

Abstract
Background and Aims
Bile acids are hepatic metabolites and have many properties considered to be relevant to the pathophysiology of NAFLD. Circulating levels of the intestinal microbiome‐modified bile acid deoxycholate are increased in cirrhosis.
Approach and Results
To further elucidate the role of bile acids and intestinal microbiota linked to bile acids in progressively severe NAFLD, a multiomic study of feces including 16S rRNA sequencing, microbial transcriptomics and metabolomics was performed in a cohort with varying phenotypes of NAFLD. Several bile acids of microbial origin derived from deoxycholic acid (DCA) (glycodeoxycholate, 7‐ketodeoxycholic acid, dehydrocholic acid) increased with disease activity and fibrosis stage. These were linked to increased expression of microbial bile salt hydrolase, bile acid operon (BaiCD) and hydroxysteroid dehydrogenases (hdhA) required for DCA and downstream metabolite synthesis providing a mechanistic basis for altered bile acid profiles with disease progression. Bacteroidetes and several genera of Lachnospiraceae family containing DCA generating genes increased with increasing disease severity, whereas several potentially beneficial microbes sensitive to antibacterial effects of DCA e.g., Ruminococcaceae were decreased. The clinical relevance of these data was confirmed in an independent cohort enrolled in a clinical trial for NASH where at entry DCA and its conjugates were associated with advanced fibrosis. In patients treated with placebo, DCA declined in those with fibrosis regression and increased in those with fibrosis progression. DCA rose further in those with compensated cirrhosis when they experienced decompensation.
Conclusions
These findings demonstrate a role for bile acids and the bile acid dependent microbiome in the development and progression of NAFLD and set the stage to leverage these findings for NASH biomarker development and for therapeutics.

INTRODUCTION
NAFLD is a heterogeneous disorder driven by complex gene‐environmental interactions.[ 1 ] The clinical‐histological spectrum of NAFLD ranges from a nonalcoholic fatty liver (NAFL) to NASH; NASH is a more active form of the disease and progresses to cirrhosis with greater frequency than NAFL.[ 2 ] The clinical course of individuals with NAFLD is characterized by variable rates of progression.[ 3 ] Similarly, responses to various therapies under investigation are also variable.[ 4 ] The multifactorial basis for this heterogeneity is under intense investigation because it is likely to explain the heterogeneity in clinical course and eventually inform strategies tailored to each individual's unique set of biological drivers of disease progression.
A major development over the last decade has been an explosion of information on the complexity of the intestinal microbiome both in terms of its composition and function.[ 5 , 6 ] There is already a body of literature evaluating the microbiome in NAFLD. These indicate a reversal of the Firmicutes to Bacteroides ratio and an increase in Proteobacteria.[ 7 ] Recent studies have also identified taxa and metabolites related to advanced fibrosis.[ 8 ] There is however substantial functional redundancy across taxa and a change in a given taxa may or may not have functional consequences. Bacterial transcriptomic data are thus needed to understand how microbial functionality changes with shifts in microbial composition. Further, the physiological outcome of these findings require confirmation by interrogation of the microbial metabolome. The relevance of these to disease development and progression require comparison of data from individuals at specific stages of disease and eventually from longitudinal datasets.
Bile acids are liver‐derived metabolites of cholesterol and have a multitude of biological effects that are relevant to NAFL biology. These include modulation of insulin sensitivity, glucose disposal, hepatic lipogenic drive, cell‐injury, inflammation and fibrosis.[ 9 ] We have previously demonstrated several changes in the circulating bile acid profile in those with NASH of varying stages including an increase in the secondary bile acid deoxycholic acid in those with advanced fibrosis.[ 10 ] However, the mechanistic basis for such changes were not known. Bile acids are also important modulators of the intestinal microbiome and the impact of altered bile acid profile on the composition of the microbiome is also not known. The clinical relevance of these changes in bile acids have also not been validated in large independent data sets or longitudinally over time.
The goal of this study was to generate a comprehensive view of the functional dysbiosis, using 16S rRNA sequencing, microbial transcriptomics and fecal metabolomics, associated with varying histological phenotypes of NAFLD with a specific focus on the role of the bile acid dependent microbiome in mediating the changes in bile acid profile associated with increasing fibrosis. The impact of the microbiome on the bile acid profile as well as the impact of changes in bile acid profile on bile acid‐sensitive microbiota were also assessed. Further, the biological and clinical relevance of changes in intestinal bile acids were validated across independent populations using both cross‐sectional as well as longitudinal serum bile acids data.
MATERIALS AND METHODS
The study was performed according to the Virginia Commonwealth University (VCU) regulations for the protection of human research subjects after the protocol was approved by the institutional review board (IRB # HM14081). All study participants gave written informed consent.
Participants recruitment
The patients were recruited at VCU Medical Center. Patients with suspected NAFLD who were undergoing a liver biopsy to further evaluate their liver disease stage were considered for this study. NAFLD was suspected by the presence of abnormal liver enzymes without evidence of other liver diseases or radiologic evidence of a fatty liver. Those with a history of alcohol use above commonly used thresholds (≥30 g/day for male patients and ≥20 g/day for female patients) or other causes of liver disease (viral hepatitis B, viral hepatitis C, primary biliary cirrhosis, sclerosing cholangitis, autoimmune hepatitis, hemochromatotis, Wilson's disease, α1‐antitrypsin deficiency, and drug‐induced liver disease) were excluded. A core biopsy was obtained after an overnight fast in all either a percutaneous or transjugular route. The presence of NAFLD was diagnosed according to standard criteria.[ 11 , 12 ] On the basis of the liver histology, two NAFLD patient groups were studied: (1) participants with nonalcoholic fatty liver (NAFL) as diagnosed by the NASH CRN criteria included regardless of BMI and presence of type 2 diabetes; (2) participants with NASH. These groups were compared to controls defined by asymptomatic status, ALT < 19 IU/I for women and <31 IU/I for men, absence of steatosis by CAP measure (<260 db/m) on fibroscan and no fibrosis on fibroscan (<6 kp).[ 2 , 3 , 4 ] In total, 75 subjects took part in the study.
Liver histology assessment
Liver biopsies were collected in 1% formalin and processed using routinely used clinical methods (see Supporting Materials for details). All liver biopsies were read by a hepato‐pathologist using the NASH CRN scoring system.[ 13 ] NAFLD was diagnosed by steatosis >5%. Steatohepatitis was diagnosed as steatosis, lobular inflammation and hepatocellular ballooning; for purposes of this analysis those with definite steatohepatitis i.e., lesions mainly in zone III were combined with those with borderline steatohepatitis as defined by the CRN system. For the population of patients with NAFLD, the NAFLD activity score (NAS) and fibrosis stages were scored as previously described by us.[ 12 , 13 ] Participants with an NAS score of ≥4 were considered as having a high NAS score. Clinically significant fibrosis was defined as having a fibrosis score of ≥2, and participants with a fibrosis score of ≥3 were defined as having advanced fibrosis.
Stool samples collection
A standardized approach based on prior studies of the stool microbiome[ 14 , 15 ] was established. Samples collected for RNA later was used for metatranscriptomic analysis, leftover biomass was used for 16S rRNA sequencing. Samples collected in empty containers were also used for metabolomic analyses. Samples were transported from the research laboratory to Second Genome (for microbiome) and Metabolon (for metabolomics) on dry ice. All sample handling was done by trained staff per federal standards for biosample collection, handling and transport.[ 15 ] All stool samples were collected within 6 months of liver biopsy or recruitment into the study. Additional details are available in the Supporting Materials.
Patient recruitment and specimen collection for the external validation study
For the bile acid signature validation study, serum data for patients with NASH with stages 2, 3, and 4 from Selonsertib and Simtuzumab Phase 2b studies[ 16 , 17 ] and placebo arm in the Phase 2b ATLAS trial[ 18 ] were included. In ATLAS, Liver biopsies, obtained at baseline screening and week 48, were evaluated in a blinded, unpaired manner by a single central pathologist. Subjects with ≥1‐stage fibrosis improvement from baseline to week 48 were defined as fibrosis improvement responders, whereas fibrosis worsening was defined with ≥1‐stage fibrosis increase from baseline to week 48. Patient recruitment and study randomization details have been previously published.[ 16 , 17 , 18 ]
Serum specimen collection
Fasting blood samples for the external validation study were collected at baseline and through week 48. Specimen collection details were previously described elsewhere.[ 18 ]
Statistical analysis
Software
The statistical analysis was performed using R software.[ 19 ] Unless stated otherwise, cutoff of 0.05 was used for significance.
Multiomics correlation
Mantel tests were performed on Bray‐Curtis dissimilarity for each pair of ‐omics data (e.g., 16S and metabolites) using R function mantel.rtest() in package ade4. Only samples with both available data types were used in the analysis.
PCo analysis of community structure
Brays–Curtis dissimilarity principal coordinates analysis (PCoA) on taxa relative abundance was used for visualization of each omics features data beta diversity.
Association with demographic and clinical covariates
The significance between individual ‐omics data types and clinical covariates was evaluated using univariate PERMANOVA tests on Bray‐Curtis dissimilarity[ 20 ] with R function adonis() in package vegan.
Identification of differentially abundant features
Microbiome and metatranscriptome pathways: The linear discriminant analysis effect size (LEfSe)[ 21 ] was used to identify enrichment in relative abundance 16S taxa and functional metatranscriptomic pathways between treatment groups (e.g., NAFL versus NASH, etc). The method uses the nonparametric Kruskal‐Wallis statistical test to compute differences among treatment groups and then a linear discriminant analysis (LDA) model to provide an effect size for the significantly different features. Alpha values of 0.05 were used for the Kruskal‐Wallis rank‐sum test, and a threshold of >2.0 for logarithmic LDA score significance. Metabolomics: Two sample Welsh t‐test was used to identify metabolites (log10 transformed data) that were significantly different each comparison group. The p values for bile acids were adjusted for the false discovery rate using R function qvalue.[ 22 , 23 ] Metatranscriptome genes: The univariate taxa differential abundance in two groups of samples was tested using a negative binomial model for the overdispersion and Poisson process intrinsic to gene count data with the adjustment for the multiple comparisons, as implemented in DESeq2 package in R.[ 24 ]
Confounder adjustment analyses
The effect of confounding factors on the significance of bile acids and 16S taxa were tested using logistic regression and random forest classification models. Details are available in the Supporting Materials.
Heatmaps of differentially abundant features
Heatmaps of differentially abundant features were constructed by taking the ratio of average expressions in two groups.
ATLAS serum bile acid statistical analysis
Kruskal‐Wallis test was used for analyses comparing healthy subjects with F2, F3, and compensated patients with cirrhosis. Wilcoxon rank‐sum test was used to compare the percent changes from baseline in serum bile acid levels between different fibrosis response groups.
RESULTS
Demographic characteristic of the patient cohort
Multiomics fecal data (16S, gene expression and derived pathways from meta‐transcriptomics, metabolomics) were obtained from patients with NAFLD (n = 23 for NAFL and n = 34 for NASH) and related to 18 controls (Figure 1A–C). The baseline demographics and clinical characteristics of the cohort are provided in Table S1 and graphically represented in Figure S1A–C. Most “Omics” measures were significantly correlated with a correlation coefficient between 0.185 and 0.729 (Figure 1B). Principal components analysis of Bray‐Curtis distances within each omics type demonstrated significant separation between disease groups i.e., control, NAFL and NASH for meta‐transcriptomic and related pathway activation data (p = 0.001 for both) but not for 16S data (Figure 1A,C) indicating that although there were overlapping distribution of the bacterial taxa across the groups, microbial functionality varied across these groups.
FIGURE 1.
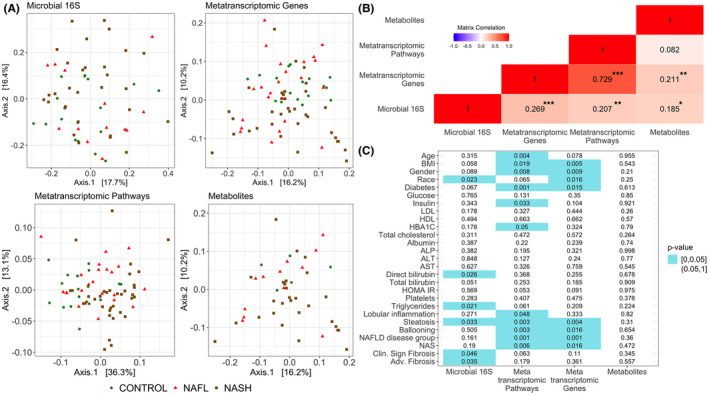
Multiomics of the NAFLD progression study. (A) Principal coordinates analyses (PCoA) Bray‐Curtis distance plots depicting the relationship between individual ‐omics type and NAFLD study groups. (B) Pairwise correlation between each ‐omics Bray‐Curtis distance matrices colored by correlation strength; for each correlation value stars represent significance strength accessed using mantel tests. Significance codes: *0.05 < p ≤ 0.1; **0.001 < p ≤ 0.05; ***p ≤ 0.001. (C) Significance values heatmap for univariate PERMANOVA tests of association between individual ‐omics data type (x axis) Bray‐Curtis distances and clinical covariates.
Association of omics measures with patient demographic profile
A PERMANOVA test was used to identify characteristics with overall distributions of microbial taxa, gene expression, pathway activation and fecal metabolites (Figure 1C). Differential foldchange distribution of significantly expressed individual genes, pathways, and metabolites are further presented in supplemental figures (Figures S2–S4). Multiple patient characteristics were related to the 16S profile of microbial taxa and microbial gene expression including demographic features (e.g., age for metatranscriptomics and sex for metatranscriptomics and 16S), laboratory parameters (triglycerides and bilirubin for 16S) and severity of several histological features (e.g., steatosis for metatranscriptomics and 16S, and NAFLD activity score (NAS) for metatranscriptomics). Fibrosis stage 2 and higher are known to be associated with poorer liver outcomes with a substantial step up in risk in stages 3 (bridging fibrosis) and 4 (cirrhosis).[ 25 ] The distribution of taxa was also significantly different within groups with or without clinically significant fibrosis (stage 2 or higher) or advanced fibrosis (stages 3–4).
Changes in fecal bile acids and bile acid dependent microbiome in NAFLD and with increasing disease activity
We have previously reported a decrease in total circulating secondary bile acids with NAFLD of increasing severity.[ 10 ] In contrast to these previously reported circulating bile acid profiles, the proportion of secondary bile acids in feces was increased in NAFLD (Figure 2A). The proportion of unconjugated bile acids was lower in NAFLD compared to controls and in those with NASH compared to NAFL (Figure 2B). Although no overall differences in glycine conjugates to taurine conjugates were seen in NAFLD versus controls, a numerically higher proportion of taurine conjugated bile acids was seen in those with high NAFLD activity scores (NAS) (Figure 2C).
FIGURE 2.
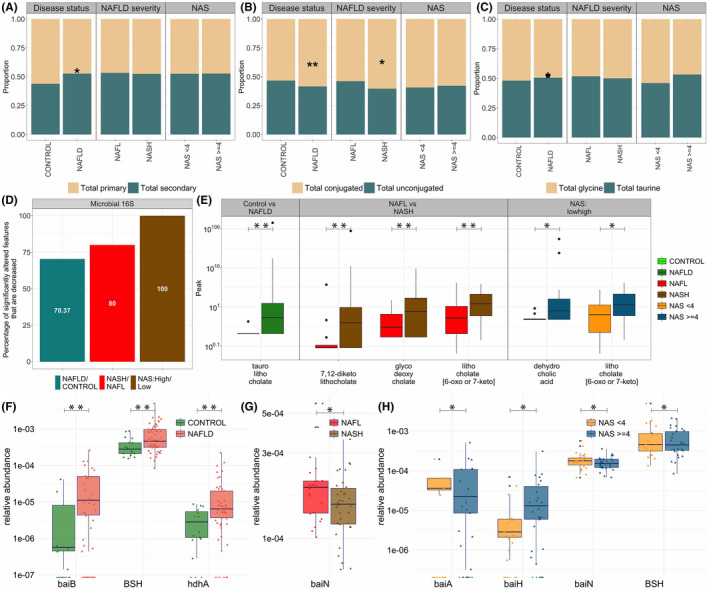
Comparison of NAFLD to control and disease activity groups: (i) NAFLD to CONTROL, (ii) NAFL to NASH, and (iii) NAS low to NAS high. (A) Proportion of total primary and total secondary bile acids. (B) Proportion of total conjugated and total unconjugated bile acids. (C) Proportion of total glycine conjugated and total taurine conjugated bile acids. (D) Proportion of features that are significantly decreased among the total number of significantly different 16S features in individual comparisons of NAFLD to control and disease activity groups. For each comparison group, proportions were calculated by dividing the number of taxa that were significantly decreased in that comparison (e.g., NAFLD vs. CONTROL) by a total number of taxa that were significantly different in that comparison group. (E) Boxplots of bile acid metabolites that are significantly different in NAFLD to control and disease activity group comparisons. (F‐H) comparison of expression of microbial genes involved in gut biotransformation of bile acids across NAFLD disease states; bai, bile acid inducible operon; BSH, bile acid hydrolase; HDH, hydroxysteroid dehydrogenase. Significantly expressed differences (p value ≤ 0.05) are identified by a * symbol; bile acid and genes in panels E–H controlled under the 11% the false discovery rate (FDR) are identified by a ** symbol.
Disease activity was determined by (1) presence of NASH rather than NAFL and (2) by a high NAS (≥4).[ 13 ] For each comparison group, the proportion of features that were decreased was defined as the number of taxa that were decreased in more severe stage of the disease (e.g., NASH in the comparison of NAFL vs. NASH) out of the total number of all taxa that were differentially expressed in that group. Among all taxa that were differentially expressed within each comparison group, 70% of differentially expressed taxa in NAFLD compared to controls were decreased, 80% of differentially expressed taxa in NASH versus NAFL and 100% of differentially expressed taxa in those with high NAS versus low NAS were decreased (Figure 2D).
Despite a general decrease in bacterial diversity, several bile acids derived from microbial metabolism increased with increasing disease activity. 7,12‐diketolithocholic acid (p = 0.035; q = 0.103), glycodeoxycholic acid (p = 0.028; q = 0.103), and lithocholic acid (p = 0.036; q = 0.103) were increased in NASH compared to NAFL, whereas dehydrocholic acid (p = 0.007; q = 0.160) and lithocholic acid (p = 0.027; q = 0.278) were increased in those with a high NAS versus low NAS indicative of specific changes in gut modified fecal bile acids that were correlated with disease activity (Figure 2E).
To obtain additional insights on the mechanistic basis for these findings, the expression of microbial genes involved in bile acid transformations was determined (Figure 2F–H). Bile salt hydrolase (BSH) is the first and rate‐limiting step in intestinal bile acid metabolism and the expression of this gene was increased in NAFLD versus controls (p = 0.001; p‐adjusted = 0.030). The major gut modification of bile acids is 7‐dehydroxylation, which is a multistep process involving bile acid inducible operons (bai) that convert primary to secondary bile acids. BaiB was increased in NAFLD versus control (p < 0.001; p‐adjusted = 0.020), whereas BaiH was increased in high NAS versus low NAS (p = 0.033; p‐adjusted = 0.176). The expression of hydroxysteroid dehydrogenase (hdhA), which mediates formation of dehydrocholic and 7,12‐diketolithocholic acid, was significantly increased in NAFLD versus controls (p = 0.001; p‐adjusted = 0.008). Together, these demonstrate both changes in the fecal bile acid profile with increasing disease activity and the underlying mechanistic changes in the gut microbiome responsible for these changes.
Changes in fecal bile acids and bile acid dependent microbiome in NAFLD and with disease severity
Both all‐cause mortality and liver‐related outcomes increase in those with stage 2 disease with a further significant step up with stages 3 and 4 respectively.[ 25 ] The changes in fecal bile acids and the bile acid dependent microbiome were thus interrogated in those with fibrosis stage 2 or higher (clinically significant fibrosis) and in those with stage 3 or 4 (advanced fibrosis).
The proportion of secondary bile acids in feces was smaller in those with clinically significant and advanced fibrosis, whereas total unconjugated bile acids were increased (Figure 3A,B). In contrast to changes seen with increasing disease activity, there was a decrease in taurine conjugated bile acids in those with clinically significant fibrosis (stage 2 and higher) as well as in those with advanced fibrosis (stages 3 or 4) (Figure 3C). Among all taxa that were differentially expressed, 75% and 73% were decreased in those with clinically significant and advanced fibrosis, respectively (Figure 3D).
FIGURE 3.
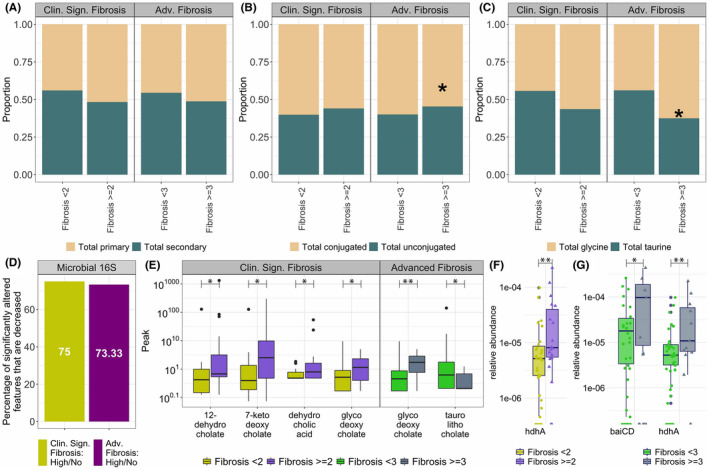
Comparison of fibrosis stage groups: (i) clinically significant fibrosis, and (ii) advanced fibrosis. (A) Proportion of total primary and total secondary bile acids. (B) Proportion of total conjugated and total unconjugated bile acids. (C) Proportion of total glycine conjugated and total taurine conjugated bile acids. (D) Proportion of features that are significantly decreased among the total number of significantly different features in individual comparisons in two fibrosis stages for the 16S data set. For each comparison group, proportions were calculated by dividing the number of taxa that were significantly decreased in that comparison (e.g., high advanced fibrosis vs. no advanced fibrosis) by a total number of taxa that were significantly different in that comparison group. (E) Boxplots of bile acid metabolites that are significantly different in comparison of fibrosis stage groups. (F‐G) comparison of expression of microbial genes involved in gut biotransformation of bile acids across NAFLD disease states; bai, bile acid inducible operon; BSH, bile acid hydrolase; HDH, hydroxysteroid dehydrogenase. Significantly expressed differences (p value ≤0.05) are identified by a * symbol; bile acid and genes in panels D–G controlled under the 11% the false discovery rate (FDR) are identified by a ** symbol.
Multiple bile acids were also altered with disease severity (Figure 3E). Several microbially‐derived metabolites of deoxycholate were increased with increasing fibrosis. 12‐dehydrocholate (p = 0.045; q = 0.159), 7‐ketodeoxycholate (p = 0.045; q = 0.159), dehydrocholic acid (p = 0.047; q = 0.159), and glycodeoxycholic acid (p = 0.039; q = 0.159) was increased in those with clinically significant fibrosis. In patients with advanced fibrosis, glycodeoxycholic acid remained elevated (p = 0.004; q = 0.097), indicative of significant changes in the gut modified fecal bile acids with more advanced disease. Interestingly, taurolithocholic acid was decreased in advanced fibrosis. This secondary bile acid has previously been reported to be increased in alcoholic hepatitis.[ 26 ]
The increase in the bile acids mentioned above was accompanied by an increased expression of microbial genes involved in bile acid transformation (Figure 3F,G). hdhA was increased in patients with clinically significant fibrosis (p = 0.002; p‐adjusted = 0.105), and was also increased in patients with advanced fibrosis (p = 0.003; p‐adjusted = 0.099). Additionally, patients with advanced fibrosis had increased expression of baiCD (p = 0.037; p‐adjusted = 0.327), which is critical for the formation of secondary bile acids especially deoxycholic acid.[ 27 , 28 ]
The fecal bile acid signature of NAFLD and its relationship to the intestinal microbiome
The levels of bile acids derived from the cytosolic Cyp7A pathway (Figure 4A, 12‐OH group) were increased to a greater degree than those from the mitochondrial 27α hydroxylase pathway (Figure 4A,B). Glycodeoxycholate was increased in patients with NASH as compared to NAFL and was also increased in patients with both clinically significant fibrosis and advanced fibrosis. These are in line with previously published data on circulating bile acids in NASH.[ 10 ] Oxidized bile acid epimers derived from microbial modification of deoxycholate were also increased with increasing disease activity and fibrosis. Together, these resulted in distinct bile acid profiles in NAFL versus NASH, NASH with high activity, and in advanced fibrosis.
FIGURE 4.
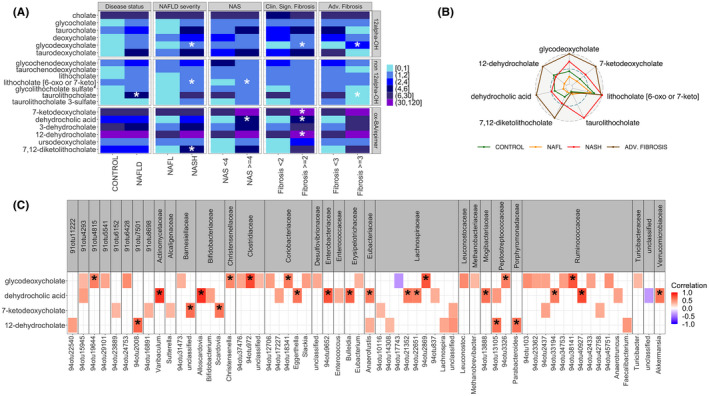
Changes in bile acids with NAFLD progression to fibrosis. (A) Heat map of bile acids relative concentrations averaged across all patients within each comparison group; 12α‐OH: 12α hydroxylated bile acids; ox‐BA/epimer: Bile acids with oxidation or epimerization at 3, 7 or 12 position. Significantly expressed differences (p value ≤ 0.05) are identified by a * symbol. (B) Radar plot for differentially expressed bile acids in control, NAFL, NASH and advanced fibrosis. Each pathway expression is normalized to the maximum across control, NAFL, and NASH patient groups. Outer dashed circle corresponds to the maximum expression of each pathway, middle dashed circle to the 0.5*maximum expression, and inner circle to the minimum expression. (C) Correlation heatmap of significantly expressed taxa with bile acids increased in clinically significant and advanced fibrosis. Only correlations with |ρ| ≥ 0.3 are highlighted in color. Correlations with p value ≤ 0.1 adjusted for the multiple comparisons are denoted by a * sign.
Next, to further evaluate the relationship of altered fecal bile acids and the microbiome, the concentrations of the key bile acids in those with more severe disease activity and fibrosis were related to the abundance of specific taxa (Figure 4C). Glycodeoxycholate was tightly correlated with multiple families including Christensenellaceae, Clostridiaceae, Coriobacteriaceae, Lachnospiraceae, Peptostreptococcaceae and Ruminoccoceae, whereas dehydrocholate levels correlated with mainly Actinomycetaceae, Bifidobacteriaceae, Coriobacteriaceae, Enterobacteriaceae, Erysipelotrichaceae, Eubacteriaceae, Lachnospiraceae, Mogibacteriaceae, Ruminococcaeae, and Verrucomicrobiaceae. A more limited set of microbiota were correlated with 7‐ketodeoxycholate and 12‐dehydrocholate—7‐ketodeoxycholate correlated with Barnesiellaceae and Bifidobacteriaceae, whereas 12‐dehydrocholate correlated with Peptostreptococcaceae and Porphyromonadaceae.
To further interrogate these relationships and their relevance to NASH of increasing severity, microbial taxa were compared across multiple disease phenotypes (Figure 5A). The abundance of the majority of differentially abundant taxa in NAFLD and in NASH versus NAFL was decreased. However, in those with clinically significant fibrosis virtually all of whom had NASH, several taxa were increased including Bacteroidaceae Bacteriodes (p = 0.015; LDA score = 4.492) and Lachnospiraceae, whereas most differentially abundant taxa in the Ruminococcaceae family were decreased including Ruminococcus (p = 0.034; LDA score = 4.091). Genera Sutterella (p = 0.023; LDA score = 3.445) and Lachnospira (p = 0.041; LDA score = 3.591) were further increased only in those with advanced fibrosis, whereas Prevotella (p = 0.032; LDA score = 3.759), Lactobacillus (p = 0.016; LDA score = 4.948), Ruminococcus (p = 0.031; LDA score = 4.278) and Turicibacter (p = 0.040; LDA score = 2.987) were decreased. Thus, only a limited set of microbiota were increased with increasing disease severity both in terms of activity and fibrosis.
FIGURE 5.

Analysis of changes in microbial genes and taxa related to bile acids within each comparison group. (A) Heatmap of the average log2 fold change for significantly expressed taxa in LEfSe pairwise comparisons of NAFLD to control and disease activity; significantly expressed taxa are identified by a * symbol. (B) Heatmap of the average log2 fold change for taxa known to contain genes involved in gut biotransformation of bile acids; significantly expressed taxa are identified by a * symbol. (C) Heatmap of the average log2 fold change for genes known to induce sensitivity or undefined changes in taxa susceptibility toward bile acids; genes with p value ≤ 0.1 are identified by a * symbol, genes with p value adjusted for the multiple comparisons value ≤ 0.1 are identified by ** symbol.
To further determine which of the differentially expressed microbiota in NASH had the ability to metabolize bile acids and potentially able to contribute to the alterations in the fecal bile acids observed, the differentially expressed taxa were screened for their ability to metabolize bile acids from existing literature (Figure 5B).[ 28 ] Multiple BSH expressing taxa including the genera Lactobacillus (p = 0.020; LDA score = 3.728) and Methanobrevibacter (p = 0.044; LDA score = 2.932) were significantly enriched in NAFLD as a whole, but the abundance of Lactobacillus (p = 0.016; LDA score = 3.948) decreased in those with advanced fibrosis. Catenibacterium, which also expresses 12α‐OH steroid dehydrogenase, was enriched (although not significantly) with increasing fibrosis. Eggerthella, containing both 3α‐ and 12α‐OH steroid dehydrogenases and BaiCD, which is critical for deoxycholate generation was increased (although not significantly) in those with more advanced fibrosis. Lachnospira (p = 0.041; LDA score = 3.591 in advanced fibrosis) and several unnamed genera within the Lachnospiraceae family containing key genes necessary to produce the differentially abundant bile acids were significantly increased in both those with clinically significant fibrosis and advanced fibrosis.
Bile acids can modify the microbiome by reducing the abundance of taxa that are sensitive to the antibiotic effects of bile acids and increasing the abundance of taxa that are dependent on bile acids for their growth. The loss of microbiota sensitive to the antibiotic effects of bile acids was interrogated by evaluating the expression of microbial genes that are in such taxa.[ 29 , 30 ] These suggested a general suppression of these genes in NAFLD versus controls, NASH versus NAFL, and with increasing fibrosis with a few exceptions. Gene expression of Endonuclease 4 (nfo) (p = 0.083; p‐adjusted = 0.410) and Penicillin‐binding protein 1A (mrcA) (p = 0.028; p‐adjusted = 0.270) were decreased in patients with NASH versus NAFL. Gene expression of DNA adenine methylase (dam) (p = 0.080; p‐adjusted = 0.270), single‐stranded‐DNA‐specific exonuclease RecJ (recJ) (p = 0.018; p‐adjusted = 0.142), and outer membrane protein TolC (tolC) (p = 0.022; p‐adjusted = 0.144) were decreased in patients with high activity. TolC was also decreased in patients with clinically significant fibrosis (p = 0.004; p‐adjusted = 0.053). These indicate substantial remodeling of the microbiome with a decrease in microbiota sensitive to bile acid–mediated growth inhibition (Figure 5C).
External validation and clinical relevance of changes in serum bile acid changes in NASH
The fecal bile acid profile demonstrated an increase in deoxycholate and related metabolites with increasing severity of disease (Figure 4B). We have previously also demonstrated an increase in circulating deoxycholate in those with increased fibrosis stage.[ 10 ] In those studies, however the deoxycholate‐metabolites such as dehydrocholate, 7‐ketodeoxycholate and 3‐dehydrocholate were not measurable. These suggested that, of the intestinally modified bile acids whose levels change with increasing fibrosis, deoxycholate rather than its downstream products is likely to be a key candidate related to disease severity.
This possibility was tested in an independent cohort of 72 patients with NASH stage 2–4 who participated in a Selonsertib and Simtuzumab Phase 2b studies, the primary results of which have been previously published.[ 16 , 17 ] Serum data at baseline was used for analysis. Compared to healthy volunteers, patients with NASH and clinically significant fibrosis had higher serum deoxycholate (DCA) levels (Figure 6A). There was also a stepwise increase in glycodeoxycholate (GDCA) and taurodeoxycholate (TDCA) further confirming the relationship of increased deoxycholate with increasing fibrosis. We further related the trajectory of fibrosis change in patients who participated in a placebo‐controlled ATLAS clinical trial for NASH (Figure 6B,C).[ 18 ] Those who had cirrhosis were further followed over time and bile acid levels measured at the time of decompensation (Figure 6B). There was a step up in both lithocholate and deoxycholate with decompensation in these individuals which reached significance for taurodeoxycholate (p < 0.03).
FIGURE 6.
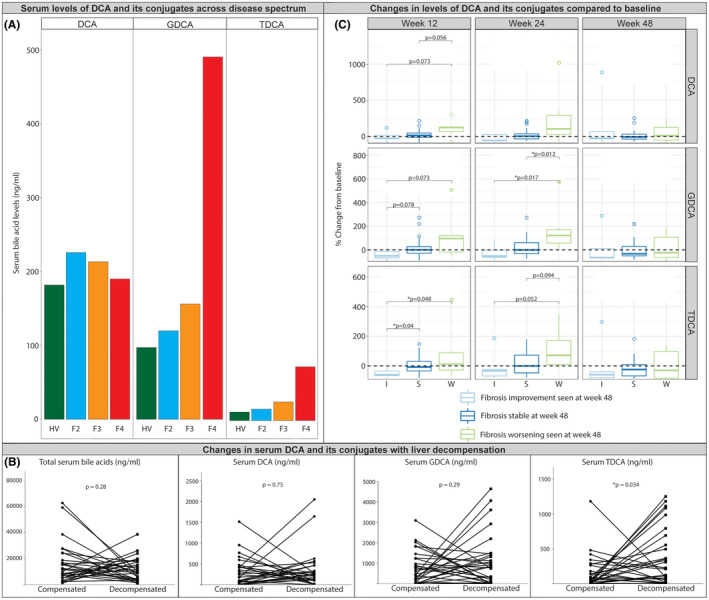
External data validation using serum biosamples. (A) Serum DCA and its conjugates across disease spectrum, seen in healthy volunteers (HV) and NASH with fibrosis stages 2–4 (F2–F4) at baseline in an independent data set from Selonsertib and Simtuzumab phase 2b studies.[ 16 , 17 ] Significant increase in G‐DCA and T‐DCA in fibrosis stage 4 (F4). (B) Changes of serum DCA and conjugates during liver decompensation in ATLAS study.[ 18 ] (C) Changes in serum DCA levels and its conjugates 12, 24, and 48 weeks from baseline over time in patients from placebo arm with improvement, stability, and worsening of fibrosis at week 48 in the ATLAS study.[ 18 ]
In general, those who experienced a one or greater fibrosis stage improvement had a trend for decreased deoxycholate and its conjugates whereas those who had worsening fibrosis had increased levels (Figure 6C). Those with unchanged fibrosis stage had relatively unchanged deoxycholate (both unconjugated and conjugated forms) levels.
DISCUSSION
The current study provides insights on the role of the bile acid dependent microbiome and changes in bile acid profile with increasingly severe NAFLD and suggests an important role for genesis of the microbially‐derived secondary bile acid deoxycholic acid in disease severity. This has several implications for the field.
First and foremost, they provide evidence relating deoxycholic acid and its conjugates as biomarkers of advanced fibrosis and disease progression. They not only increased progressively with clinically significant fibrosis (≥stage 2) to advanced fibrosis (≥stage 3) but were further increased at the time of clinical decompensation. These data generated over independent patient cohorts are further complemented by evidence from patients with NASH who were in the placebo arm of the ATLAS trial[ 18 ] where those with fibrosis improvement had decreased conjugated and unconjugated deoxycholic acid levels and those with fibrosis progression had increased levels (Figure 6). These data suggest the potential for the use of deoxycholic acid and the bile acid profile as prognostic biomarkers as well as disease‐monitoring biomarkers and support their future development for these indications.
It is known that both disease activity and fibrosis can wax and wane over time in patients with NASH.[ 12 , 31 , 32 , 33 ] The specific biological factors associated with regression are not clear. Specifically, in the NASH CRN cohort this was not a function of changes in body weight.[ 12 ] Our observation linking GDCA levels with fibrosis severity and a further step up to decompensation on one hand and increase in GDCA with fibrosis progression and decrease with fibrosis regression within the placebo arms of the ATLAS trial provides evidence for deoxycholic acid and its conjugates as drivers of disease severity and a rationale for targeting it to modify the course of the disease.
Several prior studies further provide additional support for deoxycholic acid as a driver of disease progression in NAFLD. Recent studies indicate that deoxycholic acid increase collagen synthesis by hepatic stellate cells via binding to TGR5 receptors.[ 34 ] Further, deoxycholic acid is known to perturb mitochondrial outer membranes in a concentration dependent manner which preceded mitochondrial membrane transition triggering cell death pathways.[ 35 ] We and others have previously demonstrated mitochondrial injury in NASH[ 36 , 37 , 38 ] and mitochondrial failure is a key feature of advanced liver failure requiring liver transplantation.[ 39 ] Deoxycholic acid also induced mir21/PDCD4‐mediated apoptosis in hepatocytes.[ 40 ] Given the plethora of data on the cellular mechanisms of deoxycholic acid–mediated injury, the current study focused primarily on generating evidence linking it to histological severity and clinical outcomes. Taken together, they provide deoxycholic acid as a biologically plausible and clinically relevant marker of NASH progression and potential regression.
The current study further provides a mechanistic framework and insights into the alterations in the bile acid profile across increasingly severe disease phenotypes. Although Clostridium are major producers of secondary bile acids under normal circumstances, the abundance of Clostridium was not significantly changed in advanced fibrosis (Figure 5B). On the other hand, Lactobacillus, g4otu10116 (Lachnospiraceae family) and Lachnospira emerged as major taxa that are both differentially expressed with advanced fibrosis and are known to express key bile acid metabolizing enzymes that can generate the bile acid profiles noted in this population. These data open the exciting possibility of targeted intervention to modulate these taxa to modify disease progression in a precision medicine–based approach in future studies.
Several other taxa, not directly involved with bile acid metabolism also changed with NASH progression to advanced fibrosis. The majority of these were decreased including Prevotella, multiple genera from Ruminococcaceae and Clostridiaceae families, as well as Turicibacter which have been linked to metabolic health benefits.[ 41 ] Several of these, e.g., Ruminococcaceae are sensitive to antibacterial effects of deoxycholic[ 42 ] and their abundance was inversely related to deoxycholic acid in this study. Further, numerous genes commonly expressed in microbiota sensitive to the antibacterial effects of bile acids were downregulated in advanced fibrosis as in steatohepatitis. These data provide evidence for restructuring of the microbiome with NASH development and in advanced fibrosis. The potential implications of decreased abundance of these taxa for overall metabolic health across various phenotypes of NAFLD await further elucidation.
As with many studies, there are some limitations of this work. The sample size of the multiomic study cohort was relatively modest; however, the key findings are supported by methods to minimize false discovery. The study cohorts were predominantly Caucasian and the data may or may not be generalizable to all races and ethnicities. Increased cholesterol intake is known to alter bile acids production in the liver,[ 43 ] however the detailed dietary information was not collected, and we could not characterize the role of nutrition on bile acid changes in NAFLD. Although the confounder unadjusted analyses were reported, a subanalysis adjusting for age, sex, BMI, and diabetes revealed that the conclusions hold after confounder adjustment (see Figures S5–S7). The population also had very few patients with cirrhosis and additional studies to evaluate microbial dysfunction leading to increased deoxycholic acid and its derivatives in cirrhosis due to NASH are warranted. Additionally, this was mainly a cross‐sectional study and serial changes in the microbiome will require a longitudinal study. However, the main finding of deoxycholic acid being related to fibrosis progression, more advanced disease and clinical decompensation was observed in the validation cohorts.
In summary, the current cross‐sectional study demonstrates both compositional and functional changes in the bile acid dependent microbiome and concordant changes in fecal bile acids with increasingly severe activity and fibrosis stage in NAFLD. It provides mechanistic insights on the observed increase in deoxycholic acid and its derivatives with disease severity, and lays foundation for future longitudinal studies of NAFLD. Clinical data demonstrate a strong linkage not only to development of advanced fibrosis but relate coordinate changes in deoxycholic acid and the course of fibrosis as well as with clinical decompensation. They further identify specific taxa linked to these changes and disease progression to advanced fibrosis. These data strongly support a role for the bile acid dependent microbiome in progressive NASH and set the stage to leverage these findings both for biomarker and therapeutics development.
AUTHOR CONTRIBUTIONS
Role of individual authors: Study Design: Ekaterina Smirnova, Puneet Puri, Arun J. Sanyal. Study implementation: Nicole Narayan, Mohamad S. Siddiqui, Velimir A. Luketic, Melissa J. Contos, Michael Idowu, Mulugeta Seneshaw, Hae‐Ki Min, Faridodin Mirshahi, Sherry Boyett, Arun J. Sanyal. Data analysis: Ekaterina Smirnova, Mark D. Muthiah, Nicole Narayan, Arun J. Sanyal. Results interpretation: Ekaterina Smirnova, Mark D. Muthiah, Nicole Narayan, Puneet Puri, Arun J. Sanyal, Jen‐Chieh Chuang, Andrew N Billin, Ryan S. Huss, Robert P. Myers. Manuscript write up and critical review: Ekaterina Smirnova, Mark D. Muthiah, Nicole Narayan, Mohamad S. Siddiqui, Puneet Puri, Velimir A. Luketic, Melissa J. Contos, Michael Idowu, Jen‐Chieh Chuang, Andrew N. Billin, Ryan S. Huss, Robert P. Myers, Sherry Boyett, Mulugeta Seneshaw, Hae‐Ki Min, Faridodin Mirshahi, Arun J. Sanyal.
CONFLICTS OF INTEREST
Andrew N. Billin owns stock in and is employed by Gilead Sciences. Jen‐Chieh Chuang owns stock in and is employed by Gilead Sciences and The Liver Company. Ryan S. Huss owns stock in and is employed by The Liver Company. He owns stock in Gilead Sciences. Robert P. Myers owns stock in and is employed by The Liver Company. He owns stock in Gilead Sciences. Nicole Narayan owns stock in and is employed by Second Genome. Arun J. Sanyal consults and received grants from Conatus, Gilead, Malinckrodt, Immuron, Boehringer Ingelhiem, Novartis, Bristol Myers Squibb, Merck, Lilly, Novo Nordisk, Fractyl, Siemens, Madrigal, Inventiva, and Covance. He owns stock in and consults for Genfit and Hemoshear. He consults for Intercept, Pfizer, Salix, Galectin, Sequana, Terns, Albireo, Sanofi, Jannsen, Takeda, Northsea, AMRA, Perspectum, Poxel, 89 Bio, AstraZeneca, NGM Bio, Amgen, Regeneron, Genetech, Roche, Albireo, Prosciento, Histoindex, Path AI, and Biocellvia. He received grants from Echosense‐Sandhill, OWL, and Second Genome. He received royalties from Elsevier and UptoDate. He owns stock in Sanyal Bio, Exhalenz, Durect, Indalo, Tiziana, and Rivus.
ETHICS
The study was conducted in accordance with the ethical principles expressed in the Declaration of Helsinki and the requirements of applicable federal regulations.
Supporting information
Appendix S1 Supporting Information
Smirnova E, Muthiah MD, Narayan N, Siddiqui MS, Puri P, Luketic VA, et al. Metabolic reprogramming of the intestinal microbiome with functional bile acid changes underlie the development of NAFLD. Hepatology. 2022;76:1811–1824. 10.1002/hep.32568
Ekaterina Smirnova and Mark D. Muthiah share co‐first authorship.
Funding informationNational Institutes of Health (NIH) grant NIH RO1 DK105961 (to Arun J. Sanyal); NIH grant T32 DK07150‐40 (to Arun J. Sanyal); NIH grant CTSA UL1TR002649 (to Ekaterina Smirnova) from VCU BERD Core; NIH grant CTSA 5KL2TR002648 (to Ekaterina Smirnova ) from VCU Institutional Career Development Core; and National Medical Research Council Singapore Research Training Fellowship MOH‐000193 (to Mark D. Muthiah). This work was primarily funded by NIH RO1 DK105961 and T32 DK07150‐40 to Dr. Arun Sanyal, and CTSA UL1TR002649 and VCU CCTR KL2TR002648 emerging scholar award to Dr. Ekaterina Smirnova for the proposed analyses. Samples were also collected in the VCU CCTR, which is supported by the CTSA award to VCU. These NIH institutes did not have a direct role in the design and conduct of the studies. Second genome provided in kind contribution with analysis of microbial transcriptome.
Contributor Information
Ekaterina Smirnova, Email: ekaterina.smirnova@vcuhealth.org.
Mark D. Muthiah, Email: mdcmdm@nus.edu.sg.
DATA AVAILABILITY STATEMENT
Data is available upon request from the senior author arun.sanyal@vcuhealth.org.
REFERENCES
- 1. Albhaisi S, Sanyal AJ. Gene‐environmental interactions as metabolic drivers of nonalcoholic steatohepatitis. Front Endocrinol. 2021;12:665987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328–57. [DOI] [PubMed] [Google Scholar]
- 3. Han MAT, Altayar O, Hamdeh S, Takyar V, Rotman Y, Etzion O, et al. Rates of and factors associated with placebo response in trials of pharmacotherapies for nonalcoholic steatohepatitis: systematic review and meta‐analysis. Clin Gastroenterol Hepatol. 2019;17(4):616–29.e26. [DOI] [PubMed] [Google Scholar]
- 4. Noureddin M, Muthiah MD, Sanyal AJ. Drug discovery and treatment paradigms in nonalcoholic steatohepatitis. Endocrinol Diabetes Metab. 2020;3(4):e00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. The Human Microbiome Project Consortium , Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chopyk DM, Grakoui A. Contribution of the intestinal microbiome and gut barrier to hepatic disorders. Gastroenterology. 2020;159(3):849–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R, Ajmera VH, et al. Link between gut‐microbiome derived metabolite and shared gene‐effects with hepatic steatosis and fibrosis in NAFLD. Hepatology. 2018;68(3):918–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S, et al. A universal gut‐microbiome‐derived signature predicts cirrhosis. Cell Metab. 2020;32(5):901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahmad TR, Haeusler RA. Bile acids in glucose metabolism and insulin signalling—mechanisms and research needs. Nat Rev Endocrinol. 2019;15(12):701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Puri P, Daita K, Joyce A, Mirshahi F, Santhekadur PK, Cazanave S, et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology. 2018;67(2):534–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanyal AJ. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology. 2002;123(5):1705–25. [DOI] [PubMed] [Google Scholar]
- 12. Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M, et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw Open. 2019;2(10):e1912565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–21. [DOI] [PubMed] [Google Scholar]
- 14. Gillevet P, Sikaroodi M, Keshavarzian A, Mutlu EA. Quantitative assessment of the human gut microbiome using multitag pyrosequencing. Chem Biodivers. 2010;7(5):1065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smirnova E, Puri P, Muthiah MD, Daitya K, Brown R, Chalasani N, et al. Fecal microbiome distinguishes alcohol consumption from alcoholic hepatitis but does not discriminate disease severity. Hepatology. 2020;72(1):271–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology. 2018;67(2):549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology. 2018;155(4):1140–53. [DOI] [PubMed] [Google Scholar]
- 18. Loomba R, Noureddin M, Kowdley KV, Kohli A, Sheikh A, Neff G, et al. Combination therapies including Cilofexor and Firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73(2):625–43. [DOI] [PubMed] [Google Scholar]
- 19. R Core Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 20. McArdle BH, Anderson MJ. Fitting multivariate models to community data: a comment on distance‐based redundancy analysis. Ecology. 2001;82(1):290–7. [Google Scholar]
- 21. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Storey JD. A direct approach to false discovery rates. J R Stat Soc Ser B Stat Methodol. 2002;64(3):479–98. [Google Scholar]
- 23. Storey JD, Taylor JE, Siegmund D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J R Stat Soc Ser B Stat Methodol. 2004;66(1):187–205. [Google Scholar]
- 24. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology. 2017;65(5):1557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muthiah MD, Smirnova E, Puri P, Chalasani N, Shah VH, Kiani C, et al. Development of alcohol‐associated hepatitis is associated with specific changes in gut‐modified bile acids. Hepatol Commun. 2022;6:1073–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7(1):22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heinken A, Ravcheev DA, Baldini F, Heirendt L, Fleming RMT, Thiele I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome. 2019;7(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Urdaneta V, Casadesús J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med. 2017;4:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Begley M, Gahan CGM, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29(4):625–51. [DOI] [PubMed] [Google Scholar]
- 31. Friedman S, Sanyal A, Goodman Z, Lefebvre E, Gottwald M, Fischer L, et al. Efficacy and safety study of cenicriviroc for the treatment of non‐alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp Clin Trials. 2016;47:356–65. [DOI] [PubMed] [Google Scholar]
- 32. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo‐controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67(5):1754–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ng CH, Xiao J, Lim WH, Chin YH, Yong JN, Tan DJH, et al. Placebo effect on progression and regression in NASH: evidence from a meta‐analysis. Hepatology. 2022;75:1647–61. [DOI] [PubMed] [Google Scholar]
- 34. Saga K, Iwashita Y, Hidano S, Aso Y, Isaka K, Kido Y, et al. Secondary unconjugated bile acids induce hepatic stellate cell activation. Int J Mol Sci. 2018;19(10):3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sousa T, Castro RE, Pinto SN, Coutinho A, Lucas SD, Moreira R, et al. Deoxycholic acid modulates cell death signaling through changes in mitochondrial membrane properties. J Lipid Res. 2015;56(11):2158–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanyal AJ, Campbell‐Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183–92. [DOI] [PubMed] [Google Scholar]
- 37. Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6(1):1–28. [DOI] [PubMed] [Google Scholar]
- 38. Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol. 2005;42(6):928–40. [DOI] [PubMed] [Google Scholar]
- 39. Lane M, Boczonadi V, Bachtari S, Gomez‐Duran A, Langer T, Griffiths A, et al. Mitochondrial dysfunction in liver failure requiring transplantation. J Inherit Metab Dis. 2016;39(3):427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rodrigues PM, Afonso MB, Simão AL, Borralho PM, Rodrigues CMP, Castro RE. Inhibition of NF‐κB by deoxycholic acid induces miR‐21/PDCD4‐dependent hepatocellular apoptosis. Sci Rep. 2015;5(1):17528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carrizales‐Sánchez AK, García‐Cayuela T, Hernández‐Brenes C, Senés‐Guerrero C. Gut microbiota associations with metabolic syndrome and relevance of its study in pediatric subjects. Gut Microbes. 2021;13(1):1960135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Theriot CM, Bowman AA, Young VB. Antibiotic‐induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere. 2016;1(1):e00045‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu G, Salen G, Shefer S, Tint GS, Nguyen LB, Chen TS, et al. Increasing dietary cholesterol induces different regulation of classic and alternative bile acid synthesis. J Clin Invest. 1999;103(1):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
Data is available upon request from the senior author arun.sanyal@vcuhealth.org.