

SUMMARY
Cys-tRNACys is required for translation and is typically synthesized by cysteinyl-tRNA synthetase (CysRS). However, Methanocaldococcus jannaschii synthesizes Cys-tRNACys by an indirect pathway, whereby O-phosphoseryl-tRNA synthetase (SepRS) acylates tRNACys with phosphoserine (Sep), and Sep-tRNA–Cys-tRNA synthase (SepCysS) converts the tRNA-bound phosphoserine to cysteine. We show here that M. jannaschii SepRS differs from CysRS by recruiting the m1G37 modification as a determinant for aminoacylation and by exhibiting limited discrimination against mutations of conserved nucleotides. Kinetic and binding measurements show that both SepRS and SepCysS bind the reaction intermediate Sep-tRNACys tightly, and that these two enzymes form a stable binary complex that promotes conversion of the intermediate to the product and sequesters the intermediate from binding to elongation factor EF-1αor infiltrating into the ribosome. These results highlight the importance of the protein binary complex for efficient synthesis of Cys-tRNACys.
Aminoacylation of tRNA is important for the amino acid-codon relationships of the genetic code. Typically, an aminoacyl-tRNA synthetase (aaRS) activates an amino acid with ATP and catalyzes transfer of the activated aminoacyl-adenylate to the 3′ end of its cognate tRNAs1 to synthesize an aminoacyl-tRNA (aa-tRNA) that enters the ribosome at a codon position that matches the tRNA anticodon. For each of the 20 canonical amino acids, there is a dedicated tRNA-aaRS pair. In addition, selenocysteine (Sec)2 and pyrrolysine (Pyl)3 are also decoded co-translationally, each at an internal stop codon using a specialized tRNA-aaRS pair. Amino acids that are excluded from the genetic code are believed to lack the vehicle of a tRNA-aaRS pair. An example is phosphoserine, which is created post-translationally by serine kinases to activate proteins for cell signaling.
The discovery of the natural SepRS for catalyzing O-phosphoserylation of tRNA offers the potential to gain insight into the evolution of the genetic code. SepRS was identified in methanogenic archaea, where it acylates tRNACys with Sep to synthesize Sep-tRNACys, which is converted by SepCysS to Cys-tRNACys (Fig. 1a)4. The identification of the SepRS–SepCysS pathway resolved the decade-long puzzle of the lack of a CysRS in M. jannaschii. Only two other archaea (Methanothermobacter thermautotrophicus and Methanopyrus kandleri) use the SepRS–SepCysS pathway obligatorily for synthesis of Cys-tRNACys, whereas many others (e.g., M. maripaludis, Archaeoglobus fulgidus) contain genes for both the CysRS and SepRS–SepCysS pathways5. High-resolution crystal structures of SepRS and SepCysS of A. fulgidus have been solved6–8. The structure of SepRS is most closely related to that of the class II PheRS α subunit and is unlike the class I CysRS structure. The structure of SepCysS is most similar to that of CsdB9, which is a member of the pyridoxal 5’-phosphate-dependent cysteine desulfurases that remove the thiol from a substrate cysteine and donate it to Fe-S clusters or modified nucleotides. However, despite these structures, mechanistic insights into individual reactions of the SepRS–SepCysS pathway remain obscure. Recognition of tRNA by SepRS has been recently probed7,10, the source of the enzyme was from M. maripaludis or A. fulgidus. No studies have been performed thus far on enzymes of the SepRS–SepCysS pathway that is obligate for cell growth.
Figure 1.

Aminoacylation of tRNACys transcript by M. jannaschii SepRS. (a) A scheme showing the indirect pathway for synthesis of Cys-tRNACys. (b) Sequence and cloverleaf structure of M. jannaschii tRNACys, where modification of m1G37 and mutations of U73G, G34C, C35U, A36G, G37A, A38G, G15C, and C48G are indicated by arrows. (c) Single turnover time courses of aminoacylation of the unmodified and m1G37-modified transcripts of M. jannaschii tRNACys by 10 μM SepRS, showing kapp of 0.15 s−1 and 1.3 s−1, respectively. The term ± refers to standard deviation.
The exclusive use of the SepRS-SepCysS pathway in M. jannaschii demands high efficiency synthesis of the reaction intermediate Sep-tRNACys by SepRS. A major obstacle that has complicated in vitro studies has been the poor aminoacylation activity of the M. jannaschii tRNACys transcript by the homologous SepRS, rendering the use of heterologous enzymes in some studies10. This poor activity emphasizes the importance of one or more of the many modified nucleotides that are present in archaeal tRNA. M. jannaschii tRNACys has conserved U73 and the GCA anticodon that are required for efficient aminoacylation by CysRS11, which provides a basis for comparison of tRNA recognition by SepRS and CysRS. Aminoacylation by SepRS generates the intermediate Sep-tRNACys, which must be efficiently delivered to SepCysS. If the intermediate is released from SepRS and diluted by bulk solvent, then delivery to SepCysS would be delayed and possibly intercepted by the archaeal EF1α (the homolog of bacterial EF-Tu). In most metabolic pathways that consist of multiple enzymatic steps, the intermediary products are transferred from one enzyme to the next without equilibrating with bulk solvent. Enzymes involved in successive reactions often associate as complexes to facilitate transfer of reaction intermediates12,13. The formation of a stable SepRS–SepCysS complex would provide such a mechanism to improve the overall efficiency of synthesis of Cys-tRNACys.
Here we investigate the SepRS–SepCysS pathway in M. jannaschii, using recombinant enzymes and tRNA derived from the same species. Kinetic results, obtained in both steady state and single turnover conditions, show that SepRS is distinct from CysRS in using the m1G37 modification, adjacent to the tRNA anticodon, as a determinant for efficient aminoacylation. Also, we show that SepRS exhibits limited discrimination compared to CysRS against mutations of conserved tRNA nucleotides. Importantly, we have isolated a stable binary complex of SepRS–SepCysS that effectively sequesters the highly unstable Sep-tRNACys intermediate and protects it from binding to EF-1α to facilitate the dedicated synthesis of Cys-tRNACys.
RESULTS
The m1G37 modification of M. jannaschii tRNACys
Aminoacylation by M. jannaschii SepRS (expressed and purified from E. coli) used tRNA that was 32P-labeled at A76. After aminoacylation, the labeled tRNA was degraded by P1 nuclease, resulting in a mixture of labeled Sep-AMP (product) and labeled AMP (unreacted substrate) that were resolved by TLC and quantified by image analysis. While the unfractionated M. jannaschii crude tRNA was aminoacylated to 1% (data not shown), consistent with the fraction of tRNACys in the crude14, the wild-type tRNA transcript was poorly aminoacylated. Because the great majority of G37-containing tRNAs have the m1G modification (http://lowelab.ucsc.edu/GtRNAdb), this modification was introduced to the wild-type tRNA transcript (Fig. 1b), using the MjTrm5 enzyme and the methyl donor S-adenosyl methionine15. Because the Trm5 reaction was not quantitative, the unmodified fraction was selectively cleaved by RNase H and removed by gel electrophoresis16.
The purified m1G37-tRNACys transcript was chargeable to 60% by molar excess of SepRS, whereas the unmodified transcript was chargeable to 30% (Fig. 1c). Steady-state kinetics showed that the modification improved the kcat from 0.07 s−1 to 0.24 s−1, but had little effect on the Km of tRNA (Table 1). Notably, the kcat of the modified tRNA is closer to the kcat of the native tRNACys (1.0 s−1) when assayed in the crude tRNA. Overall, the m1G37-tRNA transcript exhibited a 4.3-fold higher catalytic efficiency (kcat/Km) than the unmodified transcript, while the fully modified native tRNACys exhibited a 23-fold higher kcat/Km. Thus, while the m1G37 modification is important for aminoacylation, other site-specific modifications present in the native tRNACys are important as well. In contrast, efficient aminoacylation by bacterial and eukaryotic CysRS requires no tRNA modification11.
Table 1.
Improvement of aminoacylation of M. jannaschii tRNACys by m1G37.
| Steady-state kinetics | Single turnover kinetics | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Km (μM) | kcat (s−1) | kcat/Km (μM−1s−1) | relative | Kd (μM) | kchem (s−1) | kchem/Kd (μM−1s−1) | relative | |
|
| ||||||||
| tRNACys | 1.1 ± 0.2 | 0.07 ± 0.01 | 0.06 ± 0.01 | 1.0 | 1.4 ± 0.3 | 0.18 ± 0.03 | 0.13 ± 0.04 | 1.0 |
| m1G37-tRNACys | 0.97 ± 0.01 | 0.24 ± 0.04 | 0.25 ± 0.04 | 4.3 | 1.7 ± 0.3 | 1.3 ± 0.1 | 0.8 ± 0.1 | 6.1 |
| Native tRNACys | 0.7 ± 0.2 | 1.0 ± 0.2 | 1.4 ± 0.5 | 23 | ||||
±: standard deviation.
To better evaluate the importance of m1G37 for SepRS in terms of individual rate and equilibrium binding constants, single turnover kinetics with molar excess of SepRS relative to tRNA was performed. Kinetic titration of SepRS with the unmodified tRNA exhibited a saturating kapp of 0.18 ± 0.03 s−1, whereas titration with the m1G37-tRNA transcript showed a nearly 10-fold increase in the saturating kapp to 1.3 ± 0.1 s−1 (Supplementary Fig. 1a–d). In both cases, because the time courses showed well-fit single exponential kinetics over the full concentration range tested, the saturating kapp was defined as kchem to reflect that the chemistry is likely the rate-determining step, although this kchem might also encompass conformational rearrangements of the enzyme-tRNA complex11. The titrations also showed that the unmodified and modified transcripts had nearly identical kinetic Kds of 1.4 ± 0.3 and 1.7 ± 0.3 μM, respectively (Table 1). Because m1G37 improves both kcat and kchem to the tRNACys transcript, but little effect on Km or Kd, the modification has the ability to modulate the tRNA acceptor end activity, even though it is localized to the anticodon end. For more accurate determination of kinetics and binding analysis, all studies described below utilized purified m1G37-tRNA transcripts.
Mutational analysis of tRNA aminoacylation by SepRS
To elucidate the nucleotides necessary for aminoacylation by SepRS, mutations were introduced to the conserved U73 and the GCA anticodon of tRNACys (Fig. 1b). Also, positions 37 and 38 of the anticodon loop were evaluated because they are recognized by SepRS in the co-crystal structure7. In addition, the 15–48 base pair was of interest, because of the recent demonstration that E. coli CysRS uses indirect readout to recognize the 15–48 tertiary base pair in the tRNA elbow region17 (Fig. 1b).
The effects of mutations on the SepRS activity were examined in single turnover (Table 2) and steady state (Supplementary Table 1) kinetics with respect to M. jannaschii tRNACys and were compared with the effects of corresponding mutations in E. coli tRNACys on the E. coli CysRS activity. Although all tRNA mutants showed a reduced activity relative to the wild-type transcript, the magnitudes of reduction for the SepRS activity were markedly smaller than those for the CysRS activity. For example, U73 is the single most important nucleotide for CysRS11: the U73G substitution decreased the specificity factors kcat/Km and kchem/Kd by 105-106-fold. In contrast, this substitution decreased these factors only by 30- and 10-fold for SepRS, respectively. In the anticodon region, G34 is the most important nucleotide for CysRS. However, while the G34C substitution decreased the specificity factors for CysRS by 103-104 fold, it decreased the factors for SepRS by only 50- and 25-fold in steady state and single turnover kinetics. The contrast between CysRS and SepRS at the second and third positions of the anticodon is equally striking: while the C35U and A36G substitutions decreased the specificity factors for CysRS by 102-103-fold, each decreased the factors for SepRS by only 20–30-fold. The relative insensitivity of SepRS to mutations of nucleotide bases is also apparent at positions 37 and 38: the G37A substitution reduced the specificity factors of SepRS by ~5-fold, while the A38G substitution reduced the factors by 10–20-fold. Similarly, at the 15–48 position, while the C15-G48 substitution decreased the specificity factors of CysRS by 30–50-fold, it decreased the factors of SepRS by only 2.5–5-fold. Thus, although CysRS and SepRS both recognize conserved and non-conserved nucleotides at common positions in tRNACys, CysRS exhibits substantially stronger discrimination against mutations of these nucleotides.
Table 2.
Single turnover kinetics
| tRNA | M.j SepRS vs. M.j tRNACys(*) | E.coli CysRS vs. E coli tRNACys | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Kd (μM) |
kchem (s−1) |
kchem/Kd (μM−1s−1) | relative | kchem/Kd (μM−1s−1) | relative | |
|
| ||||||
| WT | 1.7 ± 0.3 | 1.3 ± 0.1 | 0.8 ± 0.1 | 1 | 4.8 | 1 |
| U73G | 0.6 ± 0.1 | 0.04 ± 0.02 | 0.07 ± 0.04 | 0.09 | 1.1× 10−4 | 2 × 10−5 |
| G34C | 1.5 ± 0.3 | 0.048 ± 0.003 | 0.032 ± 0.007 | 0.04 | 0.0041 | 1 × 10−3 |
| C35U | 1.6 ± 0.6 | 0.048 ± 0.008 | 0.03 ± 0.01 | 0.04 | 0.024 | 5 × 10−3 |
| A36G | 3 ± 1 | 0.11 ± 0.03 | 0.04 ± 0.02 | 0.05 | 0.07 | 0.01 |
| G37A | 1.9 ± 0.9 | 0.05 ± 0.01 | 0.03 ± 0.02 | 0.04 | Not determined | |
| A38G | 0.4 ± 0.2 | 0.03 ± 0.004 | 0.08 ± 0.04 | 0.1 | Not determined | |
| G15C-C48G | 0.4 ± 0.2 | 0.13 ± 0.02 | 0.3 ± 0.2 | 0.4 | 0.16 | 0.03 |
Transcripts of m1G37-Mj tRNACys were used except G37A, ±: standard deviation.
Release of Sep-tRNACys from SepRS assisted by SepCysS
The single turnover rate kchem (1.3 s−1) of the wild-type m1G37-tRNACys is faster than the steady-state rate of enzyme turnover kcat (0.24 s−1), suggesting the possibility that the steady-state rate might be limited by release of the Sep-tRNACys product. To test this possibility, pre-steady-state kinetic experiments were performed, in which Sep and ATP were at saturating concentrations while the ratios and concentrations of SepRS and tRNA were adjusted so as to observe a single turnover followed by a subsequent steady-state rate. The time courses indeed showed burst kinetics, exhibiting a rapid burst of synthesis of Sep-tRNACys, followed by a slower and linear phase of steady-state synthesis (Fig. 2a). Fitting the data of a representative time course to a burst equation yielded kchem of 0.89 s−1 in the first turnover and kcat of 0.23 s−1 in the steady state, similar to values determined from single turnover and steady-state analysis (Table 2).
Figure 2.
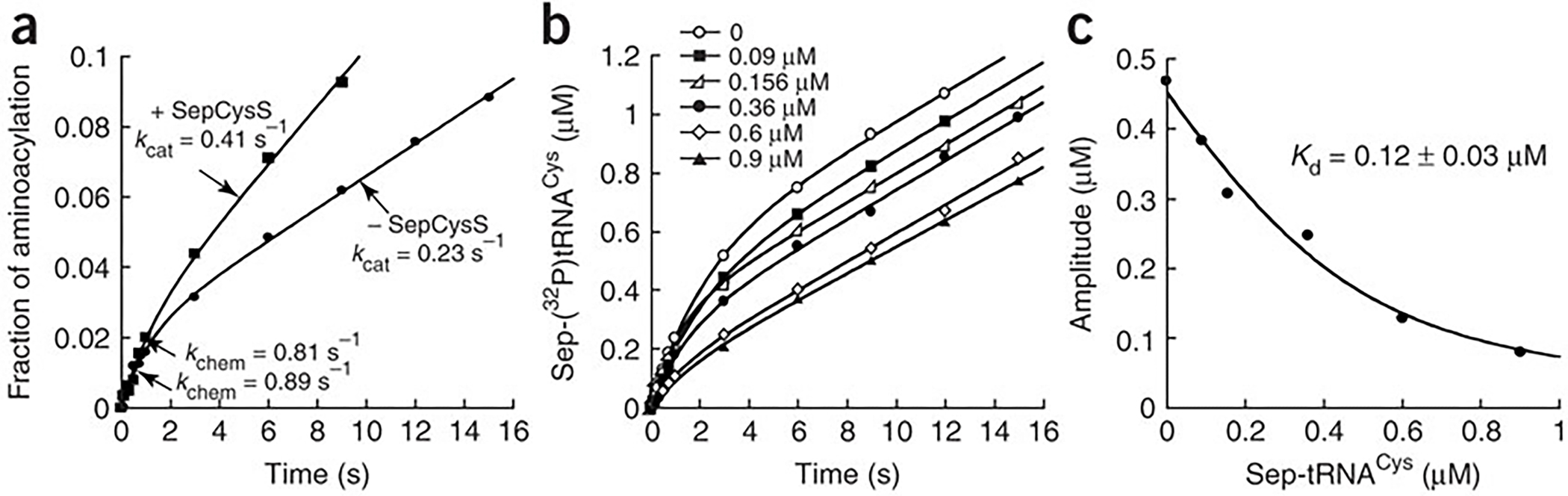
Burst kinetics of aminoacylation. (a) Pre-steady-state time course of aminoacylation of m1G37-tRNACys (7.5 μM) by SepRS (0.25 μM) in the absence or presence of SepCysS (3.12 μM). (b) Time courses of aminoacylation with increasing concentrations of Sep-tRNACys (0.09–0.9 μM) in pre-steady-state conditions of 7 μM m1G37-tRNACys and 0.5 μM SepRS. (c) Replot of the amplitude of the burst phase vs the concentration of Sep-tRNACys fit to a quadratic equation. The term ± refers to standard deviation.
The appearance of burst kinetics indicated that product release from SepRS was rate limiting. One reason could be that SepRS binds more tightly to Sep-tRNACys than to the substrate tRNACys. The Kd for Sep-tRNACys was determined by kinetics, treating it as an inhibitor of the forward reaction. Sep-tRNACys was formed from the aminoacylation reaction, pre-incubated in varying concentrations with SepRS, and the enzyme-product complexes were rapidly mixed with substrates under pre-steady-state burst conditions. The concentration-dependent effects of inhibition were clearly manifested in changes of the burst amplitude, without changes in the burst or steady-state rate (Fig. 2b). The amplitude of the burst represented the amount of uninhibited enzyme available for aminoacylation in the first turnover. A fit of the data to a quadratic equation yielded Kd of 0.12 ± 0.03 μM for the SepRS–Sep-tRNACys complex (Fig. 2c). This Kd is lower than the Kd of 1.7 ± 0.3 μM for the substrate tRNACys (Table 2) by more than 10-fold, supporting the notion that the higher affinity of the enzyme to Sep-tRNACys accounts for the slow release of this product.
The tight binding of Sep-tRNACys by SepRS might require the downstream SepCysS to help to improve SepRS turnover. To test this possibility, aminoacylation kinetics of SepRS was examined in burst conditions supplemented with SepCysS. Indeed, the addition of molar excess of SepCysS increased the steady-state kcat of SepRS by nearly 2-fold from 0.23 to 0.41 s−1, while maintaining the kchem at 0.81 s−1 of the first enzyme turnover (Fig. 2a). The improvement of the steady-state kcat arising from the addition of SepCysS reflected an effect on the release of this aa-tRNA from SepRS.
Both SepRS and SepCysS bind tightly to Sep-tRNACys
For SepCysS to assist in the release of Sep-tRNACys from SepRS, the binding affinity of SepCysS for the aa-tRNA must be at least similar to that of the SepRS–aa-tRNA complex. This was tested by equilibrium binding measurements, in which the tRNA was fluorescently labeled with pyrrolo-C (PyC18) at position 75 to specifically monitor local changes of the CCA end (Fig. 3a), which carries the Sep moiety that is recognized by SepCysS. The PyC-labeled tRNA transcript was aminoacylated by SepRS and the product Sep-PyC-tRNACys was purified by reverse phase HPLC. Fluorescent emission of the labeled Sep-PyC-tRNACys increased upon binding of SepCysS, and a titration of the enzyme revealed a binding curve (Fig. 3b, inset), showing a Kd of 0.1 ± 0.02 μM of SepCysS for the aa-tRNA (Fig. 3c). In parallel, the Kd of SepRS for the fluorescent labeled Sep-PyC-tRNACys was determined by titration to be 0.2 ± 0.08 μM (Fig. 3c), which is within 2-fold of the Kd of 0.12 ± 0.03 μM as determined from kinetic analysis (Fig. 2c). These Kd values are similar to each other, demonstrating that SepRS and SepCysS bind with similar affinity to Sep-tRNACys.
Figure 3.
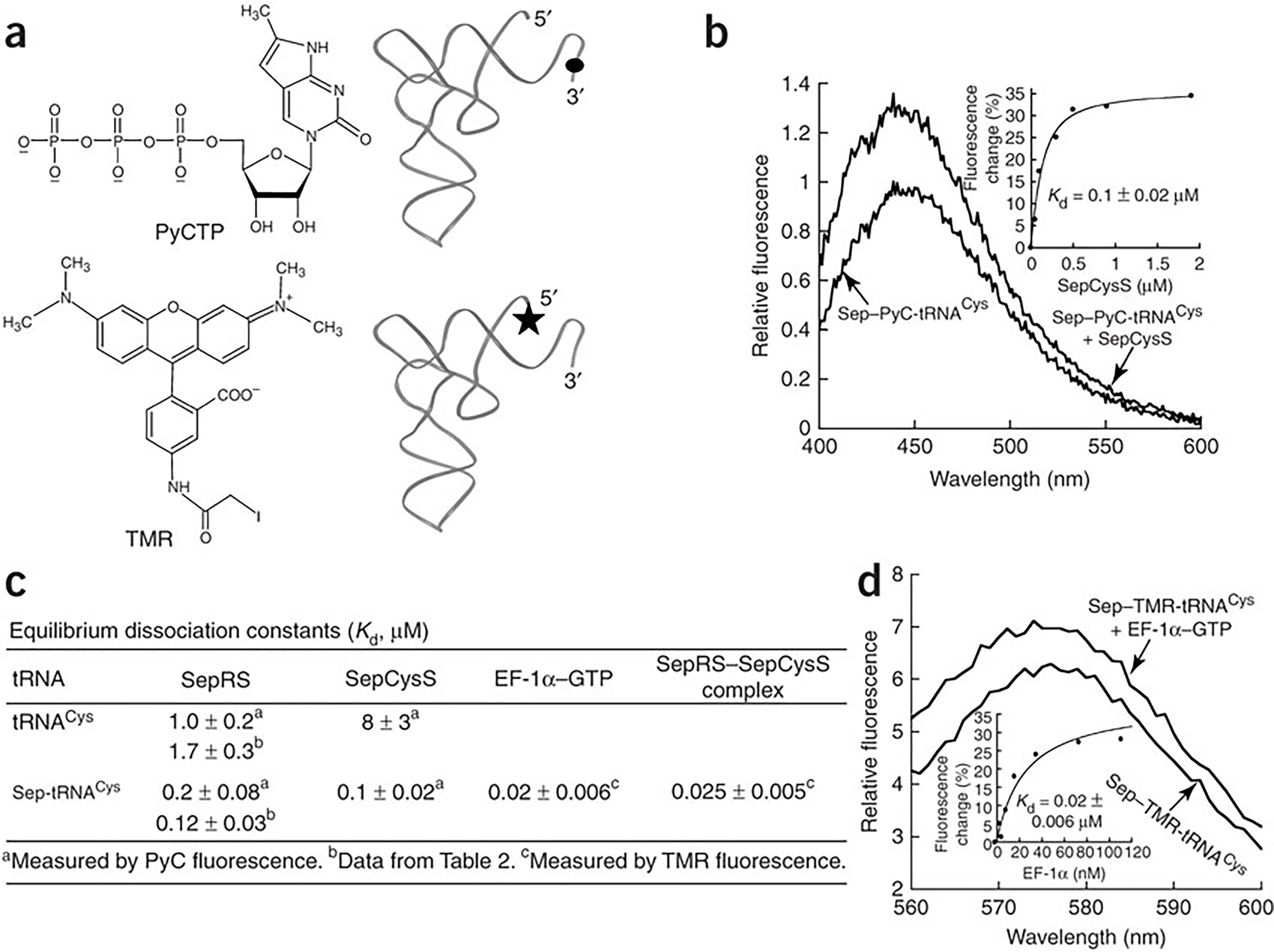
Binding of Sep-tRNACys to SepCysS and EF-1α-GTP. (a) Structures of PyCTP and TMR and their incorporation to the 3’ and 5’ ends of tRNA, respectively. (b) Fluorescence emission spectra of PyC-labeled Sep-tRNACys (0.1 μM) in the absence or presence of M. jannaschii SepCysS (0.9 μM). Inset: replot of the PyC fluorescence change vs SepCysS concentration fit to a quadratic equation. (c) Dissociation constants of tRNACys and Sep-tRNACys for SepRS, SepCysS, EF-1α and the SepRS–SepCysS complex. (d) Fluorescence emission spectra of TMR-labeled Sep-tRNACys (0.2 μM) in the absence or presence of M. jannaschii EF-1α (0.18 μM). Inset: replot of the TMR fluorescence change vs EF-1α concentration fit to a hyperbolic equation. The term ± refers to standard deviation.
Binding measurements were also performed with the PyC-labeled but uncharged tRNACys. Relative to the charged Sep-tRNACys, SepRS should have reduced affinity for the uncharged tRNACys based on the appearance of burst kinetics (Fig. 2a). The reduced affinity was also expected for SepCysS because the uncharged tRNA is not a substrate. Indeed, fluorescence titrations showed that the Kd of SepRS for the uncharged tRNA was 1.0 ± 0.2 μM (Fig. 3c), similar to the Kd of 1.7 ± 0.3 μM determined by kinetics (Table 2). The Kd of SepCysS for the uncharged tRNA was 8 ± 3 μM (Fig. 3c). Both Kd values were higher than the respective Kds of these enzymes for Sep-tRNACys.
Although Sep-tRNACys can bind tightly to SepRS or SepCysS, should it be released into solution, it would be unstable, particularly at the high temperature where M. jannaschii thrives. For example, at 60 °C in the absence of a protein-binding partner, Sep-tRNACys is rapidly deacylated with a half-life of 7.5 min (Supplementary Fig. 2). The most likely binding partner for Sep-tRNACys is the M. jannaschii elongation factor EF-1α, which would bring the charged tRNA to the ribosome. A recombinant M. jannaschii EF-1α was expressed and purified from E. coli and used for determination of Kd for Sep-tRNACys by fluorescence titration. Here, the tRNA was labeled by tetramethyl rhodamine (TMR) at the 5’ end19 (Fig. 3a). A titration of the TMR-labeled Sep-tRNACys with EF-1α-GTP showed increased fluorescence with a Kd of 0.02 ± 0.006 μM (Fig. 3d, inset), which is well within the range of the Kds of bacterial EF-Tu for correctly matched aa-tRNAs20. Thus, although Sep-tRNACys is mis-matched, it is capable of binding to EF-1α and has the potential to be brought to the ribosome.
A binary SepRS–SepCysS complex binds Sep-tRNACys
Because Sep-tRNACys has a higher affinity to EF-1α than to SepRS or SepCysS alone (Fig. 3c), the possibility of SepRS and SepCysS forming a binary complex to prevent the aa-tRNA from binding to EF-1α was tested. A pull-down assay with a metal affinity resin was used to determine if a His-tagged SepCysS would associate with a non-tagged SepRS. After extensive washes, components that remained with the resin were analyzed by SDS-PAGE for proteins and by 7M urea-PAGE for tRNA (Fig. 4a). Control experiments showed that the pull-down by the resin was specific for the His-tagged SepCysS, and that no protein or tRNA was pulled down in the absence of SepCysS (lane 5).
Figure 4.
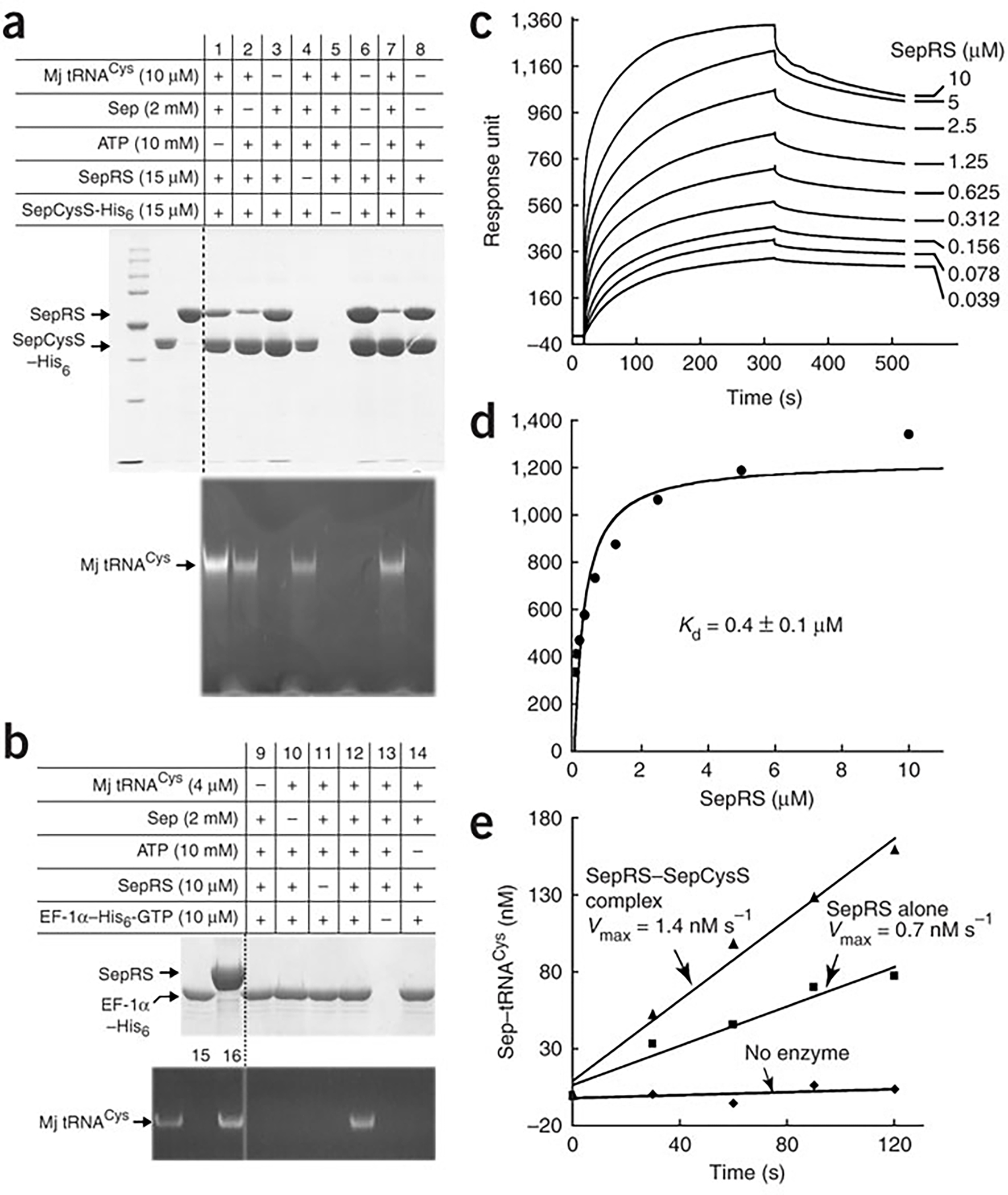
The SepRS–SepCysS binary complex. (a) Pull-down of the M. jannaschii SepRS–SepCysS binary complex analyzed by SDS-PAGE and by 12% (w/v)/7M urea PAGE. (b) Pull-down of M. jannaschii EF-1α-GTP and Sep-tRNACys. A control showing EF-1α discrimination against uncharged tRNACys and recognition of Cys-tRNACys is provided in lanes 15 and 16 respectively. (c) Kinetics of the concentration-dependent binding of SepRS to SepCysS monitored by a biosensor. (d) Replot of the response units determined from (c) vs SepRS concentrations fit to a binding equation. (e) Aminoacylation of m1G37-tRNACys by SepRS attached to a metal resin in the absence or presence of SepCysS. The term ± refers to standard deviation.
The pull-down assay succeeded in the isolation of SepRS by SepCysS, which was observed in the absence of tRNA (lanes 3, 6, 8), suggesting that the two enzymes formed a stable binary complex. The binary complex was specific to SepRS, because replacement of SepRS with E. coli CysRS eliminated the pull-down (Supplementary Fig. 3). The pull-down assay also succeeded in the isolation of a ternary complex of SepRS, SepCysS, and Sep-tRNACys, which was observed when tRNACys was present in the aminoacylation reaction (lane 7). Additionally, the pull-down assay isolated a ternary complex of SepRS, SepCysS, and the uncharged tRNACys, which was observed when the aminoacylation reaction contained tRNACys but without ATP or Sep (lanes 1, 2). This result can be explained by the observation that even SepCysS alone can bind the uncharged tRNACys under the assay condition (lane 4). Importantly, a His-tagged EF-1α was unable to pull down SepRS as a binary complex (Fig. 4b, lane 9), or as a ternary complex with the uncharged tRNACys (lanes 10, 14) or the charged Sep-tRNACys (lane 12). However, EF-1α did pull down Sep-tRNACys without co-binding with SepRS (lane 12), validating the ability of the factor to bind the aa-tRNA as determined by fluorescence titration (Fig. 3c). These results demonstrate that SepRS forms a binary complex with SepCysS, but not with EF-1α, and that the SepRS–SepCysS binary complex binds Sep-tRNACys to form a ternary complex.
The formation of the SepRS–SepCysS binary complex was verified by surface plasmon resonance using the BIAcore biosensor, which measures the binding interaction in real time. SepCysS was immobilized onto a chip, while solutions of increasing concentration of SepRS were passed over. The association of the two enzymes was monitored by an increase in response units, while the dissociation of the complex was monitored upon removal of SepRS from the binding buffer (Fig. 4c). Increasing concentrations of SepRS in the flow chamber relative to the coated chips resulted in increases in the response units (Fig. 4d), which revealed the Kd of 0.4 ± 0.1 μM for the binary complexs. This Kd is lower than the values reported for other stable protein-protein complexes measured by the same technique, such as the Kd of 2.9 μM for the human IleRS–ProRS complex21, and the Kd of 1.3 μM for the complex of M. jannaschii ProRS with a protein in C1 metabolism22.
The importance of the binary complex was evaluated by testing the SepRS activity in the complex. To preserve the stoichiometry and orientation of the complex as isolated by the pull-down assay, the enzyme activity was tested while the complex was still attached to the resin. A His-tagged SepRS was used to pull down the non-tagged SepCysS, and a control was provided by pull-down of just the His-tagged SepRS. Aminoacylation with saturating concentrations of m1G37-tRNACys showed that SepRS in the pulled-down binary complex was active, and that it exhibited a 2-fold higher Vmax than that of the control (Fig. 4e). Because these assays were done in parallel, the amounts of SepRS pulled down by the resin were roughly the same, suggesting that the Vmax effect reflected the kcat effect. Importantly, the 2-fold improvement in kcat of the binary complex on solid phase recapitulates the 2-fold effect on kcat observed by addition of SepCysS to the burst kinetics of SepRS (Fig. 2a).
The ability of Sep-tRNACys to affect the binding affinity of the SepRS–SepCysS binary complex was assessed. With the TMR-labeled Sep-tRNACys as the substrate, a saturating concentration of SepRS was used to form the SepRS–aa-tRNA complex. The fluorescence of the preformed complex increased upon addition of SepCysS (Fig. 5a) and a titration with SepCysS revealed a binding curve, from which a Kd of 0.025 ± 0.005 μM was determined for the ternary complex (Fig. 5a, inset). This Kd is interpreted as the dissociation constant of the SepRS–SepCysS binary complex in the presence of the aa-tRNA, which is lower than the Kd of 0.4 ± 0.1 μM of the binary complex in the absence of the aa-tRNA (Fig. 4d) by 16-fold. The converse experiment was also performed, by monitoring the addition of SepRS to a preformed complex of SepCysS–aa-tRNA. However, no fluorescence change was observed, indicating that the aa-tRNA stayed bound with SepCysS. These experiments suggests that the fluorescence change upon addition of SepCysS to the SepRS–aa-tRNA complex reflects transfer of the labeled aa-tRNA from SepRS to SepCysS, consistent with the notion that the binding of SepCysS prompts SepRS to release the aa-tRNA to SepCysS, as indicated by both the burst kinetics (Fig. 2a) and pull-down assay (Fig. 4e). Thus, the Kd of 0.025 ± 0.005 μM for the ternary complex (Fig. 5a) can be also interpreted as the dissociation constant of SepCysS for the aa-tRNA in the presence of SepRS, which is lower than the Kd of 0.1 ± 0.02 μM of the SepCysS–aa-tRNA complex in the absence of SepRS by 4-fold (Fig. 3c). Thus, the binary SepRS–SepCysS complex strengthens the affinity of SepCysS for the aa-tRNA.
Figure 5.
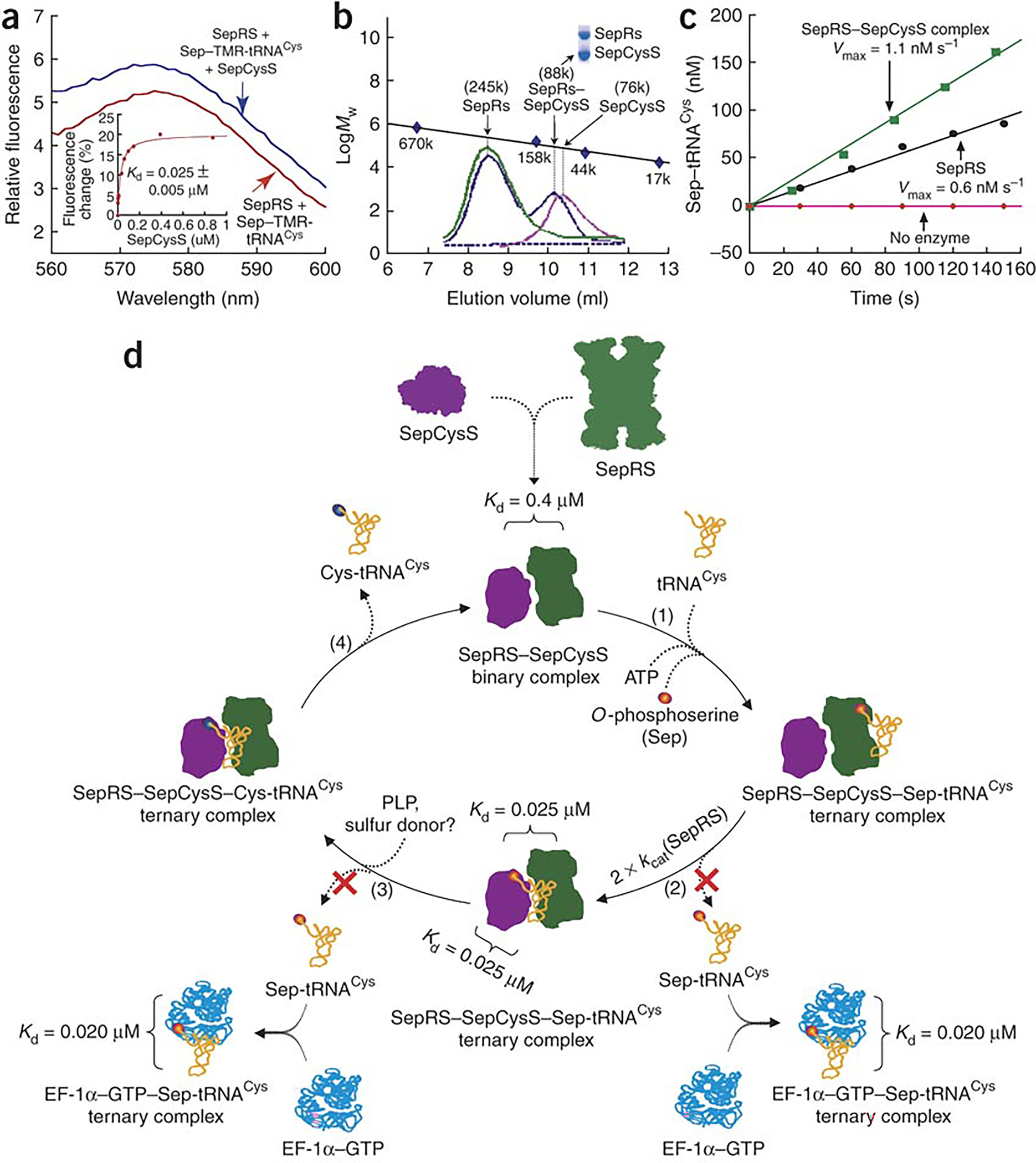
Binding of Sep-tRNACys to the SepRS–SepCysS complex. (a) Fluorescence emission spectra of TMR-labeled Sep-tRNACys (0.2 μM) with saturating SepRS (5 μM) in the absence or presence of SepCysS (0.03 μM). Inset: replot of the TMR fluorescence change vs SepCysS concentration fit to a hyperbolic equation. (b) Gel filtration of SepRS alone (green), SepCysS alone (pink), and the SepRS-SepCysS complex (blue). Mw markers were thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), and equine myoglobulin (17 kDa). (c) Aminoacylation of m1G37-M. jannaschii tRNACys by SepRS alone (5 nM from fraction 8.5) and by SepRS in the complex (5 nM from fraction 10.3). Inset: SDS-PAGE analysis of the SepRS–SepCysS complex (from fraction 10.3). (d) A scheme showing the pathway of synthesizing and sequestering Sep-tRNACys by the SepRS–SepCysS binary complex for conversion to Cys-tRNACys. The term ± refers to standard deviation.
Finally, the stoichiometry of the SepRS–SepCysS binary complex was determined by gel filtration (Fig. 5b). Analysis of SepRS alone yielded a Mw of ~245 kDa, consistent with the tetrameric structure of the enzyme6,7. Analysis of SepCysS alone yielded a Mw of ~76 kDa, consistent with the dimeric structure of the enzyme8. When molar excess of SepRS was mixed with SepCysS, gel filtration identified a single complex with a Mw of ~88 kDa, where the two enzymes co-existed at a 1:1 stoichiometry on an SDS-PAGE (inset). Aminoacylation under the kcat condition showed that the SepRS activity in the complex (Vmax = 1.1 nM/s) was 2-fold higher than that of SepRS alone (Vmax = 0.6 nM/s) per enzyme monomer (Fig. 5c). Because the high SepRS activity in the complex far exceeded the activity provided by the small amount of contaminating free SepRS, and because the 2-fold activity enhancement reproduced the 2-fold effect of SepCysS on SepRS activity in burst kinetics and in pull-down assays (Figs 2a, 4e), indicating that SepCysS is fully functional, these results suggest that both SepRS and SepCysS are active as a monomer in the complex. Thus, although the co-crystal structure of SepRS contains two tRNACys molecules per tetramer7, indicating an asymmetry of the four identical subunits, the asymmetry is resolved by forming the SepRS–SepCysS complex.
DISCUSSION
Three key features distinguish SepRS from CysRS in aminoacylation of tRNACys. First, the m1G37 modification is a determinant for SepRS: without m1G37, the effects of some mutations in the tRNA transcript are not detectable. For example, while the G34C mutation in the m1G37-transcript caused a loss of 50-fold in kcat/Km and 25-fold in kchem/Kd of aminoacylation (Table 2), these effects were not detectable in the unmodified transcript, even with elevated enzyme concentrations. Two previous studies of SepRS used unmodified tRNA transcript7,10 and thus had limited ability to monitor mutational effects. As a result, certain mutational effects such as A36G and U73G are not detectable, whereas they are readily measured in the context of the modified transcript (Table 2). The m1G37 modification is absent from bacterial tRNACys. Although present in human tRNACys, it is not required for aminoacylation by human CysRS (data not shown). In the co-crystal structure7 of A. fulgidus SepRS with the unmodified tRNACys, the N1 and N2 of G37 form H-bonds with the enzyme main chain carbonyl groups of G443 and D520, respectively, both of which are highly conserved in SepRS. The importance of m1G37 for M. jannaschii SepRS, particularly with respect to kcat and kchem of aminoacylation (Table 1), suggests that the modification alters the enzyme recognition of G37 in direct or indirect readout to modulate the activity at the active site.
Second, while SepRS and CysRS recognize common nucleotides in tRNACys, including U73, the GCA anticodon, G37, A38, and the 15–48 nculeotides, SepRS weighs these nucleotides much less than CysRS (Table 2). Also, while CysRS places the strongest emphasis on U73, SepRS lacks a clear preference for a specific nucleotide. Insights into these differences can be gleaned from the co-crystal structures of SepRS7 and CysRS17, particularly with respect to anticodon recognition. While SepRS uses antiparallel β sheets to form H-bonds with the bases of the anticodon and those of G37 and A387, CysRS uses a mixed α/β fold to contact only the anticodon bases. Importantly, while both CysRS and tRNA in the co-crystal structure exhibit major conformational rearrangements relative to their free states, such large conformational rearrangements are not seen in the SepRS co-crystal structure even with the bound amino acid substrate. The mutual rearrangements in the CysRS–tRNA complex contribute to the “induced-fit” process that can enhance aminoacylation by exploiting the intrinsic flexibility of the complex to lower the free-energy barrier for aminoacylation, and by inhibiting incorrect complexes from reaching the functional state rapidly or completely.
The third distinction between SepRS and CysRS is the rate-limiting step of aminoacylation. While the two classes of aaRSs are marked by distinct active-site motifs, they are also distinguishable by kinetics. Class I aaRSs exhibit burst kinetics, indicating that release of aa-tRNA is rate limiting23, whereas class II aaRSs lack the burst kinetics and are rate limited by the aminoacyl transfer step24 due to slow rearrangement of an active site motif. However, although SepRS possesses the class II active site, it manifests burst kinetics and has a high affinity to its product aa-tRNA (Fig. 2). These features are reminiscent of class I synthetases and suggest that the movements of the class II motifs by SepRS for tRNA aminoacylation are unconventional so as to accelerate the aminoacyl transfer step relative to release of the aa-tRNA product, which is the intermediate Sep-tRNACys for synthesis of Cys-tRNACys.
Four lines of experiments support the formation of the SepRS–SepCysS complex: pull-down, BIAcore, fluorescence binding, and gel filtration. Based on these experiments and kinetic analysis, we propose a scheme for the complex to synthesize and sequester Sep-tRNACys for facile conversion to Cys-tRNACys (Fig. 5d). In this scheme, SepRS in the complex binds tRNACys and synthesizes Sep-tRNACys (step 1). While SepRS and SepCysS in isolation both bind tightly to Sep-tRNACys (Kd = 0.1–0.2 μM), the protein complex has an even higher affinity to Sep-tRNACys (Kd = 0.025 μM). Formation of the protein complex stimulates SepRS to release Sep-tRNACys, resulting in a 2-fold increase in kcat (step 2). Because of the similar high affinity of SepRS and SepCysS to Sep-tRNACys, the released aa-tRNA can move freely to SepCysS without a thermodynamic barrier. The synthesis of Sep-tRNACys by SepRS strengthens the SepRS–SepCysS binary complex (with a decrease in Kd of the binary complex from 0.4 to 0.025 μM), and the transfer of Sep-tRNACys from SepRS to SepCysS within the binary complex strengthens the affinity of SepCysS for the aa-tRNA (with a decrease in Kd from 0.1 to 0.025 μM). This facilitates SepCysS to convert the aa-tRNA to Cys-tRNACys (step 3), which is released from the binary complex (step 4) and can be brought to the ribosome by EF-1α. Thus, the SepRS–SepCysS binary complex sequesters Sep-tRNACys and thereby protects it from rapid deacylation and from capture by EF-1α.
Formation of the SepRS–SepCysS binary complex is a unique feature of this indirect pathway. Three other indirect pathways exist to synthesize aa-tRNA for amino acids Asn, Gln, and Sec. In prokaryotes and archaea that lack AsnRS and GlnRS, the synthesis of Asn-tRNAAsn and Gln-tRNAGln is achieved by using non-discriminating AspRS and GluRS to synthesize Asp-tRNAAsn and Glu-tRNAGln, which are then converted to the correct aa-tRNA by a tRNA-dependent amidotransferase (AdT)25. The synthesis of Sec-tRNASec in bacteria is achieved by serylation of tRNASec by SerRS, followed by conversion of Ser to Sec by Sec synthase2. In eukaryotes and archaea, the conversion proceeds with phosphorylation of Ser to Sep by Sep-tRNASec kinase, followed by Sep to Sec catalyzed by SepSecS26. However, although a ternary complex of a non-discriminating AspRS, tRNAAsn, and AdT has been isolated recently27 and shown to be competent for synthesis of Asn-tRNAAsn, biochemical studies show that AspRS and AdT by themselves do not form a binary complex, and that their association is brought about only by the tRNA. This appears to be the case for all other indirect pathways, including the formation of a putative complex28 of GluRS and AdT upon binding of tRNAGln. Why then does the SepRS–SepCysS pathway form the protein binary complex? One reason is that Sep-tRNACys is more labile than the other intermediates in bulk solvent (e.g. T1/2 = 151 min for Asp-tRNAAsn at 50 °C27). More importantly, Sep-tRNACys binds to EF-1α with an affinity similar to those of the matched aa-tRNAs. Because bacterial EF-Tu is known to select aa-tRNA by seeking an affinity balance between the aminoacyl and tRNA moieties29, our result suggests that the pairing of Sep with tRNACys makes thermodynamic contributions that fall within the range of the matched aa-tRNAs for EF-1α. In contrast, the intermediates of the other three indirect pathways are incapable of binding to EF-Tu27, eliminating the possibility of being brought to the ribosome. These considerations suggest that, due to the unusual features of the Sep-tRNACys intermediate, the SepRS–SepCysS binary complex is formed to ensure that the intermediate is both stabilized and sequestered.
Structural modeling supports the sequestration of Sep-tRNACys by the SepRS–SepCysS complex from binding to EF-1α. A docking model based on the structures of SepRS-tRNA-Sep and EF-Tu-tRNACys reveals major steric clashes7, suggesting that SepRS cannot handoff Sep-tRNACys to EF-Tu. In contrast, a docking model8 based on the structures of SepRS-tRNA-Sep and SepCysS demonstrates the feasibility of a ternary complex, where the two enzyme are juxtaposed to allow the catalytic site of SepCysS to interact with the aminoacylation domain of SepRS, enabling direct transfer of the 3’ end of tRNACys from SepRS to SepCysS. The 1:1 stoichiometry of the protein complex suggests that SepRS and SepCysS each dissociate from their respective oligomeric states to form a new interface between their monomers. The dynamics of this protein complex and the ternary complex with Sep-tRNACys must involve conformational rearrangements of all partners.
The formation of the SepRS–SepCysS complex suggests that, although phosphoserine has the dedicated SepRS and can be carried on a tRNA in the form of Sep-tRNACys, it has little chance of entering the genetic code through that charged tRNA. Instead, phosphoserine is synthesized post-translationally at selected positions as part of the protein phosphorylation network. However, phosphoserine is an intermediate for cysteine synthesis by the SepRS–SepCysS pathway, and an intermediate for selenocysteine synthesis in the eukaryotic and archaeal Sec pathway. Thus, the expansion of the genetic code to include cysteine and selenocysteine appears to have co-evolved with the pathways of biosynthesis of these amino acids30.
METHODS
Recombinant proteins of M. jannaschii.
We amplified the ORFs MJ1660 (SepRS), MJ1678 (SepCysS) and MJ0324 (EF-1α) by PCR and inserted them into pET-22b(+) at NdeI and BamHI, at NdeI and SacI, and at NdeI and XhoI, respectively. Recombinant proteins were expressed in E. coli BL21(DE3)-RIL and purified by a heat step (75 °C, 20 min), followed by binding to the Talon resin (CloneTech). The un-tagged versions were expressed from plasmids that contained a site-specifically introduced stop codon (by QuikChange) placed before the sequence for the His-tag, and were purified on Q sepharose FF (Amersham), followed by an FPLC monoQ column. Protein concentrations were determined by the Bradford assay and, for SepRS, adjusted by active site titration.
tRNA preparation.
Unfractionated crude tRNA was isolated from frozen M. jannaschii cells, while transcripts of M. jannaschii tRNACys were synthesized by transcription and purified14. Modification of transcripts with m1G37 by MjTrm5 was as described16. The 3’ end of all transcripts was repaired by E. coli CCA enzyme with [α-32P]-labeled or unlabeled ATP to ensure homogeneity. Concentrations of unlabeled transcripts were determined by aminoacylation plateaus.
Aminoacylation by SepRS.
Aminoacylation was assayed at 60 °C in 30 mM NaAc (pH 6.0), 20 mM KCl, 10 mM MgCl2, 5 mM DTT, 200 μM Sep, and 10 mM ATP with varying tRNA and SepRS concentrations. Reaction aliquots were removed periodically, mixed with 2 volumes of P1 nuclease (0.1 mg per mL in 50 mM NaAc, pH 5.3, 1 mM ZnCl2, 50 mM NaCl) at 25 °C for 30 min, and resolved by TLC on PEI cellulose plates31. Steady-state kinetics was determined by fitting initial rates vs substrate concentrations to the Michaelis-Menten equation. Single turnover and pre-steady-state kinetics were performed on the KinTek RQF-3 instrument at 60 °C. Reactions were quenched by 0.4 M NaAc and 0.1% (w/v) SDS and analyzed by the TLC assay. To determine the Kd and kchem of a tRNA, aminoacylation reactions were performed in single turnover conditions of 0.1–10 μM of SepRS at 10-fold molar excess of tRNA. To test the effect of SepCysS on the burst kinetics of SepRS, 0.5 μM SepRS and 6.25 μM SepCysS were pre-incubated, and rapidly mixed with 15 μM tRNA and saturating concentrations of Sep and ATP. To determine the Kd for Sep-tRNACys, aminoacylation of 1.8–18 μM of unlabeled tRNA by 10 μM SepRS was performed at 60 °C for 3 min after which the reaction was diluted 10-fold and incubated for another 15 min to reach equilibrium. The charged tRNA was then mixed with 14 μM labeled tRNACys to initiate pre-steady-state aminoacylation23.
Protein binary complex.
Aminoacylation reactions for the pull-down assay were as above for 20 min and were gently mixed with 30 μL of Ni-NTA (Qiagen) in 20 mM HEPES, pH 7.5, 10 mM NaCl, 1 mM MgCl2, 1 mM DTT, 10% (v/v) glycerol for 1 hr at 25 °C. After 5 washes of the resin with 4 mM imidazole in the same buffer, the pull-down components were eluted by 800 mM imidazole, precipitated, and analyzed. To examine the EF-1α–GTP–Sep-tRNACys ternary complex, EF-1α was activated at 37 °C for 3 hours in 50 mM HEPES, pH 8.0, 200 mM NH4Cl, 20 mM MgCl2, 5 mM DTT, 1.5 mM GTP, 0.1 mg per ml pyruvate kinase and 1.8 mM phosphoenolpyruvate. The pull-down of EF-1α from the aminoacylation reaction was by the same procedure as that for the SepRS–SepCys complex. The biosensor measurements were performed at 25 °C. SepCysS was immobilized to the surface of a CM5 chip by amine coupling to ~1000 response units. An identical immobilization procedure was performed to another flow cell without SepCysS. Increasing concentrations of SepRS in 10 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 5 mM β-Me were injected over the SepCysS-coated surface at 20 μL per min to ensure a maximum response. The surface was regenerated by one pulse of 5 sec of 50 mM NaOH after each injection. After correction for non-specific binding to the surface, the maximum response unit vs SepRS concentration was fit to a hyperbolic equation to derive the Kd. Gel filtration analysis of the protein binary complex (pre-formed at 180 μM SepRS and 90 μM SepCysS) was performed on the FPLC Superose 12 column in 20 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5 mM β-Me, and 10% (v/v) glycerol at a flow rate of 0.5 mL per min.
Fluorescence binding measurements.
The TMR-labeled tRNACys was prepared as described19, while the PyC-labeled tRNA was prepared by using the Bst CCA enzyme to add PyCTP and ATP to a transcript that terminated at C74. Labeled tRNAs were aminoacylated by SepRS under single-turnover condition and labeled Sep-tRNAs were isolated by HPLC using a C8 reverse phase column. Binding measurements of TMR-labeled Sep-tRNACys were conducted in 20 mM HEPES, pH 7.5, 10 mM MgCl2, 5 mM DTT for the SepRS–SepCysS binary complex and in 50 mM HEPES, pH 7.5, 50 mM KCl, 100 mM NH4Cl, 10 mM MgCl2, 5 mM DTT, 1.5 mM GTP for EF-1α by excitation at 541 nm and emission at 560–600 nm. Binding measurements of PyC-labeled Sep-tRNACys for SepCysS were performed in 20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 5 mM DTT by excitation at 352 nm and monitoring emission at 400–600 nm. All measurements were performed at room temperature.
Supplementary Material
ACKNOWLEDGMENTS
Supported by NIH grant GM066267 to YMH. We thank Sangbumn Kim, Tom Christian, and Howard Gamper for assistance with experiments, John Perona and Howard Gamper for comments on the manuscript.
REFERENCES
- 1.Ibba M & Soll D Aminoacyl-Trna Synthesis. Annu Rev Biochem 69, 617–650 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Leinfelder W, Zehelein E, Mandrand-Berthelot MA & Bock A Gene for a novel tRNA species that accepts L-serine and cotranslationally inserts selenocysteine. Nature 331, 723–5 (1988). [DOI] [PubMed] [Google Scholar]
- 3.Srinivasan G, James CM & Krzycki JA Pyrrolysine encoded by UAG in Archaea: charging of a UAG-decoding specialized tRNA. Science 296, 1459–62. (2002). [DOI] [PubMed] [Google Scholar]
- 4.Sauerwald A et al. RNA-dependent cysteine biosynthesis in archaea. Science 307, 1969–72 (2005). [DOI] [PubMed] [Google Scholar]
- 5.O’Donoghue P, Sethi A, Woese CR & Luthey-Schulten ZA The evolutionary history of Cys-tRNACys formation. Proc Natl Acad Sci U S A 102, 19003–8 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamtekar S et al. Toward understanding phosphoseryl-tRNACys formation: the crystal structure of Methanococcus maripaludis phosphoseryl-tRNA synthetase. Proc Natl Acad Sci U S A 104, 2620–5 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukunaga R & Yokoyama S Structural insights into the first step of RNA-dependent cysteine biosynthesis in archaea. Nat Struct Mol Biol 14, 272–9 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Fukunaga R & Yokoyama S Structural insights into the second step of RNA-dependent cysteine biosynthesis in archaea: crystal structure of Sep-tRNA:Cys-tRNA synthase from Archaeoglobus fulgidus. J Mol Biol 370, 128–41 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Lima CD Analysis of the E. coli NifS CsdB protein at 2.0 A reveals the structural basis for perselenide and persulfide intermediate formation. J Mol Biol 315, 1199–208 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Hohn MJ, Park HS, O’Donoghue P, Schnitzbauer M & Soll D Emergence of the universal genetic code imprinted in an RNA record. Proc Natl Acad Sci U S A 103, 18095–100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu C et al. Kinetic Quality Control of Anticodon Recognition by a Eukaryotic Aminoacyl-tRNA Synthetase. J Mol Biol 367, 1063–78 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hrazdina G & Wagner GJ Metabolic pathways as enzyme complexes: evidence for the synthesis of phenylpropanoids and flavonoids on membrane associated enzyme complexes. Arch Biochem Biophys 237, 88–100 (1985). [DOI] [PubMed] [Google Scholar]
- 13.Amunts A, Drory O & Nelson N The structure of a plant photosystem I supercomplex at 3.4 A resolution. Nature 447, 58–63 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Lipman RS, Sowers KR & Hou YM Synthesis of cysteinyl-tRNA(Cys) by a genome that lacks the normal cysteine-tRNA synthetase. Biochemistry 39, 7792–8 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Christian T, Evilia C, Williams S & Hou YM Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol 339, 707–19 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Hou YM, Li Z & Gamper H Isolation of a site-specifically modified RNA from an unmodified transcript. Nucleic Acids Res 34, e21 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauenstein S, Zhang CM, Hou YM & Perona JJ Shape-selective RNA recognition by cysteinyl-tRNA synthetase. Nat Struct Mol Biol 11, 1134–41 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Tinsley RA & Walter NG Pyrrolo-C as a fluorescent probe for monitoring RNA secondary structure formation. Rna 12, 522–9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan B, Weidemaier K, Yip WT, Barbara PF & Musier-Forsyth K Intra-tRNA distance measurements for nucleocapsid proteindependent tRNA unwinding during priming of HIV reverse transcription. Proc Natl Acad Sci U S A 96, 459–64 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanderson LE & Uhlenbeck OC Exploring the specificity of bacterial elongation factor Tu for different tRNAs. Biochemistry 46, 6194–200 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Rho SB et al. A multifunctional repeated motif is present in human bifunctional tRNA synthetase. J Biol Chem 273, 11267–73 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Lipman RS, Chen J, Evilia C, Vitseva O & Hou YM Association of an Aminoacyl-tRNA Synthetase with a Putative Metabolic Protein in Archaea. Biochemistry 42, 7487–96 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Zhang CM, Perona JJ, Ryu K, Francklyn C & Hou YM Distinct Kinetic Mechanisms of the Two Classes of Aminoacyl-tRNA Synthetases. J Mol Biol 361, 300–11 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Guth EC & Francklyn CS Kinetic discrimination of tRNA identity by the conserved motif 2 loop of a class II aminoacyl-tRNA synthetase. Mol Cell 25, 531–42 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ibba M, Francklyn C & Cusack S The Aminoacyl-tRNA Synthetases, (Landes Bioscience, Georgetown, Texas, 2005). [Google Scholar]
- 26.Yuan J et al. RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc Natl Acad Sci U S A 103, 18923–7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailly M, Blaise M, Lorber B, Becker HD & Kern D The transamidosome: A dynamic ribonucleoprotein particle dedicated to prokaryotic tRNA-dependent asparagine biosynthesis. Mol Cell 26, 228–239 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Oshikane H et al. Structural basis of RNA-dependent recruitment of glutamine to the genetic code. Science 312, 1950–4 (2006). [DOI] [PubMed] [Google Scholar]
- 29.LaRiviere FJ, Wolfson AD & Uhlenbeck OC Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science 294, 165–8. (2001). [DOI] [PubMed] [Google Scholar]
- 30.Wong JT Question 6: coevolution theory of the genetic code: a proven theory. Orig Life Evol Biosph 37, 403–8 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Wolfson AD, Pleiss JA & Uhlenbeck OC A new assay for tRNA aminoacylation kinetics. Rna 4, 1019–23. (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.