

Abstract
Phosphodiesterase (PDE) inhibitors have been safely and effectively used in the clinic and increase the concentration of intracellular cyclic nucleotides (cAMP/cGMP). These molecules activate downstream mediators, including the cAMP response element-binding protein (CREB), which controls neuronal excitability and growth responses. CREB gain of function enhances learning and allocates neurons into memory engrams. CREB also controls recovery after stroke. PDE inhibitors are linked to recovery from neural damage and to stroke recovery in specific sites within the brain. PDE2A is enriched in cortex. In the present study, we use a mouse cortical stroke model in young adult and aged male mice to test the effect of PDE2A inhibition on functional recovery, and on downstream mechanisms of axonal sprouting, tissue repair, and the functional connectivity of neurons in recovering cortex. Stroke causes deficits in use of the contralateral forelimb, loss of axonal projections in cortex adjacent to the infarct, and functional disconnection of neuronal networks. PDE2A inhibition enhances functional recovery, increases axonal projections in peri-infarct cortex, and, through two-photon in vivo imaging, enhances the functional connectivity of motor system excitatory neurons. PDE2A inhibition after stroke does not have an effect on other aspects of tissue repair, such as angiogenesis, gliogenesis, neurogenesis, and inflammatory responses. These data suggest that PDE2A inhibition is an effective therapeutic approach for stroke recovery in the rodent and that it simultaneously enhances connectivity in peri-infarct neuronal populations.
SIGNIFICANCE STATEMENT Inhibition of PDE2A enhances motor recovery, axonal projections, and functional connectivity of neurons in peri-infarct tissue. This represents an avenue for a pharmacological therapy for stroke recovery.
Keywords: functional connectivity, GCaMP, neural repair, neurogenesis, phosphodiesterase, two photon
Introduction
Treatment for ischemic stroke continues to be based on reperfusion of the infarcted tissue (Lees et al., 2010), leaving the majority of patients ineligible because of its time-sensitive nature. Stroke is the leading cause of adult disability in the United States (Virani et al., 2020), highlighting the need for molecular-based pharmacotherapies that enhance recovery after the initial insult and that can be given over a longer window after the infarct. Rodent models of stroke have demonstrated the progressive morphologic and transcriptional changes that take place in peri-infarct neurons (Carmichael, 2016) and that some of these changes, such as axonal sprouting, correlate with recovery in the rodent (Benowitz and Carmichael, 2010; Li et al., 2010, 2015).
The cyclic nucleotides cAMP and cGMP are important regulators of brain responses to disease and injury, including vascular remodeling, immune cell modulation, and axonal growth (Knott et al., 2017). Many of these molecules are also tied to the field of learning and memory, mediated by their activation of protein kinases and transcription of cAMP response element (CRE) gene products (Stevens, 1994; Kida et al., 2002; Sakamoto et al., 2011). CRE-binding protein (CREB) is a well known mediator of this pathway, facilitating immediate-early genes, neuronal excitatory signaling systems, long-term potentiation, and memory formation (Stevens, 1994; Kida et al., 2002; Sakamoto et al., 2011). The level of active, phosphorylated CREB (pCREB) decreases the threshold of excitability for a neuron, such that neurons with the highest levels of pCREB during a behavior have the highest probability of being incorporated into the neuronal representation of that behavior (Kida et al., 2002; Zhou et al., 2009; Kida and Serita, 2014; Joy et al., 2019). CREB was recently shown to directly enhance stroke recovery in a mouse model: overexpression of CREB in a subset of peri-ischemic neurons enhances axonal sprouting within these cells and enhances behavioral recovery (Caracciolo et al., 2018). Blocking activation of CREB-enhanced neurons after stroke removes this enhanced recovery: mice with stroke that have enhanced recovery because of increased CREB levels deteriorate back to a baseline stroke impairment if CREB-induced neurons are selectively inhibited. These studies indicate that CREB exquisitely controls recovery, as CREB neuronal circuits can be dialed up or dialed down to either increase or decrease recovery after stroke.
Intracellular phosphodiesterase (PDE) enzymes degrade cAMP/cGMP, attenuating the signal for CREB activation. Eleven families of PDE enzymes exist, each with unique cAMP/cGMP affinity and tissue expression profiles (Delhaye and Bardoni, 2021). PDE inhibitors are safely used in the clinic for the treatment of vascular claudication (PDE3i; Packer et al., 1991), COPD exacerbations (PDE4i; Lipworth, 2005), and erectile dysfunction (PDE5i; Ghofrani et al., 2006). Given the link between cyclic nucleotides, CREB, and memory formation, PDE inhibition paradigms have been investigated as a therapeutic approach for neural repair (Knott et al., 2017). For example, the PDE4 inhibitor Rolipram promotes axonal regeneration and recovery in spinal cord injury (Nikulina et al., 2004). Several phase I–III trials are ongoing for PDE3, PDE4, and PDE5 inhibitors for neurologic disease.
The PDE2 family of enzymes are active against cAMP and cGMP and highly expressed in the brain (Delhaye and Bardoni, 2021). PDE2 inhibition has been shown to enhance memory in mouse models of aging and memory function (Domek-Łopacińska and Strosznajder, 2008; Reneerkens et al., 2013), and activate CREB signaling (Chen et al., 2019). PDE2A-T1 is a novel PDE2A-specific inhibitor that increases cGMP, without increasing cAMP, in a dose-dependent manner in the rat cortex [Compound 20 (used in the study by Mikami et al., 2017)]. We tested the effects of this PDE2A inhibitor in a mouse stroke model on axonal sprouting, behavioral function, and two-photon imaging of neuronal networks. These studies show that PDE2A inhibition promotes the recovery of sensorimotor function after stroke in young adult and aged mice, and mediates increased projections and functional networks in tissue adjacent to stroke. PDE2A inhibition promotes the association of neuronal activity in brain networks in cortical areas toward the more normal, nonstroke state.
Materials and Methods
Experimental design and statistical analysis.
Studies were designed in a manner consistent with Stroke Therapy Academic Industry Roundtable and RIGOR guidelines [Stroke Therapy Academic Industry Roundtable (STAIR), 1999; Landis et al., 2012; Lapchak et al., 2013]. Sample sizes for each analysis were determined using power analyses based on expected variances and group differences. Allocation of mice into treatment groups was performed randomly, and behavior experiments were performed with the experimenter blind to treatment group. Statistical analyses were performed with Prism 6.0 (GraphPad Software). Group sizes, statistical tests, and p values for each experiment are reported in Table 1.
Table 1.
Statistics
| Figure | Group size (n) | Statistical test | Statistical values |
|||||
|---|---|---|---|---|---|---|---|---|
| Two-way ANOVA | Ordinary | |||||||
| 2 A | 12–14 | Two-way ANOVA multiple comparisons, Tukey's multiple-comparisons test | α | 0.05 | ||||
| Source of variation | Percentage of total variation | p value | p value summary | Significant? | ||||
| Interaction | 12.31 | <0.0001 | **** | Yes | ||||
| Time | 22 | <0.0001 | **** | Yes | ||||
| Treatment | 17.65 | <0.0001 | **** | Yes | ||||
| ANOVA table | SS (type III) | df | MS | F (dfn,dfd) | p value | |||
| Interaction | 0.4271 | 9 | 0.04745 | F(9,192) = 5.507 | p < 0.0001 | |||
| Time | 0.7635 | 3 | 0.2545 | F(3,192) = 29.53 | p < 0.0001 | |||
| Treatment | 0.6125 | 3 | 0.2042 | F(3,192) = 23.69 | p < 0.0001 | |||
| Residual | 1.655 | 192 | 0.008618 | |||||
| 2 B | 7–9 | Two-way ANOVA multiple comparisons, Tukey's multiple-comparisons test | Mixed-effects model (REML) | Matching: stacked | ||||
| Assume sphericity? | No | |||||||
| α | 0.05 | |||||||
| Fixed effects (type III) | p value | F (dfn,dfd) | Geisser–Greenhouse's epsilon | |||||
| Row factor | <0.0001 | F(3.581,92.22) = 28.67 | 0.8954 | |||||
| Column factor | <0.0001 | F(3,27) = 26.32 | ||||||
| Row factor × column factor | <0.0001 | F(12,103) = 10.86 | ||||||
| Random effects | SD | Variance | ||||||
| Subject | 2.502 | 6.261 | ||||||
| Residual | 2.433 | 5.919 | ||||||
| 2 C | 8–9 | Two-way ANOVA, Sidak's multiple-comparisons test | Mean difference | 95.00% CI of difference | Summary | Adjusted p value | ||
| Sidak's multiple-comparisons test | ||||||||
| Stroke + vehicle – stroke + 3.0 mg/kg PDE2A-T1 | 0 | |||||||
| Baseline | 22.05 | −0.3310 to 44.44 | ns | 0.0542 | ||||
| 1 Week | 20.71 | 3.226–38.19 | ** | 0.0175 | ||||
| 5 Weeks | 22.03 | 1.602–42.45 | * | 0.0322 | ||||
| 9 Weeks | ||||||||
| Mean 1 | Mean 2 | Mean difference | SE of difference | N1, N2, t, df | ||||
| Test details | ||||||||
| Stroke plus vehicle – stroke plus 3.0 mg/kg PDE2A-T1 | 0 | 0 | 0 | 0 | 9, 8 | |||
| Baseline | 43.59 | 21.54 | 22.05 | 7.775 | 9, 8, 2.837, 13.2 | |||
| 1 Week | 33.56 | 12.85 | 20.71 | 6.174 | 9, 8, 3.354, 14, 81 | |||
| 5 Weeks | 35.07 | 13.04 | 22.03 | 7.204 | 9, 8, 3.057, 14.7 | |||
| 9 Weeks | ||||||||
| 3 C | 3–4 | Unpaired two-tailed t test | F (dfn,dfd) | 76.71, 2, 3 | p value | 0.0053 | ||
| 3 D | 5–6 | Unpaired two-tailed t test | F (dfn,dfd) | 2.477, 4, 5 | p value | 0.3468 | ||
| 3 E | 4 | Unpaired two-tailed t test | F (dfn,dfd) | 4.696, 3, 3 | p value | 0.2362 | ||
| 3 F | 4 | Unpaired two-tailed t test | F (dfn,dfd) | 5.063, 3, 3 | p value | 0.2158 | ||
| 3 G | 4 | Unpaired two-tailed t test | F (dfn,dfd) | 1.511, 11, 11 | p value | 0.5051 | ||
| 3 H | 3 | Unpaired two-tailed t test | F (dfn,dfd) | 2.223, 2, 2 | p value | 0.6205 | ||
| 4 A | 4–5 | Two-way ANOVA multiple comparisons, Tukey's multiple-comparisons test | ANOVA table | SS | df | MS | F statistic | p value |
| Time × stroke/drug condition | 96,557,499 | 12 | 8,046,458 | F(12,60) = 9.432 | p < 0.0001 | |||
| Time factor | 8,724,740 | 4 | 2,181,185 | F(2.619,39.28) = 2.557 | p = 0.0763 | |||
| Stroke/drug factor | 23,360,987 | 3 | 7,786,996 | F(3,15) = 3.719 | p = 0.0351 | |||
| Subject | 31,407,485 | 15 | 2,093,832 | F(15,60) = 2.454 | p = 0.0072 | |||
| Residual | 51,186,802 | 60 | 853,113 | |||||
| 4 B | 4–5 | Two-way ANOVA multiple comparisons, Tukey's multiple-comparisons test | ANOVA table | SS | df | MS | F statistic | p value |
| Time – stroke/drug condition | 8214 | 12 | 684.5 | F(12,60) = 6.795 | p < 0.0001 | |||
| Time factor | 1229 | 4 | 307.2 | F(2.504,37.56) = 3.050 | p = 0.0486 | |||
| Stroke/drug factor | 5183 | 3 | 1728 | F(3,15) = 16.42 | p < 0.0001 | |||
| Subject | 1578 | 15 | 105.2 | F(15,60) = 1.044 | p = 0.4254 | |||
| Residual | 6044 | 60 | 100.7 | |||||
| 4 C | 4–5 | Functional connectivity maps, no statistics, visual presentation | ||||||
MS, Mean sum of squares; REML, restricted maximum likelihood; SS, sum of squares; dfn, df numerator; dfd, df denominator.
Mice.
All procedures were performed in accordance with National Institutes of Health Animal Protection Guidelines. Ten- to 12-week-old male C57BL/6 mice from The Jackson Laboratory were used for young adult studies. The 18-month-old male C57BL/6 mice obtained from the National Institute on Aging were used for aged studies. Animals were housed under pathogen-free conditions in a light-controlled environment with a reverse light cycle, with free access to irradiated pellets (PicoLab Rodent Diet 20, LabDiet) and sterilized, acidified water. Housing temperatures were maintained at between 69°F and 70°F and at standard humidity between 30% and 70%.
PDE2A-T1 preparation.
Lyophilized PDE2A-T1, a PDE2A-specific inhibitor [Compound 20 (used in the study by Mikami et al., 2017)] was obtained from Takeda Pharmaceutical Company.
PDE2A-T1 was solubilized stepwise in 10% DMSO, 10% Cremophor EL, 30% PEG 400, 10% propylene glycol, and 40% H2O. At each step, the PDEA-T1 solution was vortexed. After the addition of H2O, the solution was vortexed until completely translucent. Fresh PDE2A-T1 solution was prepared twice weekly. Once daily, 50 µl of either vehicle or 3.0 mg/kg PDE2A-T1 solution was injected intraperitoneally into test subjects. Daily injections were initiated 5 d after stroke.
Drug delivery.
Daily intraperitoneal injections of drug or vehicle were initiated 5 d after stroke induction. PDE2A-T1 was injected at 3.0 mg/kg. No adverse health effects were observed over the course of drug treatment.
Stroke production.
Mice were anesthetized with 2.5% isoflurane and placed in a stereotaxic apparatus. Heads were shaved and aseptically prepared. Skulls were exposed using a midline incision. Each mouse was injected with 200 µl of a sterile solution of 10 mg/ml Rose Bengal in PBS. After 5 min, a 200 mV light source was shone focally onto forelimb motor cortex (anteroposterior, 0.0 mm; mediolateral, 1.5 mm; dorsoventral, 0.0 mm) for 15–18 min. The skull was rinsed using sterile solution, and the incision was closed. Body temperature was monitored and maintained at 37°C (±3°C) during the procedure. For the head-fixed behavioral and in vivo imaging studies, stroke was produced with a laser (laser (CLD1010LP, ThorLabs; laser diode LP520-MF100, ThorLabs), as described previously (Latifi et al., 2020), with a wavelength of 520 nm.
5-Ethynyl-2′deoxyuridine preparation.
A stock solution of 5 mg/ml of 5-ethynyl-2′deoxyuridine (EdU) was prepared in potable water. EdU, along with the antibiotic trimethoprim–sulfamethoxazole, was added to mouse drinking water on days 3–10 after stroke, for a final EdU concentration of 200 µg/ml. Water was refreshed every 48 h.
BDA injections.
Ten weeks after stroke, the neuroanatomical tracer biotinylated dextran amine (BDA; molecular weight, 10,000; Thermo Fisher Scientific) was injected into motor cortex ipsilateral to the cortical stroke site. Following anesthesia with 2.5% isoflurane and placement in a stereotaxic apparatus, a burr hole was made in the skull 0.0 mm anterior and 1.5 mm lateral to bregma. Using a Hamilton syringe and automated injection apparatus, 0.3 µl of BDA was infused at a rate of 0.03 µl/min, 0.75 mm ventral to the surface of the brain. Body temperature was monitored and maintained at 37°C (±3°C) during the procedure. Animals were killed 1 week after BDA injections.
In vivo imaging surgeries and virus.
Adeno-associated virus 9 (AAV9) with GCaMP6s under the CaMKII promoter was purchased from AddGene (catalog #107790-AAV9 (previously called AV-9-PV4365 from Penn Vector Core, University of Pennsylvania, Philadelphia, PA). These AAV9 particles drive expression of GCaMP6s in CaMKII-expressing cells, primarily pyramidal neurons. Each viral order was delivered as a volume of 100 μl of at least 1 × 1013 viral genomes/ml titer. This volume was aliquoted to 5 μl volumes, which could be individually thawed on the day of use. The following three injections were delivered: one 500 nl injection to the primary motor cortex (M1) and two to the secondary motor areas (M2). Coordinates for these areas were based on the Allen Adult Mouse Atlas (http://atlas.brain-map.org/) defined the primary motor area and the secondary motor area. After craniotomy and viral injection, mice were allowed to recover while the GCaMP expression ramps up. GCaMP6s expression was typically observed to begin 21 d after surgery, but may take up to 6 weeks. Photothrombotic stroke was applied 6–8 weeks after surgery and at least three baseline imaging and behavioral sessions (not including habituation). Craniotomy and imaging procedures are published and used without modification (Latifi et al., 2020).
Imaging and data processing.
Two-photon imaging of mouse cortical neurons in stroke followed our published approach (Latifi et al., 2020), with the experimental timeline of imaging before and after stroke (Fig. 1A). Briefly, a resonant scanning two-photon microscope (Neurolabware) with a Chameleon III laser (Coherent) at a fixed wavelength of 920 nm was used. The 8 kHz resonant raster scanner equates to 512 lines at 30 Hz bidirectionally, which results in image capture at 15.42 Hz. A dichroic mirror (Semrock) is used to filter the captured light and allow for imaging corresponding to GCaMP6s signals. The objective used was a 16× water-based lens (0.8 numerical aperture; Nikon; Fig. 1B). Image capture was relayed to Scanbox, an acquisition software that runs through MATLAB (https://scanbox.org/).
Figure 1.
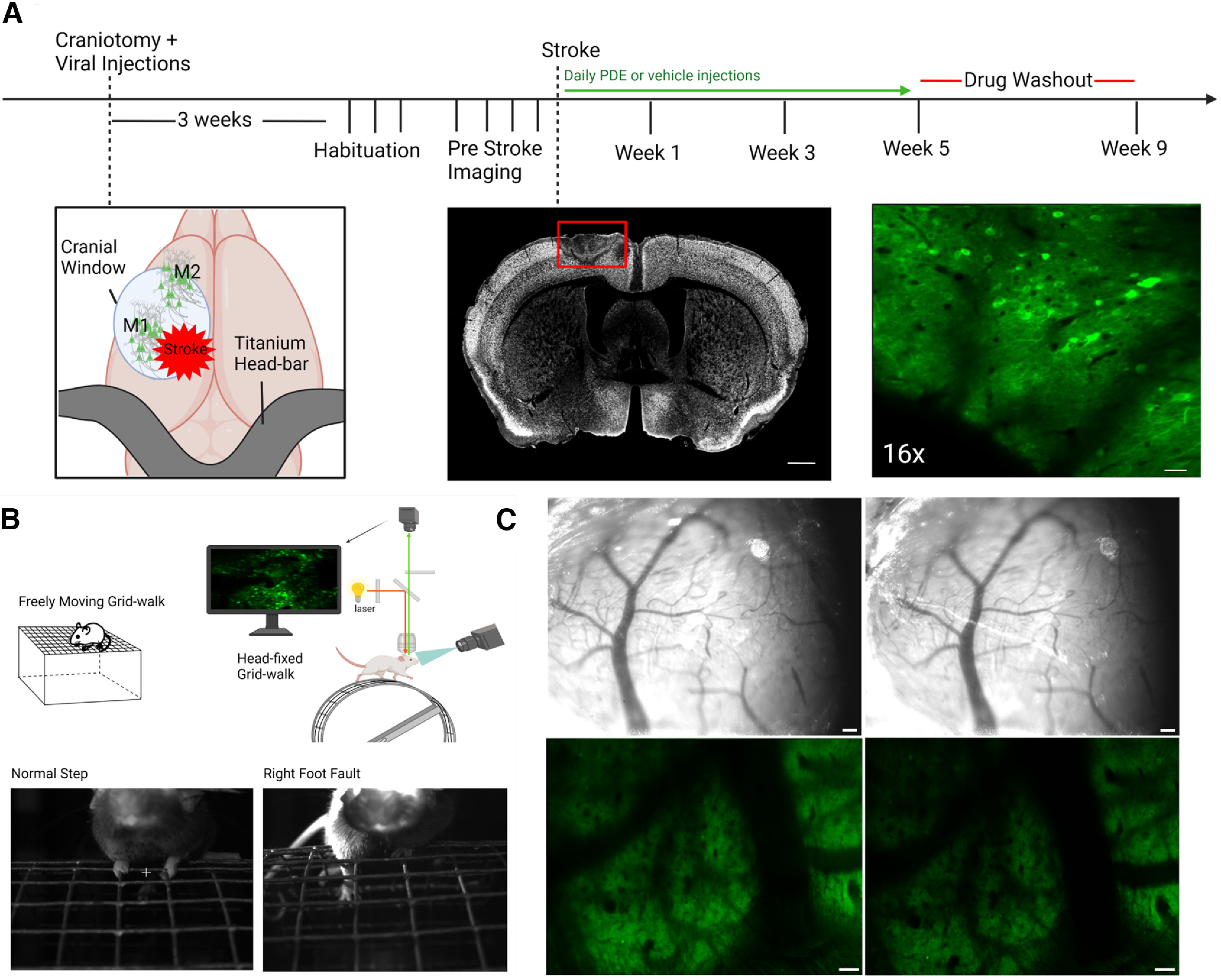
Experimental setup and timeline. A, Schematic of AAV viral injections at M1 and M2 during craniotomy and the application of a 4 mm coverglass over the region; approximate location of stroke is designated. Right, photo of imaging window showing CGaMP6s-labeled neurons. Top, Timeline for the imaging studies, starting with craniotomy/injections, through imaging, stroke, and drug delivery; time since craniotomy is indicated on the timeline. Middle, Photomicrograph coronal section through the stroke with NeuN stain, 14 d after stroke. Red rectangle indicates the stroke core and the loss of live neurons in this region. Scale bars, 1 mm. Left, Imaging window of GcaMP6s-transduced neurons in green. Scale bar, 50 µm. B, Behavioral and imaging setup. Schematic of mouse on grid for gait assessment. Schematic of the two-photon light path and integration with the head-fixed mouse for calcium transient imaging. Bottom, Images show correctly placed step and a foot fault. C, Long-term clarity of coverglass and imaging window, 1 (left column) and 3 (right column) months after craniotomy. Scale bar, 100 µm.
Custom-written MATLAB software with ImageJ (NIH) are used to analyze extracted data, First, raw imaging files are converted to .tif files. Regions of interest (ROIs) related to neuronal cell bodies with an intensity >30% of background intensity (darkest region of a video) are selected using a semiautomated algorithm (Latifi et al., 2020). These ROIs are also size constrained from 100 to 250 μm2. The output map is achieved with all potential ROIs and their fluorescent traces, and each ROI is manually inspected for its morphology and fluorescent traces. Any ROIs that were suspicious of overexpressing GCaMP6s, such as the expression of GCaMP in the nucleus, are excluded from the data analysis. Fluorescence intensity is converted to ΔF/F values, which were calculated by subtracting the average fluorescence of the ROI from the current fluorescence at each particular frame, then dividing by the average fluorescence. Calcium transient peaks are detected by applying a MATLAB smoothing and peak detection function to the waveform. Peaks that are not greater than the root mean square of ΔF/F are not considered calcium transients. Amplitudes of each transient are calculated as the peak of the transient from baseline ΔF/F, and frequency was calculated by dividing the number of identified Ca2+ transient peaks and dividing by seconds being imaged, resulting in the number of transients per time (hertz).
A deconvolution algorithm is applied to all ΔF/F traces to remove the nonphysiological slow decay of viral-expressing GcaMP6s signals and to sharpen the calcium transients (Yaksi and Friedrich, 2006). The Pearson product-moment correlation coefficient (PCC) for each deconvolved trace is calculated about all other deconvolved paths, which correspond to all different ROIs. Thus, PCC is a measure of the correlation between one neuron and another neuron in the population. Next, a Monte-Carlo simulation is applied that randomly shifts each deconvolved ΔF/F trace in time by 0.060 s up to 180 s. The back end of the trace is relocated to the beginning of the trace. The PCC value for each time-shifted trace is recalculated to all nonshifted deconvolved traces. The random time-shifted simulations are repeated 100 times for each trace, indicating an average number of 801,000 comparisons per recording (the average number of neurons per field of view was 90; thus, 100 × 90 × 89 = 801,000 comparisons). The distribution of all observed PCC values is then generated. The 99th percentile of this distribution is used as a threshold to define a “significant PCC value” for each trace. The original PCC values are compared with this threshold, and only values exceeding this threshold are defined as significant. A table of all significant connections and the two ROIs that are “functionally connected” to each other is generated. The average PCC value of these significant connections is calculated and is referred to as the average PCC for the particular recording. For each mouse subject, there are at least three baseline imaging sessions before the experimental variable is applied—in this case, a stroke to the underlying cortex. The average PCC value for each baseline video is noted, and an aggregate average is calculated, which is referred to as the “threshold” for a particular mouse.
Generating functional connectivity measures.
Once the threshold for a particular mouse is determined, a custom-written MATLAB code is applied that uses the threshold as a cutoff for all videos for a particular mouse. All significant PCC values (defined above) above the threshold were defined as the functional connectivity (FC) of the recordings. These are the values that are above the average of the baseline PCC values, signifying them as “functional connections” of this network over the time series. The FC maps are then generated representing nodes (connected neurons) and edges (FC).
After the assessment of FC, network density (ND), defined as the ratio of measured edges (FC) to the number of total possible edges (FC), is calculated for each mouse. ND provides the measurement of correlativity for comparing data-points across mouse subjects and several time points:
Behavioral assays.
The grid-walking test to assay motor control of the contralateral forelimb was performed as described previously (Li et al., 2010; Li et al., 2015; Clarkson et al., 2011; Joy et al., 2019). Mice were placed on a suspended wire grid and video recorded while walking freely for 5 min each. The total grid area was 32 × 20 cm, with 1 × 1 cm squares. Videos were subsequently analyzed offline by evaluators blinded to the treatment groups. The total number of steps taken and the total number of foot faults by the stroke-affected forelimb, defined as the total number of slips through the grid, were recorded. The number of foot faults was reported as percentages of the total number of steps taken and normalized to prestroke baseline performance.
An adapted version of the grid walking was created for two-photon imaging. The same grid was used in the non-head-fixed foot fault task, and was cut into ∼7-inch-wide sections and fit into the backs of a 9-inch-wide section. A further adaptation was added to facilitate accurate analysis of behavioral function: a small DC motor was connected to the axle of the wheel such that the grid could be driven forward at an adjustable rate. This adaptation forces the mouse to move forward. Infrared lights and infrared-sensitive cameras were placed facing the mouse and from the side of the mouse to record locomotion while all lights are turned off for two-photon imaging. These cameras captured video at 30 Hz.
Immunohistochemistry.
Brains were perfused with 4% paraformaldehyde, cryoprotected, and frozen. Tissue sections were incubated in primary antibodies overnight at 4°C, following permeabilization and blocking. The following primary antibodies were used: rat anti-GFAP (Thermo Fisher Scientific); goat anti-IBA-1 (Abcam); rat anti-Glut-1 (Abcam); rabbit anti-Olig2 (Millipore); rabbit anti-NeuN (Abcam); and rat anti-CD31 (BD Biosciences). Secondary antibodies were as follows: Alexa Fluor 488 donkey anti-rabbit, Alexa Fluor 488 donkey anti-rat, Alexa Fluor 647 donkey anti-rabbit, Alexa Fluor 647 donkey anti-rat, and Alexa Fluor 647 streptavidin (Jackson ImmunoResearch). Confocal z-stack images from peri-infarct and contralesional striatum, as well as confocal z-stack images of ipsilesional cortex, were obtained at 40× magnification. The number of cells and the colocalization of nuclear cell markers through the z-stack were quantified using Imaris (Bitplane) Sample size was four to five animals per group.
Brains were perfused with 4% paraformaldehyde, cryoprotected, and frozen. Tissue sections were incubated in primary antibodies overnight at 4°C, following permeabilization and blocking. The following primary antibodies were used: rat anti-GFAP (Thermo Fisher Scientific); goat anti-IBA-1 (Abcam); rat anti-Glut-1 (Abcam); rabbit anti-Olig2 (Millipore); rabbit anti-NeuN (Abcam); and rat anti-CD31 (BD Biosciences). Secondary antibodies were as follows: Alexa Fluor 488 donkey anti-rabbit, Alexa Fluor 488 donkey anti-rat, Alexa Fluor 647 donkey anti-rabbit, Alexa Fluor 647 donkey anti-rat, and Alexa Fluor 647 streptavidin (Jackson ImmunoResearch). Confocal z-stack images from peri-infarct and contralesional striatum, as well as confocal z-stack images of ipsilesional cortex, were obtained at 40× magnification. The number of cells and colocalization of nuclear cell markers through the z-stack were quantified using Imaris (Bitplane; n = 4–5 animals/group).
Axonal sprouting analysis.
Following BDA injection and tissue harvesting, brain sections were stained for BDA using a streptavidin-conjugated fluorophore. Confocal z-stack images of each full section were obtained at 20× magnification, and maximum intensity projection images were analyzed. Using ImageJ, integrated densities were recorded from various brain regions, including white matter, cortex, and striatum of both hemispheres. After background subtraction, intensity data of each section were normalized to the integrated density of the BDA injection site of the section. Mean integrated density across six sections was calculated and plotted for each animal.
Nissl staining and infarct analysis.
Coronal sections were run through ascending alcohol solutions (50%, 75%, 95%, and 100%), placed in a 1:1 alcohol/chloroform solution for 45 min, rehydrated in descending alcohols, rinsed in distilled water, and finally stained in a cresyl violet solution. Using ImageJ and StereoInvestigator (MBF Bioscience), the volumes of both ipsilesional and contralesional hemispheres or cortices were calculated for a full series of coronal sections through the striatal strokes or cortical strokes, respectively. Values were plotted as ratios of the ipsilesional to contralesional volumes.
Results
Daily PDE2A inhibition enhances motor recovery after stroke
The PDE2A isoform is preferentially expressed in forebrain sites of cortex and hippocampus (Lakics et al., 2010; Stephenson et al., 2012). Photothrombotic stroke was delivered to the primary motor cortex to test the role of this region-specific PDE2A isoform in stroke recovery (Fig. 1A), using young adult mice to induce behavioral deficits on forelimb tasks, in a manner identical to those of previously published studies (Li et al., 2010; Li et al., 2015; Clarkson et al., 2011; Overman et al., 2012; Caracciolo et al., 2018; Joy et al., 2019). In the present study, a dose that elevates cortical cGMP and enhances memory function (Mikami et al., 2017), 3 mg/kg PDE2A-T1, was delivered (intraperitoneally) starting 5 d after photothrombotic stroke. The grid-walking task measures forelimb placement on a challenging grid during exploratory gait, is sensitive to contralateral deficits and recovery after stroke, and can be used both in freely moving mice and in head-fixed mice in a two-photon imaging system (Li et al., 2010; N. Li et al., 2015; Clarkson et al., 2011; Overman et al., 2012; Caracciolo et al., 2018; Joy et al., 2019). Treatment with PDE2A-T1 improved performance on the grid-walking task 1 and 9 weeks after stroke (Fig. 2A; *p = 0.0478 and p = 0.0452, respectively; n = 12–15/group). Improvement was quantified as a reduced number of step errors taken with the impaired forepaw relative to stroke and vehicle control. These results demonstrate PDE2A-augmented recovery in the early and chronic phases of recovery after stroke.
Figure 2.

PDE2A inhibition enhances motor recovery after stroke. A, In young adult mice receiving cortical stroke, PDE2A-T1 reduces gait abnormalities in the grid-walking task with affected forelimb 1 and 9 weeks after stroke (*p = 0.0478 and p = 0.0452, respectively; n = 12–15/group). B, In young adult mice in a head-fixed position on a grid, PDE2A-T1 reduces gait abnormalities with affected forelimb at weeks 1 and 3, before washout of the drug (*p = 0.0347, **p = 0.023, ***p = 0.009, #p = 0.0425; n = 7–9). C, PDE2A-T1 reduces gait abnormalities in aged mice 1 and 9 weeks after stroke (*p = 0.0175, **p = 0.0322; n = 8–9/group).
The grid-walking task was adapted for head fixation of the mouse (Fig. 1B), allowing simultaneous recording of the behavior and of calcium transients in two-photon in vivo neuronal imaging. Prestroke foot-fault rate was ∼3.3% across all groups, demonstrating similar proficiency in the head-fixed task as the original. At 1 week poststroke, the foot-fault rate increased to close to 20% (p = 0.0023 vs sham plus vehicle, n = 7–9) in vehicle-injected mice and 13.8% in stroke plus PDE2A-T1 mice compared with sham plus vehicle (p = 0.0347). Similar to their freely walking counterparts, PDE2A-T1-injected mice demonstrated lower mean rates of affected forelimb placement errors at 3 and 5 weeks after stroke compared with stroke plus vehicle (at 3 weeks, p = 0.009; at 5 weeks, 0.0425). To further test the role of PDE2A inhibition, drug injection was stopped after 5 weeks, and a final washout imaging session was recorded at week 9 poststroke. After 4 weeks of washout, drug-injected mice had plateaued but retained their recovery near 10% foot faults, while the Vehicle-Injected Stroke group continued to spontaneously improve and was statistically indistinguishable from the Stroke + PDE2A-T1 group (p = 0.9993). This result suggests that PDE2A-T1 needs to be continuously delivered at least at these time intervals of 2 months after stroke to foster behavioral recovery after stroke. Overall, the behavioral results from daily PDE2A inhibition demonstrate positive effects on the recovery of forelimb function in the early and the chronic phases of stroke recovery.
PDE2A inhibition improves stroke recovery and enhances axonal projections in aged mice after stroke
Axonal sprouting is the formation of new patterns of projections from tissue adjacent to the infarct and is causally associated with stroke functional recovery (Benowitz and Carmichael, 2010; Overman et al., 2012; Li et al., 2015a,b). Inhibition of another isoform of PDE, PDE10A, which is largely restricted to the striatum, enhances axonal sprouting and functional recovery in striatal stroke (Birjandi et al., 2021). Stroke is a disease of aging, and aged individuals recover less well from stroke (Li et al., 2010; Tennant et al., 2015). To further test the mechanism of action of PDE2A inhibition, aged mice (age, 19 months) were given this same photothrombotic stroke in forelimb motor cortex, and the behavioral effect of PDE2A-T1 was tested. PDE2A-T1-treated mice made fewer foot faults with the affected forepaw compared with mice receiving stroke and vehicle at 5 and 9 weeks after stroke (p = 0.0175, p = 0.0322; n = 8–9/group; Fig. 2C). These results confirm a functional recovery effect in stroke in aged mice, in parallel with that seen in young adult mice. To measure axonal sprouting, cohorts of aged mice received a stroke to the motor cortex (Fig. 3A) and were subsequently injected with the neuroanatomical tracer BDA ipsilateral to the stroke in adjacent peri-infarct cortex 9 weeks later, and the density of BDA-positive axons was assessed in the peri-infarct cortex (Birjandi et al., 2021). This measure correlates with direct quantification of axonal profiles in brain tissue (Omura et al., 2015). The time point of 9 weeks was selected as it was the end point for the behavioral studies in which behavioral recovery with PDE2A-T1 was observed (Fig. 2C). In these cohorts, PDE2A-T1 demonstrates increased density of peri-infarct axonal labeling (p = 0.0299; n = 4-5; Fig. 3B,C), indicating that PDE2A inhibition increases axonal projections in the tissue adjacent to the infarct after stroke.
Figure 3.
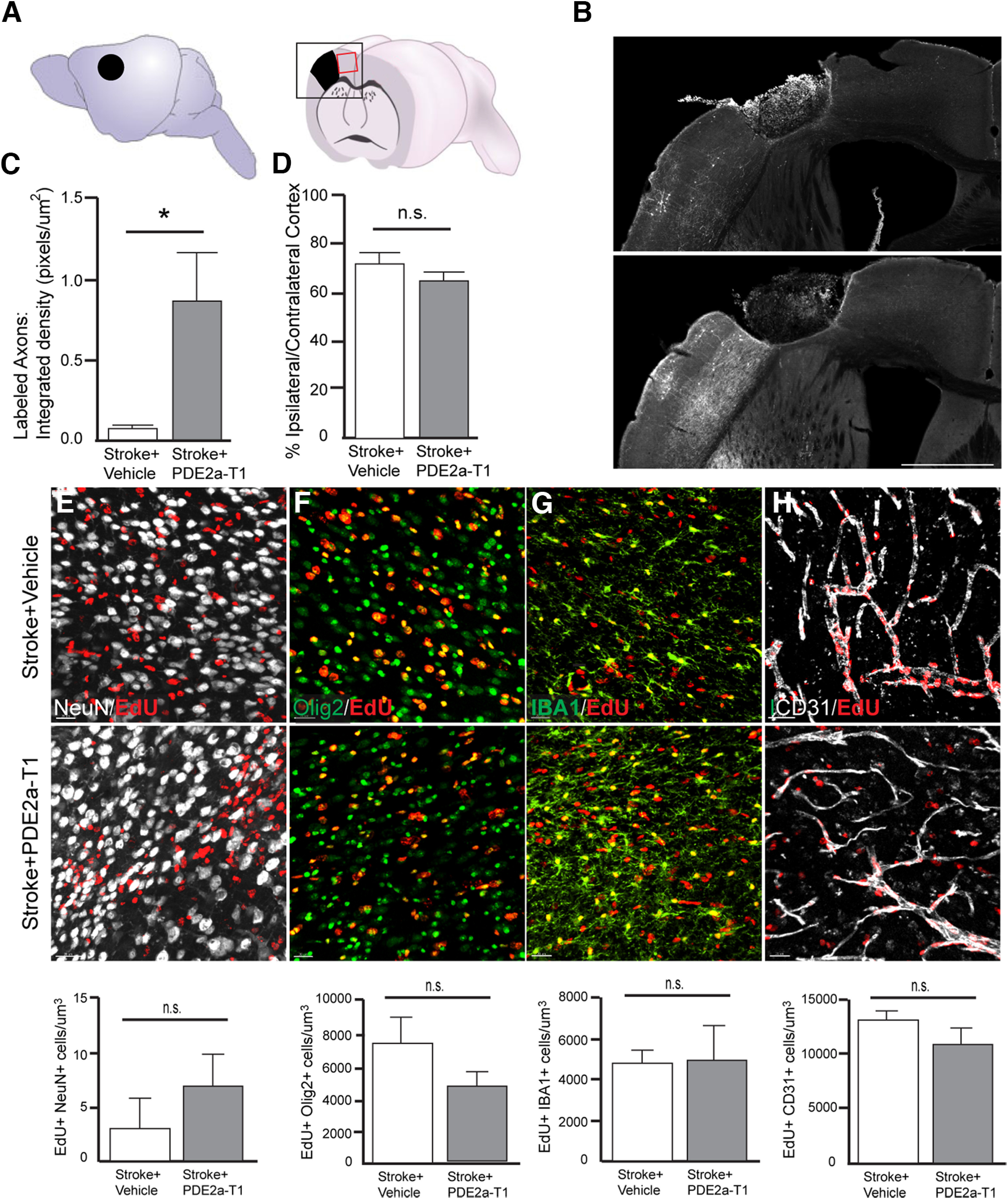
Axonal sprouting and tissue repair with PDE2A inhibition. A, Schematic lateral view of mouse brain with stroke site and cutaway coronal view of mouse brain with stroke. The black boxed area indicates a region of photomicrographs in B. The red boxed area indicates the region shown in E–H. B, Photomicrographs of BDA-labeled axons from the motor cortex anterior to the stroke site. White fibers are labeled axons. Scale bar, 1 mm. C, Quantification of labeled axons in peri-infarct cortex (*p = 0.0299; n = 4–5). E–H, Photomicrographs of EdU and cell-specific label for neurons (E), oligodendrocyte lineage cells (F), microglia/macrophages (G), and endothelial cells (H). Bottom graphs are quantification of double-labeled cells. n.s., Not statistically significant. N = 4 animals/group.
PDE2A-T1 inhibition does not influence infarct size, angiogenesis, or progenitor responses
In addition to axonal sprouting, PDE2A inhibition may also improve behavioral recovery by enhancing intrinsic elements of tissue repair or by acting as a neuroprotective agent. The late start of this PDE2A-T1 regimen, 5 d after stroke, suggests that it is past a period of providing neuroprotection; however, this has not been tested. To assess the degree of neuroprotection of PDE2A-T1 in stroke, mice received stroke to the primary motor cortex and then received daily injections of 3 mg/kg PDE2A-T1 or vehicle as in the behavioral recovery studies starting 5 d after stroke, until 9 weeks after stroke. Infarct size was quantified and was not significantly different between stroke and stroke plus PDE2A-T1 (p = 0.0937; n = 5–6; Fig. 3D), indicating that PDE2A inhibition does not decrease infarct size and is not acting as a neuroprotective agent.
To assess the effects of PDE2A inhibition on angiogenesis and the main progenitor cell in cortex, oligodendrocyte progenitor cells (OPCs), and for newly derived neurons, drinking water was dosed with EdU from poststroke day 3 until day 10. This is the major time frame of newly proliferating endothelial cells, OPCs, and neuronal progenitors after stroke (Sozmen et al., 2016). EdU was colocalized with cell-specific or lineage-specific markers NeuN, Olig2, and CD31. PDE2A modulates microglial function in neurodegenerative disease models (Hunter et al., 2021). IBA-1 was used to assess microglial reactivity. Numbers of colabeled neurons, oligodendrocytes, microglia, and endothelial cells were measured in the peri-ischemic tissue of mice receiving daily administrations of PDE2A-T1 or saline after stroke (Fig. 3E–H; p = 0.3946, 0.2052, 0.9035, 0.2553; n = 4–5/group). No statistically significant effect of PDE2A inhibition on colabeled cells was observed, suggesting that any effects of the drug were not because of the changes in neuronal or glial progenitors, angiogenesis, or microglial inflammation.
Daily PDE2A inhibition enhances functional connectivity and ND
CREB modulation, which is downstream from cAMP/cGMP, enhances coactivation of neurons into coactive units in its effects on learning and memory (Stevens, 1994), providing a possible functional mechanism for enhanced axonal connectivity after stroke. To investigate PDE-mediated changes to neuronal network dynamics, the activity of neurons in peri-infarct cortex was imaged with in vivo two-photon approaches. AAV9-CaMKII-GCaMP6s was injected into the secondary motor area/rostral forelimb motor area adjacent to stroke and neuronal activity was measured before stroke and for the period of behavioral recovery after stroke. Mice were habituated on the head-fixed grid-walk treadmill to simultaneously record cortical neuronal activity and forelimb function. The field of GCaMP6s-expressing cortical neurons was stereotaxically located and imaged during each session. Cohorts of mice received stroke or control (no stroke) and were then further split into groups receiving PDE2A-T1 or vehicle starting 1 d after stroke. These are the same cohorts of mice used for the head-fixed grid-walking behavioral results (Fig. 2B). The same neuronal populations were located and imaged at weeks 1, 3, and 5, after which drug injection was discontinued, and a final imaging session was conducted at week 9 poststroke after this drug washout.
GCaMP6s fluorescent traces were extracted from individual excitatory neurons, and the correlation of their activity was calculated. Neuronal pairs with significant correlation of calcium activity spikes were deemed functionally connected (Fig. 4A). FC is defined as the number of neuronal pairs with PCC values above the baseline and is a measure of correlativity in the cortical network. FC in excitatory neurons in cortex adjacent to stroke decreases after stroke and partially recovers after 30 d (Latifi et al., 2020), correlating with the same time period of behavioral recovery. The ND of each recording is a normalization of FC based on the total possible number of neuronal pairs (Fig. 4B). This function is thus independent of the number of neurons in the network. The number of neurons from each of the four groups were not significantly different, before and after stroke induction (data not shown). In control groups receiving sham and vehicle injections, FC and ND remained constant and were not significantly different across time points (p > 0.4814, p > 0.9137). In the group Stroke + Vehicle, FC was significantly lower at week 1 (*p = 0.0246) and week 3 (**p = 0.0098) after stroke, consistent with other reports (Latifi et al., 2020). There was also significant recovery of FC in this Stroke + Vehicle group from week 1 to week 9 after stroke (p = 0.0229). In contrast, the Stroke + PDE2A-T1 group did not significantly decline in FC at 1 week after stroke (p = 0.0547) and significantly increased at 3 weeks after stroke, compared with the Stroke + PDE2A-T1 group at baseline prestroke (p < 0.0001). In directly assessing the effect of PDE2A-T1 on FC with stroke, excitatory neurons in premotor cortex in the Stroke + PDE2A-T1 group are significantly more functionally connected than in those in the Stroke + Vehicle group at week 3 after stroke (p < 0.0001; Fig. 4A). ND of the functionally connected neuronal network in the Stroke + Vehicle group (Fig. 4B) was similarly decreased at week 1 (*p = 0.0496) and week 3 (***p = 0.0006). However, this decrease in ND did not occur after stroke with PDE2A-T1 treatment, and in fact PDE2A-T1 treatment in stroke significantly increased ND at week 3 (p = 0.0154) compared with the prestroke baseline. This is a similar increase in the connectivity of neurons in the tissue near the stroke site as seen in the FC measure. In a direct comparison of stroke plus PDE2A-T1 with stroke plus vehicle, PDE2A-T1 increased ND at week 1 (p = 0.0425) and week 3 (p = 0.0009). Finally, during week 9, after 4 weeks of drug washout, all groups had returned to baseline and were indistinguishable from one another.
Figure 4.
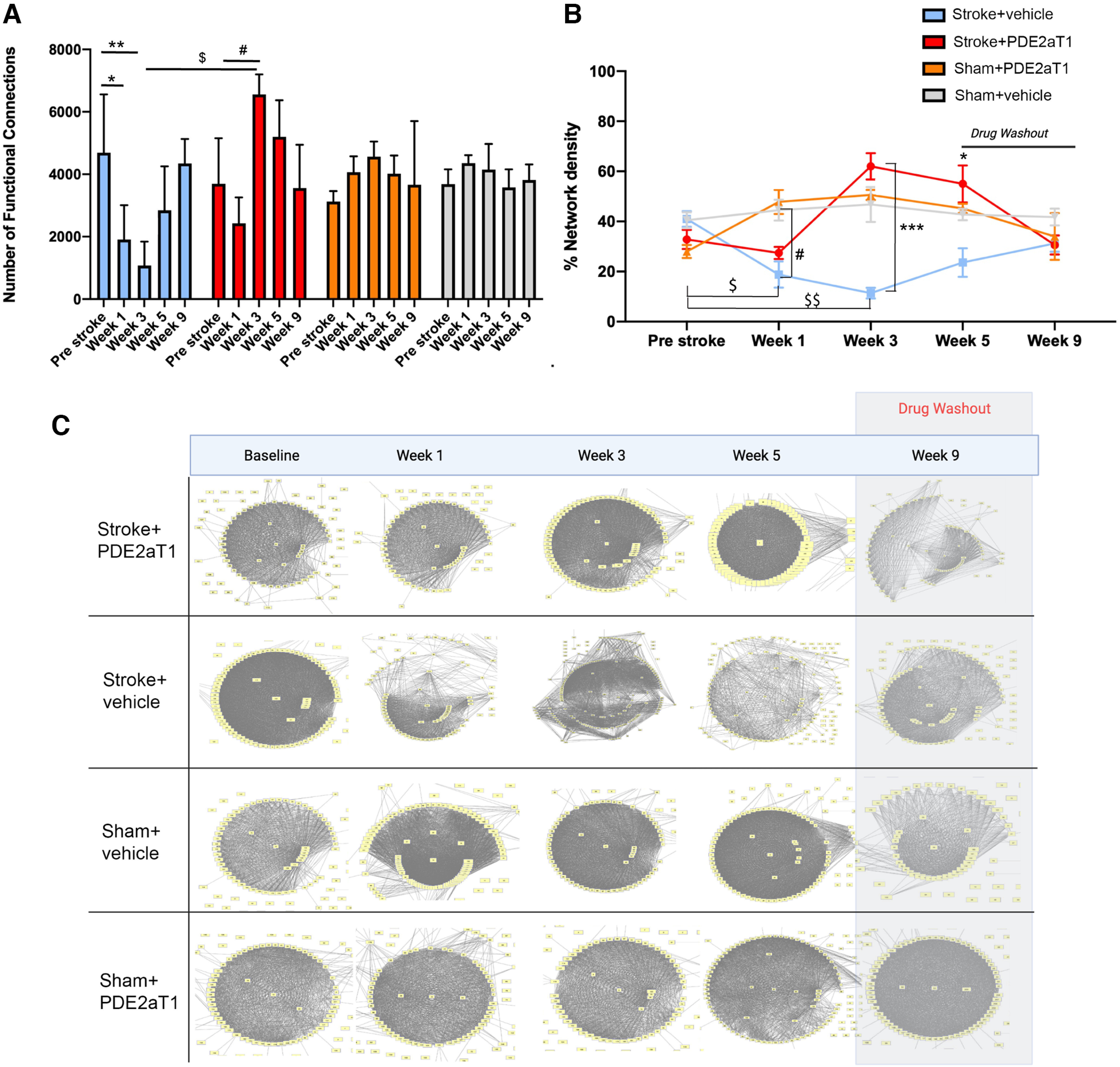
Functional connectivity of neuronal networks with PDE2A inhibition. FCs were defined as the PCC between two neurons above the defined baseline. Significant values are relative to the “Pre Stroke” condition for each group (n = 4–5 mice/group). A, The number of FCs averaged across animals for each group. Stroke + Vehicle group: *p = 0.0246 (week 1), **p = 0.098 (week 3); Stroke + PDE2A-T1 group: **p = 0.0092 (week 3); Sham + PDE2A-T1 group: *p = 0.0425 (week 3); Stroke + PDE2A-T1 group, week 3 versus week 1, ***p < 0.0001. B, Percentage of network density, calculated by dividing FCs by total possible connections between neurons in the cortical field; ***p = 0.0009 (Stroke + PDE2A-T1 group vs Stroke + Vehicle group at 3 weeks). *p = 0.0425 (Stroke + PDE2A-T1 group vs Stroke + Vehicle group at 1 week). #p = 0.0347 (Stroke + Vehicle group vs Sham + vehicle at 1 week). Note that results for the Stroke + PDE2A-T1 group are not significantly different at 1 week after stroke compared with the Sham + Vehicle group (p = 0.587). $p = 0.0496 (Stroke + Vehicle group at week 1 vs Stroke + Vehicle at baseline). $$p = 0.0006 (Stroke + Vehicle at week 3 vs Stroke + Vehicle at baseline). C, Functional connectivity maps of the motor cortex during spontaneous recovery after stroke. FC maps were generated for an individual mouse representative of each group with neurons graphed as nodes and FCs represented as edges.
Quantification of measures of FC and ND establish that PDE2A-T1 enhances the coactivation of neurons into groups, preventing the disassociation of this normal pattern of cortical activity that occurs with stroke in the absence of any intervention (Latifi et al., 2020; Fig. 4A,B). Visualization of individual cortical networks in mice, using a mapping approach in which each node is an excitatory neuron and edges are the significant, coactive patterns across neurons, supports these group-level quantifications (Fig. 4C). Neurons are tightly active together in networks before stroke, but deteriorate in weeks 1 and 3 after stroke, with many neurons showing no significant connectivity. By week 9, functional connectivity recovers and resembles that seen in the prestroke condition. This pattern of functional disassociation is not seen with drug treatment.
Discussion
Inhibition of PDE2A enhances functional recovery after stroke in a mouse stroke model, in both young adult and aged mice. This effect is associated with increased axonal projections in the cortex adjacent to the infarct and enhanced functional connectivity in excitatory neurons in this area. PDE2A inhibition is not associated with other aspects of tissue repair after stroke, such as neurogenesis, gliogenesis, or angiogenesis, or in a modulation of inflammatory responses. PDE2A inhibition has an early effect on stroke recovery in young adult mice. Two days after drug administration started, there is a significant difference in motor function in the affected forelimb. This early effect is also seen with other drugs that affect CREB (Joy et al., 2019) or excitatory signaling (Clarkson et al., 2011) after stroke. This early effect may be because of a rapid increase in synaptic responsiveness in otherwise depressed neuronal circuits (Joy and Carmichael, 2021) and appears to differ with respect to age. PDE2A inhibitors are in clinical trials for other diseases, and these data suggest that PDE2A inhibition is a tractable pharmacological avenue for stroke recovery.
PDEs are divided into three groups, based on their specificity to cyclic nucleotides, as follows: specific to cAMP (PDE4, PDE7, and PDE8), specific to cGMP (PDE5, PDE6, and PDE9), and hydrolyzing both cAMP and cGMP (PDE1, PDE2, PDE3, PDE10, and PDE11; Delhaye and Bardoni, 2021). By hydrolyzing cAMP and cGMP, PDEs degrade the signaling that leads to the activation of protein kinase A and protein kinase C and their downstream targets, such as CREB (Akiyama et al., 2016). PDEs thus terminate intracellular signaling cascades that lead to downstream activation of growth factors, neuronal excitability systems, axonal elongation and dendritic spine morphogenesis (Shelly et al., 2010; Akiyama et al., 2016; Delhaye and Bardoni, 2021). PDE isoforms have different regional localizations. Within the brain, PDE10 is maximally expressed in the striatum/basal ganglia, and PDE2 is the most abundant PDE isoform in the hippocampus and cortical regions (Farmer et al., 2020). Both striatum and cortex are common sites of human stroke (Bogousslavsky et al., 1988). Inhibition of PDE10A enhances recovery in striatal stroke in the mouse, but not in cortical stroke (Birjandi et al., 2021), indicating that this regional expression pattern can be exploited to produce specific pharmacological recovery based on stroke subtype. Clinical trials are under way or have been completed with PDE2A inhibitors in migraine (Delhaye and Bardoni, 2021) and in phase 1 pharmacodynamics (ClinicalTrials.gov: NCT01981499, NCT02584569).
In the present studies, a dose of PDE2A was used that has previously demonstrated target engagement and activation of cGMP signaling. It is a dose used to enhance memory function (Mikami et al., 2017). This effect and this intracellular pathway have the following important cellular actions relevant to stroke recovery: axonal sprouting through elongation and guidance (Shelly et al., 2010; Akiyama et al., 2016); enhanced synaptic plasticity through effects on the morphology and growth of dendritic spines, which fluctuate after stroke and are associated with stroke recovery and motor learning (Jones and Schallert, 1992; Mostany et al., 2010); and through increases in cellular excitability and allocation of neurons into functional units for motor, sensory, or cognitive actions (Joy et al., 2019). PDE2A inhibition induces CREB activity (Chen et al., 2019), and these data place PDE2A inhibition at a crucial node in the experimental evidence for poststroke recovery.
A wide range of cellular inputs leads to intracellular signaling cascades that converge on downstream CREB activation. In this way, CREB couples experience-dependent neuronal activation with gene transcription and long-term cellular and molecular changes associated with plasticity, learning, and memory (Glazewski et al., 1999; Barth et al., 2000). CRE-dependent transcription has additional roles in the allocation of neurons into a memory trace or engram, defined as the cluster of neurons that coactivate to encode or store a particular memory (Hsiang et al., 2014; Sano et al., 2014; Park et al., 2016; Lisman et al., 2018; Miyashita et al., 2018). CREB might allocate neurons into other types of task-specific engrams or circuits, such as the motor circuit for a particular limb movement, or the sensory circuit for a particular type of visual stimulus (Dombeck et al., 2009; Hira et al., 2013; Tonegawa et al., 2015; Caracciolo et al., 2018). Critically, CREB mediates reorganization of these task-specific neuronal circuits in response to altered input or experience (Barth et al., 2000; Mower et al., 2002). This reorganization role is particularly relevant to the context of repair after stroke. Stroke causes task-specific neuronal engrams to be disrupted, either directly via the death of participating neurons or indirectly via functional disconnection or lack of coincident firing of surviving neurons (Benowitz and Carmichael, 2010; Zhang et al., 2017; Chi et al., 2018). Stimulation of CREB signaling after stroke enhances functional recovery (Caracciolo et al., 2018; Joy et al., 2019), possibly by enhancing neuronal allocation into recovering motor circuits after stroke (Joy and Carmichael, 2021).
PDE2A-T1 treatment in cortical stroke increases the axonal projections in peri-infarct cortex. This pattern of increased axonal projections is consistently associated with functional recovery in studies of cortical strokes spanning motor regions (Li et al., 2010; Li et al., 2015; Caracciolo et al., 2018; Joy et al., 2019). Axonal sprouting within motor or somatosensory systems has been repeatedly demonstrated in many rodent and primate stroke models to correlate with functional recovery (Li et al., 2010; Li et al., 2015; Caracciolo et al., 2018; Joy et al., 2019). With PDE2A inhibition, this pattern could represent the sprouting of new axonal projections into adjacent cortical regions spared after stroke, which suggests a possible takeover by these surviving regions of motor functions lost because of stroke (Hsiang et al., 2014; Caracciolo et al., 2018; Miyashita et al., 2018). In mechanistic studies, treatments that induce this pattern of axonal sprouting in mouse stroke models increase functional recovery, and treatments that block this pattern of axonal sprouting block functional recovery (Overman et al., 2012; Li et al., 2015). Alternatively, the increased density of axonal projections observed in both the striatal (Birjandi et al., 2021) and cortical stroke models may represent a selective sparing of projections in response to PDE inhibition after stroke. Future studies might use multiple axonal tracers conjugated to different fluorophores to distinguish between prestroke axonal projections and those that sprout after stroke.
The potential enhancement of poststroke axonal sprouting after PDE inhibition suggests a possible induction of growth programs within peri-infarct neurons in response to drug treatment (Carmichael et al., 2005; Li and Carmichael, 2006; Li et al., 2010). An initial induction of neuronal growth programs within surviving neurons is part of the endogenous cellular response of the brain to stroke (Carmichael, 2006; Li and Carmichael, 2006). This growth state comprises transcriptional changes in genes with known functions relevant to axonal sprouting and cortical circuit reorganization (Carmichael et al., 2005; Li et al., 2010, 2015). However, this progrowth state is limited: 21 d after ischemic injury, in the transcriptional profiles of peri-infarct sprouting neurons much of the early growth programs are turned off and replaced with upregulation of genes that maintain the stimulated axonal growth (Li et al., 2010). The axonal sprouting observed in response to PDE inhibition suggests a potential action of these PDE inhibitors in enhancing or reopening this axonal progrowth state. This is likely to occur through the action of CREB, with known roles in governing plasticity during development, learning, and memory, and following multiple models of neurologic disease (Glazewski et al., 1999; Barth et al., 2000; Kida et al., 2002; Sakamoto et al., 2011; Middei et al., 2012; Sano et al., 2014; Caracciolo et al., 2018; Miyashita et al., 2018). Consistent with this hypothesis, viral upregulation of CREB in peri-infarct excitatory neurons after cortical stroke activates genes with roles in nervous system development and tissue development (Caracciolo et al., 2018). This suggests a role for CREB in mediating a progrowth state similar to, but distinct from, developmental growth.
Two-photon GCaMP6s imaging was used in the present study to record calcium transients from populations of CaMKII-expressing premotor neurons near the stroke site in the motor cortex. Stroke has an inhibitory effect on the coactivation of excitatory neurons—their FC–into circuits normally active during motor behavior (Latifi et al., 2020). In the current study, the degree of FC, and its normalized value ND, were calculated as measurements of correlation in the motor areas near the stroke site. We report similar negative effects of stroke to the FC of premotor excitatory networks at weeks 1 and 3, which were indistinguishable by week 5, as in previous work (Latifi et al., 2020). As in the prior study, these changes were not because of a change in the number of neurons recorded, or in significant changes in their amplitude or frequency of activity.
The behavioral experiments were designed to use distinct testing approaches and drug effects, and this is both a benefit to, and a limitation of, the study. This has now been included in the Discussion section. Aged and young adult mice were tested in freely moving motor function on the grid-walking test (Fig. 2A,C). These tests show an effect of PDE2A inhibition on motor function after stroke at early (7 d) and late (9 weeks) time points after stroke in young adult (Fig. 2A), and late after stroke in aged adults (Fig. 2C). It is interesting that this early effect of PDE2A inhibition is not seen in aged mice and bears further study as to the effects of age in stroke recovery.
To further define the mechanism of action of the PDE2A inhibitor, an additional behavioral study was performed on young adult mice with a head-fixed grid-walking test, for simultaneous two-photon in vivo imaging. Also, in this additional test, the effect of drug washout was studied, with 4 weeks of no PDE2A inhibitor after the 5 week poststroke time point. This additional behavioral test has a limitation, compared with the young adult animal (Fig. 2A) and aged animal (Fig. 2C), which are behavioral results in freely moving mice. For two-photon imaging, the mice cannot be freely moving, so this behavioral test is on head-fixed mice that walk on a rotating wheel with a grid. This additional two-photon behavioral test also showed enhanced recovery with PDE2A inhibition at early (7 d) and late (5 weeks) time points. Interestingly, after the washout period, there was no difference in motor recovery level between stroke plus PD2A inhibition and stroke alone. This lack of effect with drug washout, however, may be because of spontaneous recovery in this stroke model, which is a limitation of small cortical strokes in young adult mice.
We report here, for the first time, a positive effect of drug treatment on FC of the premotor network in the first month after stroke. Furthermore, drug washout from week 5 to week 9 eliminated any positive FC effects of PDE2A inhibition, indicating a transient increase in neuronal coactivity with the drug. These data demonstrate that PDE2A inhibition is a positive stimulus for FC, rescues the FC deficit because of stroke, and requires continued drug exposure for its effect.
Footnotes
This work was supported by research grants from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and Takeda Pharmaceutical Company Limited. Takeda Pharmaceutical Company Limited personnel had no role in data collection, analysis, or interpretation.
K.S. and H.I. are employees of Takeda Pharmaceutical Company Limited. As experts in PDE2A research, these two authors had a role in experimental design but no role in data analysis or interpretation. The authors declare no other competing financial interests.
References
- Akiyama H, Fukuda T, Tojima T, Nikolaev VO, Kamiguchi H (2016) Cyclic nucleotide control of microtubule dynamics for axon guidance. J Neurosci 36:5636–5649. 10.1523/JNEUROSCI.3596-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, McKenna M, Glazewski S, Hill P, Impey S, Storm D, Fox K (2000) Upregulation of cAMP response element-mediated gene expression during experience-dependent plasticity in adult neocortex. J Neurosci 20:4206–4216. 10.1523/JNEUROSCI.20-11-04206.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Carmichael ST (2010) Promoting axonal rewiring to improve outcome after stroke. Neurobiol Dis 37:259–266. 10.1016/j.nbd.2009.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birjandi SZ, Abduljawad N, Nair S, Dehghani M, Suzuki K, Kimura H, Carmichael ST (2021) Phosphodiesterase 10A inhibition leads to brain region-specific recovery based on stroke type. Transl Stroke Res 12:303–315. 10.1007/s12975-020-00819-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogousslavsky J, Van Melle G, Regli F (1988) The lausanne stroke registry: analysis of 1,000 consecutive patients with first stroke. Stroke 19:1083–1092. 10.1161/01.STR.19.9.1083 [DOI] [PubMed] [Google Scholar]
- Caracciolo L, Marosi M, Mazzitelli J, Latifi S, Sano Y, Galvan L, Kawaguchi R, Holley S, Levine MS, Coppola G, Portera-Cailliau C, Silva AJ, Carmichael ST (2018) CREB controls cortical circuit plasticity and functional recovery after stroke. Nat Commun 9:2250. 10.1038/s41467-018-04445-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST (2006) Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann Neurol 59:735–742. 10.1002/ana.20845 [DOI] [PubMed] [Google Scholar]
- Carmichael ST (2016) Emergent properties of neural repair: elemental biology to therapeutic concepts. Ann Neurol 79:895–906. 10.1002/ana.24653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael ST, Archibeque I, Luke L, Nolan T, Momiy J, Li S (2005) Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol 193:291–311. 10.1016/j.expneurol.2005.01.004 [DOI] [PubMed] [Google Scholar]
- Chen L, Cui S, Yu H, Li G, Liu N, Wu Q, Zhang HT, O'Donnell JM, Wang G, Xu Y (2019) Reduced phosphodiesterase-2 activity in the amygdala results in anxiolytic-like effects on behavior in mice. J Psychopharmacol 33:568–576. 10.1177/0269881119832753 [DOI] [PubMed] [Google Scholar]
- Chi NF, Ku HL, Chen DY, Tseng YC, Chen CJ, Lin YC, Hsieh YC, Chan L, Chiou HY, Hsu CY, Hu CJ (2018) Cerebral motor functional connectivity at the acute stage: an outcome predictor of ischemic stroke. Sci Rep 8:16803. 10.1038/s41598-018-35192-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Overman JJ, Zhong S, Mueller R, Lynch G, Carmichael ST (2011) AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci 31:3766–3775. 10.1523/JNEUROSCI.5780-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhaye S, Bardoni B (2021) Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders. Mol Psychiatry 26:4570–4582. 10.1038/s41380-020-00997-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Graziano MS, Tank DW (2009) Functional clustering of neurons in motor cortex determined by cellular resolution imaging in awake behaving mice. J Neurosci 29:13751–13760. 10.1523/JNEUROSCI.2985-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domek-Łopacińska K, Strosznajder JB (2008) The effect of selective inhibition of cyclic GMP hydrolyzing phosphodiesterases 2 and 5 on learning and memory processes and nitric oxide synthase activity in brain during aging. Brain Res 1216:68–77. 10.1016/j.brainres.2008.02.108 [DOI] [PubMed] [Google Scholar]
- Farmer R, Burbano SD, Patel NS, Sarmiento A, Smith AJ, Kelly MP (2020) Phosphodiesterases PDE2A and PDE10A both change mRNA expression in the human brain with age, but only PDE2A changes in a region-specific manner with psychiatric disease. Cell Signal 70:109592. 10.1016/j.cellsig.2020.109592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghofrani HA, Osterloh IH, Grimminger F (2006) Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov 5:689–702. 10.1038/nrd2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazewski S, Barth AL, Wallace H, McKenna M, Silva A, Fox K (1999) Impaired experience-dependent plasticity in barrel cortex of mice lacking the alpha and delta isoforms of CREB. Cereb Cortex 9:249–256. 10.1093/cercor/9.3.249 [DOI] [PubMed] [Google Scholar]
- Hira R, Ohkubo F, Ozawa K, Isomura Y, Kitamura K, Kano M, Kasai H, Matsuzaki M (2013) Spatiotemporal dynamics of functional clusters of neurons in the mouse motor cortex during a voluntary movement. J Neurosci 33:1377–1390. 10.1523/JNEUROSCI.2550-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiang HLL, Epp JR, van den Oever MC, Yan C, Rashid AJ, Insel N, Ye L, Niibori Y, Deisseroth K, Frankland PW, Josselyn SA (2014) Manipulating a “cocaine engram” in mice. J Neurosci 34:14115–14127. 10.1523/JNEUROSCI.3327-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter M, Spiller KJ, Dominique MA, Xu H, Hunter FW, Fang TC, Canter RG, Roberts CJ, Ransohoff RM, Trojanowski JQ, Lee VMY (2021) Microglial transcriptome analysis in the rNLS8 mouse model of TDP-43 proteinopathy reveals discrete expression profiles associated with neurodegenerative progression and recovery. Acta Neuropathol Commun 9:140. 10.1186/s40478-021-01239-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Schallert T (1992) Overgrowth and pruning of dendrites in adult rats recovering from neocortical damage. Brain Res 581:156–160. 10.1016/0006-8993(92)90356-e [DOI] [PubMed] [Google Scholar]
- Joy MT, Carmichael ST (2021) Encouraging an excitable brain state: mechanisms of brain repair in stroke. Nat Rev Neurosci 22:38–53. 10.1038/s41583-020-00396-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joy MT, Ben Assayag E, Shabashov-Stone D, Liraz-Zaltsman S, Mazzitelli J, Arenas K, Abduljawad N, Kliper E, Korczyn AD, Thareja NS, Kesner EL, Zhou M, Huang S, Silva TK, Katz N; Natan M, Bornstein NM, Silva AJ, Shohami E, Carmichael ST (2019) CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell 176:1143–1157.e13. 10.1016/j.cell.2019.01.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida S, Serita T (2014) Functional roles of CREB as a positive regulator in the formation and enhancement of memory. Brain Res Bull 105:17–24. 10.1016/j.brainresbull.2014.04.011 [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, De Ortiz SP, Peña de Ortiz S, Kogan JH, Chevere I, Masushige S, Silva AJ (2002) CREB required for the stability of new and reactivated fear memories. Nat Neurosci 5:348–355. 10.1038/nn819 [DOI] [PubMed] [Google Scholar]
- Knott EP, Assi M, Rao SNR, Ghosh M, Pearse DD (2017) Phosphodiesterase inhibitors as a therapeutic approach to neuroprotection and repair. Int J Mol Sci 18:696. 10.3390/ijms18040696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakics V, Karran EH, Boess FG (2010) Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 59:367–374. 10.1016/j.neuropharm.2010.05.004 [DOI] [PubMed] [Google Scholar]
- Landis SC, et al. (2012) A call for transparent reporting to optimize the predictive value of preclinical research. Nature 490:187–191. 10.1038/nature11556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA, Zhang JH, Noble-Haeusslein LJ (2013) RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res 4:279–285. 10.1007/s12975-012-0209-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latifi S, Mitchell S, Habibey R, Hosseini F, Donzis E, Estrada-Sánchez AM, Nejad HR, Levine M, Golshani P, Carmichael ST (2020) Neuronal network topology indicates distinct recovery processes after stroke. Cereb Cortex 30:6363–6375. 10.1093/cercor/bhaa191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees KR, et al. (2010) Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet 375:1695–1703. 10.1016/S0140-6736(10)60491-6 [DOI] [PubMed] [Google Scholar]
- Li N, Chen TW, Guo Z, V, Gerfen CR, Svoboda K (2015) A motor cortex circuit for motor planning and movement. Nature 519:51–56. 10.1038/nature14178 [DOI] [PubMed] [Google Scholar]
- Li S, Carmichael ST (2006) Growth-associated gene and protein expression in the region of axonal sprouting in the aged brain after stroke. Neurobiol Dis 23:362–373. 10.1016/j.nbd.2006.03.011 [DOI] [PubMed] [Google Scholar]
- Li S, Overman JJ, Katsman D, Kozlov SV, Donnelly CJ, Twiss JL, Giger RJ, Coppola G, Geschwind DH, Carmichael ST (2010) An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat Neurosci 13:1496–1504. 10.1038/nn.2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Nie EH, Yin Y, Benowitz LI, Tung S, Vinters HV, Bahjat FR, Stenzel-Poore MP, Kawaguchi R, Coppola G, Carmichael ST (2015) GDF10 is a signal for axonal sprouting and functional recovery after stroke. Nat Neurosci 18:1737–1745. 10.1038/nn.4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipworth B (2005) Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet 74:193–229. [DOI] [PubMed] [Google Scholar]
- Lisman J, Cooper K, Sehgal M, Silva AJ (2018) Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat Neurosci 21:309–314. 10.1038/s41593-018-0076-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middei S, Spalloni A, Longone P, Pittenger C, O'Mara SM, Marie H, Ammassari-Teule M (2012) CREB selectively controls learning-induced structural remodeling of neurons. Learn Mem 19:330–336. 10.1101/lm.025817.112 [DOI] [PubMed] [Google Scholar]
- Mikami S, Kawasaki M, Ikeda S, Negoro N, Nakamura S, Nomura I, Ashizawa T, Kokubo H, Hoffman ID, Zou H, Oki H, Uchiyama N, Hiura Y, Miyamoto M, Itou Y, Nakashima M, Iwashita H, Taniguchi T (2017) Discovery of a novel series of pyrazolo[1,5-a]pyrimidine-based phosphodiesterase 2A inhibitors structurally different from N-((1S)-1-(3-fluoro-4-(trifluoromethoxy)phenyl)-2-methoxyethyl)-7-methoxy-2-oxo-2,3-dihydropyrido[2,3-b]pyrazine-4(1H)-carboxamide (TAK-915), for the treatment of cognitive disorders. Chem Pharm Bull (Tokyo) 65:1058–1077. 10.1248/cpb.c17-00564 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Kikuchi E, Horiuchi J, Saitoe M (2018) Long-term memory engram cells are established by c-Fos/CREB transcriptional cycling. Cell Rep 25:2716–2728.e3. 10.1016/j.celrep.2018.11.022 [DOI] [PubMed] [Google Scholar]
- Mostany R, Chowdhury TG, Johnston DG, Portonovo SA, Carmichael ST, Portera-Cailliau C (2010) Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci 30:14116–14126. 10.1523/JNEUROSCI.3908-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower AF, Liao DS, Nestler EJ, Neve RL, Ramoa AS (2002) cAMP/Ca2+ response element-binding protein function is essential for ocular dominance plasticity. J Neurosci 22:2237–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulina E, Lille Tidwell J, Dai HN, Bregman BS, Filbin MT (2004) The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U|S|A 101:8786–8790. 10.1073/pnas.0402595101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, et al. (2015) Robust axonal regeneration occurs in the injured CAST/Ei Mouse CNS. Neuron 86:1215–1227. 10.1016/j.neuron.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW, Carmichael ST (2012) A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U|S|A 109:E2230–E2239. 10.1073/pnas.1204386109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U, Kukin ML, Mallis GI, Sollano JA, Shannon J, Tandon PK, DeMets DL (1991) Effect of oral milrinone on mortality in severe chronic heart failure. N Engl J Med 325:1468–1475. 10.1056/NEJM199111213252103 [DOI] [PubMed] [Google Scholar]
- Park S, Kramer EE, Mercaldo V, Rashid AJ, Insel N, Frankland PW, Josselyn SA (2016) Neuronal Allocation to a Hippocampal Engram. Neuropsychopharmacology 41:2987–2993. 10.1038/npp.2016.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reneerkens OAH, Rutten K, Bollen E, Hage T, Blokland A, Steinbusch HWM, Prickaerts J (2013) Inhibition of phoshodiesterase type 2 or type 10 reverses object memory deficits induced by scopolamine or MK-801. Behav Brain Res 236:16–22. 10.1016/j.bbr.2012.08.019 [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Karelina K, Obrietan K (2011) CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem 116:1–9. 10.1111/j.1471-4159.2010.07080.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano Y, Shobe JL, Zhou M, Huang S, Shuman T, Cai DJ, Golshani P, Kamata M, Silva AJ (2014) CREB regulates memory allocation in the insular cortex. Curr Biol 24:2833–2837. 10.1016/j.cub.2014.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, Poo MM (2010) Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science 327:547–552. 10.1126/science.1179735 [DOI] [PubMed] [Google Scholar]
- Sozmen EG, Rosenzweig S, Llorente IL, DiTullio DJ, Machnicki M, Vinters HV, Havton LA, Giger RJ, Hinman JD, Carmichael ST (2016) Nogo receptor blockade overcomes remyelination failure after white matter stroke and stimulates functional recovery in aged mice. Proc Natl Acad Sci U|S|A 113:E8453–E8462. 10.1073/pnas.1615322113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson DT, Coskran TM, Kelly MP, Kleiman RJ, Morton D, O'Neill SM, Schmidt CJ, Weinberg RJ, Menniti FS (2012) The distribution of phosphodiesterase 2A in the rat brain. Neuroscience 226:145–155. 10.1016/j.neuroscience.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF (1994) CREB and memory consolidation. Neuron 13:769–770. 10.1016/0896-6273(94)90244-5 [DOI] [PubMed] [Google Scholar]
- Stroke Therapy Academic Industry Roundtable (STAIR) (1999) Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 30:2752–2758. [DOI] [PubMed] [Google Scholar]
- Tennant KA, Kerr AL, Adkins DL, Donlan N, Thomas N, Kleim JA, Jones TA (2015) Age-dependent reorganization of peri-infarct “premotor” cortex with task-specific rehabilitative training in mice. Neurorehabil Neural Repair 29:193–202. 10.1177/1545968314541329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonegawa S, Liu X, Ramirez S, Redondo R (2015) Memory engram cells have come of age. Neuron 87:918–931. 10.1016/j.neuron.2015.08.002 [DOI] [PubMed] [Google Scholar]
- Virani SS, et al. (2020) Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation 141:e139–e596. 10.1161/CIR.0000000000000757 [DOI] [PubMed] [Google Scholar]
- Yaksi E, Friedrich RW (2006) Reconstruction of firing rate changes across neuronal populations by temporally deconvolved Ca2+ imaging. Nat Methods 3:377–383. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang Y, Wang L, Sang L, Yang J, Yan R, Li P, Wang J, Qiu M (2017) Disrupted structural and functional connectivity networks in ischemic stroke patients. Neuroscience 364:212–225. 10.1016/j.neuroscience.2017.09.009 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ (2009) CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci 12:1438–1443. 10.1038/nn.2405 [DOI] [PMC free article] [PubMed] [Google Scholar]