

Abstract
Discovery of the genetic components underpinning fundamental and disease-related processes is being rapidly accelerated by combining efficient, programmable genetic engineering with phenotypic readouts of high spatial, temporal, and/or molecular resolution. Microscopy is a fundamental tool for studying cell biology, but its lack of high-throughput sequence readouts hinders integration in large-scale genetic screens. Optical pooled screens using in situ sequencing provide massively scalable integration of barcoded lentiviral libraries (e.g., CRISPR perturbation libraries) with high-content imaging assays, including dynamic processes in live cells. The protocol uses standard lentiviral vectors and molecular biology, providing single-cell resolution of phenotype and engineered genotype, scalability to millions of cells, and accurate sequence reads sufficient to distinguish >106 perturbations. In situ amplification takes ~2 days, while sequencing can be performed in ~1.5 hours per cycle. The image analysis pipeline provided enables fully parallel automated sequencing analysis using a cloud or cluster computing environment.
INTRODUCTION
Cells use discrete components organized in time and space to carry out the functions of life. Assaying cell functions in a format amenable to high-throughput genetic screening rapidly accelerates the discovery of underlying genes and will provide insight into how cellular processes go awry in disease. While CRISPR-based tools have demonstrated tremendous success in scalable, programmable modulation of gene activity, quantifying functional phenotypes (e.g., activation of a signaling pathway or cell behavioral changes in response to environmental stimuli) in high throughput remains a major limiting factor in performing screens. Selection of cell populations based on fitness1–3 or fluorescence-activated cell sorting (FACS)4 remains the workhorse of large-scale pooled screening, although the cellular phenotypes that can be measured by these techniques are limited. Recently, pooled screens with single-cell molecular profiling have emerged as an exciting route to capture high-dimensional cell states, enabling multiple phenotypes to be defined across different biological pathways in a single dataset5–10 (Fig. 1a), albeit at a scale restricted by cost. Fluorescence microscopy, including live-cell and high-content imaging, is widely used to assay diverse cell functions but has lacked methods for integration in scalable pooled genetic screens. While arrayed imaging screens have been applied at large scales11–16, they, unlike pooled screens, require generating and handling large numbers of individual genetic perturbations and cell populations, limiting flexibility in assay design and requiring access to automated liquid-handling infrastructure.
Figure 1.
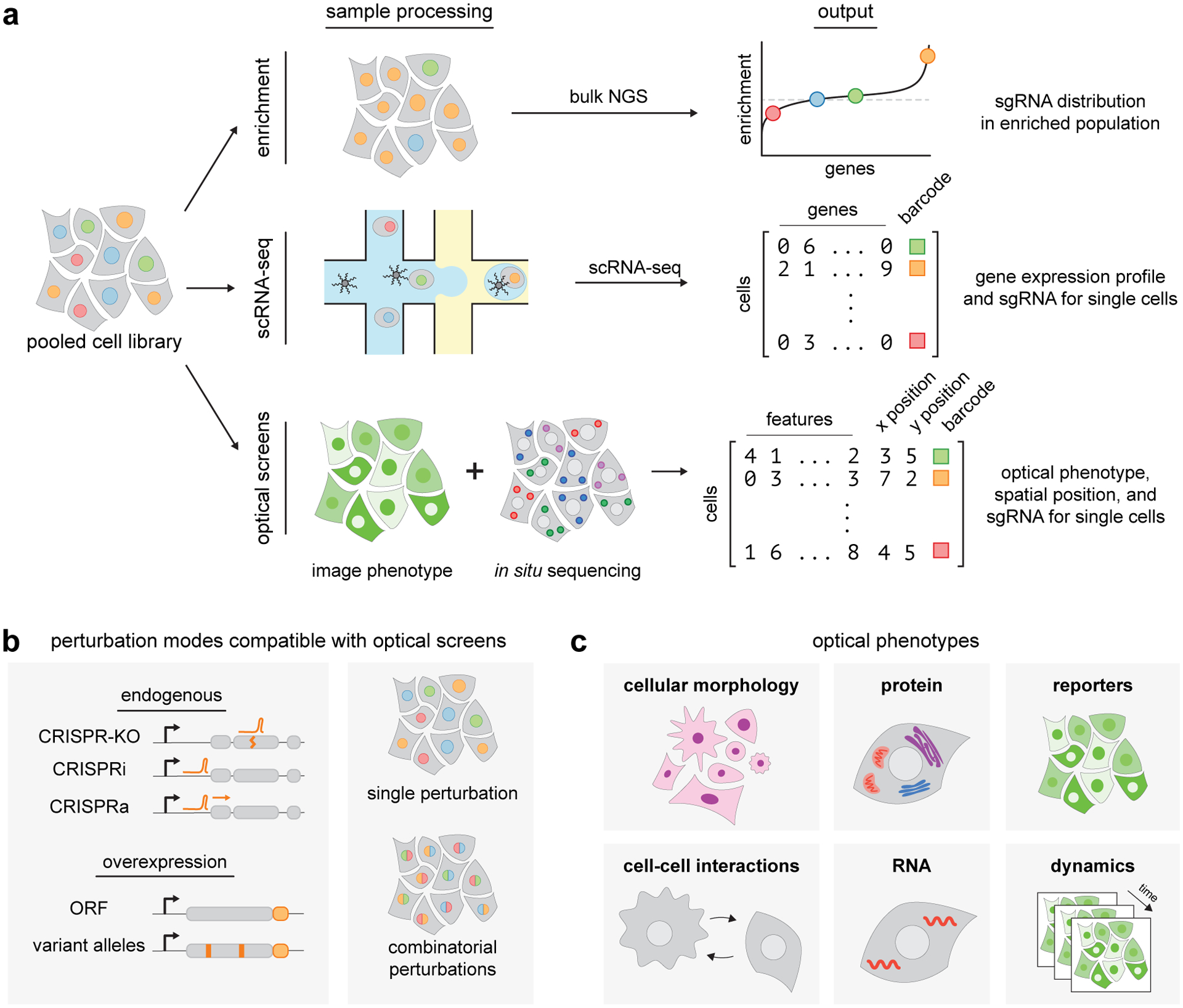
Pooled screening approaches and applications of optical pooled screens. (a) In pooled screening, a population of cells is subjected to a library of genetic perturbations, such as guide RNAs for CRISPR screens. Enrichment, single-cell profiling, and optical-based assays are three common approaches for phenotypic readout. Enrichment-based screens determine population-level changes in perturbation abundance by bulk next-generation sequencing (NGS) following an applied selection. Single-cell profiling and optical screens do not require an enrichment step and instead rely on information-rich phenotypic measurements. Single-cell assays pair perturbation barcodes to a cell phenotype at single-cell resolution, such as cell transcriptome for single-cell RNA sequencing-based screens. Through in situ sequencing, optical pooled screens pair image-based phenotypes with perturbation barcodes, also at single-cell resolution. (b) Optical screens are compatible with multiple perturbation modalities, including CRISPR-based perturbations of endogenous genomic loci and exogenous overexpression of barcoded transgenes. The single-cell readout enables both single and combinatorial perturbation screens. (c) Optical screens enable rich phenotypic measurements, including cellular morphology, cell-cell interactions, dynamic behaviors, and abundance and localization of endogenous protein and RNA molecules and exogenous reporters.
Image-based cell assays
Light microscopy offers a plethora of options for monitoring the phenotypic state of cells, many of which provide information complementary to molecular measurements of RNA or protein abundances. Fixation of cells followed by fluorescent labeling with antibodies, RNA or DNA hybridization probes, or small-molecule affinity reagents allows measuring spatial distributions in ~5 distinct channels, with many additional channels potentially available using sequential detection approaches17–20. In living cells, the abundance and localization of protein and RNA molecules can be visualized by genetic fusion or binding to fluorescent reporters. Additionally, fluorescent reporters can relate a wide range of biochemical states in living cells, ranging from ion concentrations and membrane potentials to kinase activity21–24. Time-lapse imaging can track cells longitudinally, enabling high-resolution measurements of dynamic variables, such as the time to activate or relax a signaling response25–28. Image-based assays can also employ mixtures of cell types that are optically distinguishable (e.g., by reporter or marker expression) to more accurately model a physiological environment or identify interactions between cells29–31 (Fig. 1c). While image phenotype measurements can often be quantified via straightforward metrics, such as mean fluorescence intensity or cross-correlation between channels, machine learning techniques have shown significant enhancements in classifying cell behaviors by extracting meaningful features from pixel-level raw data or higher-level descriptors32–35.
Due to their versatile nature, image-based cell assays have been successfully used for a wide variety of genetic screens with arrayed perturbations, including genome-wide RNA interference (RNAi) and targeted CRISPR-based screens characterizing genes involved in mitosis and cell cycle progression11,36, membrane trafficking12, autophagy13, viral and bacterial infection14–16,37, and cellular morphology38. However, the complexity and cost of performing large-scale arrayed genetic screens have limited their feasibility for many applications and potential users as they require expensive or customized automation to deliver precise amounts of each perturbation, culture individual cell populations, and image phenotypic assays at scale. Maintaining arrayed cell populations is particularly challenging with assays that require longer periods between perturbation and phenotypic measurement (e.g., assays requiring cell differentiation after perturbation) as differential perturbation efficiency and fitness effects accumulated over time can increase well-to-well variability and biological noise, in addition to the burden of maintaining a large number of wells in culture over time. CRISPR-based perturbations especially suffer from this limitation as they typically require more time to modulate target gene activities than RNAi – often several days. Additionally, new arrayed perturbation libraries are expensive to synthesize and will therefore lag technical developments in fast-moving fields such as CRISPR perturbations. For these reasons, comprehensive CRISPR-based imaging screens have been largely impractical. The application of image phenotype measurements in the context of pooled screening significantly decreases the complexity of image-based screens, facilitates their extension to new application areas and users, and potentially reduces biological noise as all cells experience the same culture and assay conditions.
Approaches to pooled screening
Pooled genetic screens fundamentally require a method to link genotype, either the perturbation identity or the actual genetic alteration, to phenotype. In enrichment-based methods, this is achieved by stably preserving the genotype within each cell and applying next-generation sequencing (NGS) to measure the abundance of genotypes before and after enrichment. As genotypes are not uniquely traceable to individual cells, the resulting enrichments reflect either population- or lineage-averaged phenotypes depending on the experimental approach1–3,39,40. By contrast, screens read out via single-cell molecular profiling methods, such as single-cell RNA sequencing or mass cytometry, simultaneously capture a genotype and phenotype for each cell, identifying the former via either a proxy barcode or the perturbation sequence itself5–10. Rather than averaged per-genotype enrichments, these screens yield a rich data matrix of cells by phenotypic features in which each cell is labeled by genotype (Fig. 1a).
Development of optical pooled screens
In this work, genotype is linked to phenotype at single-cell resolution by sequencing cellular perturbation identity in situ, within fixed cells. Phenotypic data acquired by methods such as immunostaining or live-cell imaging are aligned to sequenced perturbations on a cell-by-cell basis, providing a matrix of individual genotyped cells by phenotypic features. Perturbation identities are deduced from mRNA containing either the single-guide RNA (sgRNA) sequence itself or a short barcode, analogous to barcode capture in pooled single-cell RNA sequencing screens. Barcodes are read out in fixed cells via padlock-based in situ sequencing41,42, a process involving padlock probe hybridization and gap-filling, rolling circle amplification (RCA), and in situ sequencing by synthesis (SBS) (Fig. 2). We carefully optimized the in situ sequencing protocol in adherent cells, adding glutaraldehyde fixation after reverse transcription of cDNA and maximizing gap-fill reaction efficiency by titrating dNTP concentration43. These optimizations improve both the number and brightness of sequencing reads, enabling high-throughput optical pooled screening with perturbations successfully identified for a large fraction of cells when sequenced with 10✕ magnification. In previous work, we show that >80% of identified sequencing reads over 12 cycles of SBS exactly match the designed set of library sequences, allowing pooled screening with genetic libraries containing thousands of perturbations43.
Figure 2.
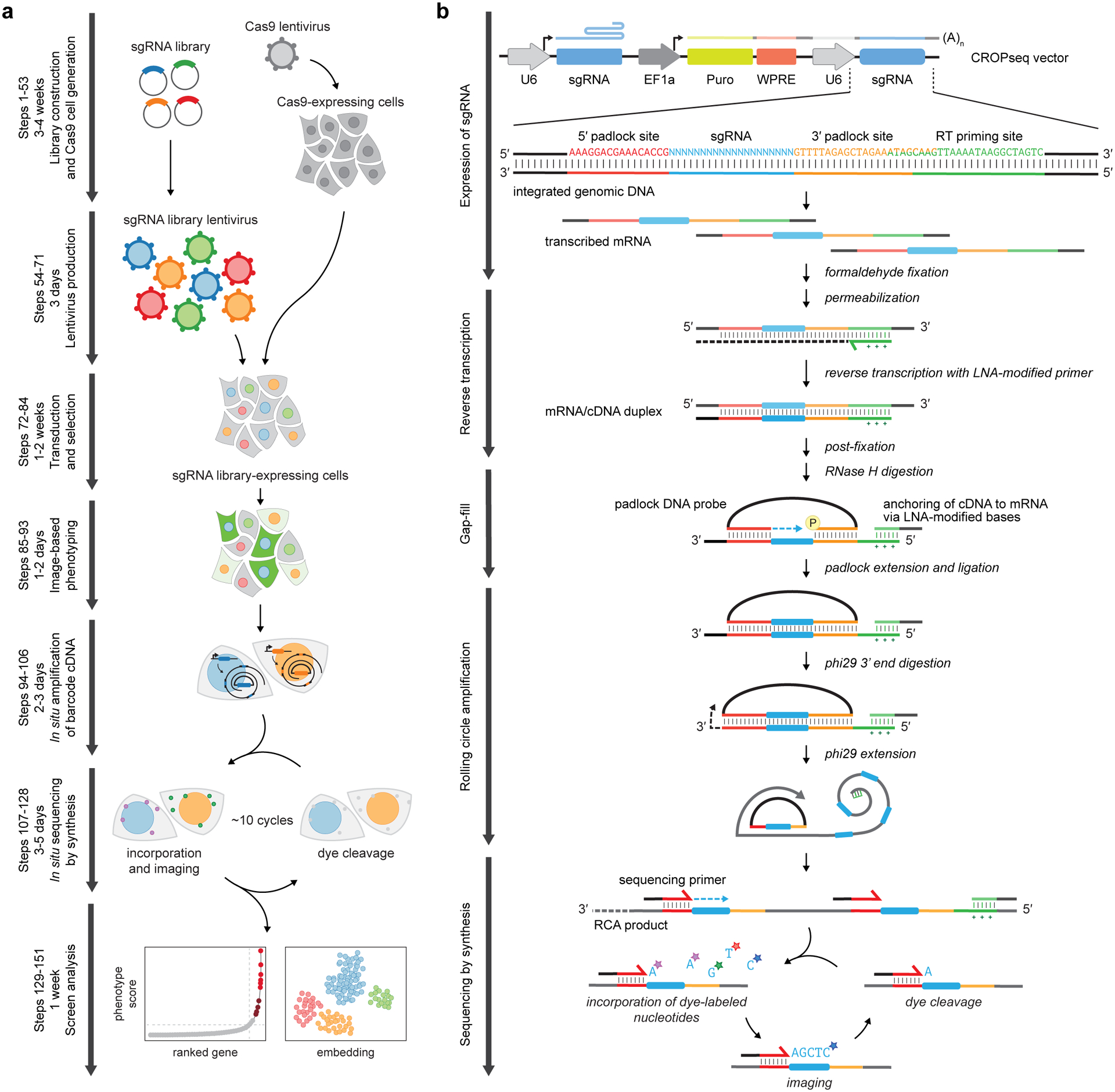
Overview of optical pooled screening. (a) Experimental workflow. First, a pooled sgRNA library is designed, packaged into lentivirus, and delivered into Cas9-expressing cells. A live-cell or fixed-cell imaging assay is performed to generate an optical phenotypic profile of individual cells. The sgRNA spacer sequences in each cell are then amplified and read out by in situ sequencing by synthesis (SBS), consisting of cycles of dye incorporation, imaging, and cleavage. Finally, sgRNA-encoded perturbations are mapped to phenotypic scores at the single-cell level, with candidate genes identified through various statistical approaches. (b) Schematic of the in situ SBS process. The sgRNA is expressed as a polyadenylated mRNA transcript from an integrated copy of the CROPseq vector. After fixation and permeabilization, a locked nucleic acid (LNA)-modified primer is used to reverse transcribe a cDNA copy of the sgRNA sequence. After glutaraldehyde and formaldehyde post-fixation, the mRNA is digested and a padlock probe is hybridized to cDNA regions flanking the sgRNA sequence. The padlock probe is then extended and ligated to copy the sgRNA sequence into a single-stranded circularized DNA. This circularized DNA serves as a template for rolling circle amplification with Phi29 polymerase, the amplified product of which contains tandem repeats of the sgRNA spacer sequence. These sequences are read out by successive cycles of SBS.
Comparison with alternative methods
Several recent technologies enable pooled imaging screens in bacterial and mammalian systems based on in situ optical barcoding of genetic perturbations or physical retrieval of relevant cell subpopulations. Barcoding methods have relied on in situ sequencing, as in the optical pooled screening approach presented here, or iterative fluorescence in situ hybridization (FISH) to map genetic perturbations44–47. While in situ sequencing achieves higher imaging throughput and direct detection of CRISPR sgRNAs using the standard CROP-seq vector48, FISH requires higher imaging magnification and long barcodes with multiple hybridization sites, necessitating bespoke library cloning methods and random pairing of perturbations and barcodes. However, FISH provides higher detection sensitivity, which may be useful in mammalian cell types with low barcode expression. Thus far FISH has been the only RNA detection method applied to bacterial systems44,46.
Approaches that physically retrieve subpopulations of cells are an extension of enrichment-based pooled screening. Cells with phenotypes of interest are identified by imaging a pooled cell library, and then are physically retrieved by either photoactivation of individual cells followed by FACS selection49–52 or using magnetic manipulation to select cells grown on microraft arrays53. Unlike optical barcoding methods in which a phenotype and genotype are assigned for each cell in the population, subpopulation retrieval approaches measure bulk enrichment of perturbations in a few pre-defined phenotypic bins, limiting the characterization of cell-level heterogeneity. However, physically separating cells of interest enables subsequent phenotypic measurements of cells from the screen, such as deep molecular profiling of relevant cell states and additional functional assays not compatible with the conditions of the screen.
Limitations
While optical pooled screens are promising for a wide range of model systems and phenotypic assays, several criteria must be met for a screen to be feasible. First, in situ sequencing must be validated in the model system of interest (initially demonstrated for seven cancer cell lines43, see Supplementary Figure 1); guidelines are provided here for validation of additional cell models (Experimental design and Box 1). For efficient screening, RCA amplicons (sequencing “spots”) must be detected in a sufficient fraction of cells, and background fluorescence from non-specific binding of SBS dyes must be low enough to enable accurate sequence mapping across cycles. The number of spots detected per cell is constrained by expression of barcode mRNA from a single lentiviral integration, as well as inefficiencies of in situ reverse transcription and padlock detection, all of which may depend on cell type. While cancer cell lines suitable for high-content imaging frequently produce good results without any protocol optimization, some cell types (e.g., differentiated stem cells, neurons) may require tailored reagents (e.g., alternative promoters to increase barcode mRNA levels).
Box 1 |. Cell models and perturbation vectors.
Critical choices in developing cell-based high-throughput screening applications include defining which model cell populations to use and how to deliver genetic perturbations to those cells. In addition to the six cell lines initially used for optical pooled screening, the method can be extended to many other cell-based models and applications. Requirements for additional cell models include adherence to an imaging surface, which may be achieved via surface coatings for cells that do not themselves adhere, and the ability to detect barcode mRNA using in situ sequencing. The key metric to evaluate when testing a new cell model is the fraction of cells containing an adequate number of sequencing spots that match an expected group of sequences (see Step 46 for validation experiment design and Supplementary Figure 1 for example data across several cell lines). This metric may be optimized by changing culture conditions, promoters to increase Pol II driven expression of barcode mRNA, and/or using a fluorescent reporter gene and FACS to select highly expressing cell subpopulations while maintaining sufficient library representation (typically >300 cells per library element). An additional issue to evaluate when using a new cell line is whether barcode mRNA molecules are adequately fixed within the cell of origin; this is identified by the presence of many sequencing spots outside the boundaries of cells when inspecting images. Diffusion into neighboring cells could disrupt accurate identification of cell perturbations and may require alternative strategies for fixing mRNA to the cell matrix, such as biotin-streptavidin linkage of the RT primer (also see the troubleshooting solutions in Table 3 for the listed problem of sequencing spots outside the boundaries of cells in step 114).
The most physiologically relevant cell model choice for a given application may not be optimum for optical pooled screening due to low in situ perturbation detectability, difficulty of cell culture, or other factors. A common approach is to complete a primary, large-scale screen using a tractable cell line that demonstrates relevant phenotypes and then validate any candidate hits with a more physiologically relevant system. When evaluating a potential screening cell model, positive control perturbations should be tested to determine if the resulting phenotypic state is measurably distinct from that of a wild-type or null perturbation cell population.
For CRISPR-based screening, sgRNA delivery using the CROPseq lentiviral vector is recommended to enable direct readout of sgRNA sequences. The sgRNAs can be delivered to any Cas9-expressing cell line, with inducible Cas9 expression allowing more control of perturbation timing. Optionally, a clonal Cas9 cell line may be used to maximize the uniformity of the underlying cell population (see Step 53). For applications in which direct sequencing of genetic perturbations is not possible (e.g., ORF screening), a linked, Pol II-expressed barcode sequence can be used to identify perturbations in situ. Cloning perturbation libraries for this approach often requires two separate Golden Gate assembly steps with distinct restriction enzymes: first the insertion of the linked perturbation and barcode into an empty vector, followed by the insertion of the necessary promoter and other elements between the perturbation and the barcode. During lentivirus production with a linked perturbation barcode approach, it is important to limit unwanted intermolecular recombination that may result in barcode swapping if the perturbation and barcode are far apart in the vector design. This can be achieved by co-packaging the perturbation library with a non-integrating carrier vector57, although this reduces the effective viral titer and is most suitable for smaller perturbation libraries.
Cells that do not grow in a monolayer or that have morphologies that are difficult to segment (e.g., highly polarized cell lines like WI-38) may also cause difficulties in cell barcode assignment. Segmentation issues can be addressed by optimizing culture conditions (e.g., surface treatment, cell density), by including a specific stain for demarcating cells (e.g., CellMask stains, Thermo Fisher Scientific, cat. nos. H32714, C10046), or by using segmentation algorithms such as CellPose54 that perform well on a wide range of morphologies. An option to use CellPose for nuclear and cytoplasmic segmentation is integrated into the analysis pipeline in this work (see Supplementary Figure 1 and the CRITICAL flag after Step 133).
Compared to arrayed screens, pooled lentiviral screens are inherently limited in monitoring non-cell autonomous phenotypes (e.g., changes in paracrine signaling), as neighboring cells have heterogeneous genotypes. Furthermore, the low copy number integration of genetic perturbations necessary for pooled lentiviral transduction limits the expression level of perturbations, which may result in reduced gene activity modulation, e.g., incomplete gene knock-out in CRISPR-based screens. Particularly for optical pooled screens, the required cost and time scales with the surface area imaged, hence screening for rare phenotypes or with large or sparsely plated cells necessitates longer imaging times and alternative approaches should be considered. For typical cell models and phenotypes, optical pooled screening is cost-effective for screens with a few hundred perturbations or more, while smaller experiments may be better suited to image-based arrayed screening.
Finally, optical pooled screening requires automated and efficient imaging hardware (Box 2) as well as high-throughput analysis of the imaging data. Python scripts are provided for both sequence and simple phenotype analyses. While basic knowledge of command-line tools should be sufficient for sequencing analysis using these scripts, scoring cell phenotypes may require more in-depth custom image analysis or integration with tools such as CellProfiler55. The wet lab and image acquisition sections of the protocol are accessible to researchers with general experience in molecular biology and fluorescence microscopy.
Box 2 |. Microscope hardware.
Optical pooled screens can be read out with standard commercially-available imaging hardware, such as an epifluorescence microscope equipped with an automated stage, sensitive camera (e.g., sCMOS), broadband or multispectral light source, and adaptive focus control. One of the key considerations when designing or adapting a system is selecting an illuminator and optical filters to achieve sufficient spectral separation of the sequencing channels. We provide illuminator and filter examples for both broadband and multi-laser excitation (Table 2). However, other arrangements may be validated by (1) visually assessing a sample image with all four sequencing channels, aiming for controlled crosstalk between nucleotide channels, and (2) performing a multi-cycle sequencing experiment and quantifying the mapping rate of reads to expected barcode sequences. Note that a moderate level of spectral crosstalk can be tolerated and corrected in the analysis stage (Fig. 3a-b).
Imaging throughput is another priority and is largely determined by stage scanning speed, filter switching times, and communication overhead between the computer and hardware components. Additionally, efficient illumination and detection using a low-magnification, high-NA objective and well-designed optical filters will maximize the signal-to-noise for a given exposure time. Hardware lag times and communication overhead contribute appreciably to the overall acquisition time and can be minimized by choosing fast components and implementing triggered acquisitions (see examples in Tables 1 and 2). If stage movement is faster than filter switching and highly reproducible, all positions of the sample can be imaged one channel at a time; the provided image analysis pipeline automatically aligns channels using the background cell staining of the sequencing dyes. If the exposure time is a limiting factor in the overall acquisition, increasing illumination intensity can boost screen throughput but must be carefully managed to avoid sample damage (e.g., by using oxygen-scavenger systems to avoid photodamage). Additionally, 2-color SBS chemistries can be used to further reduce imaging time, photodamage, and free up phenotyping channels56, at the cost of less accurate base calling in later cycles due to the smaller color space.
Experimental design
The design of an optical pooled screen begins with careful consideration of the biological process, available cell models, and possible phenotypic assays, with a preference for the simplest screen design that can identify relevant genetic components at sufficient statistical power. When using an unvalidated cell model, it is important to validate in situ sequencing compatibility by experimental side-by-side comparison to a validated cell line and perform any necessary optimizations before proceeding further (see Step 46 and Box 1).
This protocol focuses on the common example of CRISPR-Cas9 knockout screening, but the optical pooled screening approach and most procedure elements are generalizable to other screening modalities, including CRISPRi, CRISPRa, cDNA overexpression, and endogenous gene tagging56 (Fig. 1b). For CRISPR-based screening, we recommend using a Cas9-expressing cell line validated for high perturbation efficiency; optionally, generating a clonal cell line can ensure consistent perturbation efficiency while also reducing biological noise in a screen. Pooled sgRNAs can then be separately delivered by the CROPseq lentiviral vector to enable direct in situ sequencing of a Pol II-transcribed copy of the sgRNA sequence. For screen designs in which direct perturbation sequencing is not feasible, an alternative is to use a lentiviral perturbation vector containing a short, linked barcode sequence, which must be carefully employed to avoid swapping of barcodes between perturbations43,57 (Box 1).
After a compatible cell model and perturbation strategy have been selected, the phenotyping assay should be validated. A wide variety of phenotype measurements are compatible with optical pooled screening, and it may be advantageous to consider multiplexing several together. Up to three fluorescent channels that do not overlap with the sequencing dye spectra are available for phenotype imaging. Typically, nuclei are identified via a DNA counterstain, while non-specific binding of sequencing dyes to the cell is sufficient to segment the cell body. Additional staining strategies can further expand the range of available channels (Box 3). For phenotype assessment, the screening perturbation vector should be transduced at a relevant multiplicity of infection (MOI, 0.05–0.1) with a manageable number of positive controls (e.g., sgRNAs targeting genes known to affect the phenotype under study) and negative controls (e.g., non-targeting sgRNAs) in an arrayed format. In addition to validating the perturbation method using approaches such as indel sequencing or qPCR3, statistically comparing the resulting phenotypic states of control perturbations can guide the selection of a phenotypic assay and other screening parameters such as time point and additional chemical perturbations if applicable. Also at this stage compatibility of the phenotype assay with in situ sequencing should be confirmed, in particular evaluating cross-talk between fluorescent phenotype stains and the SBS dye channels and the optimum placement of phenotype imaging amongst other steps in the overall screening workflow (Box 3). As a final screening preparation step, a pilot pooled screen should be completed with a limited number of control genetic perturbations and many cells per perturbation (>1,000) to test all phenotype and in situ sequencing components along with the analytical pipeline.
Box 3 |. Approaches to optical phenotyping.
One of the most compelling reasons to use microscopy for genetic screens is the variety of imaging assays that can be used to study cellular phenotypes. Immunofluorescence, fixed- and live-cell fluorescent reporter assays, small-molecule stains, and in situ nucleic acid detection are highly customizable approaches that can be used to probe molecular abundance and localization, cell state and morphology, cell-cell interactions, and temporal dynamics. Each of these approaches can be combined with optical pooled screens for bespoke phenotypic analysis but require careful integration with the in situ sequencing workflow.
When combining fixed-cell phenotyping with optical pooled screens, it is critical to interleave the protocols in a way that accurately recapitulates imaging phenotypes while preserving the perturbation information. In situ sequencing efficiency can be reduced when phenotypes are stained and imaged before RCA. However, as certain cellular structures or labeling methods can be disrupted by steps of the in situ detection protocol, it may be necessary to stain and image before the RT or padlock detection steps. In general, phenotype imaging should be completed before the first SBS cycle. Performing phenotyping steps after fixation but prior to reverse transcription can cause significant loss of in situ sequencing reads due to RNA degradation; if this operating order is necessary, minimize handling time and include RNase inhibitor in staining and imaging solutions. It is recommended to test each of the different workflow options for fixed-cell phenotyping shown below, evaluating both the resulting phenotype image quality and the in situ sequencing spot count and brightness.
Box 3 – Figure.
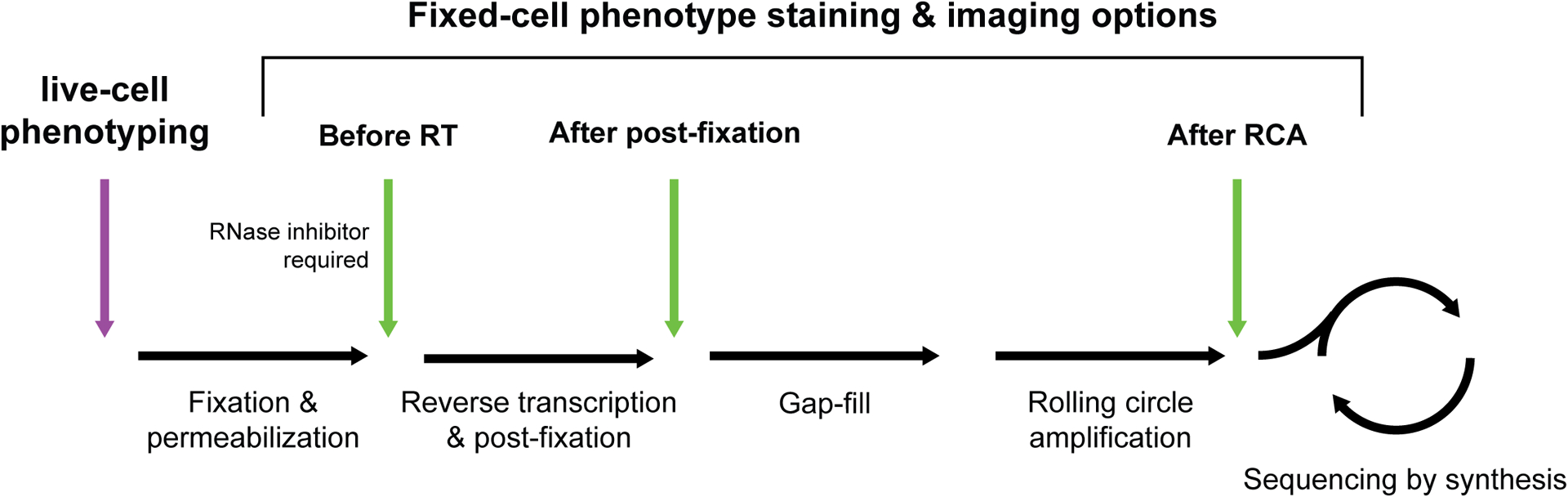
Options for phenotype acquisition between in situ amplification steps.
The ability to multiplex stains is a key feature of fluorescence microscopy and can greatly increase the information content of experiments. However, it is important to avoid fluorophores that interfere with in situ base calling. If only a few phenotypic labels are needed, phenotyping can be completed using fluorophores that do not overlap with in situ sequencing channels, such as DAPI, FITC, or Alexa Fluor 750. Note that a separate stain for cell segmentation is not often necessary, as the sequencing dye background uniformly stains the cytoplasm and nucleus and is used as the input for segmentation. If additional labels are needed, fluorophores that do overlap sequencing channels may be used if concentrations are titrated low enough to avoid interference with in situ sequence identification. Alternatively, destainable imaging probes enable signal removal after phenotype imaging17–20 and some live-cell stains are removed during permeabilization (e.g., CellMask Plasma Membrane stain, Thermo Fisher Scientific, cat. no. C10046) to re-establish a low background for SBS imaging.
Optical pooled screens with live-cell phenotype imaging can directly probe dynamics in a way that is not possible with other pooled screening modalities, enabling measurement of event duration, frequency, amplitude, and population synchrony. A key challenge when designing live-cell optical pooled screens is to sufficiently sample dynamics, which generally involves tradeoffs between sampling frequency, total number of cells imaged, and image quality. We recommend first determining an imaging frequency that properly samples all relevant processes and is fast enough to enable cell tracking, then identifying imaging parameters (e.g., laser power and exposure time) that provide adequate image quality with acceptable photodamage, and finally running a test acquisition to estimate how many cells can be imaged under these conditions. The resulting total number of cells should be allocated amongst genetic perturbations in the pooled experiment such that each perturbation is represented sufficiently for observing expected effect sizes (see Experimental design).
When designing a large-scale optical pooled screen, there is a tradeoff to consider between the number of perturbations to include and the number of cells measured per perturbation for a given scale of total cell population size. Completing a pilot screen enables estimating the number of cells per perturbation necessary to distinguish positive and negative control populations, as well as the associated imaging and hands-on time (spreadsheet calculator provided in Supplementary Table 1). When designing the perturbation library itself, separate subpools can be combined in one oligo array synthesis order to enable efficient screening of variable library scales and gene sets.
Following the completion of the included procedure, the proposed functions of candidate hit genes within the biological process of interest should be validated. For cases in which low cell counts per perturbation were achieved in an initial optical pooled screen, it may be useful to validate the screen phenotype in an arrayed format. Alternatively, top-scoring genes can be analyzed in a “secondary” pooled screen with a larger number of cells and perturbations per gene to identify candidate genes for further study. In addition to phenotype confirmation, orthogonal approaches to those in the screen should be used to characterize gene function. Techniques will vary widely for different applications but may include different perturbation modalities, different phenotypic assays, or characterization of functional interactions with known biological pathway components.
MATERIALS
BIOLOGICAL MATERIALS
HEK293FT cell line (https://scicrunch.org/resolver/CVCL_6911; Thermo Fisher Scientific, cat. no. R70007)
HeLa cell line (https://scicrunch.org/resolver/CVCL_0030; ATCC, cat. no. CCL-2) ! CAUTION Cell lines should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
REAGENTS
Custom sgRNA library cloning
Custom oligonucleotide library, sequences designed in Steps 1–4 (Agilent, cat. no. G7222A or similar)
- sgRNA lentiviral delivery vector such as:
- CROPseq-Guide-Puro (Addgene, cat. no. 86708)
- CROPseq-Guide-Zeo (Addgene, cat. no. 127173)
- LentiGuide-BC-EF1a-Puro (Addgene, cat. no. 127170)
PCR primer pairs to amplify oligo libraries for cloning (Integrated DNA Technologies, see Supplementary Table 2 for oligo sequences)
KAPA HiFi HotStart ReadyMix (Roche, cat. no. KK2602)
FastDigest Esp3I (Thermo Fisher Scientific, cat. no. FD0454)
2% E-gel (Thermo Fisher Scientific, cat. no. G401002)
100 bp DNA Ladder (New England Biolabs, cat. no. N3231S)
1 kb DNA Ladder (New England Biolabs, cat. no. N3232S)
QIAquick PCR purification kit (Qiagen, cat. no. 28104)
Zymoclean Gel DNA recovery kit (Zymo Research, cat. no. D4008)
Rapid Ligase buffer (Enzymatics/Qiagen Beverly, cat. no. B1010)
BSA, molecular biology grade (New England Biolabs, cat. no. B9000S)
UltraPure DNase/RNase-Free Distilled Water (Thermo Fisher Scientific, cat. no. 10977023)
T7 ligase (Enzymatics/Qiagen Beverly, cat. no. L6020L)
Agencourt Ampure XP SPRI beads (Beckman-Coulter, cat. no. A63881)
100% ethanol (VWR, cat. no. 89125-172)
Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, cat. no. Q32851)
sgRNA plasmid amplification
Endura electrocompetent cells (Lucigen, cat. no. 60242-2)
Recovery Media (Lucigen, cat. no. 80026-1)
LB media (Thermo Fisher Scientific, cat. no. 10855001)
Ampicillin, 100 mg/ml, sterile filtered (Sigma-Aldrich, cat. no. A5354)
LB Agar Plates, Carbenicillin-100 (Teknova, cat. no. L1010)
Plasmid Plus midi prep kit (Qiagen, cat. no. 12943)
Next-generation sequencing
- NGS validation primers for CROPseq-puro vector (Integrated DNA technologies)
- NGS_CROPseq-puro_P5: ACACGACGCTCTTCCGATCTtcttgtggaaaggacgaaac
- NGS_CROPseq-puro_P7: CTGGAGTTCAGACGTGTGCTCTTCCGATCTaagcaccgactcggtgccac
- TruSeq indexing PCR primers, NNNNNNNN = index sequence (Integrated DNA technologies)
- NGS_TruSeq_P5 (Forward): AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCT
- NGS_TruSeq_P7 (Reverse): CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTC
- See Supplementary Table 3 for full oligo sequences with indices.
Jumpstart ReadyMix (Sigma-Aldrich, cat no. P2893-400RXN)
MiniSeq High Output Reagent Kit, 150 cycle (Illumina, cat. no. FC-420-1002)
PhiX Control Kit v3 (Illumina, cat. no. FC-110-3001)
Sodium hydroxide solution, 10 N (Sigma-Aldrich, cat. no. 72068-100ML) ! CAUTION Concentrated sodium hydroxide is corrosive.
Tris, pH 7.0 (Thermo Fisher Scientific, cat. no. AM9850G)
Mammalian cell culture
DMEM, high glucose, GlutaMAX supplement, pyruvate (Thermo Fisher Scientific, cat. no. 10569010)
Penicillin–streptomycin, 100X (Thermo Fisher Scientific, cat. no. 15140122)
FBS, premium grade (VWR, cat. no. 97068-085)
TrypLE Express, no phenol red (Thermo Fisher Scientific, cat. no. 12604021)
Dulbecco’s PBS (DPBS; Thermo Fisher Scientific, cat. no. 14190250)
Cas9 cell line creation and validation
Blasticidin S HCl, 10 mg/mL (Thermo Fisher Scientific, cat. no. A1113903)
lentiCas9-Blast (Addgene, cat. no. 52962)
pXPR_011 (Addgene, cat. no. 59702)
Lentivirus production and titer
Opti-MEM I reduced serum medium (Thermo Fisher Scientific, cat. no. 31985062)
pMD2.G (Addgene, cat. no. 12259)
psPAX2 (Addgene, cat. no. 12260)
Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, cat. no. L3000015)
Polybrene (hexadimethrine bromide; Sigma-Aldrich, cat. no. 107689-10G)
Puromycin dihydrochloride (Thermo Fisher Scientific, cat. no. A1113803)
Lentiviral transduction
Calcium chloride solution (Sigma-Aldrich, cat. no. 21115-1ML)
MgCl2, 1 M (Thermo Fisher Scientific, cat. no. AM9530G)
UltraPure 0.5M EDTA, pH 8.0 (Thermo Fisher Scientific, cat. no. 15575020)
Triton X-100 (Sigma-Aldrich, cat. no. T9284-1L)
UltraPure 1 M Tris-HCI Buffer, pH 7.5 (Thermo Fisher Scientific, cat. no. 15567027)
Proteinase K (Sigma-Aldrich, cat. no. P2308-25MG)
Zymo DNA Clean & Concentrator kit (e.g., VWR, cat. no. 77001-152)
Live-cell Phenotyping
p65-mNeonGreen (Addgene, cat. no.127172)
DMEM, high glucose, HEPES, no phenol red (Thermo Fisher Scientific, cat. no. 21063029)
L-Glutamine, 200 mM (Thermo Fisher Scientific, cat. no. 25030081)
Hoechst 33342 Solution, 20 mM (Thermo Fisher Scientific, cat. no. 62249)
Recombinant human TNF-ɑ (Invivogen, cat. code rcyc-htnfa)
Recombinant human IL-1β (Invivogen, cat. code rcyec-hil1b)
Fixed-cell Phenotyping
Rabbit anti-p65 antibody (Cell Signaling Technology, cat. no. 8242; https://scicrunch.org/resolver/AB_10859369)
Goat anti-rabbit IgG Alexa Fluor 488 (Cell Signaling Technology, cat. no. 4412; https://scicrunch.org/resolver/AB_1904025)
BSA (VWR, cat. no. 97061-422)
DAPI (Sigma-Aldrich, cat no. D9542-10MG)
In situ amplification & sequencing
70% ethanol (VWR, cat. no. 76212-358) ! CAUTION Ethanol is highly flammable.
32% Paraformaldehyde (Electron Microscopy Sciences, cat. no. 15714) ! CAUTION Paraformaldehyde is flammable and toxic; follow institutional guidelines and handle in a fume hood.
10% Glutaraldehyde (Electron Microscopy Sciences, cat. no. 16120) ! CAUTION Glutaraldehyde is toxic; follow institutional guidelines and handle in a fume hood.
RNase-free 20X SSC buffer (Ambion, cat. no. AM9763)
RNase-free 10X PBS buffer (Ambion, cat. no. AM9625)
Glycerol (Sigma-Aldrich, cat. no. G5516)
RevertAid H minus Reverse Transcriptase (Thermo Fisher Scientific, cat. no. EP0452)
Locked nucleic acid (LNA)-modified RT primer (Qiagen, cat. no. 339413, see Supplementary Table 4 for sequences corresponding to common vectors)
Ribolock RNase inhibitor (Thermo Fisher Scientific, cat. no. EO0384)
RNase H (Enzymatics, cat. no. Y9220L)
TaqIT DNA polymerase (Enzymatics, cat. no. P7620L)
Ampligase (Lucigen, cat. no. A3210K)
5’-phosphorylated padlock probe (Integrated DNA Technologies, see Supplementary Table 4 for sequences corresponding to common vectors)
Phi29 DNA polymerase (Thermo Fisher Scientific, cat. no. EP0091) CRITICAL For alternate sources, enzymatic activity in situ should be carefully validated before substituting.
dNTP mix (New England Biolabs, cat. no. N0447L)
BSA, molecular biology grade (New England Biolabs, cat. no. B9000S)
Tween-20 Solution, 10% (VWR, cat. no. 100216-360)
Sequencing primer (Integrated DNA Technologies, see Supplementary Table 4 for sequences corresponding to common vectors)
MiSeq Reagent Nano kit v2 with PR2 buffer (Illumina, cat. no. MS-103-1003) CRITICAL The protocol and optical filter designs are optimized for the sequencing chemistry and 4-color encoding system used in the Miseq Reagent kits v2. ! CAUTION MiSeq Reagent 4 (cleavage mix) emits a strong odor; it is recommended to handle in a fume hood.
Isopropyl alcohol (e.g., VWR, cat. no. BDH1133-4LP) ! CAUTION Isopropyl alcohol is highly flammable.
EQUIPMENT
Axygen 8-Strip PCR Tubes (Fisher Scientific, cat. no. 14-222-250)
Axygen PCR plates, 96 well (VWR, cat. no. PCR-96M2-HS-C)
Eppendorf LoBind protein or genomic microcentrifuge tubes, 1.5 mL (VWR, cat. no. 80077-232)
Eppendorf LoBind protein or genomic microcentrifuge tubes, 2 mL (VWR, cat. no. 80077-226)
Falcon tubes, polypropylene, 15 mL (Corning, cat. no. 352097)
Falcon tubes, polypropylene, 50 mL (Corning, cat. no. 352070)
Filtered sterile pipette tips (e.g., Rainin)
DynaMag-96 Side Skirted plate magnet (Thermo Fisher Scientific, cat. no. 12027)
VWR Bacti Cell Spreaders (VWR, cat. no. 60828-688)
14 mL round-bottom culture tubes (e.g., VWR cat. no. 60819-761)
Electroporation cuvettes (BioRad, cat. no. 165-2093)
Gene Pulser Xcell Microbial System (Bio-Rad, cat. no. 1652662)
Nunc EasYFlask 225 cm2, filter cap, 70-mL working volume (T225 flask; Thermo Fisher Scientific, cat. no. 159934)
Stericup vacuum filtration system, 0.22 μm (Millipore, cat. no. S2GVU11RE)
0.45 μm low protein-binding syringe filters (e.g., Sigma-Aldrich, cat. no. CLS431220-50EA)
Disposable Syringes with Luer-Lok Tip (Fisher Scientific, cat. no. 14-829-45)
Falcon tissue culture plate, 6 wells (Corning, cat. no. 353224)
Falcon tissue culture plate, 12 wells (Corning, cat. no. 353043)
Glass-bottom tissue culture plate, 24 wells (VWR, cat. no. 82050-898)
Glass-bottom tissue culture plate, 6 wells (Cellvis, cat. no. P06-1.5H-N)
Cellometer SD100 Counting Chambers (Nexcelom Bioscience, cat. no. CHT4-SD100-002)
Cell counter (e.g., Cellometer Image Cytometer; Nexcelom Bioscience)
Microscope with phase contrast imaging capabilities for observing cell cultures (e.g., Nikon Eclipse Ts2)
Thermocycler with programmable temperature stepping functionality, 96 well (e.g., Applied Biosystems Veriti, cat. no. 4375786)
Flat-top thermocycler (e.g., Thermo Fisher Scientific, cat. no. 4484078)
Desktop microcentrifuges (e.g., Eppendorf, cat. nos. 5424 and 5804)
E-Gel electrophoresis device (Invitrogen, cat. no. G8100)
Blue-light transilluminator and orange filter goggles (SafeImager 2.0; Invitrogen, cat. no. G6600)
Qubit Assay Tubes (Thermo Fisher Scientific, cat. no. Q32856)
Qubit Fluorometer (Thermo Fisher Scientific, cat. no. Q33216)
MiniSeq System (Illumina, cat. no. SY-420-1001)
Benchtop centrifuge with plate carriers, e.g., Avanti J-15R (Beckman Coulter, cat. no. B99516)
Lens cleaning tissue (Thorlabs, cat. no. MC-5)
Plate sealing roller (Bio-Rad, cat. no. MSR0001)
Aluminized foil microseals (Bio-Rad, cat. no. MSF1001)
Fiji58, a distribution of ImageJ, available at https://imagej.net/Fiji
Micro-Manager59, available at https://micro-manager.org, or other microscope control software
Epifluorescence microscope with a motorized stage and the recommended excitation & emission wavelength capabilities: ~405 nm excitation, 410–480 nm emission (DAPI); ~480 nm excitation, 500–540 nm emission (GFP, FITC); ~540 nm excitation, 558–586 nm emission (Cy3, MiSeq G); ~575 nm excitation, 603–627 nm emission (Alexa Fluor 594, MiSeq T); ~635 nm excitation, 659–701 nm emission (Cy5, MiSeq A); ~660 nm excitation, 698–766 nm emission (Cy7, MiSeq C); ~750 nm excitation, 765–875 nm emission (Alexa Fluor 750, optional for extending phenotype imaging channels). Recommended objective lenses are plan apochromatic 10X air NA 0.45 (e.g., Nikon, cat. no. MRD00105) and 20X air NA 0.75 (e.g., Nikon, cat. No. MRD00205). Recommended microscope components and examples of compatible systems are in Tables 1 and 2; see Box 2 for further discussion of microscope system design and validation.
Table 1 |.
Recommended microscope components
| Component | Recommended Options | Description |
|---|---|---|
| Microscope body | Nikon Ti2 | Large field-of-view and fast stage reduce acquisition time; note that custom sizing of filters may be required |
| Control software | Micro-Manager | Micro-Manager is open source and freely available; however, NIS Elements allows more flexibility, such as easier definition of imaging sites, improved alignment and autofocusing options, and saving of imaging workflows using the JOBS module |
| NIS Elements with JOBS module | ||
| Camera | Iris 9 (Teledyne Photometrics); Sensor: 12.58 × 12.58 mm; 2960 × 2960 pixels |
The goal is to have a sensor area large enough to take advantage of the full field-of-view of the microscope. For cameras with smaller sensor areas, a 0.7X relay lens coupler can be used (e.g., Nikon, cat. no. MQD42075), although this decreases resolution and may result in vignetting. |
| ORCA-Flash4.0 v3 (Hamamatsu Photonics); Sensor: 13.31 × 13.31 mm; 2048 × 2048 pixels | ||
| ORCA-Fusion (Hamamatsu Photonics); Sensor: 14.98 × 14.98 mm; 2304 × 2304 pixels | ||
| Objective Lenses | 10X air NA 0.45 (e.g., Nikon, cat. no. MRD00105) | For SBS and some phenotype image acquisition |
| 20X air NA 0.75 (e.g., Nikon, cat. No. MRD00205) | Optional for phenotype image acquisition |
Table 2 |.
Examples of compatible microscope excitation and emission configurations
| Example system with a broadband light source | Example system with a multispectral laser light source, external filter wheel, and triggered acquisition | Notes | |
|---|---|---|---|
| Approximate acquisition time for one field-of-view of imaging with DAPI and SBS channels | 5300 ms | 610 ms | |
| Light source | Sola SE FISH 2 light engine (Lumencor) | CELESTA or CELESTA quattro light engine (Lumencor) | The Sola is more economical but has reduced power, requiring longer exposures especially for base C. CELESTA is available with different laser line configurations; only 405 nm, 545 nm, and 635 nm are strictly needed for sequencing. |
| External filter wheel | N/A | HS-625 (Finger Lakes Instrumentation) or Lambda 10-B (Sutter Instrument) | Enables fast emission filter switching |
| Components for external hardware triggering | N/A | NIS-Elements Hardware Triggering module, compatible NI-DAQ card |
Reduces communication overhead in synchronizing components |
| Multiband dichroic mirror | N/A | ZT408/473/545/635/750rpc (Chroma) or Di01-R488/543/635 (Semrock) |
When using an external emission filter wheel, a multiband dichroic mirror is required in the microscope body filter turret. The first option works for all suggested SBS and phenotype channels but is expensive. The second option is more economical but does not work for DAPI or near-infrared channels. |
| DAPI filter set | LED-DAPI-A filter set (Semrock) | 405 nm laser line excitation | |
| ZET408/473/545/635/750m (Chroma) or any compatible single-band emission filter | |||
| GFP filter set | GFP-1828A filter set (Semrock) | 473 nm laser line excitation | For phenotype imaging |
| Same multiband filter as for DAPI or any compatible single-band emission filter | |||
| MiSeq G filter set | FF01–534/20 (Semrock) | 545 nm laser line excitation | |
| FF552-Di02 (Semrock) | |||
| FF01–572/28 (Semrock) | FF01–565/24 (Semrock) | ||
| MiSeq T filter set | FF03–575/25 (Semrock) | 545 nm laser line excitation | |
| FF596-Di01 (Semrock) | |||
| FF01–615/24 (Semrock) | FF01–624/40 (Semrock) | ||
| MiSeq A filter set | FF01–635/18 (Semrock) | 635 nm laser line excitation | |
| FF652-Di01 (Semrock) | |||
| FF01–680/42 (Semrock) | FF01–676/29 (Semrock) | ||
| MiSeq C filter set | FF01–661/20 (Semrock) | 635 nm laser line excitation | |
| FF695-Di01 (Semrock) | |||
| FF01–732/68 (Semrock) | FF01–732/68 (Semrock) | ||
| near-IR filter set | N/A | 750 nm laser line excitation | Optional for extending phenotype fluorophore options (e.g., Alexa Fluor 750). |
| FF765-Di01 (Semrock) | |||
| ET820/110 (Chroma custom) |
REAGENT SETUP
80% (vol/vol) ethanol
For SPRI cleanup washes, prepare 80% ethanol immediately before use by diluting 100% ethanol in UltraPure water.
D10 medium
For culturing HEK 293FT and HeLa cells, prepare D10 medium by supplementing DMEM with GlutaMAX, 10% (vol/vol) FBS, and 1X penicillin–streptomycin. Filter the medium through a 0.22 μm filter and store at 4°C for up to 1 month.
Live-cell imaging medium
For live-cell phenotyping, use DMEM containing high glucose, L-glutamine, and HEPES but without phenol red. Combine with 10% (vol/vol) FBS and 1X penicillin–streptomycin. Filter the medium through a 0.22 μm filter and store at 4°C for up to 1 month.
gDNA extraction solution
As described in ref.60, combine 1 mM CaCl2, 3 mM MgCl2, 1 mM EDTA, 1% (wt/vol) Triton X-100, 10 mM Tris buffer (pH 7.5), and 0.2 mg/mL Proteinase K for gDNA extraction from cells. Aliquot the solution and store at −20°C for up to 1 year.
PBS, 1X
Dilute RNase-free 10X PBS in UltraPure water. Store at room temperature (18–22°C) for up to 1 year.
PBS-T
Prepare 1X PBS with 0.05% (vol/vol) Tween-20 in UltraPure water. Store at room temperature for up to 1 year.
SSC, 2X
Dilute RNase-free 20X SSC in UltraPure water. Store at room temperature for up to 1 year.
DAPI, 200 ng/mL
To prepare a stock solution of 20 mg/mL DAPI, resuspend 10 mg of DAPI in 500 μL of 2X SSC. Dilute the stock solution in 2X SSC to make a 200 ng/mL DAPI working solution for staining. Store the stock solution at 4°C in the dark for up to 1 year, and the working solution at room temperature for up to 2 months.
Fixation solution
Prepare 4% (vol/vol) paraformaldehyde (PFA) in 1X PBS immediately before use.
Post-fixation solution
Prepare 3% (vol/vol) PFA and 0.1% (vol/vol) glutaraldehyde in 1X PBS immediately before use.
Immunofluorescence blocking buffer
Dissolve 3% (wt/vol) BSA in 1X PBS. Once dissolved, filter through a 0.22 μm filter, and store at 4°C for up to 1 year.
50% (vol/vol) glycerol
Dilute glycerol in UltraPure water. Store at room temperature for up to 1 year.
EQUIPMENT SETUP
Python package installation
Download the Python package and follow installation instructions from https://github.com/feldman4/OpticalPooledScreens61. Briefly, using a terminal, change the current working directory to the downloaded folder and initialize a Python virtual environment:
$ python3 -m venv venv
Then, install the necessary dependencies:
$ sh install.sh
PROCEDURE
Designing a custom sgRNA library Timing 2–5 weeks; 2 d hands-on
-
1Prepare the following input files for designing an sgRNA library and corresponding oligo pool order; examples of each file are provided in the GitHub repository. Note that an oligo pool order can contain multiple subpools, each targeting different sets of genes. Each subpool can be independently retrieved from the full oligo pool using dialout PCR, which specifically amplifies oligos of interest using orthogonal primer pairs complementary to designed flanking sequences62.
- An input sgRNA table, consisting of a list of gene IDs and corresponding sgRNA sequences to select from. This table is often a compilation of publicly available CRISPR sgRNA libraries.
- A gene list, with one gene ID per row in a text file. There should be one gene list file for each subpool design. For non-targeting sgRNAs, a null gene ID of −1 is used. Non-targeting sgRNAs can be included in the same subpool as targeting sgRNAs, or designed as a separate subpool to enable modular combination with other subpools. In either case, cells carrying non-targeting sgRNAs should make up 5% or more of the final pool of cells.
- A pool design spreadsheet, with one row for each subpool. Several parameters are specified for each gene set:
pool name of the oligo pool dialout dialout PCR primer set, corresponds to the primer sequences in the kosuri_dialout_primers.csv table and Supplementary Table 2. These primer sequences are derived from Kosuri, et al.62 design name of the subpool gene set, corresponds to the gene list text file name group sgRNAs from subpools within the same group will be designed to have unique 5’ sgRNA sequence prefixes to enable the option of experimentally combining these subpools prefix_length the desired minimum read length for 5’-to-3’ in situ sequencing to distinguish all library elements with a given minimum edit distance between prefixes. Usually a prefix length of 12 is long enough for large libraries up to ~80,000 sgRNAs. Smaller libraries can often use a much shorter prefix, reducing the number of necessary in situ sequencing cycles for demultiplexing the sgRNA identities. edit_distance the minimum Levenshtein edit distance between all pairs of prefixes. A minimum Levenshtein distance of 2 is generally recommended to enable detection of single base insertion, deletion, or substitution errors. num_genes total number of genes in the subpool, matching the number of gene IDs in the corresponding gene list sgRNAs_per_gene number of desired targeting sgRNAs per gene duplicate_oligos for oligo array synthesis it may be advantageous to synthesize multiple spots with the same oligo sequence to achieve a narrower distribution of oligo representation and/or to match the supplier’s synthesis scale
-
2
Generate a custom sgRNA library design by following the Jupyter notebook in the GitHub repository, “example_notebooks/example_pool_design.ipynb.” This notebook provides detailed instructions on guide selection and library design.
-
3
Validate that the desired number of sgRNAs per gene are present in the final design table, and that the majority of the selected sgRNAs are highly ranked in the source libraries. If this is not the case, increase the prefix_length parameter in the pool design spreadsheet from Step 1 for the deficient subpools and repeat the sgRNA selection steps as necessary. Ensure that non-targeting control sgRNAs are present in each subpool, or a separate subpool of non-targeting sgRNAs is designed with barcode prefixes that are compatible with each targeting sgRNA subpool.
-
4
Order the final oligo pool from a DNA synthesis vendor, such as Agilent, Twist Bioscience, or Genscript. Synthesis and delivery may take between 1–5 weeks.
Cloning a custom sgRNA library Timing: 1 d
-
5PCR amplification of pooled oligo library. Throughout the sgRNA library cloning protocol, refer to the table below for the recommended number of reactions at each step for a genome-scale library of 60,000 sgRNAs, and scale the number of reactions according to the size of the custom library.
Steps Cloning process Number of reactions 6–9 PCR amplification of pooled oligo library 2 10–13 Restriction digest of plasmid backbone 6 14–15 Golden Gate assembly 20 -
6Amplify the library from the oligo pool using the forward and reverse dialout primers corresponding to the subpool designs from Steps 1–4 (see Supplementary Table 2 for primer sequences). Prepare a reaction mix using the ratios below:
Component Amount per reaction (μL) Final concentration Oligo pool from Step 4 1 0.2 ng/μL Forward dialout primer, 100 uM 0.15 0.3 μM Reverse dialout primer, 100 uM 0.15 0.3 μM KAPA HiFi HotStart ReadyMix, 2X 25 1X UltraPure water 23.7 Total 50
^ CRITICAL STEP: To minimize PCR amplification errors, use a high-fidelity polymerase, such as KAPA HiFi (KAPA Biosystems), Q5 (New England Biolabs), or Herculase II Fusion (Agilent).
-
7Divide the master mix into 50 μL reactions and run the PCRs using the following cycling program:
Cycle number Denature Anneal Extend 1 95°C, 3 min 2–21 98°C, 20 sec 65°C, 15 sec 72°C, 15 sec 22 72°C, 1 min
^ CRITICAL STEP To reduce amplification biases, limit the number of PCR cycles to 18–20.
-
8
Pool the PCR reactions that used the same dialout primer set and purify the product using the QIAquick PCR Purification Kit according to the manufacturer’s directions.
-
9
Run an aliquot of the purified PCR product on a 2% E-Gel EX Agarose Gel for 10 min along with a 100 bp ladder to confirm there is a single band at about 200 bp.
-
10Restriction digest of plasmid backbone. Digest the library plasmid backbone (sgRNA delivery vector) with FastDigest Esp3I (BsmBI). Set up the master mixes as below:
Component Amount per reaction (μL) Final concentration Library plasmid backbone 1 50 ng/μL FastDigest Esp3I, 1 unit/μL 1 0.05 units/μL FastDigest buffer, 10X 2 1X UltraPure water 16 Total 20 -
11
Divide the master mix into 20-μL reactions and incubate at 37°C for 15 min.
-
12
Pool the restriction digest reactions and run the entire volume on a 2% E-Gel EX agarose gel for 10 min along with a 1 kb ladder. For CROPseq-Guide-Puro, the band for the excised 1,885-bp filler sequence should be visible.
-
13
Gel extract the plasmid backbone (8,329 bp for CROPseq-Guide-Puro) using the Zymoclean Gel DNA Recovery Kit according to the manufacturer’s protocol. Quantify the product using a Qubit dsDNA HS kit, aiming for a yield of 500 ng or more of digested plasmid per reaction.
-
14Golden Gate assembly. Set up a master mix for the Golden Gate reactions on ice, according to the ratios below.
Component Amount per reaction (μL) Final concentration Rapid Ligase Buffer, 2X 25 1X BSA, 20 mg/mL 0.25 0.1 mg/mL FastDigest Esp3I, 1 unit/μL 1 0.02 units/μL T7 Ligase, 3 × 106 units/mL 0.25 15 × 103 units/mL sgRNA library insert from Step 8 15 ng 0.3 ng/μL Digested library plasmid backbone from Step 13 100 ng 2 ng/μL UltraPure water up to 50 μL Total 50 -
15Divide the master mix into 50-μL reactions and run the following cycling program:
Cycle number Digest Ligate 1–15 37°C, 5 min 20°C, 5 min -
16
SPRI cleanup. Pool cloning reactions. Purify and concentrate the sgRNA library using Agencourt AMPure XP DNA SPRI beads. The following is the standard SPRI cleanup protocol, but we elute in a small fixed volume regardless of input volume.
^ CRITICAL STEP The following cleanup steps remove components from the Golden Gate reaction that can interfere with electroporation.
-
17
Vortex SPRI beads thoroughly. Mix equal volumes of the Golden Gate reaction from Step 15 and SPRI beads. Distribute 100 μL aliquots into wells of a 96-well plate and incubate at room temperature for 5 min.
-
18
Put the plate on a magnetic plate rack and allow beads to settle. Aspirate the supernatant.
-
19
Remove the plate from the magnet and fully resuspend each well in 100 μL 80% ethanol. Place the plate back onto the rack to allow beads to settle.
-
20
Repeat 80% ethanol washes three more times. Pool SPRI reactions during these washes until beads are combined in one well for each plasmid library pool.
-
21
After the last wash, place the plate on the rack, allow the beads to settle and carefully aspirate all supernatant (using a 10 μL pipette to remove residual ethanol).
-
22
Allow the plate to dry completely at room temperature for 5–10 min.
-
23
Remove the plate from the magnet and elute sgRNA library by resuspending in 10 μL water.
-
24
Place the plate back on the magnetic rack. Aspirate and move supernatant (containing plasmid DNA) to another clean well. Allow residual beads to settle on the magnetic rack.
-
25
Aspirate and collect supernatant from each library pool to a separate Eppendorf tube (each library was pooled into a single well in Step 20). Quantify the plasmid DNA using a Qubit dsDNA HS kit; a minimum of 50 ng of product is recommended for electroporation in the following steps.
^ CRITICAL STEP Ensure that no SPRI beads are present in the final sgRNA library plasmid product. Residual SPRI beads will interfere with electroporation.
Amplification of pooled sgRNA library Timing 2 d
-
26
Pooled sgRNA library transformation. Thaw Lucigen recovery media at room temperature and Lucigen Endura electrocompetent cells on ice for 10 minutes. Pre-chill Eppendorf tubes and electroporation cuvettes on ice. Pre-warm 2 standard LB agar plates (100-mm Petri dish, ampicillin) for calculating electroporation efficiency at 37 °C.
-
27
For higher-complexity (e.g. genome-scale) libraries, perform electroporation using 50 μL of electrocompetent cells. For smaller libraries, aliquot cells into Eppendorf tubes (25 μl per tube).
-
28
Add up to 500 ng of DNA in up to 10 μl of water per 25 μL of cells, mixing with a pipette tip.
^ CRITICAL STEP Be careful not to introduce any bubbles when working with electrocompetent cells (can lead to electrical arcing during the electroporation process).
-
29
Transfer cells and DNA to an electroporation cuvette and incubate on ice for 5 min.
-
30Electroporate using the manufacturer’s recommended settings:
- Voltage: 1800 V
- Capacitance: 10 μF
- Resistance: 600 Ω
- Cuvette: 1 mm
-
31
Immediately following electroporation, add 2 mL of recovery media and transfer cells to a 14 mL culture tube.
-
32
Allow the bacterial culture to recover in an incubated shaker (37°C, 225 rpm) for 1 h.
-
33
Move the culture to a 250 mL Erlenmeyer flask with 50 mL LB media containing 100 μg/mL ampicillin. Mix well.
-
34
Prepare 1/105 and 1/106 dilutions of the culture for calculating transformation efficiency. Add 5 μL from the 50 mL culture to 995 μL of LB, and plate 100 μL (10−5 dilution) and 10 μL (10−6 dilution) on a prewarmed LB-ampicillin plate.
-
35
Incubate plates at 37°C overnight.
-
36
Incubate the liquid culture in a shaker (37°C, 225 rpm) overnight (16 hours).
-
37
Calculate electroporation efficiency. Count colonies on the LB-Amp plates and multiply by the dilution factor to estimate the total number of colonies. Aim to have 300–1000x as many colonies as library elements to maintain full library representation.
? TROUBLESHOOTING
-
38
Use a plasmid purification kit (e.g. Qiagen Plasmid Plus Midi Kit) to isolate the sgRNA library DNA and quantify using a Qubit dsDNA HS kit, commonly yielding 200 μg or more of plasmid.
PAUSE POINT Purified plasmid libraries can be stored at −20°C for at least 1 year.
Next-generation sequencing of the amplified sgRNA library Timing 2–3 d
-
39Library PCR for NGS (PCR1). For libraries in the CROPseq-puro backbone, use the provided P5 and P7 NGS primer sequences to amplify the sgRNA locus. Set up the PCR reaction as follows:
Component Amount per reaction (μL) Final concentration JumpStart, 2X 25 1X NGS_CROPseq-puro_P5 primer, 10 μM 0.75 0.15 μM NGS_CROPseq-puro_P7 primer, 10 μM 0.75 0.15 μM Pooled sgRNA library plasmid from Step 38 100 ng 2 ng/μL UltraPure water up to 50 μL Total 50 Run the following thermocycling program:Cycle number Denature Anneal Extend 1 95°C, 5 min 2–19 95°C, 20 sec 55°C, 30 sec 72°C, 30 sec 20 72°C, 4 min To minimize amplification errors, high-fidelity enzymes, such as Kapa HiFi or Q5 may be used; error rates will be lower than for Taq (JumpStart).
-
40Indexing PCR for NGS (PCR2). Append sample-specific indices in a second PCR using TruSeq indexing primers. It is helpful to validate the sequences of both the plasmid products and the dialout PCR, indexing each uniquely. Set up the reaction as follows:
Component Amount per reaction (μL) Final concentration JumpStart, 2X 25 1X NGS_TruSeq_P5 index primer, 5 μM 1.25 0.125 μM NGS_TruSeq_P7 index primer, 5 μM 1.25 0.125 μM PCR1 product from Step 39 OR dialout PCR from Step 8 1 UltraPure water up to 50 μL Total 50 Run the following thermocycling program:Cycle number Denature Anneal Extend 1 95°C, 5 min 2–15 95°C, 20 sec 55°C, 30 sec 72°C, 30 sec 16 72°C, 4 min -
41
Pool the indexed PCR2 reactions and run the product on a 2% E-Gel EX agarose gel for 10 min. The expected amplicon length is about 250 bp.
-
42
Gel extract the PCR samples using the Zymoclean Gel DNA Recovery Kit according to the manufacturer’s protocol.
-
43
Quantify the purified product using the Qubit dsDNA HS Assay Kit according to the manufacturer’s instructions; approximately 20 ng of product is needed for sequencing.
-
44
Sequence the samples on the Illumina MiniSeq or similar, following the Illumina user manual. Acquire 8 cycles each of index reads 1 and 2, along with a minimum of 60 cycles of read 1. We recommend aiming for a coverage of 300–1000 reads per sgRNA in the library; adding in a 5–10% PhiX control is recommended to improve library diversity and sequencing quality.
-
45NGS analysis. Extract sgRNA sequences from FASTQ files and build a histogram of counts for each sgRNA using the bash scripts provided in the GitHub repository. In a terminal, change the working directory to the folder of demultiplexed FASTQ files and run:
$ bash <path/to/OpticalPooledScreens>/scripts/fastq2hist.sh
Calculate a skew ratio (here defined as the ratio between the number of NGS reads assigned to the 90th and 10th percentile sgRNAs) to assess the uniformity of sgRNA representation in the plasmid pool. Compare the sequenced sgRNAs to the expected list of sgRNAs to determine a mapping rate and dropout rate. These steps may be completed by following the “example_notebooks/example_ngs_analysis.ipynb” Jupyter notebook in the provided GitHub repository. Recommended targets for library quality are a skew ratio less than 10 and dropout of less than 1% of sgRNA sequences.
? TROUBLESHOOTING
Validation of in situ sequencing in cell line of interest Timing 1 week
-
46
When performing in situ sequencing in a new cell line, barcode detection should be validated before attempting a pilot or full-scale screen. Transduce a small pool of 5 sgRNAs into the cell line of interest as well as a previously-validated cell line (for in situ detection only, not necessary to engineer cells to also express Cas9; see Steps 54–71 for lentivirus production and transduction). Perform 4 cycles of in situ sequencing starting at Step 85 and determine the percentage of reads that correspond to sgRNAs in the designed pool for both the new and validated cell line. A successful validation will achieve 70–80% of reads or higher matching a designed sequence (compared to the expectation of (number of barcodes)/(4^(number of cycles)), or 2% by chance). Similarly, 80% of cells or more should have at least one identified in situ sequencing read to enable efficient screening. To estimate the in situ phenotype-to-genotype mapping accuracy of a cell line, a frameshift reporter system can be used as previously described43. See Box 1 for guidelines on optimizing the performance of a new cell line and Supplementary Figure 1 for example validation data.
Optional: Reporter cell line creation and validation Timing 1–2 weeks
-
47
If using a reporter cell line for screening (e.g. p65-mNeonGreen), prepare reporter virus and transduce as described in Steps 54–71.
-
48
Use FACS to select cells expressing the reporter. First, run a sample of the parental cell line lacking expression of the reporter fluorescent protein as a negative control. Then sort the reporter cell line: excluding cells in the same range as the negative control or any high fluorescence outliers, select cells with the top 15–30% of fluorescent protein expression levels.
-
49
After 4–10d outgrowth, repeat FACS-based selection of cells (again select the top 15–30% excluding outliers) to select for cells with a narrower range of reporter expression.
PAUSE POINT Selected reporter cell lines can be frozen and stored long-term according to the parent cell line manufacturer’s protocol.
Cas9 cell line creation and validation Timing 2–4 weeks
-
50
Transduce the desired screening cell line with lentiCas9-Blast, following Steps 54–71 for lentivirus production and transduction.
-
51
Select with blasticidin (we typically select for 7d at 10 ug/mL in cancer cell lines but these parameters may need to be optimized for individual cell lines of interest).
-
52
Validate Cas9 activity (minimum >=85% for screening, recommend >=95%) using pXPR_011, a reporter vector expressing GFP and an sgRNA targeting GFP, as previously described43,63.
-
53
Optional: Select Cas9 clone for screening. A Cas9 clone can yield increased reproducibility and Cas9 activity for screening. Seed single Cas9 cells in 96-well plate by flow cytometry or limiting dilution, allow to grow out for 2–3 weeks passaging every 2–3 days. Assess clone activity by pXPR_011 assay.
PAUSE POINT Validated Cas9 cell lines can be frozen and stored long-term according to the parent cell line manufacturer’s protocol.
Lentivirus production and titer Timing 1 week
! CAUTION This protocol generates replication-incompetent lentivirus, which should be handled carefully to avoid exposure. Contact your biosafety office about institutional guidelines and any required training for working with lentivirus.
-
54
Preparation of cells for transfection. Seed HEK 293FT cells in 6-well dishes at a density of 106 cells/well in a total volume of 2 mL/well of D10 medium, aiming for 80% confluency after 16–20 hr.
^ CRITICAL STEP Overconfluent cells will result in a reduced transfection efficiency. Lentivirus production can alternatively be completed in 15 cm tissue culture dishes to accommodate virus volumes necessary for large transductions; scale up listed volumes 10-fold for lentivirus production.
-
55Lentivirus plasmid transfection. The following day, once the cells reach an optimal confluency of 80–90%, transfect HEK 293FT cells according to the following protocol, using 2:3:4 mass ratio of pMD2.G:psPAX2:sgRNA library (transfer plasmid). For each sgRNA library, prepare a plasmid mix as outlined below and vortex to mix.
Component Amount per well of 6-well plate Opti-MEM 166 μL pMD2.G (lentiviral helper plasmid) 704 ng psPAX2 (lentiviral helper plasmid) 1056 ng sgRNA plasmid library from Step 38 1408 ng P3000 reagent 12 μL -
56Prepare the Lipofectamine reagent mix as follows and invert to mix:
Component Amount per well of 6-well plate Opti-MEM 166 μL Lipofectamine 3000 12 μL -
57
Add the plasmid mix to the Lipofectamine mix, gently pipette, and incubate at room temperature for 10 min.
-
58
Pipette 400 μL of the transfection mix from Step 57 to each well of the 6-well plate from Step 54. Gently swirl to mix and return to the incubator.
-
59
After 4 h, replace with 2 mL of prewarmed D10 medium.
-
60
Harvest lentivirus. 36–48 h after the transfection, harvest the lentivirus by collecting the supernatant and filtering through a 0.45 μm low-protein binding syringe filter to remove cellular debris.
-
61
Optional: Purify the lentivirus using ultracentrifugation or PEG-based precipitation64; this step is critical for cell types that respond adversely to FBS.
-
62
Aliquot the filtered virus and store at −80°C. At least two aliquots are required: one for titering and another for a large-scale transduction.
PAUSE POINT Filtered lentivirus supernatant can be stored at −80°C for up to 1 year. Avoid freeze-thaw cycles, as this can significantly affect viral titer.
-
63
Lentivirus transduction and titering by spinfection. Determine lentiviral titer using the screening cell line in the following steps. First, thaw an aliquot of lentivirus at room temperature.
-
64
Trypsinize and count Cas9-expressing cells of interest.
-
65
Seed cells into 4 wells of a 12-well plate in 1 mL/well of D10 medium, with 8 μg/mL polybrene. Aim for 90–95% confluency immediately after spinfection. For HeLa cells, typically this corresponds to 106 cells/mL.
^ CRITICAL STEP Optimal infection conditions (cell density, polybrene concentration, +/− spinfection) may depend on the cell type used.
-
66
Add 0 μL, 1μL, 5 μL, and 25μL of lentivirus to separate wells and mix thoroughly by pipetting.
-
67
Spinfect by centrifuging plates at 1,000g for 2 h at 33°C. After the spinfection, move cells to 37°C, 5% CO2 incubator.
-
68
After 3 h, replace with 1 mL of prewarmed D10 medium.
-
69
24–48 h after spinfection, trypsinize and move 1/4 and 1/40 of cells from each condition to separate wells of a new 12-well plate in D10 medium. Add the appropriate antibiotic (e.g. puromycin for CROPseq-puro) to the higher density well from each condition. It is recommended to optimize the antibiotic concentration by performing a kill curve as described in Steps 40–41 of the Procedure in ref.3 and determine the lowest concentration that kills all uninfected cells in 2–7 days.
-
70
Two days after adding the antibiotic, check for complete killing of cells in untransduced control wells and count cells in the +/− antibiotic conditions.
^ CRITICAL STEP For antibiotics other than puromycin, more than 2 days may be necessary to kill all untransduced cells. This should be determined by performing a kill curve prior to transduction.
-
71For each condition, calculate:
- Multiplicity of infection (MOI) = 0.1*(cell count from higher seeding density well with antibiotic)/(cell count from lower seeding density well without antibiotic)
- Viral titer = total colonies / virus volume
? TROUBLESHOOTING
Lentiviral transduction Timing 1 week
-
72
Library transduction. The following steps describe how to scale up the transduction linearly (to preserve the cell number to virus volume ratio) to achieve a suitable number of colonies (aiming for 300–1000x the library complexity). Based on Step 71, select the virus volume closest to 5–10% MOI and use the viral titer calculated for that volume in the following step.
^ CRITICAL STEP It is important to maintain cell lines with sufficient cellular representation for a given library. As a general guideline, aim to keep >300 cells per library element at all times to avoid bottlenecking. In addition, minimize the number of passages from the time of infection to screening to minimize the effects of positive or negative selection. Clone-specific effects of CRISPR perturbations are not uncommon; performing screen replicates starting from lentiviral transduction is preferred.
-
73Perform the library transduction, repeating Steps 63–68, this time using a volume of virus and total number of input cells yielding an MOI of 0.05–0.1 and number of transduced colonies equal to 300–1000x the library complexity:
- Virus volume = (target number of transduced colonies)/(viral titer)
- Total number of input cells = (target number of transduced colonies)/(target MOI)
-
74
24 h after spinfection, move all cells in the same condition to a T225 flask with appropriate antibiotic.
-
75
Two days after adding the antibiotic, count colonies in a few sample fields of view on a phase contrast microscope.
-
76
Estimate the total number of colonies in the entire T225 flask by multiplying the average colonies per field-of-view by the ratio of the flask surface area to the microscope field-of-view area. Make sure appropriate cellular representation has been achieved (300–1000x the library complexity).
? TROUBLESHOOTING
-
77
Expand the cell culture so that removing sufficient cells for NGS does not bottleneck the population diversity (always maintaining >300 cells per library element).
-
78
NGS validation of cell library representation. The following protocol proceeds directly from cell lysis to PCR without purification. This works well for all library scales but can become cumbersome once the cell numbers get large (~107 cells). Alternatively, genomic DNA can be extracted using a commercial kit (e.g. Zymo Quick-DNA Midiprep Plus Kit) and quantified before loading 2.5 μg into a 50 μL PCR (PCR1) instead of lysate in Step 82.
-
79
Trypsinize and count cells. Remove 300–1000X as many cells as the library complexity into another tube. Wash the cells twice with PBS and pellet them by centrifuging at 500g for 5 minutes at 4°C.
-
80
Resuspend cells in gDNA extraction solution at a concentration of 1e6 cells per 100 μl and aliquot into a PCR plate, 100 μl per well.
-
81Lyse cells and extract genomic DNA by running the following thermocycling program:
Cycle number Condition 1 65°C, 10 min 2 95°C, 15 min -
82Library PCR for NGS (PCR1). For libraries in the CROPseq-puro backbone, use the provided P5 and P7 NGS primers to amplify the sgRNA locus. Set up the PCR reaction as follows. For large libraries, this can be a very large number of reactions. For example, for a 60,000 perturbation library, aiming for 300-fold coverage would require 1.8e7 cells, which requires at least 1.8 mL of cell lysate or 144 × 50 μL PCR reactions.
Component Amount per reaction (μL) Final concentration JumpStart, 2X 25 1X NGS_CROPseq-puro_P5 primer, 10 μM 0.75 0.15 μM NGS_CROPseq-puro_P7 primer, 10 μM 0.75 0.15 μM Cell lysate from Step 81 12.5 ul 2 ng/μL UltraPure water up to 50 μL Total 50 Run the following thermocycling program:Cycle number Denature Anneal Extend 1 95°C, 5 min 2–29 95°C, 20 sec 55°C, 30 sec 72°C, 30 sec 30 72°C, 4 min
^ CRITICAL STEP The cell lysate can be very viscous at this step. Pipette carefully. Additionally, it is recommended to perform two replicates from PCR1 onwards in order to assess whether cell sampling is adequate.
-
83
Pool all PCR1 reactions for a given sample, mix thoroughly, and perform a column cleanup (e.g. with the Zymo DNA Clean and Concentrator Kit) to concentrate the PCR product. Quantify the concentration of purified PCR1 product using the Qubit dsDNA HS Assay kit; 100 ng of purified product is used in the following step.
-
84
Complete indexing PCR, sequencing, and NGS analysis as specified in Steps 40–45, using 100 ng of purified PCR product from Step 83 as the PCR template for Step 40. Evaluate the resulting skew ratio and sgRNA dropout rate and compare to the plasmid library sequencing results to assess whether the transduced cell library achieved adequate representation of perturbations (skew ratio < 10, dropout of <1% of sgRNA sequences).
? TROUBLESHOOTING
Image-Based Phenotyping Timing 2–3 d
^ CRITICAL Image-based screens can be performed with live-cell or fixed-cell phenotyping. Presented below are examples of both live-cell (Step 88) and fixed-cell (Step 99) phenotyping protocols for a screen measuring p65 translocation in HeLa cells. For fixed-cell p65 screens, imaging and antibody staining was performed after the post-fixation step (as specified in Step 99). However, phenotype staining and imaging may be performed at other steps, including phenotyping after RCA (after Step 104). See Box 3 for further discussion of phenotyping considerations and guidelines.
-
85
Optional: some cells may require culture plate pretreatment for adherent cell growth.
-
86
Seed the cell library from Step 77 in glass-bottom multi-well plates 48 hours prior to fixation or live-cell imaging, aiming for the maximum confluency compatible with the phenotyping assay (often 80–90%) at 48 hours post seeding. Typically 7e4 HeLa cells/well in 0.5 mL D10 media are plated in 24-well plates, or 4e5 HeLa cells/well in 2 mL D10 media in 6-well plates. Adjust numbers based on cell size and doubling time.
^ CRITICAL STEP A protocol video available at https://youtu.be/TEqMbMjS1tA includes a detailed demonstration of Steps 86–128.
? TROUBLESHOOTING
-
87
Optional: prior to fixation or live-cell imaging, perform stimulus if applicable for the screen of interest. For p65 translocation, TNF or IL1b was added to a final concentration of 30 ng/mL 40 minutes prior to fixation or immediately before live-cell imaging.
-
88
Optional: live cell phenotyping. Stain cells stably expressing a p65-mNeonGreen reporter with 0.1 μg/mL Hoechst 33342 in live cell imaging medium in a cell culture incubator for 2 hours. Stimulate the stained cells with a final concentration of 30 ng/mL of either TNF-α or IL-1β by spiking the cytokines directly into the imaging media. Immediately load the plate onto an automated live-cell microscope with environmental control (37°C, 5% CO2); no dye or stimulant washout is performed. Acquire images of the Hoechst nuclear stain and p65-mNeonGreen with 5X or greater magnification at 20 minute intervals over 6 hours. If the center of well A1 was not part of the live imaging grid, acquire an image of the Hoechst channel at this stage position for future stage alignment immediately after finishing live-cell imaging, then quickly proceed with fixation in Step 89 and continue with the remainder of the protocol.
-
89
After 48 hours of cell culture, addition of any stimuli of interest, and live-cell imaging if applicable, remove media and fix cells with freshly prepared 4% PFA in PBS (fixation solution) for 30 minutes at room temperature.
-
90
Wash the cells three times with RNase-free PBS. All wash steps in the remainder of the protocol are performed with 1 mL/well for 6-well plates, 0.5 mL/well for 24-well plates unless otherwise noted.
-
91
Aspirate PBS and permeabilize with 70% ethanol for 30 minutes at room temperature.
-
92
Successively add and remove 1 mL PBS+ 0.05% Tween (PBS-T) until ethanol concentration is titrated below 1% (six exchanges) to avoid dehydration.
-
93
Wash 3X with PBS-T.
In situ amplification of sgRNA sequences Timing: 2d
-
94Assemble RT reaction mix in the order listed below on ice and add to wells. Use a total volume of 750 μl/well for 6-well plates, or scale to 190 μl/well for 24-well plates.
Component Amount per well (μl) Final UltraPure water 533 RevertAid RT buffer, 5X 150 1X dNTPs, 10mM each 19 250μM LNA-modified RT primer, 100μM; see Supplementary Table 4 for oligo sequence 7.5 1μM BSA, molecular biology grade, 20 mg/mL 7.5 0.2 mg/mL Ribolock RNase inhibitor, 40 units/μL 15 0.8 units/μL RevertAid H minus Reverse Transcriptase, 200 units/μL 18 4.8 units/μL Total volume 750 -
95
Incubate overnight at 37°C, shaking gently at 135 rpm. If time is limiting, this reaction may run for as little as 6 hours depending on the experiment.
^ CRITICAL STEP It is important to avoid dehydration of any wells during this incubation. Fill all empty wells and spaces between wells with RNase-free water to minimize evaporation. Also, cover the plate with an aluminum foil microseal and secure it with a plate sealing roller to ensure a tight seal.
? TROUBLESHOOTING
-
96
Return plate to room temperature and wash six times with PBS-T.
^ CRITICAL STEP These wash steps are important to remove excess dNTPs, as dNTP concentration in the gap-fill reaction is around 1000X lower than the reverse transcription reaction. Excess dNTPs or TaqIT polymerase can inhibit the gap-fill reaction by displacing the padlock.
-
97
Perform a post-fixation with freshly diluted 3% paraformaldehyde + 0.1% glutaraldehyde in PBS (post-fixation solution) for 30 minutes at room temperature.
-
98
Wash three times with PBS-T.
-
99
Optional: fixed-cell phenotyping can be performed with either a fluorescent reporter (option A) or via immunofluorescence (option B).
^ CRITICAL For fixed-cell p65 screens, imaging and antibody staining was performed after the post-fixation step (here). However, phenotype staining and imaging may be performed at other steps, including phenotyping after RCA (after Step 104). See Box 3 for further discussion.
- Fixed-cell phenotyping of a reporter cell line
- For a fixed fluorescent reporter (e.g. p65-mNeonGreen), add 200 ng/mL DAPI in 2X SSC to each well and image. p65 nuclear translocation can be accurately quantified at 10X but dim reporters or more subtle phenotypic effects typically require 20X magnification or higher. Be sure to acquire an image of the DAPI channel at the center position of well A1 for future stage alignment.
- Fixed-cell immunofluorescence phenotyping
- Incubate cells with primary anti-rabbit p65 at 1:400 in 3% BSA immunofluorescence blocking buffer for 1 hr at room temperature.
- Perform three quick washes with PBS-T.
- Incubate with secondary goat anti-rabbit AF488 at 1:1000 for 45min in 3% BSA blocking buffer.
- Perform three quick washes with PBS-T.
- Add 200 ng/mL DAPI in 2X SSC to each well and acquire phenotype images. Be sure to acquire an image of the DAPI channel at the center position of well A1 for future stage alignment.
-
100Assemble the gap-fill reaction mix in the order listed below on ice and add to wells. Use a total volume of 600 μl/well for 6-well plates, or scale to 125 μl/well for 24-well plates.
Component Amount per well (μl) Final UltraPure water 422 Ampligase buffer, 10X 60 1X dNTPs, 10μM each 3 50nM Padlock probe, 100μM; see Supplementary Table 4 for oligo sequence 0.6 0.1μM BSA, molecular biology grade, 20 mg/mL 6 0.2 mg/mL RNase H, 5 units/μL 48 0.4 units/μL TaqIT DNA polymerase, 50 units/μL 0.24 0.02 units/μL Ampligase, 5 units/μL 60 0.5 units/μL Total volume 600
^ CRITICAL STEP dNTPs should be freshly diluted at 1:1000 before adding, as excess dNTPs inhibit the reaction.
-
101
Load plate onto a flat-top thermocycler for 5 minutes at 37°C, then 90 minutes at 45°C.
? TROUBLESHOOTING
-
102
Wash three times with PBS-T.
-
103Assemble RCA reaction mix in the order listed below on ice and incubate overnight at 30°C. If time is limiting, this reaction may run for as little as 6 hours depending on the experiment. Use a total volume of 600 μl/well for 6-well plates, or scale to 125 μl/well for 24-well plates.
Component Amount per well (μL) Final UltraPure water 399 Glycerol, 50% 60 5% Phi29 buffer, 10X 60 1X dNTPs, 10mM each 15 250 μM BSA, molecular biology grade, 20 mg/mL 6 0.2 mg/mL Phi29 DNA polymerase, 10 units/μL 60 1 unit/μL Total volume 600
^ CRITICAL STEP Phi29 is unstable and temperature-sensitive. Be sure to store at −20°C and take to the bench in a cooler or on ice quickly. Prepare the master mix with all components except phi29 and chill on ice before adding. Once the master mix is complete, add to the sample quickly (within a few minutes) as the enzyme may be less stable once diluted.
? TROUBLESHOOTING
-
104
Wash three times with PBS-T.
PAUSE POINT After RCA, samples should be stable for several weeks when stored at 4°C between cycles of in situ sequencing.
-
105
Dilute the sequencing primer (see Supplementary Table 4) to 1 μM in 2X SSC buffer and incubate in the wells for 30 minutes at 37°C.
-
106
Wash three times with PBS-T.
In situ sequencing-by-synthesis Timing 1.5–4 h per cycle; 1 h hands-on
^ CRITICAL The time required for in situ sequencing-by-synthesis is highly dependent on microscope speed; see Box 2 and Tables 1 and 2.
-
107
Pre-warm flat-top thermocycler to 60°C.
-
108
Add 500 μL incorporation mix (MiSeq Nano kit v2 reagent 1) at room temperature; move plate to flat-top thermocycler and incubate for 5 minutes at 60°C. Use 500 μl/well of incorporation mix for 6-well plates, 125 μl/well for 24-well plates.
^ CRITICAL STEP Do not incubate longer than 5 minutes, as extending incorporation can greatly increase background staining of dye-labeled nucleotides.
? TROUBLESHOOTING
-
109
Remove plate from flat-top thermocycler. For 6-well plates, add 1 mL/well MiSeq Nano kit PR2 buffer to dilute incorporation mix before aspirating, then quickly add and remove 1 mL/well PR2 six additional times at room temperature. For 24-well plates, use 0.5 mL/well of PR2.
^ CRITICAL STEP The incorporation mix is very concentrated. Washing is needed to remove dyes that stick to the cells, which is the main contribution to background noise in sequencing. If multiple washes are not performed quickly, further washing at high temperature may substantially increase the background.
-
110
Wash five times with PR2 at 60°C, 5 minutes for each wash. Between each heated wash, remove plate from thermocycler and replace well contents with fresh PR2.
^ CRITICAL STEP This step removes background fluorescence from non-specific binding of dye-labeled nucleotides to the glass or cells. If background is a problem during imaging, it may be helpful to increase the number and duration of washes, especially in later sequencing cycles.
? TROUBLESHOOTING
-
111
Exchange into 200 ng/mL DAPI in 2X SSC, with at least 2 mL/well for 6-well plates, 0.5 mL/well for 24-well plates. Allow 5 minutes for DAPI to bind DNA.
-
112
Clean the bottom of the plate with isopropyl alcohol and lens cleaning tissue, visually inspect to ensure there are no remaining streak marks from cleaning.
-
113
Load the plate on the microscope stage, ensuring that the plate is well-seated on the stage to prevent movement during imaging.
-
114
Acquire SBS images using a 10X objective lens. Provided below in Steps 115–123 are detailed instructions for microscopes controlled with Micro-Manager, although many other systems can be used. See Box 2 and Tables 1 and 2 for details on microscope system configuration. The protocol video (https://youtu.be/TEqMbMjS1tA) also includes a detailed demonstration of image acquisition and stage alignment (starting at 10:47).
^ CRITICAL STEP After the first cycle, the plate must be carefully aligned for imaging each subsequent cycle. While alignment refinement will be done computationally, it is important to minimize cycle-cycle alignment differences in order to prevent data loss. For the most straightforward data analysis, the stage alignment, magnification, and imaging grid should also match the phenotype images.
? TROUBLESHOOTING
-
115
Open Micro-Manager 2.0.
-
116
Select “Plugins” > “Acquisition Tools” > “HCS Site Generator.” Select a plate format (typically 6-well for screening). Custom formats can be created if necessary.
-
117
Calibrate and align the plate. If this is the first cycle of phenotype or sequencing imaging, follow option A, otherwise use option B.
- Acquiring a calibration image for future stage alignment
- Manually move the stage to the center of well A1 in live mode (found by identifying the point equidistant from well edges) and calibrate the stage position by selecting “Calibrate XY…”
- Acquire and save an image of the DAPI channel at this position for future cycle alignment.
- Aligning to an existing calibration image
- Move the stage to the approximate center of well A1 either manually or by using the “Go to” option of the stage position list found under “Tools” > ”Multi-Dimensional Acquisition” > “Multiple Positions (XY)” > “Edit Position List.”
- Open the calibration image of the center of well A1 from the phenotype imaging or the first cycle of sequencing by clicking “ImageJ” > “Open.”
- With the DAPI channel selected, press “Image” > “Adjust” > “Threshold,” adjust to select nuclei and click “Apply” > “OK.”
- Select the thresholded nuclei by pressing “Edit” > “Selection” > “Create Selection.”
- Add the selected nuclei to the ROI manager by pressing “Image” > “Overlay” > “Add Selection.”
- Press “Live” with the DAPI channel active and manually move the stage until the nuclei match the selection from the first cycle.
- Press “Calibrate XY…”, select well A1, and press “OK” to recalibrate the center of the well.
- Check calibration by moving the stage, then returning to the center site using the stage position list. Ensure that the live DAPI image still matches the thresholded selection from the calibration image.
-
118
Define imaging sites by specifying the number of rows, columns, and spacing (μm) and selecting spacing rule “Equal XY” and site visit order “Snake.” For 6-well plates, a 21×21 grid with 1280 μm spacing at 10X objective magnification is recommended. Spacing depends on microscope configuration; in general it is recommended to use imaging sites with 10–15% overlap to minimize data loss upon alignment).
-
119
Select wells for imaging by clicking “Select” and highlighting desired wells, then clicking “Build MM List.” Micro-Manager also supports custom stage position lists to more efficiently image circular wells.
-
120
Set up acquisition by pressing “Tools” > “Multi-Dimensional Acquisition,” and checking “Multiple Positions (XY),” “Autofocus,” “Channels,” and “Save Images” (Saving format: Image stack file). To acquire images for sequencing, use 2×2 binning at 10X magnification (approximately 1.2 μm/pixel) and a 16-bit depth. Z-Stacks are typically not necessary at 10X magnification.
-
121Select the appropriate channel group under channels and select ‘New’ to add channels. Check “Use?” for DAPI and the four sequencing channels and define the appropriate settings. A negative Z-offset in the DAPI channel of −0.1 to −0.5 μm from the sequencing channels is sometimes used to maintain focus. The following exposure settings are recommended for broadband light source systems, such as presented in Table 2, although these will depend on microscope configuration. Use the listed acquisition order for ease of input into the downstream computational workflow, otherwise pre-process images to place channels in this order prior to analysis. To determine exposure and illumination power settings, ensure that the majority of signal is within 50% of the bit depth and the signal-to-noise-ratio for in situ spots or nuclei is at least 3 (ideally 5). See Box 2 for further discussion of microscope system design and validation.
DAPI (10% power) 50 ms MiSeq G 200 ms MiSeq T 200 ms MiSeq A 200 ms MiSeq C 800 ms
^ CRITICAL STEP It is recommended to use a low DAPI exposure (illumination intensity and duration) to minimize photodamage.
-
122
Ensure that the “Multi-Dimensional Acquistion” window summary reflects the expected number of imaging sites, channels, and channel configurations and that the computer has sufficient storage space available for the listed “total memory” requirement (10–20 MB per imaging site depending on camera configuration, ~40 GB for one cycle of sequencing across a full 6-well plate).
-
123
Press “Acquire!”
PAUSE POINT Samples should be stable for several weeks when stored at 4°C between cycles of sequencing.
-
124
After imaging, add cleavage mix (MiSeq Nano kit v2 reagent 4) and incubate at 60°C for 6 minutes. Use 600 μl/well of cleavage mix for 6-well plates, 125 μl/well for 24-well plates. Cleavage occurs quickly at 60°C. Imaging the sample after cleavage and washing should show no detectable fluorescence from reads.
? TROUBLESHOOTING
-
125
Quickly wash three times with PR2 at room temperature.
-
126
Wash three times with PR2 at 60°C for 1 minute. Between each heated wash, remove plate from thermocycler and replace well contents with fresh PR2.
? TROUBLESHOOTING
-
127
Wash three times with PR2 at room temperature.
-
128
Repeat incorporation, imaging, and cleavage of subsequent bases (Steps 107–127) until the necessary number of cycles for resolving all barcodes in the given library are complete (defined by the prefix_length parameter chosen during library design in Steps 1–3).
Analysis Timing 1 week
^ CRITICAL The analysis pipeline is implemented primarily in Python, with command line execution using simple scripts or the Snakemake workflow manager65, which is automatically installed as a dependency of the provided Python package. For the following steps, the GitHub repository must be downloaded and installed as detailed in EQUIPMENT SETUP, and for each specified command <path/to/OpticalPooledScreens> should be replaced with the actual file path to the downloaded repository. All commands must be executed from within the corresponding virtual environment, which is activated from the command line by:
$ source <path/to/OpticalPooledScreens>/venv/bin/activate
Extensive example data is publicly available from Cell-IDR66, with functions and instructions for access provided in the GitHub repository along with an example Snakemake pipeline. See Supplementary Figure 2 for an overview of the analysis pipeline.
-
129Create a folder for the experiment and copy the necessary files from the downloaded code repository by changing the working directory of the command line interface to the experiment folder and then running:
$ bash \ <path/to/OpticalPooledScreens>\ /scripts/setup_experiment_directory.sh
The copied files include the Snakemake workflow (“OpticalPooledScreens.smk”), corresponding config file for defining analysis parameters (“config.yaml”), and a spreadsheet for defining how images are pre-processed in the following steps (“input_files.xlsx”).
! CAUTION Microscope software varies in how raw image files are formatted and saved. Thus we include the following initial file formatting steps (Steps 130–133), including setting standard filenames. File formatting starts from *.tiff images, each containing data from no more than one field-of-view. For image files where multiple fields of view are saved within the same file (e.g., sometimes the default when using NIS Elements), these large files should be split into one *.tiff file per field-of-view before proceeding. Some software will save individual fluorescent channels of the same field-of-view as separate image files. In that case, step 132 concatenates these channels to generate one image file per cycle for each field-of-view.
-
130
Move all raw input images into the “input” folder created by the set-up script.
-
131
Open and edit the “input_files.xlsx” spreadsheet in the experiment folder such that each raw input *.tiff file has a unique line in the spreadsheet, with corresponding fields specifying the image magnification (“10X”, “20X”, etc.), cycle, well, tile, and channel. The “snakemake filename” field should automatically fill based on the manually-entered fields.
^ CRITICAL STEP File paths should be relative to the experiment parent directory, e.g., “input/raw/<filename>.tiff”. The “channel” column is used for sorting the order of channels when concatenating files containing only a single fluorescent channel. If all channels are present in the same input *.tiff file, “ALL” should be entered in the channel field. Otherwise the channel column entry should be one of: “DAPI”, “G”, “T”, “A”, or “C”.
-
132From within the experiment folder, execute the ops.io.format_input function to format the input files:
$ python -m ops.io format_input input_files.xlsx --n_jobs=<number of parallelized jobs>
-
133
When formatting is complete, verify that file names and locations are as expected from the “input_files.xlsx” table, and that a “input/well_tile_list.csv” file was also created with one line for each well and tile in the entire experiment. This table will be used later as an input to the full analysis workflow.
^ CRITICAL STEP For each dataset, several parameters need to be set in the Snakemake config file before running the full pipeline: “THRESHOLD_READS” (for identifying sequencing reads), and “THRESHOLD_DAPI,” “THRESHOLD_CELL,” and “NUCLEUS_AREA” (for the default morphological cell and nuclear segmentation method). An alternative method for segmentation, based on the CellPose algorithm54, can be chosen by changing the “SEGMENT_METHOD” parameter from “cell_2019” to “cellpose” and setting “DIAMETER” to the expected cell size in pixels, which may be estimated by running the CellPose GUI in calibration mode (typical values are 20–35 μm converted into pixel units). Note that CellPose segmentation may improve accuracy at the cost of increased computation time (see Supplementary Figure 1). Users may also supply their own segmentations by replacing the masks created in the Snakemake pipeline. Steps 134–140 explain a simple approach for choosing parameters using Fiji for the default morphological segmentation method, while Steps 141–145 describe how to select “THRESHOLD_READS” using Snakemake.
-
134
Open an example formatted input image (from Step 132) in Fiji.
-
135
Make a duplicate of the DAPI channel (“Image” > “Duplicate”, then set “Channels” to 1, click “OK”).
-
136
Open the threshold tool (“Image” > “Adjust” > “Threshold”), and choose a threshold value that separates most nuclei from background by moving the slider. Enter this value as “THRESHOLD_DAPI” in the Snakemake config file (“config.yaml”) using a text editor.
-
137
Separate connected nuclei by watershed transformation (“Process” > “Binary” > “Watershed”).
-
138
Measure the area of the resulting regions (“Analyze” > “Analyze Particles…”, make sure “Display Results” is checked, click “OK”) and plot the distribution (with the “Results” window open, run “Results” > “Distribution…”, make sure “Area” is the selected parameter, click “OK”). This distribution is a rough approximation of the nuclear sizes. Choose a minimum area that excludes the smallest particle sizes that represent spurious segmentation errors, and a maximum value that is ~30% greater than the largest particle. Enter these values in the “config.yaml” file for “NUCLEUS_AREA”.
-
139
For cell body segmentation, cells are identified using the background signal from non-specific SBS dye accumulation. Returning back to the original input image opened in Fiji in Step 134, make a duplicate of the SBS channels (“Image” > “Duplicate”, then set “Channels” to include only the SBS channels, typically 2–5, and click “OK”), and take the mean across these channels (“Image” > “Stacks” > “Z project…”, set “Start slice” to 1, “Stop slice” to 4, select “Average Intensity”, click “OK”).
-
140
Repeat Step 136 for this average SBS intensity image to choose a threshold for cell segmentation and enter this value in “config.yaml” for “THRESHOLD_CELL”. Note that multiple values of these segmentation parameters can be tested for a few example images using the actual segmentation functions by running the Snakemake pipeline using a config file with the MODE parameter set to “paramsearch_segmentation,” similar to Steps 141–144. This will output one image containing possible cell segmentations for each combination of provided nuclear segmentation parameters; see the “example_config_paramsearch_segmentation.yaml” file in the “resources” folder of the code repository for an example.
^ CRITICAL STEP The optimum value of “THRESHOLD_READS” varies between experiments. The goal in choosing a “THRESHOLD_READS” value is to identify all true sequencing spots while not including too many spots that are low intensity or the result of spurious noise. A straightforward way to achieve this goal is to vary the “THRESHOLD_READS” value for a few example tiles and evaluate the resulting percentage of reads mapping to an expected barcode sequence (“mapping rate”), as demonstrated in the following steps.
-
141
Edit the “MODE” parameter in “config.yaml” to be “paramsearch_read-calling” and “INCLUDE_WELLS_TILES” to be a list of [well, tile] pair lists, including 2–3 example tiles per well.
-
142
Confirm the values of “THRESHOLD_DAPI”, “NUCLEUS_AREA”, and “THRESHOLD_CELL” in “config.yaml” to be those chosen in Steps 134–140. “THRESHOLD_READS” can be set to a list of values to test, although if it is not set as a list, a reasonable default range of values will be used for the parameter search. See the “example_config_paramsearch_read-calling.yaml” file in the “resources” folder of the code repository for an example config file for this step.
-
143Run the Snakemake pipeline using the edited config file:
$ snakemake --cores <number of cores> \ -s OpticalPooledScreens.smk \ --configfile config.yaml
-
144
Once Snakemake is finished, inspect the “nuclei.tif” and “cells.tif” images output to the “PROCESS_DIRECTORY” folder defined in the config file using Fiji and compare to the input images to confirm segmentation performance. Adjust “THRESHOLD_DAPI”, “NUCLEUS_AREA”, and “THRESHOLD_CELL” in “config.yaml” if necessary and re-run the Snakemake pipeline.
-
145
Open the “paramsearch_read-calling.summary.pdf” plots output to the defined “PROCESS_DIRECTORY” folder and select a value of “THRESHOLD_READS” where the read mapping rate begins to plateau (typical values are between 50 and 300, see Fig. 3c for an example plot). The values plotted here are also output in the “paramsearch_read-calling.summary.csv” table.
Figure 3.
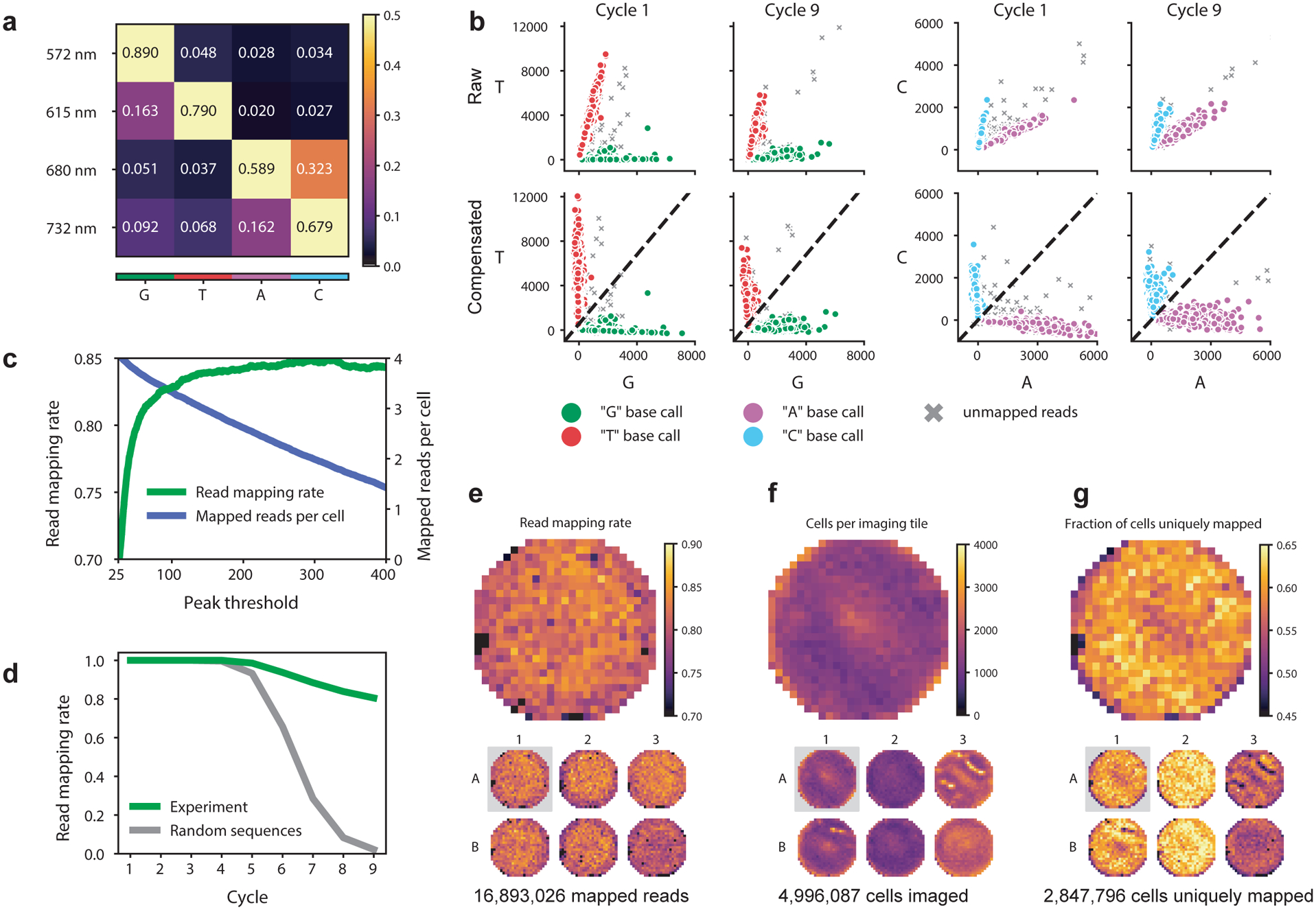
Technical performance and quality control of in situ sequencing by synthesis (SBS). Data are from a screen in A549 cells with a CROPseq-puro library of 5,738 sgRNAs43. (a) Example compensation matrix used for correcting spectral cross-talk between SBS imaging channels. (b) Spectral compensation of the two-channel combinations with the most cross-talk (T vs G and C vs A) at the first and last cycle of an SBS experiment. Mapped reads are those with barcode sequences exactly matching expected sequences from the designed sgRNA library. Dotted lines in the compensated plots demarcate the decision boundary for base calling. (c) Plotting read mapping rate and mapped reads per cell against increasing thresholds on the peak parameter demonstrate that most non-mapping reads are excluded by thresholding this value. (d) Longer read lengths provide increased confidence of mapped reads representing true sequencing spots from barcode mRNA. Plotting plate heatmaps of quality control metrics such as read mapping rate (e), total cells imaged (f), and fraction of cells with reads mapping to one expected barcode sequence (g) is useful for evaluating the quality of an experiment and identifying potential issues.
? TROUBLESHOOTING
-
146
Edit “config.yaml” to set “MODE” to “process”, “INCLUDE_WELLS_TILES” to “all” and “THRESHOLD_READS” to the value chosen in Step 145, and provide a file path to a table of the designed barcode sequences from Step 3 as the “BARCODE_TABLE” parameter.
-
147
Edit the “NUCLEUS_PHENOTYPE_FEATURES” and “CELL_PHENOTYPE_FEATURES” lists in “config.yaml” to extract the desired phenotype features for the nuclear and cell segmentation masks respectively. This is specific to individual applications, with functionality provided for some common use cases. A full list of defined features can be found in the “ops/features.py” file of the Python package and can be easily extended.
-
148From within the experiment directory, run the Snakemake pipeline on the full set of images with the updated configuration file:
$ snakemake --cores <number of cores> \ -s OpticalPooledScreens.smk \ --configfile config.yaml
Results from the Snakemake workflow are output to the “PROCESS_DIRECTORY” folder defined in the config file and include:*.log.tif Aligned and Laplacian-of-Gaussian filtered SBS data *.nuclei.tif Labeled segmentation mask of nuclei *.cells.tif Labeled segmentation mask of cells *.bases.csv Table of extracted raw intensity for each base channel for each spot *.reads.csv Table of extracted read sequences for each spot *.cells.csv Table of called reads for each segmented cell (includes two most common barcode sequences for each cell) *.phenotype.csv Table of user-defined phenotype features extracted for each segmented cell combined.csv Merged table of barcode calls and phenotype features for every processed tile
^ CRITICAL STEP Often the full plate analysis pipeline is run in a computing environment with many available CPUs (e.g., in a Google Cloud VM). Snakemake also provides capability for executing a workflow on a remote compute cluster. For around 10 cycles of SBS data, expect the pipeline to take about 3 minutes per field-of-view with the default segmentation algorithm on a single CPU (~1 hour for a full 6 well plate using 96 CPUs).
-
149
If phenotype and sequencing fields-of-view were acquired using the same imaging grid and magnification, and the same segmentation masks were used for SBS and phenotype extraction, the “combined.csv” table output from the Snakmake pipeline provides all the necessary data for downstream analysis. If phenotype and sequencing acquisitions were completed using different imaging layouts, magnification, microscopes, and/or segmentation masks, the phenotype and sequencing data must be aligned before proceeding. This can be done by using the geometry of neighboring nuclei segmentations in each dataset to match corresponding fields-of-view and individual cell identities. The primary assumption that must hold for this approach to be successful is that cells have not moved relative to each other between phenotype and sequencing acquisitions, which may not be true if many cells detach from the plate or fixation was poorly-timed in a live-cell experiment. Additionally, segmentation masks must be approximately the same, as the centroids of segmented objects are used as landmarks for alignment. Functions for performing this more complex dataset alignment are available in the “ops/triangle_hash.py” file of the python package.
^CRITICAL STEP If a screen design necessitates separate microscopes or different magnification between phenotype and SBS images, it is highly recommended to test aligning datasets with example acquisitions before attempting a full screen.
-
150
The quality of an experiment can be evaluated by computing several common metrics, including read mapping rate, cell mapping rate, and average number of mapped barcodes per cell (Fig. 3). Often it is useful to plot these metrics on a per-field-of-view basis to understand well- and plate-level variability and identify potential issues. The “ops/qc.py” file of the code repository includes functions for producing many of these plots.
? TROUBLESHOOTING
-
151
Analysis of phenotypic differences between perturbed cell populations can be highly experiment-dependent. In general, phenotypic features should be normalized relative to a population of negative control cells (e.g., those expressing non-targeting sgRNAs) within each well to reduce batch effects. If individual phenotype features are known to be informative a priori, aggregated sgRNA and/or gene-knockout scores can be computed using summary statistics, being careful to account for phenotype differences between screen replicates and/or sgRNAs targeting the same gene (e.g., taking the median across replicates or sgRNAs). If many image features are measured and none are expected to individually represent functional phenotypes of interest, a high-dimensional profiling approach can be used34. Alternatively, individual cells can be classified or clustered into known or learned phenotype categories using machine learning approaches, with the fraction of cells from each perturbation falling into each category compared statistically. Null distributions for calculating p values can be generated using a permutation procedure or by bootstrapping non-targeting sgRNA phenotype measurements. Significance should be determined using the Benjamini-Hochberg procedure for a given false-discovery rate or a similar approach to handling multiple hypothesis testing. Thresholds for calling hits can also be determined by calculating z-scores and/or by comparing negative and positive control perturbations. Existing methods for ranking genes from other screening modalities may also be helpful, such as RIGER or MAGeCK67,68. In addition to computational analysis of screen phenotypes, visual assessment of cell images is an important step in evaluating results.
TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
Table 3 |.
Troubleshooting table
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 37 | Insufficient colonies after library transformation (less than 300x library complexity) | Inefficient golden gate reactions and/or electroporation | Change to a fresh batch of reagents (T7 ligase, Esp3I, and electrocompetent cells are most critical) |
| Confirm transformation efficiency with pUC19 control plasmid | |||
| Input DNA mass for electroporation too low. | Increase the number of golden gate reactions to increase input DNA mass (see Step 5), carefully purify assembled DNA and quantify with Qubit prior to transformation | ||
| 45 | Representation of sgRNAs in plasmid library is skewed (skew ratio > 10), many expected sgRNA sequences are missing or are present in the wrong subpool | Jackpot effect from PCR amplification of oligo pool | Run multiple oligo pool PCR amplifications, use qPCR to ensure the reactions are halted during the exponential amplification phase, and pool the PCR products |
| Inefficient golden gate reactions and/or electroporation | Follow troubleshooting solutions for Step 37 | ||
| Insufficient representation (<300x) of library elements as input to any earlier step (oligo pool PCR, golden gate reactions, electroporation, NGS library prep) | Increase the number of oligo pool PCR and golden gate reactions (see Step 5), ensure adequate DNA mass input for electroporation, and increase the coverage of NGS reads per library element (see Step 44) | ||
| Oligo pool library design errors | Ensure the recommended subpool dialout primers are used and these match the designed oligo PCR handle sequences | ||
| 71 | Titer of lentivirus batch is low | Transfection of and/or virus production by HEK 293FT cells inefficient | Use a fresh batch of low-passage HEK 293FT cells and replace transfection reagents |
| Verify plasmid transfection and viral transduction efficiencies with a high-titer control fluorescent protein vector (e.g., Addgene #25999) by fluorescence imaging or flow cytometry | |||
| 76 | Yield of successfully transduced cells is low compared to the library complexity | Inefficient viral transduction | Make a fresh batch of lentivirus with improved titer or increase the spinfection scale to achieve the necessary library representation (see Steps 72–73) |
| Miscalculation of viral titer | Re-titer the lentivirus batch and make a new batch if titer is low | ||
| 84 | Representation of sgRNA sequences in the cell library is skewed (skew ratio > 10) | Input plasmid library was skewed | Compare cell library skew to plasmid library skew and address as described in troubleshooting for Step 45 |
| Spinfection inefficient or of insufficient scale | Follow troubleshooting solutions for Step 76 and ensure target representation of >300 cells/perturbation in spinfection scale calculation (see Steps 72–73) | ||
| sgRNA sequences targeting essentials genes have decreased representation as compared to the plasmid library | Cells carrying sgRNAs targeting essential genes are dying | Filter out depleted guides or genes in screen analysis. If essential genes are important for a specific application, use an inducible Cas9 system to allow generation of the cell library prior to gene perturbation and optimize the time between Cas9 induction and screen readout. If essential gene depletion occurs prior to induction, inducible expression may be leaky and the cell line should be re-validated with control sgRNAs | |
| 86 | Plating of cells on imaging plates is uneven or too dense | Cells are not fully resuspended after trypsinization | Minimize cell clumps by optimizing enzymatic detachment, pipette-mixing the cell suspension, and filtering through a cell strainer if necessary |
| Too many cells are seeded per well | Test multiple cell plating densities and methods of plating (e.g., gently swirling plate in a “figure 8” motion during plating). The best density and method for plating may vary by cell type, but poor plating can result in many problems such as poor phenotype images and/or in situ sequencing quality. | ||
| Cells are not distributed evenly across the wells during plating | |||
| 95, 101, 103, 108, 110, 124, 126 | Evaporation during heated enzymatic or washing steps | Plate not fully sealed | Carefully seal the plate with a fresh foil seal before each incubation |
| Fill empty wells and spaces between wells with water to limit evaporation | |||
| Place a flat and moderately heavy item on top of the plate to keep the seal from detaching | |||
| 114 | Low sequencing spot brightness in the first cycle of SBS | RCA reaction inefficient or too short | Use a fresh batch of Phi29 polymerase and buffer; Phi29 polymerase is thermo-sensitive and should be stored appropriately, freeze-thaw cycles minimized, and RCA reaction mix assembled quickly on ice |
| Run the RCA reaction for at least 16 hours | |||
| Cell plating is too dense | Test at lower cell densities and see troubleshooting for step 86. | ||
| High SBS background in the first cycle of SBS | Non-specifically bound dye-labeled nucleotides | Repeat post-incorporation washes (Step 110), proceeding rapidly between steps as demonstrated in the protocol video (https://youtu.be/TEqMbMjS1tA) | |
| Background from phenotype stains | Image sequencing channels prior to first cycle incorporation to confirm origin of background and consider altering phenotyping approach following guidelines in Box 3 to reduce background | ||
| High SBS dye background within cells in any cycle of sequencing (especially the “C” channel) | Insufficient washing before and/or after incorporation steps | Repeat heated post-incorporation washes (Step 110) and increase the number and duration of these washes for future cycles and plates | |
| Wells drying out during solution exchanges | Carefully avoid plate drying by performing buffer exchange quickly | ||
| Irreversible sticking of dye to cells or build-up of background over many cycles | Eliminate imaging of the DAPI channel in cycles after cycle 1 to avoid UV exposure-associated damage to DNA, using the SBS stain background to align subsequent cycles. The extent of background staining may vary by cell-type | ||
| Incorrect sample storage | Store plate at 4C between SBS cycles (stable for weeks) | ||
| Many sequencing spot appear to be outside of the boundaries of any cell | Diffusion of barcode mRNA or DNA from improper fixation, post-fixation, or a cell type-specific effect | Use fresh batches of PFA and glutaraldehyde | |
| Ensure an LNA-modified reverse transcription primer is used | |||
| While fixation and post-fixation conditions were uniformly applicable to the tested adherent cell lines, different sample types may require alternative strategies to fix cDNA to cell matrix such as biotin-streptavidin linkage | |||
| Subpopulation of cells without sequencing spots | Incomplete antibiotic selection after lentiviral infection with perturbation library | Optimize concentration and duration of antibiotic selection using fresh antibiotic and parental cell line | |
| Repeat selection steps with fresh antibiotic or re-infect and select starting from the parental cell line | |||
| Highly variable sequencing spot counts within or between wells | Evaporation during earlier steps, especially after ethanol permeabilization | In addition to troubleshooting solutions for Step 95, ensure reagent exchanges in the well plate are completed quickly to avoid dehydration of samples | |
| review proper ethanol removal procedure after permeabilization (Step 92) in the protocol video (see 06:38 at https://youtu.be/TEqMbMjS1tA). | |||
| Regions of high cell density | Follow troubleshooting solutions for Step 86 to reduce cell density and/or uniformity | ||
| Low average sequencing spot count per cell (<= 2) | Inefficient in situ enzymatic reactions | Use fresh batches of reagents (reverse transcription and gap-fill enzymes, dNTPs, PFA and glutaraldehyde) | |
| Excess dNTPs during gap-fill reaction | Increase PBS-T washes prior to gap-fill reaction and ensure dNTPs are properly diluted | ||
| Cell plating is too dense | See troubleshooting solutions for Step 86 and optimize cell density to avoid over-confluency | ||
| Sequencing spot count may vary by cell type | If an un-validated cell line is being used, barcode expression may be lower or full protocol may need to be re-optimized, including choice of promoter or by using a CROPseq vector that incorporates a fluorescent protein and flow-sorting high-expressing cells (see Box 1) | ||
| Decrease in spot fluorescence intensity over sequencing cycles | Photodamage from excess DAPI exposure | Use the minimum DAPI exposure (excitation intensity and duration) needed to acquire alignable images. If possible, avoid imaging DAPI at magnification higher than 10X after RCA. | |
| Some fields-of-view out of focus | Adaptive focus control not tracking focal plane across the plate due to well plate quality, imaging solution volume, or because the bottom of the well plate is dirty | Increase volume of imaging media as some infrared autofocus systems may confuse the solution meniscus for plate surface | |
| Thoroughly and carefully clean the bottom of the well plate using lens paper and isopropyl alcohol, allow to air dry | |||
| Check for plate flatness and use recommended glass-bottomed plates if possible | |||
| Contact microscope representatives for system-specific help | |||
| Fields-of-view from successive cycles of sequencing are misaligned by >10% of the field-of-view size | Incorrect plate alignment during plate loading | Practice carefully aligning the plate to a standard location each time it is loaded onto the stage; use a spring-clamped plate holder to ensure minimal displacement | |
| Microscope control software issues | Contact relevant microscope representatives for system-specific help | ||
| Poor stage repeatability | Test the ability of the stage to return to the same location and contact relevant microscope representatives for system-specific help | ||
| 145, 150 | Rate of in situ sequencing reads mapping to expected barcode sequences is low (<70%) | Sequencing spot brightness too low | Inspect sequencing images and follow relevant troubleshooting solutions from Step 114 |
| SBS dye background staining too high | |||
| Fields-of-view from successive cycles of sequencing are not aligned | |||
| Microscope setup not sufficiently separating SBS dye emission | Validate successful read mapping using a given microscope configuration on a technical experiment (e.g., frameshift reporter43, also see Box 2) | ||
| 150 | Many cells have reads mapping to multiple expected barcode sequences | MOI for sgRNA lentiviral infection is too high |
Make a fresh batch of lentivirus or re-titer the current batch |
| Restrict screen analysis to cells mapping to only a single barcode (if MOI is very high and spot count and/or read mapping rate is low, this can lead to many false-positive cells) | |||
| Poor cell segmentation | Adjust cell segmentation parameters, compare morphological and CellPose segmentation results, or use an algorithm optimized for a given cell type or experimental condition |
TIMING
Steps 1–4, designing a custom sgRNA library: 2–5 weeks; 2 d hands-on
Steps 5–25, cloning a custom sgRNA library: 1 d
Steps 26–38, amplification of pooled sgRNA library: 2 d
Steps 39–45, next-generation sequencing of the amplified sgRNA library: 2–3 d
Step 46, validation of in situ sequencing in cell line of interest: 1 week
Steps 47–49, optional: reporter cell line creation and validation: 1–2 weeks
Steps 50–53, Cas9 cell line creation and validation: 2–4 weeks
Steps 54–71, lentivirus production and titer: 1 week
Steps 72–84, lentivirus transduction: 1 week
Steps 85–93, image-based Phenotyping: 2–3 d
Steps 94–106, in situ amplification of sgRNA sequences: 2 d
Steps 107–128, in situ sequencing-by-synthesis: 1.5–4 h per cycle; 1 h hands-on
Steps 129–151, analysis: 1 week
ANTICIPATED RESULTS
Here we provide example screening results for fixed- and live-cell pooled CRISPR knockout screens for p65 translocation43. Despite low levels of spectral cross-talk between sequencing channels, the rate of reads mapping to expected barcode sequences should remain high as a result of spectral compensation (Fig. 3a-c). Read mapping rates typically decrease gradually across later sequencing cycles, but remain stable through at least 9–12 cycles to enable analysis of large barcode libraries (Fig. 3d, ref.43). Additionally, some plate- and well- level variability is acceptable when evaluating quality metrics (Fig. 3e-g). Due to the heterogeneity of actual genetic alterations induced in individual cells and the variable efficiencies of sgRNAs, phenotype variability between individual cells and sgRNAs targeting the same gene is expected. However, the population behavior will identify phenotypes of interest if appropriate statistical power is achieved (Fig. 4).
Figure 4.

Anticipated results. (a) Example images from a CRISPR knockout screen for regulators of NFkB activation in A549 cells43. Sequencing and phenotyping data (p65 localization) are shown for a single field of view. White outlines in sequencing images represent individual cells; colored outlines in the phenotype image represent clusters of neighboring cells with identical sgRNA assignments (scale bar, 50 μm). (b) Distribution of per-cell nuclear translocation scores after TNFɑ stimulation for non-targeting sgRNAs and sgRNAs targeting TNFRSF1A (TNFa receptor), MAP3K7 (downstream NFkB regulator), and IL1R1 (not involved in TNFa signaling). All sgRNAs were downsampled to a maximum of 100 cells to more easily compare distributions. (c) Kinetics of NFkB activation from a separate live-cell optical pooled screen performed in HeLa cells43. In unperturbed cells, p65 translocates from the cytoplasm to the nucleus ~45 min after stimulation, followed by a slower, partial relaxation back to the cytoplasm. Top, translocation traces for individual cells mapped to sgRNAs targeting TNFRSF1A, RIPK1, and TNFAIP3 (negative regulator of TNFa signaling). The gray curve indicates the average of cells assigned to non-targeting controls. Middle, per-sgRNA averages, with error bars indicating standard error of the mean (data downsampled to 300 cells per sgRNA). Bottom, per-gene distributions at fixed time points of all cells mapped to sgRNAs targeting the respective gene (green) or non-targeting sgRNAs (gray). The single-cell resolution of optical pooled screens reveals distribution features of perturbation effects, including bimodality and distribution width.
Supplementary Material
Supplementary Table 1. Spreadsheet calculator for estimating the scale of an optical pooled screen.
Supplementary Table 2. Oligo pool dialout primer sequences.
Supplementary Table 3. Primer sequences for NGS library preparation.
Supplementary Table 4. Oligo sequences for in situ amplification of sgRNA sequences.
Supplementary Figure 1. Cell line comparison. (a) Suitability of example cell lines for optical pooled screening. NFkB screens were published in ref.43 (b) Example cell and nuclear segmentation using CellPose, a generalist algorithm based on a deep learning model trained on diverse cell and image types. For each cell line, the left panel shows the exact RGB input provided to CellPose, and the right panel shows segmentations using the “cyto” and “nuclei” models in single-model mode (scale bar, 100 μm). All cell lines were analyzed identically, varying only in a single “diameter” parameter automatically generated by the CellPose “cyto” calibration model. Segmentation of a 1.2mm × 1.2mm field of view took ~4 min on 1 CPU.
Supplementary Figure 2. Image analysis pipeline overview. Flowchart demonstrating the major steps of the screen processing pipeline. Input data are represented by rounded shapes; image and tabular data are in green and purple, respectively.
ACKNOWLEDGMENTS
The authors thank Emily Botelho for research support; Kelsey Caetano-Anolles for critical reading of the manuscript; Russell Walton for assistance with figures; Pulse Media Inc. for producing the video; Anthony Garrity, Jonathan Schmid-Burgk, and Anja Mezger for contributions to the development of the protocol; Iain Cheeseman, J.T. Neal, Anne Carpenter, Feng Zhang, and Aviv Regev for helpful discussions; and all members of the Blainey Lab for continued collaboration and support. This work was supported by two grants from the National Human Genome Research Institute (HG009283 and HG006193). L.F. was supported by a National Defense Science and Engineering Graduate Fellowship. R.C. was supported by a Fannie and John Hertz Foundation Fellowship and an NSF Graduate Research Fellowship.
Related links
Key reference using this protocol
Feldman D. et al. Optical Pooled Screens in Human Cells. Cell 179, 787–799.e17 (2019): https://doi.org/10.1016/j.cell.2019.09.016
Footnotes
CODE AVAILABILITY
The Python package and associated resources for the sgRNA library pool design and image analysis pipeline are freely available at https://github.com/feldman4/OpticalPooledScreens61 under the terms of the MIT license.
COMPETING INTERESTS
P.C.B. is a consultant to and/or equity holder in companies in the life sciences industries including 10X Genomics, GALT, Celsius Therapeutics, Next Generation Diagnostics, Cache DNA, and Concerto Biosciences. The Broad Institute and MIT have filed U.S. patent applications on work described here and may seek to license the technology.
DATA AVAILABILITY
The dataset used to produce Figures 3 and 4 was originally published in ref.43 and is publicly available from Cell-IDR66 (idr0071, experiment C).
REFERENCES
- 1.Shalem O et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang T, Wei JJ, Sabatini DM & Lander ES Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joung J et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parnas O et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 162, 675–686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixit A et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adamson B et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaitin DA et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167, 1883–1896.e15 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Wroblewska A et al. Protein Barcodes Enable High-Dimensional Single-Cell CRISPR Screens. Cell 175, 1141–1155.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubin AJ et al. Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell 176, 361–376.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mimitou EP et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neumann B et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collinet C et al. Systems survey of endocytosis by multiparametric image analysis. Nature 464, 243–249 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Orvedahl A et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karlas A et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463, 818–822 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Mercer J et al. RNAi Screening Reveals Proteasome- and Cullin3-Dependent Stages in Vaccinia Virus Infection. Cell Rep 2, 1036–1047 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Agaisse H et al. Genome-Wide RNAi Screen for Host Factors Required for Intracellular Bacterial Infection. Science 309, 1248–1251 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Saka SK et al. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol 37, 1080–1090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Codeluppi S et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 15, 932–935 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Lin J-R, Fallahi-Sichani M & Sorger PK Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun 6, 8390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gut G, Herrmann MD & Pelkmans L Multiplexed protein maps link subcellular organization to cellular states. Science 361, (2018). [DOI] [PubMed] [Google Scholar]
- 21.Regot S, Hughey JJ, Bajar BT, Carrasco S & Covert MW High-Sensitivity Measurements of Multiple Kinase Activities in Live Single Cells. Cell 157, 1724–1734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hochbaum DR et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 11, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y et al. An Expanded Palette of Genetically Encoded Ca2+ Indicators. Science 333, 1888–1891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin MZ & Schnitzer MJ Genetically encoded indicators of neuronal activity. Nat. Neurosci 19, 1142–1153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson DE et al. Oscillations in NF-κB Signaling Control the Dynamics of Gene Expression. Science 306, 704–708 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Cohen-Saidon C, Cohen AA, Sigal A, Liron Y & Alon U Dynamics and Variability of ERK2 Response to EGF in Individual Living Cells. Mol. Cell 36, 885–893 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Albeck JG, Mills GB & Brugge JS Frequency-Modulated Pulses of ERK Activity Transmit Quantitative Proliferation Signals. Mol. Cell 49, 249–261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lahav G et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet 36, 147–150 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Biederer T & Scheiffele P Mixed-culture assays for analyzing neuronal synapse formation. Nat. Protoc 2, 670–676 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Scheiffele P, Fan J, Choih J, Fetter R & Serafini T Neuroligin Expressed in Nonneuronal Cells Triggers Presynaptic Development in Contacting Axons. Cell 101, 657–669 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Varadarajan N et al. A high-throughput single-cell analysis of human CD8+ T cell functions reveals discordance for cytokine secretion and cytolysis. J. Clin. Invest 121, 4322–4331 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moen E et al. Deep learning for cellular image analysis. Nat. Methods 16, 1233–1246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grys BT et al. Machine learning and computer vision approaches for phenotypic profiling. J. Cell Biol 216, 65–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caicedo JC et al. Data-analysis strategies for image-based cell profiling. Nat. Methods 14, 849–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caicedo JC, McQuin C, Goodman A, Singh S & Carpenter AE Weakly Supervised Learning of Single-Cell Feature Embeddings. in 9309–9318 (2018). [DOI] [PMC free article] [PubMed]
- 36.Strezoska Ž et al. High-content analysis screening for cell cycle regulators using arrayed synthetic crRNA libraries. J. Biotechnol 251, 189–200 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Kim HS et al. Arrayed CRISPR screen with image-based assay reliably uncovers host genes required for coxsackievirus infection. Genome Res. 28, 859–868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Groot R, Lüthi J, Lindsay H, Holtackers R & Pelkmans L Large-scale image-based profiling of single-cell phenotypes in arrayed CRISPR-Cas9 gene perturbation screens. Mol. Syst. Biol 14, e8064 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michlits G et al. CRISPR-UMI: single-cell lineage tracing of pooled CRISPR–Cas9 screens. Nat. Methods 14, 1191–1197 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Schmierer B et al. CRISPR/Cas9 screening using unique molecular identifiers. Mol. Syst. Biol 13, 945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ke R et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–860 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Larsson C, Grundberg I, Söderberg O & Nilsson M In situ detection and genotyping of individual mRNA molecules. Nat. Methods 7, 395–397 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Feldman D et al. Optical Pooled Screens in Human Cells. Cell 179, 787–799.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawson MJ et al. In situ genotyping of a pooled strain library after characterizing complex phenotypes. Mol. Syst. Biol 13, 947 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camsund D et al. Time-resolved imaging-based CRISPRi screening. Nat. Methods 17, 86–92 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Emanuel G, Moffitt JR & Zhuang X High-throughput, image-based screening of pooled genetic-variant libraries. Nat. Methods 14, 1159–1162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, Lu T, Emanuel G, Babcock HP & Zhuang X Imaging-based pooled CRISPR screening reveals regulators of lncRNA localization. Proc. Natl. Acad. Sci 116, 10842–10851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Datlinger P et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasle N et al. High-throughput, microscope-based sorting to dissect cellular heterogeneity. Mol. Syst. Biol 16, e9442 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan X et al. High-content imaging-based pooled CRISPR screens in mammalian cells. J. Cell Biol 220, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee J et al. Versatile phenotype-activated cell sorting. Sci. Adv 6, eabb7438 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanfer G et al. Image-based pooled whole-genome CRISPRi screening for subcellular phenotypes. J. Cell Biol 220, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wheeler EC et al. Pooled CRISPR screens with imaging on microraft arrays reveals stress granule-regulatory factors. Nat. Methods 17, 636–642 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stringer C, Wang T, Michaelos M & Pachitariu M Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021). [DOI] [PubMed] [Google Scholar]
- 55.McQuin C et al. CellProfiler 3.0: Next-generation image processing for biology. PLOS Biol. 16, e2005970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reicher A, Koren A & Kubicek S Pooled protein tagging, cellular imaging, and in situ sequencing for monitoring drug action in real time. Genome Res. 30, 1846–1855 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feldman D, Singh A, Garrity AJ & Blainey PC Lentiviral co-packaging mitigates the effects of intermolecular recombination and multiple integrations in pooled genetic screens. bioRxiv 262121 (2018) doi:10.1101/262121. [Google Scholar]
- 58.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edelstein AD et al. Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmid-Burgk JL et al. Highly Parallel Profiling of Cas9 Variant Specificity. Mol. Cell 78, 794–800.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feldman D & Funk L Pooled genetic perturbation screens with image-based phenotypes, OpticalPooledScreens, doi: 10.5281/zenodo.5002684, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kosuri S et al. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nat. Biotechnol 28, 1295–1299 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doench JG et al. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol 32, 1262–1267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kutner RH, Zhang X-Y & Reiser J Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc 4, 495–505 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Köster J & Rahmann S Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Williams E et al. Image Data Resource: a bioimage data integration and publication platform. Nat. Methods 14, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo B et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci 105, 20380–20385 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Spreadsheet calculator for estimating the scale of an optical pooled screen.
Supplementary Table 2. Oligo pool dialout primer sequences.
Supplementary Table 3. Primer sequences for NGS library preparation.
Supplementary Table 4. Oligo sequences for in situ amplification of sgRNA sequences.
Supplementary Figure 1. Cell line comparison. (a) Suitability of example cell lines for optical pooled screening. NFkB screens were published in ref.43 (b) Example cell and nuclear segmentation using CellPose, a generalist algorithm based on a deep learning model trained on diverse cell and image types. For each cell line, the left panel shows the exact RGB input provided to CellPose, and the right panel shows segmentations using the “cyto” and “nuclei” models in single-model mode (scale bar, 100 μm). All cell lines were analyzed identically, varying only in a single “diameter” parameter automatically generated by the CellPose “cyto” calibration model. Segmentation of a 1.2mm × 1.2mm field of view took ~4 min on 1 CPU.
Supplementary Figure 2. Image analysis pipeline overview. Flowchart demonstrating the major steps of the screen processing pipeline. Input data are represented by rounded shapes; image and tabular data are in green and purple, respectively.
Data Availability Statement
The dataset used to produce Figures 3 and 4 was originally published in ref.43 and is publicly available from Cell-IDR66 (idr0071, experiment C).