 1H), 6.98 (d, J = 9.0 Hz, 1H), 6.54 (s, 1H), 4.57−4.35 (m, 2H), 3.74 (d, J = 11.8 Hz, 1H), 3.16 (d, J = 11.8 Hz, 1H), 1.92 (s, 3H). HRMS m/z [M + Na]+ for C24H19N5O4S calculated 496.1055, found 496.1062.
9-(Azidomethyl)-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21j).
Following the procedure described for the synthesis of 21a, compound 20j,k (15.0 mg, 0.036 mmol) gave 21j,k as a white/yellowish solid (6.0 mg, 30%; Rf = 0.2, TLC eluent = 20% methanol in dichloromethane; purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 30/70 → 50/50 for 40 min, flow rate = 5.0 mL/min, Rt = 26.8 min). 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 8.28 (d, J = 8.8 Hz, 1H), 7.92 (d, J = 1.7 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 9.0 Hz, 1H), 7.70−7.60 (m, 1H), 7.55 (t, J = 7.9 Hz, 1H), 7.37 (dd, J = 7.9, 2.2 Hz, 1H), 7.05 (d, J = 9.0 Hz, 1H), 6.54 (s, 1H), 4.65 (s, 2H), 3.77 (d, J = 11.8 Hz, 1H), 3.17 (d, J = 11.8 Hz, 1H). HRMS m/z [M + H]+ for C22H15N7O3S calculated 458.1035, found 458.1034.
Data for 9-(azidomethyl)-7-bromo-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]-diazepine-2,4(3H)-dione triethylamine salt (21k).
2.0 mg, 9%; Rf = 0.2, TLC eluent = 20% methanol in dichloromethane; purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 30/70 → 50/50 for 40 min, flow rate = 5.0 mL/min, Rt = 33.4 min. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 8.35 (d, J = 8.7 Hz, 1H), 8.10 (s, 1H), 7.86 (d, J = 7.7 Hz, 1H), 7.77 (dd, J = 8.8, 1.7 Hz, 1H), 7.69 (s, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.41−7.36 (m, 1H), 7.35 (s, 1H), 6.54 (s, 1H), 4.76 (s, 2H), 3.84 (d, J = 11.9 Hz, 1H), 3.18 (d, J = 11.9 Hz, 1H). MS m/z [M + H]+ for C22H14N7O3SBr calculated 536.0135, 538.0115, found 536.0, 538.0. Purity − 93.1% at 254 nm
7-Phenyl-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)-phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21l).
Following the procedure described for the synthesis of 21a, compound 20l (17.0 mg, 0.039 mmol) gave 21l as a white/yellowish solid (18 mg, 80%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 30/70 → 45/55 for 40 min, flow rate = 5.0 mL/min, Rt = 33.8 min). 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 8.34 (d, J = 8.5 Hz, 1H), 7.85−7.65 (m, 3H), 7.59 (ddd, J = 8.2, 6.9, 1.1 Hz, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.49−7.36 (m, 4H), 7.34−7.25 (m, 2H), 6.87 (s, 1H), 3.85 (d, J = 11.8 Hz, 1H), 3.21 (d, J = 11.9 Hz, 1H). HRMS m/z [M + H]+ for C27H18N4O3S calculated 479.1178, found 479.1172.
7-(Phenylethynyl)-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione (21m).
Following the procedure described for the synthesis of 21a, compound 20m (17 mg, 0.037 mmol) gave 21m as a white/yellowish solid (10 mg, 45%, Rf = 0.30, TLC eluent = 10% methanol in dichloromethane). 1H NMR (400 MHz, DMSO-d6) δ 11.08 (s, 1H), 8.43−8.28 (m, 2H), 7.86 (d, J = 7.8 Hz, 1H), 7.83−7.74 (m, 2H), 7.70 (d, J = 2.1 Hz, 1H), 7.65 (dd, J = 6.6, 3.0 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.46−7.37 (m, 3H), 7.27 (s, 1H), 6.54 (s, 1H), 3.84 (d, J = 11.9 Hz, 1H), 3.20 (d, J = 11.9 Hz, 1H). HRMS m/z [M + H]+ for C29H18N4O3S calculated 503.1178, found 503.1170.
7-(Pyridin-3-ylethynyl)-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione (21n).
Following the procedure described for the synthesis of 21a, compound 20n (15 mg, 0.033 mmol) gave 21n as a white/yellowish solid (9 mg, 40%, Rf = 0.15, TLC eluent = 10% methanol in dichloromethane). 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 8.85 (d, J = 2.2 Hz, 1H), 8.58 (dd, J = 4.9, 1.7 Hz, 1H), 8.38 (ddd, J = 17.8, 6.7, 4.1 Hz, 2H), 8.07 (dt, J = 7.9, 2.0 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.80 (qd, J = 7.0, 3.6 Hz, 2H), 7.69 (d, J = 2.4 Hz, 0H), 7.58 (t, J = 7.9 Hz, 1H), 7.48−7.37 (m, 1H), 7.32 (s, 2H), 6.54 (s, 1H), 3.85 (d, J = 11.9 Hz, 1H), 3.20 (d, J = 11.9 Hz, 1H). HRMS m/z [M + H]+ for C29H18N4O3S calculated 504.1130, found 504.1137.
7-(Hex-1-yn-1-yl)-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione (21o).
Following the procedure described for the synthesis of 21a, compound 20o (12 mg, 0.027 mmol) gave 21o as a white/yellowish solid (6 mg, 46%, Rf = 0.10, TLC eluent = 10% methanol in dichloromethane). 1H NMR (400 MHz, DMSO-d6) δ 10.97 (s, 1H), 8.34−8.24 (m, 1H), 8.24−8.15 (m, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.73 (qd, J = 7.5, 3.6 Hz, 2H), 7.66 (t, J = 1.9 Hz, 1H), 7.56 (t, J = 7.9 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.05 (s, 1H), 5.94 (s, 1H), 3.79 (d, J = 11.9 Hz, 1H), 3.17 (d, J = 11.8 Hz, 1H), 1.60−1.48 (m, 2H), 1.48−1.35 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H). HRMS m/z [M + H]+ for C27H22N4O3S calculated 483.1491, found 483.1496.
7-Ethyl-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21p).
Following the procedure described for the synthesis of 21a, compound 20p (10.0 mg, 0.026 mmol) gave 21p as a white/yellowish solid (6.5 mg, 47%, purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 30/70 → 50/50 for 40 min, flow rate = 5.0 mL/min, Rt = 27.7 min). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.25 (d, J = 8.2 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 7.7 Hz, 1H), 7.73−7.58 (m, 3H), 7.54 (t, J = 7.9 Hz, 1H), 7.37 (dd, J = 7.9, 2.2 Hz, 1H), 6.85 (s, 1H), 3.74 (d, J = 11.7 Hz, 1H), 3.15 (d, J = 11.8 Hz, 1H), 2.91 (ddq, J = 22.5, 14.9, 7.4 Hz, 2H), 1.12−1.06 (m, 3H). HRMS m/z [M + H]+ for C23H18N4O3S calculated 431.1178, found 431.1178.
10-(Aminomethyl)-5-(3-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21q).
The azido compound 21g (15 mg, 0.027 mmol) was dissolved in THF-H2O (9:1), and the mixture was treated with trimethylphosphine (PMe3, 7.0 μL, 0.068 mmol). After stirring the reaction mixture for 5 h at rt, the solvent was rotary evaporated and the residue was purified by RP-HPLC to afford 21q as a light yellow solid (11.7 mg, 82%, Rf = 0.1, TLC eluent = 20% methanol in dichloromethane; purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 20/80 → 30/70 for 40 min, flow rate = 5.0 mL/min, Rt = 23.2 min). 1H NMR (400 MHz, DMSO-d6) δ 8.28 (s, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 9.0 Hz, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.59−7.51 (m, 2H), 7.42 (d, J = 8.1 Hz, 1H), 7.01 (d, J = 9.0 Hz, 1H), 4.09 (d, J = 2.8 Hz, 2H), 3.74 (d, J = 11.8 Hz, 1H), 3.19 (d, J = 11.8 Hz, 1H). HRMS m/z [M + Na]+ for C22H17N5O3S calculated 454.0950, found 454.0950.
5-(3-Methyl-5-(5-thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)dione (21r).
Following the procedure described for the synthesis of 21a, compound 20r (34 mg, 0.091 mmol) gave 21r as a white solid (20 mg, 53%, Rf = 0.3, TLC eluent = 20% methanol in dichloromethane). 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 8.28 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.76−7.65 (m, 3H), 7.61 (t, J = 7.5 Hz, 1H), 7.54 (s, 1H), 7.37 (s, 1H), 7.02 (d, J = 9.0 Hz, 1H), 3.76 (d, J = 11.8 Hz, 1H), 3.18 (dd, J = 11.8, 1.6 Hz, 1H), 2.40 (s, 3H). HRMS m/z [M + H]+ for C22H17N4O3S calculated 417.1021, found 417.1029.
5-(4-(5-Thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)pyridin-2-yl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21s).
Following the procedure described for the synthesis of 21a, compound 20s (25 mg, 0.069 mmol) gave 21s as a white solid (10 mg, 29%, Rf = 0.3, TLC eluent = 20% methanol in dichloromethane; purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 10/90 → 45/55 for 40 min, flow rate = 5.0 mL/min, Rt = 28.2 min). 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 8.49 (d, J = 5.1 Hz, 1H), 8.25 (d, J = 8.5 Hz, 1H), 8.13 (s, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 5.1 Hz, 1H), 7.68 (dd, J = 12.8, 8.5 Hz, 2H), 7.60 (t, J = 7.5 Hz, 1H), 6.96 (d, J = 8.9 Hz, 1H), 3.80 (d, J = 11.8 Hz, 1H), 3.19 (d, J = 11.8 Hz, 1H). HRMS m/z [M + H]+ for C20H13N5O3S calculated 404.0817, found 404.0816.
5-(5-(5-Thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)pyridin-3-yl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21t).
Following the procedure described for the synthesis of 21a, compound 20t (25 mg, 0.069 mmol) gave 21t as a white solid (19 mg, 54%, Rf = 0.3, TLC eluent = 10% methanol in dichloromethane; purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 10/90 → 45/55 for 40 min, flow rate = 5.0 mL/min, Rt = 32.3 min). 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 8.96 (d, J = 1.9 Hz, 0H), 8.56 (d, J = 2.5 Hz, 1H), 8.28 (d, J = 8.5 Hz, 1H), 8.02 (t, J = 2.2 Hz, 1H), 7.95 (d, J = 7.9 Hz, 1H), 7.74 (d, J = 9.0 Hz, 1H), 7.69 (dd, J = 8.6, 6.4 Hz, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.06 (d, J = 9.0 Hz, 1H), 3.79 (d, J = 11.9 Hz, 1H), 3.21 (d, J = 11.9 Hz, 1H). HRMS m/z [M + H]+ for C20H13N5O3S calculated 404.0817, found 404.0816.
5-(2-(5-Thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)pyridin-4-yl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21u).
To a solution of 20u (10 mg, 0.028 mmol) in anhydrous acetonitrile (1 mL) at 0 °C was added DBU (8 μL, 8.22 mg, 0.054 mmol), and the mixture was stirred for 10 min. Thiocarbonyldiimidazole (9.6 mg, 0.054 mmol) was added, and the resulting solution was stirred for 2 h at rt. The volatiles were removed under vacuum, and the residue was purified by RP-HPLC (linear gradient of CH3CN-10 mM TEAA in H2O 20/80 → 40/60 for 40 min, flow rate = 5.0 mL/min, Rt = 34.0 min) to give 21u as a white solid (10 mg, 75%, Rf = 0.3, TLC eluent = 10% methanol in dichloromethane): 1H NMR (400 MHz, DMSO) δ 10.95 (s, 1H), 8.93 (s, 1H), 8.69 (d, J = 5.3 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 7.97 (dd, J = 8.1, 1.4 Hz, 1H), 7.76 (d, J = 9.0 Hz, 1H), 7.73−7.58 (m, 3H), 7.41 (dd, J = 5.3, 2.2 Hz, 1H), 7.06 (d, J = 8.9 Hz, 1H), 3.82 (d, J = 11.9 Hz, 1H), 3.23−3.16 (m, 1H). HRMS m/z [M + H]+ for C20H13N5O3S calculated 404.0817, found 404.0818.
5-(6-(5-Thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)pyridin-2-yl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Triethylamine Salt (21v).
Following the procedure described for the synthesis of 21a, compound 20v (25 mg, 0.069 mmol) gave 21v as a white solid (19 mg, 54%, Rf = 0.3, TLC eluent = 10% methanol in dichloromethane; purified by RP-HPLC, linear gradient of CH3CN-10 mM TEAA in H2O 20/80 → 40/60 for 40 min, flow rate = 5.0 mL/min, Rt = 35.9 min). 1H NMR (400 MHz, DMSO-d6) δ 10.94 (s, 1H), 8.24 (d, J = 8.4 Hz, 1H), 8.12 (t, J = 7.8 Hz, 1H), 7.92 (d, J = 7.7 Hz, 2H), 7.76−7.64 (m, 3H), 7.60 (t, J = 7.4 Hz, 1H), 6.95 (d, J = 9.0 Hz, 1H), 6.52 (s, 1H), 3.78 (d, J = 11.8 Hz, 1H), 3.22−3.13 (m, 1H). HRMS m/z [M + H]+ for C20H13N5O3S calculated 404.0817, found 404.0818.
5-(3-(5-Thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)phenyl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione Sodium Salt (5).
Compound 21a (10.0 mg, 25 μmol) was suspended in 1:1 acetonitrile–water, and the mixture was treated with 0.1 M NaOH solution (250 μL). The solution turned clear after shaking, and particles/turbidity, if any, was filtered off using Nylon Acrodisc filters. The clear solution was frozen and lyophilized to obtain 5 as a pale-yellow solid (10.7 mg, quantitative yield). 1H NMR (400 MHz, D2O) δ 8.10 (d, J = 8.5 Hz, 1H), 7.75−7.58 (m, 4H), 7.48 (dt, J = 18.4, 7.7 Hz, 2H), 7.39 (d, J = 9.1 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 6.82 (d, J = 9.0 Hz, 1H), 3.67 (d, J = 12.2 Hz, 1H), 3.36 (d, J = 11.9 Hz, 1H). Compounds 22b and 22c were prepared in the same way and used as such without further analysis. Both sodium and triethylamine salts were water-soluble.
Biological Assays.
The cell lines stably transfected with the functional receptors (CHO-K1 for hP2RX1, X2/X3, X3, or HEK-293 for hP2X4R) were used to analyze the antagonism at hP2XRs by various synthesized analogues of 5 using a fluorescent Ca2+-sensitive dye (Fluo-8 NW) or a luminescence Ca2+-sensitive photoprotein as readout, using a FLIPRTETRA to measure the emitted light. The assays of the test compounds were performed in a 384 format by Axxam SpA (Milan, Italy). The assays hP2RX2/P2X3 and hP2RX4 used a Ca2+-sensitive dye (Fluo-8 NW No Wash Calcium Assay Kit, AAT Bioquest, Sunnyvale, CA, cat.# 36316) with fluorescent measurement, and the hP2RX1 and hP2RX3 assays used coelenterazine (preloaded in the cells during incubation for 3 h at 37 °C, Biosynth AG, Staad, Switzerland, cat.# C-7001) and a Ca2+-responsive photoprotein with a signal of luminescence. For hP2RX1 and hP2RX3 assays, the cells were seeded at 10,000 cells/well in 384-well plates in complete medium (25 μL/well). Following the removal of medium and preloading with coelenterazine in fresh medium (10 μM, 30 μL per well), the test compound was added to each well (10 μL per well) and the emitted light was recorded. After 30 min incubation, the reference agonist (at ~EC80) was added and the luminescence was again recorded. For hP2RX2/P2X3 and hP2RX4 assays, the cells were seeded at 10,000 cells/well in 384-well plates in complete medium (25 μL/well). After 24 h, the medium was removed and replaced with calcium-sensitive dye (Fluo-8 NW, 20 μL/well) in assay buffer. After 1 h incubation at rt in the dark, the test compound was added to each well (10 μL per well) and the fluorescence was recorded. After 30 min, the reference agonist (at ~EC80) was added and the fluorescence was again recorded.
The following standard P2XR ligands were used (prepared at 10 mM in water and stored in aliquots at −20 °C): agonist α,β-Me-ATP: (Sigma-Aldrich, cat.# M6517); P2X4R agonist CTP (Sigma-Aldrich, cat.# C1506); antagonist TNP-ATP: (Tocris Bio-Techne SRL, Milan, Italy, cat.# 2464). The assay buffer consisted of Standard Tyrode’s Buffer: (in house solution), 130 mM NaCl, 5 mM KCl, 2 mM CaCl2, 5 mM NaHCO3, 1 mM MgCl2, 20 mM HEPES, pH 7.4. For hP2X1, hP2RX2/P2X3, and hP2X3, α,β-Me-ATP was used at 100, 31.6, 10, 3.16, 1, 0.316, 0.1, and 0.0316 μM. For hP2X4R, CTP was used at 100, 31.6, 10, 3.16, 1, 0.316, 0.1, and 0.0316 μM. The reference agonist (at ~EC80) was 1 μM α,β-Me-ATP for hP2X1 and hP2X3, 1 μM α,β-Me-ATP for hP2X/P2X3, and 10 μM CTP for hP2X4R.
In Vivo Experiments.
Experimental Design and Animals.
We used age- and weight-matched male and female C57B/6 (wild-type) WT mice generated in our breeding colony at the UConn Health animal facility. The mice were fed standard chow diet and water ad libitum. Standard housing conditions were maintained at a controlled temperature with a 12 h light/dark cycle. All experiments were approved by the Institutional Animal Care and Use Committee of University of Connecticut Health and conducted in accordance with the U.S. National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
A total of 34 young (8- to 12-week-old male mice) and 56 middle-aged (11–12 months, old both male and female) mice were randomly divided into sham, vehicle (Alzet minipump containing 1× PBS, Alzet Osmotic Pumps, Cupertino, CA), 21u (Alzet minipump containing 0.5–3 mg/kg/day for 3 days) or 22c (Alzet minipump containing 5.0 mg/kg/day for 3 days) groups. These mice except sham were then subjected to Alzet minipump implantation immediately after initiation of reperfusion. For infarct volume analysis study, all of the mice were euthanized after 3 days to isolate brain and kept in a deep freezer until further processing. In a long-term study group, the mice survived for 35 days post stroke and were subjected to functional behavioral determination on a weekly or a one-time basis. After completion of behavioral assays, all of the mice were euthanized and perfused with 4% paraformaldehyde for atrophy measurement. Three mice were excluded from the study due to death (two vehicle- and one 22c-treated) in young adult group, and 14 mice were excluded due to death from the middle-aged group, corresponding to six vehicle- and eight 21u-treated mice (6 at a dose of 3 mg/kg and 2 at a dose of 1.5 mg/kg).
Middle Cerebral Artery Occlusion (MCAO).
We induced transient focal cerebral ischemia by a 60 min right MCAO under isoflurane anesthesia followed by reperfusion for either 3 or 35 days as described previously.46 Briefly, we proceeded with midline ventral neck incision and unilateral right MCAO using a 6.0 silicone rubber-coated monofilament (size 602145/602245; Doccol Corporation, Sharon, MA) placed 10–11 mm away from the bifurcation point of the internal carotid artery through an external carotid artery stump. Rectal temperatures were monitored and maintained at 37 ± 0.5 °C with the help of a heating pad. We used laser Doppler flowmetry to measure cerebral blood flow and to confirm occlusion and reperfusion. All animals were fed wet mash and given injectable normal saline (1% v/w daily) for maximum 1 week or until sacrifice after surgery to ensure adequate nutrition for chronic endpoints.
Measurement of the Cerebral Infarct Volume.
The isolated brains were cut into five equal coronal sections and stained with TTC (1.5% solution in phosphate-buffered saline (PBS)) for 20 min and then fixed in 10% buffered formaldehyde solution. The stained brain slices were digitally photographed, and the infarct area of each brain was measured in a blinded manner using an image analysis software, SigmaScan Pro 5 (Systat, Palo Alto, CA). The infarct volume was calculated by Swanson’s method to correct for edema.48 The total volumes of both contralateral and ipsilateral hemisphere and the volumes of the striatum cortex in both hemispheres were measured, and the infarct percentage was calculated as % contralateral structure to avoid mis-measurement secondary to edema.
Cresyl Violet Staining for Tissue Atrophy.
After completion of the behavioral task, all of the animals were sacrificed at 35 days after stroke surgery. The mice were deeply anesthetized with an overdose of Avertin (250 mg/kg i.p.) and underwent by trans-cardiac perfusion using cold PBS followed by 4% paraformaldehyde. Brains were then fixed overnight and placed in cryoprotectant (30% sucrose in 1× PBS) for 48–72 h before processing. Brains were then cut into 30 μm free-floating sections using a freezing microtome, and every eighth slice was mounted and stained with cresyl violet. These 30 μm sections were then used for tissue atrophy calculation. Loss of brain tissue was determined by measuring the amount of tissue atrophy in the ipsilateral hemisphere. Tissue atrophy was calculated using the following formula: % tissue atrophy = 100 − [(total ipsilateral tissue/total contralateral tissue) × 100] [49].
Sensory Motor Deficit Tests.
We used the open-field test (OFT) and rotarod test to measure spontaneous locomotor activity/anxiety-like behavior and motor balance coordination, respectively, at baseline and post-MCAO days 2, 7, 14, 21, and 28. Being a nonstressful test, OFT was performed prior to the rotarod test, with a time gap of 1 h between the two tests.
Open-Field Test (OFT).
OFT measures exploratory behavior and general activity in rodents. Briefly, the mice were positioned at the corner of a clear acrylic box (16″ × 16″) to enable them to explore the box for roughly 10 min. Their locomotor activity was then quantified in terms of total number of breaks in the beam by a computer-operated, open-field photo beam activity system (San Diego Instruments, San Diego, CA). The percentage of beam breaks in the center zone (16″/3″ × 16″/3″) was then calculated in comparison to the total, signifying a measure of anxiety-like behavior. Importantly, OFT tests serve to analyze the pattern of behavior at several time points without hindrance by habituation.50
Rotarod Test.
Motor coordination in rodents was examined by the rotarod test. Mice were placed on a rotating cylindrical rod that accelerated from 4 to 40 rotations per min, for 5 min duration. There were two trials per subject including a 30 min break in between. The latency of falling from the rotating rod per trial (in seconds) was recorded, and mean latency was used for comparing the two groups.51
Novel Object Recognition Task (NORT).
The NORT is used to evaluate cognition, particularly recognition memory, in rodent models of CNS disorders. This test measured the time spent by a mouse exploring a novel object than a familiar one. This preference assessed intact recognition memory as detailed earlier.49 Briefly, the mice were placed in the behavioral room for acclimatization for 1 h. During habituation, the animals were allowed to explore an empty arena for at least 10 min. After habituation, the animals were exposed to the familiar arena with two identical objects placed at an equal distance for 10 min (trial phase). If the total time of exploration of these objects was greater than 20 s, these mice qualified for the experimental test, which was conducted 24 h after the trial. One of the objects from the trial was replaced with a novel object. The mice were then again allowed to explore the test arena for 10 min. The experiment was recorded and analyzed using Any Maze Software (Stoelting Co., Wood Dale, IL) by a trained observer. A discrimination index (DI) was calculated using the formula DI = (TN − TF)/(TN + TF), where TN is the time spent exploring the novel object and TF si the time spent exploring familiar objects. The NORT was performed on days 28 after stroke and analyzed by an experimenter blinded to treatment.
Calcium Influx Assay on Human Monocyte-Derived Macrophages.
Human monocytes were isolated from fresh whole blood obtained from healthy subjects after informed consent using EasySep Direct Human Monocyte Isolation Kit (Stemcell, Inc., Vancouver, BC, Canada, Cat # 19669). Pure monocytes were then supplemented with 5 ng/mL of Human GM-CSF (GenScript, Cat# Z02695) and cultured in RPMI 1640 media (90% RPMI 5%FSB+ 1% antibiotics) for 5–7 days till they become macrophage-like cells (and they show processes). Prior to Ca2+ influx assay, these human monocytes (Hu-Mo)-derived macrophages (Hu-Mø) were harvested and seeded (104 cells/plate) in a μ-Dish 35 mm (35 mm cell culture imaging dish, Ibidi, Gräfelfing, Germany, Cat# 81156) for at least 24 h. On the day of experiment, the medium was replaced with imaging buffer Hank’s balanced salt solution (HBSS) + 20% HEPES (v/v) as described in the Ca2+ influx assay provider protocol (Calbryte 590 Am ATT Bioquest, https://www.aatbio.com/products/calbryte-590-am). Briefly, a calcium indicator, Calbryte 590 AM (5 μM/mL), was loaded in each dish and incubated for 60 min followed by washing with HBSS buffer. Ca2+ imaging was performed in the presence or absence of 21u at the indicated concentrations in the presence of ATP (50 μM) using a Zeiss LSM880 laser scanning confocal microscope. Acquired images were then processed with MetaMorph Microscopy Automation and Image Analysis Software to measure Ca2+ activity signals and peak amplitude.
Statistics.
Data in vivo experiment were represented as mean ± S.D. Significance was determined using Student’s t-test (for two groups) or one-way ANOVA (for more, the two groups followed by Tukey’s post hoc test) for comparing the experimental groups between vehicle and drug treatment after stroke (GraphPad Prism Software, Inc., San Diego, CA). *p < 0.05 were considered statistical significance.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIDDK Intramural Research Program (ZIADK31127 to K.A.J.), NIH grant HL48225 (to B.T.L.), and American Heart Association grant 18CDA34110011 and NINDS grant 1R01NS125405-01A1 (to R.V.). The authors thank Robert O’Connor (NIDDK) for 2D-NMR and John Lloyd (NIDDK) for HRMS measurements. They thank Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health’s Psychoactive Drug Screening Program (Contract # HHSN-271-2008-00025-C) for screening data.
ABBREVIATIONS USED
- BDNF
brain-derived neurotrophic factor
- CHO
Chinese hamster ovary
- CV
cresyl violet
- HBSS
Hanks’ balanced salt solution
- HMDM
human monocyte-derived macrophage
- HEK-293
human embryonic kidney 293
- NORT
Novel Object Recognition Test
- NOS
nitric oxide synthase
- PDSP
NIMH Psychoactive Drug Screening Program
- TTC
2,3,5-triphenyltetrazolium chloride
- VNUT
vesicular nucleotide transporter
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01197.
Additional synthetic details, spectral and HPLC data, off-target binding inhibition data, and pharmacological data (PDF)
Molecular Formula Strings (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.2c01197
The authors declare no competing financial interest.
Contributor Information
Kiran S. Toti, National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, Maryland 20892-0810, United States.
Rajkumar Verma, Department of Neuroscience, UConn School of Medicine, Farmington, Connecticut 06032, United States.
Michael J. McGonnigle, Department of Neuroscience, UConn School of Medicine, Farmington, Connecticut 06032, United States
Daylin Gamiotea Turro, Department of Neuroscience, UConn School of Medicine, Farmington, Connecticut 06032, United States.
Zhiwei Wen, National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, Maryland 20892-0810, United States.
Sarah A. Lewicki, National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, Maryland 20892-0810, United States
Bruce T. Liang, Calhoun Cardiology Center, UConn School of Medicine, Farmington, Connecticut 06032, United States
Kenneth A. Jacobson, National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, Maryland 20892-0810, United States.
REFERENCES
- (1).Antonioli L; Blandizzi C; Fornai M; Pacher P; Lee HT; Haskó G P2X4 receptors, immunity, and sepsis. Curr. Opin. Pharmacol 2019, 47, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Stokes L; Layhadi JA; Bibic L; Dhuna K; Fountain SJ P2X4 receptor function in the nervous system and current breakthroughs in pharmacology. Front. Pharmacol 2017, 8, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dunton CL; Purves JT; Hughes FM; Jin H; Nagatomi J Elevated hydrostatic pressure stimulates ATP release which mediates activation of the NLRP3 inflammasome via P2X4 in rat urothelial cells. Int. Urol. Nephrol 2018, 50, 1607–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yu W; Hill WG; Robson SC; Zeidel ML Role of P2X4 receptor in mouse voiding function. Sci. Rep 2018, 8, No. 1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Xu J; Bernstein AM; Wong A; Lu XH; Khoja S; Yang XW; Davies DL; Micevych P; Sofroniew MV; Khakh BS P2X4 receptor reporter mice: sparse brain expression and feeding-related presynaptic facilitation in the arcuate nucleus. J. Neurosci 2016, 36, 8902–8920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fois G; Föhr KJ; Kling C; Fauler M; Wittekindt OH; Dietl P; Frick M P2X4 receptor re-sensitization depends on a protonation/deprotonation cycle mediated by receptor internalization and recycling. J. Physiol 2018, 596, 4893–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bertin E; Deluc T; Pilch KS; Martinez A; Pougnet JT; Doudnikoff E; Allain AE; Bergmann P; Russeau M; Toulmé E; Bezard E; Koch-Nolte F; Séguéla P; Lévi S; Bontempi B; Georges F; Bertrand SS; Nicole O; Boué-Grabot E Increased surface P2X4 receptor regulates anxiety and memory in P2X4 internalization-defective knock-in mice. Mol. Psychiatry 2021, 26, 629–644. [DOI] [PubMed] [Google Scholar]
- (8).Bernier LP; Ase AR; Boue-Grabot E; Seguela P P2X4 receptor channels form large noncytolytic pores in resting and activated microglia. Glia 2012, 60, 728–737. [DOI] [PubMed] [Google Scholar]
- (9).Schneider M; Prudic K; Pippel A; Klapperstück M; Braam U; Müller CE; Schmalzing G; Markwardt F Interaction of purinergic P2X4 and P2X7 receptor subunits. Front. Pharmacol 2017, 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Di Virgilio F; Sarti AC Microglia P2X4 receptors as pharmacological targets for demyelinating diseases. EMBO Mol. Med 2018, 10, No. e9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zabala A; Vazquez-Villoldo N; Rissiek B; Gejo J; Martin A; Palomino A; Perez-Samartín A; Pulagam KR; Lukowiak M; Capetillo-Zarate E; Llop J; Magnus T; Koch-Nolte F; Rassendren F; Matute C; Domercq M P2X4 receptor controls microglia activation and favors remyelination in autoimmune encephalitis. EMBO Mol. Med 2018, 10, No. e8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Matsumura Y; Yamashita T; Sasaki A; Nakata E; Kohno K; Masuda T; Tozaki-Saitoh H; Imai T; Kuraishi Y; Tsuda M; Inoue K A novel P2X4 receptor-selective antagonist produces antiallodynic effect in a mouse model of herpetic pain. Sci. Rep 2016, 6, No. 32461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Srivastava P; Cronin CG; Scranton VL; Jacobson KA; Liang BT; Verma R Neuroprotective and neuro-rehabilitative effects of acute purinergic receptor P2X4 (P2X4R) blockade after ischemic stroke. Exp. Neurol 2020, 329, No. 113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Masuda T; Ozono Y; Mikuriya S; Kohro Y; Tozaki-Saitoh H; Iwatsuki K; Uneyama H; Ichikawa T; Salter MW; Tsuda M; Inoue K Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat. Commun 2016, 7, No. 12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Malcangio M Spinal mechanisms of neuropathic pain: Is there a P2X4-BDNF controversy? Neurobiol. Pain 2017, 1, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Long T; He W; Pan Q; Zhang S; Zhang D; Qin G; Chen L; Zhou J Microglia P2X4R-BDNF signalling contributes to central sensitization in a recurrent nitroglycerin-induced chronic migraine model. J. Headache Pain 2020, 21, No. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ozaki T; Muramatsu R; Sasai M; Yamamoto M; Kubota Y; Fujinaka T; Yoshimine T; Yamashita T The P2X4 receptor is required for neuroprotection via ischemic preconditioning. Sci. Rep 2016, 6, No. 25893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bowler JW; Bailey JR; North AR; Surprenant A P2X4, P2Y1 and P2Y2 receptors on rat alveolar macrophages. Br. J. Pharmacol 2003, 140, 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang L; Feng X; Hu B; Xia Q; Ni X; Song Y P2X4R promotes airway remodeling by acting on the phenotype switching of bronchial smooth muscle cells in rats. Purinergic Signalling 2018, 14, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yoshida K; Ito M; Sato N; Obayashi K; Yamamoto K; Koizumi S; Tanaka S; Furuta K; Matsuoka I Extracellular ATP augments antigen-induced murine mast cell degranulation and allergic responses via P2X4 receptor activation. J. Immunol 2020, 204, 3077–3085. [DOI] [PubMed] [Google Scholar]
- (21).Yoshida K; Tajima M; Nagano T; Obayashi K; Ito M; Yamamoto K; Matsuoka I Co-stimulation of purinergic P2X4 and prostanoid EP3 receptors triggers synergistic degranulation in murine mast cells. Int. J. Mol. Sci 2019, 20, 5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yang R; Beqiri D; Shen JB; Redden J; Dodge-Kafka K; Jacobson KA; Liang BT P2X4 receptor-eNOS signaling pathway in cardiac myocytes as a novel protective mechanism in heart failure. Comput. Struct. Biotechnol. J 2015, 13, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bragança B; Correia-de-Sá P Resolving the ionotropic P2X4 receptor mystery points towards a new therapeutic target for cardiovascular diseases. Int. J. Mol. Sci 2020, 21, 5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Csóka B; Németh ZH; Szabó I; Davies DL; Varga ZV; Pálóczi J; Falzoni JS; Di Virgilio F; Muramatsu R; Yamashita T; Pacher P; Haskó G Macrophage P2X4 receptors augment bacterial killing and protect against sepsis. JCI Insight 2018, 3, No. e99431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hernandez-Olmos V; Abdelrahman A; El-Tayeb A; Freudendahl D; Weinhausen S; Müller CE N-substituted phenoxazine and acridone derivatives: structure-activity relationships of potent P2X4 receptor antagonists. J. Med. Chem 2012, 55, 9576–9588. [DOI] [PubMed] [Google Scholar]
- (26).Abdelrahman A; Namasivayam V; Hinz S; Schiedel AC; Köse M; Burton M; El-Tayeb A; Gillard M; Bajorath J; de Ryck M; Müller CE Characterization of P2X4 receptor agonists and antagonists by calcium influx and radioligand binding studies. Biochem. Pharmacol 2017, 125, 41–54. [DOI] [PubMed] [Google Scholar]
- (27).Werner S; Mesch S; Hillig RC; ter Laak A; Klint J; Neagoe I; Laux-Biehlmann A; Dahllöf H; Bräuer N; Puetter V; Nubbemeyer R; Schulz S; Bairlein M; Zollner TM; Steinmeyer A Discovery and characterization of the potent and selective P2X4 inhibitor N-[4-(3-chlorophenoxy)-3-sulfamoylphenyl]-2-phenylaceta-mide (BAY-1797) and structure-guided amelioration of its CYP3A4 induction profile. J. Med. Chem 2019, 62, 11194–11217. [DOI] [PubMed] [Google Scholar]
- (28).Nagata K; Imai T; Yamashita T; Tsuda M; Tozaki-Saitoh H; Inoue K Antidepressants inhibit P2X4 receptor function: a possible involvement in neuropathic pain relief. Mol. Pain 2009, 5, 1744–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yamashita T; Yamamoto S; Zhang J; Kometani M; Tomiyama D; Kohno K; Hidetoshi Tozaki-Saitoh H; Inoue K; Tsuda M Duloxetine inhibits microglial P2X4 receptor function and alleviates neuropathic pain after peripheral nerve injury. PLoS One 2016, 11, No. e0165189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tian M; Abdelrahman A; Baqi Y; Fuentes E; Azazna D; Spanier C; Densborn S; Hinz S; Schmid R; Müller CE Discovery and structure relationships of salicylanilide derivative as potent, non-acidic P2X1 receptor antagonists. J. Med. Chem 2020, 63, 6164–6178. [DOI] [PubMed] [Google Scholar]
- (31).Coddou C; Sandoval R; Hevia MJ; Stojilkovic SS Characterization of the antagonist actions of 5-BDBD at the rat P2X4 receptor. Neurosci. Lett 2019, 690, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ase AR; Honson NS; Zaghdane H; Pfeifer TA; Seǵueĺa P Identification and characterization of a selective allosteric antagonist of human P2X4 receptor channels. Mol. Pharmacol 2015, 87, 606–616. [DOI] [PubMed] [Google Scholar]
- (33).Latapiat V; Rodríguez FE; Godoy F; Montenegro FA; Barrera NP; Huidobro-Toro JP P2X4 receptor in silico and electrophysiological approaches reveal insights of ivermectin and zinc allosteric modulation. Front. Pharmacol 2017, 8, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Dhuna K; Felgate M; Bidula SM; Walpole S; Bibic L; Cromer BA; Angulo J; Sanderson J; Stebbing MJ; Stokes L ginsenosides act as positive modulators of P2X4 receptors. Mol. Pharmacol 2019, 95, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Asatryan L; Ostrovskaya O; Lieu D; Davies DL Ethanol differentially modulates P2X4 and P2X7 receptor activity and function in BV2 microglial cells. Neuropharmacology 2018, 128, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang Z; Mei M; Wang Q; Guo R; Liu P; Wang Y; Zhang Z; Wang L Role of dehydrocorybulbine in neuropathic pain after spinal cord injury mediated by P2X4 receptor. Mol. Cells 2019, 42, 143–150. Correction in: Mol. Cells. 2019, 42(4), 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kawate T; Carlisle Michel J; Birdsong WT; Gouaux E Crystal structure of the ATP-gated P2X4 ion channel in the closed state. Nature 2009, 460, 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hattori M; Gouaux E Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 2012, 485, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sakuma S; Takahashi T; Ushioda M; Imai T; Inoue K Diazepinedione derivatives. US2011319610A1, 2011.
- (40).Sathunuru R; Rao UN; Biehl E Facile, high-yield, regioselective synthesis of ortho-nitrophenols using cerium (IV) ammonium nitrate. ARKIVOC 2003, 15, 124–133. [Google Scholar]
- (41).Joshi AV; Baidoosi M; Mukhopadhyay S; Sasson Y Nitration of phenol and substituted phenols with dilute nitric acid using phase-transfer catalysts. Org. Process Res. Dev 2003, 7, 95–97. [Google Scholar]
- (42).Jagtap A; Gharpure M; Shinde N; Patil N; Shah C; Raut C; Krishnmurthy D An improved process for the preparation of Clobazam and its intermediate. WO2016151464A1, 2016.
- (43).Wolfe JP; Buchwald SL Palladium-catalyzed amination of aryl iodides. J. Org. Chem 1996, 61, 1133–1135. [DOI] [PubMed] [Google Scholar]
- (44).Sherwood J; Clark JH; Fairlamb IJS; Slattery JM Solvent effects in palladium catalysed cross-coupling reactions. Green Chem. 2019, 21, 2164–2213. [Google Scholar]
- (45).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FRC; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Verma R; Cronin CG; Hudobenko J; Venna VR; McCullough LD; Liang BT Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain, Behav., Immun 2017, 66, 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Verma R; Harris NM; Friedler BD; Crapser J; Patel AR; Venna V; McCullough LD Reversal of the detrimental effects of post-stroke social isolation by pair-housing is mediated by activation of BDNF-MAPK/ERK in aged mice. Sci. Rep 2016, 6, No. 25176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Swanson RA; Morton MT; Tsao-Wu G; Savalos RA; Davidson C; Sharp FR A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab 1990, 10, 290–293. [DOI] [PubMed] [Google Scholar]
- (49).Verma R; Friedler BD; Harris NM; McCullough LD Pair housing reverses post-stroke depressive behavior in mice. Behav. Brain Res 2014, 269, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Moy SS; Nikolova VD; Riddick NV; Baker LK; Koller BH Preweaning sensorimotor deficits and adolescent hypersociability in Grin1 knockdown mice. Dev. Neurosci 2012, 34, 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Truong DT; Venna VR; McCullough LD; Fitch RH Deficits in auditory, cognitive, and motor processing following reversible middle cerebral artery occlusion in mice. Exp. Neurol 2012, 238, 114–121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.