

Abstract
The AIDS pandemic is still of importance. HIV-1 and HIV-2 are the causative agents of this pandemic, and in the absence of a viable vaccine, drugs are continually required to provide quality of life for infected patients. The HIV capsid (CA) protein performs critical functions in the life cycle of HIV-1 and HIV-2, is broadly conserved across major strains and subtypes, and is underexploited. Therefore, it has become a therapeutic target of interest. Here, we report a novel series of 2-pyridone-bearing phenylalanine derivatives as HIV capsid modulators. Compound FTC-2 is the most potent anti-HIV-1 compound in the new series of compounds, with acceptable cytotoxicity in MT-4 cells (selectivity index HIV-1 > 49.57; HIV-2 > 17.08). However, compound TD-1a has the lowest EC50 in the anti-HIV-2 assays (EC50 = 4.86 ± 1.71 μM; CC50 = 86.54 ± 29.24 μM). A water solubility test found that TD-1a showed a moderately increased water solubility compared with PF74, while the water solubility of FTC-2 was improved hundreds of times. Furthermore, we use molecular simulation studies to provide insight into the molecular contacts between the new compounds and HIV CA. We also computationally predict drug-like properties and metabolic stability for FTC-2 and TD-1a. Based on this analysis, TD-1a is predicted to have improved drug-like properties and metabolic stability over PF74. This study increases the repertoire of CA modulators and has important implications for developing anti-HIV agents with novel mechanisms, especially those that inhibit the often overlooked HIV-2.
Keywords: HIV, capsid, phenylalanine derivatives, protein-protein interaction
1. Introduction
Human immunodeficiency virus (HIV), causing acquired immune deficiency syndrome (AIDS), remains one of the most serious global problems threatening human health [1]. HIV is comprised two main types, HIV type 1 (HIV-1) and HIV type 2 (HIV-2), which have similar modes of replication, transmission, and clinical symptomatology, but HIV-2 is less widespread and has reduced infectivity and transmissibility [2,3]. By simultaneously targeting different steps of the HIV life cycle, combined antiretroviral therapy (cART) has achieved remarkable success in reducing overall morbidity and mortality [4]. However, in the absence of a viable vaccine, taking these antiviral drugs is a life-long commitment. With the time people are required to remain on their regimens, drug resistance and side effects caused by cART represent real challenges [5]. One way to circumvent the problem of drug resistance is to develop new anti-HIV drugs with new targets and modes of action [6].
HIV capsid (CA) performs critical functions throughout the virus’s life cycle, making it an important pharmacological target [7,8]. The mature HIV-1 capsid is a lattice composed of ~ 1500 CA monomers which self-assemble into the typical fullerene cone structure composed of 12 pentamers and ~ 250 hexamers (Figure 1) [9,10]. Each CA monomer contains an N-terminal domain (NTD), a C-terminal domain (CTD), and a flexible linker connecting the two domains. During the formation of pentamers and hexamers, adjacent monomers are connected by NTD-NTD interactions and NTD-CTD interactions. In contrast, pairs of subunits between hexamers are connected by CTD-CTD interactions [11,12]. Both genetic and pharmacological studies have demonstrated that the stability of the capsid is fine-tuned such that any increase or decrease has deleterious effects on the replication of the virus [13].
Figure 1.

Monomeric and assembled structure of the HIV-1 capsid protein.
Research on HIV capsid modulators has revealed several vulnerable regions of the CA protein structure [14,15,16,17,18,19,20]. Despite multiple pockets to which CA modulators could bind, particular attention has been given to a pocket formed between two adjacent protomers within the hexamer: the interprotomer pocket. This interprotomer pocket is broadly conserved across strains and the binding site for critical host cell factors such as cleavage and polyadenylation specific factor 6 (CPSF6) [21,22,23,24,25] and nucleoporins 153 and 358 (NUP153, NUP358) [23,24,26,27,28,29,30,31,32,33]. Despite the high conservation within the NTD-CTD, affinity alterations across clades have been observed for CPSF6 and NUP153 peptides targeting this site, and a complete loss of binding of NUP153 peptide (residue 1407 to 1423) to HIV-1 CA from clade A1 [34]. This points towards slight alterations of nuclear translocation mechanisms across clades. In addition, this essential interprotomer pocket is also targetable by small molecules, as evidenced by its being the binding site for the two most studied CA compounds; PF-3450074 (PF74) and GS-6207 (Figure 2A) [35]. PF74 is a peptidomimetic reported by Pfizer [36], which is proven to inhibit HIV-1 replication with a bimodal mechanism. PF74 stabilizes the CA core structure post-infection, thus inhibiting the uncoating process and HIV-1 reverse transcription in the early stage. In the late-stage, PF74 destabilizes CA, causing aberrant virus morphologies that do not undergo maturation [20,35,36,37]. The co-crystal structure revealed the binding mode of PF74, in which the benzyl group of phenylalanine is essential for compound binding, and the indole is oriented towards the NTD of the adjacent subunit, forming a key cation-π interaction with Lys70 (Figure 2B) [38]. Even though the mechanism of PF74 is novel, inferior metabolic stability precludes further studies [20]. GS-6207 (Lenacapavir), by Gilead Science, is a compound derived from the heavy decoration of PF74 and that shows robust antiviral activity in MT-4 cells in the low picomolar range (EC50 = 105 pM) [39]. GS-6207 stabilizes the capsid, leading to a buildup of intact core in the cytoplasm, similar to PF74. Because it binds to the same pocket as PF74 (Figure 2C), GS-6207 prevents adequate binding of NUP153 and CPSF6 to the CA [40]. GS-6207 has stable metabolic stability, allowing for once every six month injection therapy, which is vital to reducing toxicity, reducing patient stigma, and improving medication adherence [12]. This drug has been approved by the EC (European Community) and is the first drug to target CA available in the clinic.
Figure 2.

(A) Structures of PF74 and GS-6207; (B) Binding mode of PF74, PDB ID: 5HGL; (C) Binding mode of GS-6207, PDB ID: 6V2F. The two HIV-1 CA protomers are highlighted in cyan and pink. Cation-π interactions are shown in green dotted lines and hydrogen bonds in yellow dotted lines.
Although GS-6207 has many advantages, its synthetic strategy is complicated, and the cost is high, probably making it of consequence for patients in wealthy countries only. Moreover, drug-resistant strains have also already appeared in clinical trials [41]. Therefore, developing a new generation of drugs targeting CA that address these inadequacies is imperative.
To address this need, we proceeded to modify PF74 and replace the indole with 2-pyridone. The rationale for this change is to form cation-π interactions with Lys70 and Arg173 on both sides, thereby increasing the affinity and potency of the compounds. Moreover, the carbonyl of 2-pyridone is expected to form hydrogen bonds with Lys70 and Arg173 of the adjacent subunit. The extended substituted phenyl groups are expected to form more abundant interactions with the amino acids of the adjacent subunit CTD, while phenyl groups and 2-pyridone are connected by an amide (Figure 3).
Figure 3.

The design rationale of the 2-pyridone-containing compounds.
Herein, we report the design, synthesis, and mechanism study of 2-pyridone-bearing phenylalanine derivatives as novel HIV capsid modulators. The antiviral activities of all synthesized compounds were tested in MT-4 cells infected by HIV-1IIIB or HIV-2 ROD, and structure-activity relationships were established. We performed docking studies to gain insights into the binding modes of representative compounds and to understand the biological potencies. Finally, the drug-like properties and metabolic stability of representative compounds and PF74 were computationally predicted.
2. Chemistry
As shown in Scheme 1, starting from commercially available 4-methoxy-N-methylaniline (1), the target compounds were prepared via a concise and well-established synthetic route, as outlined below. Treating of 1 with Boc-L-phenylalanine or Boc-3,5-difluoro-L-phenylalanine and 2-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU) in N,N-diisopropylethylamine (DIEA) and dichloromethane afforded 2a or 2b, followed by removal of tert-butyloxycarbonyl (Boc) protection resulted in the formation of free amine 3a or 3b. The intermediate 4a or 4b was obtained by acylation of 3a or 3b with bromoacetic acid in dichloromethane solution. The nucleophilic substitution of 2-pyridone with 4a or 4b in THF resulted in TC-1 or FTC-1. We then removed the Boc group and obtained TC-2 or FTC-2. Finally, triethylamine (TEA), TC-2 or FTC-2 were added, and benzoyl chloride was substituted by different groups in dichloromethane to afford the compounds TD-1a-1l. Another target compound, TD-1m, was prepared by a hydrogenation reduction of the nitro group of TD-1l.
Scheme 1.

Preparation of TC-1-2, FTC-1-2 and TD-1a-1m. Reagents and conditions: (i) Boc-L-phenylalanine or Boc-3,5-difluoro-L-phenylalanine, HATU, DIEA, dichloromethane, 0 °C to r.t.; (ii) trifluoroacetic acid, dichloromethane, r.t.; (iii) bromoacetic acid, HATU, DIEA, dichloromethane, 0 °C to r.t.; (iv) tert-butyl (2-oxo-1,2-dihydropyridin-4-yl)carbamate, NaH, THF, 0 °C to r.t.; (v) trifluoroacetic acid, dichloromethane, r.t.; (vi) substituted benzoyl chloride, TEA, DCM, 0 °C to r.t.: (vii) H2, Pd/C, DCM.
3. Results and Discussion
3.1. In Vitro Anti-HIV Assays and SAR Analysis for HIV-1 Potency
The antiviral activities and cytotoxicities of the target compounds were tested using MT-4 cells infected by HIV-1 IIIB or HIV-2 ROD. Table 1 shows the EC50 and CC50 values of each compound. PF74 was utilized as the control drug in this assay.
Table 1.
Anti-HIV Activity and Cytotoxicity in MT-4 Cells Infected with HIV-1 IIIB and HIV-2 ROD.
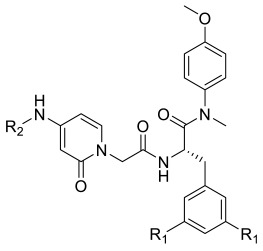
| |||||||
|---|---|---|---|---|---|---|---|
| Compound ID | R1 | R2 | EC50 a (μM) | CC50 b (μM) | SI c | ||
| HIV-1 IIIB | HIV-2 ROD | HIV-1 | HIV-2 | ||||
| FTC-2 | F | H | 5.36 ± 0.98 | 15.56 ± 1.06 | >265.69 | >49.57 | >17.08 |
| TC-2 | H | H | 29.07 ± 4.23 | 19.72 ± 6.42 | 261.13 ± 33.14 | 8.98 | 13.24 |
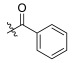
| TD-1a | H |
|
16.81 ± 0.91 | 4.86 ± 1.71 | 86.54 ± 29.24 | 5.15 | 17.81 |
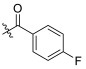
| TD-1b | H |
|
>34.80 | 16.22 ± 17.16 | 34.80 ± 5.75 | N.D.d | 2.15 |
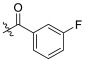
| TD-1c | H |
|
18.90 ± 2.66 | 6.59 ± 1.63 | 61.65 ± 8.44 | 3.26 | 9.36 |
| TD-1d | H |
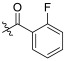
|
17.88 ± 2.68 | 7.92 ± 0.83 | 81.42 ± 22.66 | 4.55 | 10.28 |
| TD-1e | H |
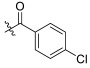
|
>25.48 | >25.48 | 25.48 ± 2.90 | N.D. | N.D. |
| TD-1f | H |
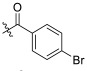
|
>23.51 | >23.51 | 23.51 ± 2.79 | N.D. | N.D. |
| TD-1g | H |
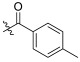
|
25.60 ± 7.51 | 20.48 ± 8.49 | 58.27 ± 4.85 | 2.28 | 2.85 |
| TD-1h | H |
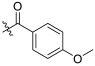
|
29.62 ± 4.34 | 10.67 ± 3.66 | 53.13 ± 5.12 | 1.79 | 4.98 |
| TD-1i | H |
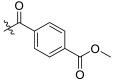
|
19.09 ± 5.08 | 15.25 ± 0.55 | 96.17 ± 19.83 | 5.04 | 6.31 |
| TD-1j | H |
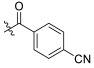
|
16.77 ± 0.78 | 17.83 ± 2.79 | 137.19 ± 22.50 | 8.18 | 7.69 |
| TD-1k | H |
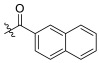
|
>27.59 | >27.59 | 27.59 ± 3.18 | N.D. | N.D. |
| TD-1l | H |
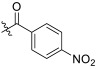
|
>31.87 | >31.87 | 31.87 ± 1.73 | N.D. | N.D. |
| TD-1m | H |
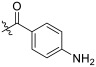
|
>189.23 | 17.14 ± 2.49 | 189.23 ± 2.19 | N.D. | 11.04 |
| PF74 | - | - | 1.08 ± 0.24 | 5.76 ± 2.14 | >293.75 | >271.99 | >51.00 |
a EC50: the concentration of the compound required to achieve 50% protection of MT-4 cells against the HIV-induced cytotoxicity effect, determined in at least triplicate against HIV in MT-4 cells; b CC50: the concentration of the compound required to reduce the viability of uninfected cells by 50%, determined in at least triplicate against HIV in MT-4 cells; values were averaged from at least three independent experiments; c SI: selectivity index, the ratio of CC50/EC50. d N.D.: not determined.
The majority of the newly designed compounds showed anti-HIV activities in MT-4 cells, in which FTC-2 exhibited the best anti-HIV-1 activity with an EC50 value of 5.36 ± 0.98 μM. Meanwhile, TD-1a was the most potent compound inhibiting HIV-2 replication (EC50 = 4.86 ± 1.71 μM), surpassing the parental compound PF74. In addition, both compounds showed acceptable cytotoxicity in MT-4 cells (FTC-2 selectivity index HIV-1 > 49.57; HIV-2 > 17.08, TD-1a selectivity index HIV-1 = 5.15; HIV-2 = 17.81).
As shown in Table 1, fluorine substituting R1 enhanced the anti-HIV-1 activity (FTC-2 > TC-2). The 2-pyridone bearing side chain did not show the expected activity increase for the compounds when testing the anti-HIV-1 activity. In contrast, modifying the amino group of TC-2 did result in potency improvement for most compounds (TD-1a, TD-1c, TD-1d, TD-1g, TD-1i, TD-1j) over the parental TC-2. Nevertheless, different substituents of R2 have different effects on the anti-HIV-1 activity, although the activities of most substituted benzene compounds were reduced. When R2 is carbonyl benzene substituted by fluorine, the ortho-substitution was superior to the meta- and para-substitution, where the para-substitution lost its anti-HIV-1 potency (TD-1d > TD-1c > TD-1b). The law of loss of effectiveness for para-substitution also applies to some other weak electron-withdrawing substituents, such as chlorine, bromine, and the nitro group (TD-1e, TD-1f, TD-1l). However, when the para-position was substituted with strong electron-withdrawing groups (TD-1i, TD-1j), the compounds showed comparable or slightly weakened activity than the unsubstituted counterparts. In conclusion, for this series of compounds, the substitution of R1 by fluorine and the substitution of R2 by a robust electron-withdrawing group in the para-position substituting or unsubstituted phenylcarbonyl group were beneficial in enhancing the anti-HIV-1 activity.
For anti-HIV-2 efficacy, this series of compounds showed enhanced activity compared to PF74. Fluorine substituting R1 enhanced anti-HIV-2 activity (FTC-2 > TC-2), and modifying the amino group substantially improved potency. Another aspect of the anti-HIV-1 experiment was that the compound was the most potent when R2 was substituted with an unsubstituted phenylcarbonyl group (TD-1a). When the R2-benzene of TD-1a was replaced by fluorine, the activities were reduced. However, unlike in the anti-HIV-1 experiment, the unsubstituted compounds did not lose their activity, and the activity laws differed (meta > ortho > para). When the electron-donating group is substituted for the para-position, the activity decreases to a small extent compared to the electron-withdrawing group. In conclusion, compared to targeting HIV-1, this series of compounds has a stronger ability to inhibit HIV-2 replication that is superior to PF74.
3.2. Solubility of Representative Compounds
To test the changes in water solubility when two more amides or its mimics were introduced to the target compounds, which may lower the solubility of the target compounds, we performed water solubility assays (Table 2). In the HPLC assay, the peak area of PF74 saturated aqueous solution under pH 2 is much smaller than the minimum value of its standard curve (7.03 mAu, 1.12 μg/mL). Therefore, we cannot calculate its water solubility. It was illustrated that the solubility of compounds varied greatly under different pH. TD-1a had a moderately increased water solubility compared with PF74, while the water solubility of FTC-2 was improved by a factor of hundreds, which may be due to the introduction of its arylamine group.
Table 2.
Solubility of Representative Compounds.
| Compounds | Solubility (μg/mL) a | ||
|---|---|---|---|
| pH 2 | pH 7 | pH 7.4 | |
| TD-1a | 2.69 ± 0.52 | 5.07 ± 2.60 | 8.66 ± 0.41 |
| FTC-2 | 366.57 ± 58.39 | 261.19 ± 130.46 | 436.58 ± 51.87 |
| PF74 | <1.12 b | 1.44 ± 0.10 c | 1.91 ± 0.56 |
a n = 3. b all of the data were smaller than the minimum value of the standard curve. c n = 2, one of the data was smaller than the minimum value of the standard curve.
3.3. Binding Mode of FTC-2 within the Interprotomer Pocket
To shed light on the binding mode of representative compounds to the HIV-1 capsid, molecular docking of the most potent compound, FTC-2, was performed using Schrödinger 2022-1 software (Schrödinger, New York, NY, USA).
As shown in Figure 4, FTC-2 had two main binding modes in the docking study, in which the difference was mainly the orientation of the 2-pyridone moiety. When the carbonyl group of 2-pyridone faced the inside of the binding pocket, its carbonyl formed hydrogen bonds with Arg173 and Lys70, but the original cation-π interactions of Lys70 and Arg173 were absent (Figure 4A). On the contrary, if the carbonyl group of the 2-pyridone moiety is kinked “upwards” towards the solvent, it results in the formation of cation-π interactions sandwiched by Lys70 and Arg173 (Figure 4B). In addition, both conformations formed a hydrogen bond with the main chain carbonyl of Tyr169. Although there were two binding modes and the glide scores of the two modes were almost the same, the interaction of FTC-2 with the critical amino acid Lys70 was weakened compared to PF74, and the hydrogen bond with Gln63 was also lost, which may be one of the reasons for the decreased potency.
Figure 4.

The two highest populated binding modes of FTC-2 within the CTD-NTD interprotomer pocket of HIV-1 CA. (A) The binding mode when the carbonyl group of 2-pyridone faced the inside of the binding pocket. (B) The binding mode when the carbonyl group of 2-pyridone faced the outside of the binding pocket. The two HIV-1 CA protomers are highlighted in cyan and pink. Cation-π interactions are shown in green dotted lines and hydrogen bonds in yellow dotted lines.
3.4. Computational Assessment of Drug-like Properties and Metabolic Stability
PF74 suffers from several problems that limit its clinical use that are primarily related to its drug-like properties. Therefore, we sought to analyze the new compounds’ predicted ADME properties (absorption, distribution, metabolism, and excretion) and compare them with PF74 (Figure 5). To accomplish this comparison, we used in silico prediction of drug-like metrics of the results as implemented in the oral non–central nervous system (CNS) drug profile in StarDrop 7 (Optibrium, Ltd., Cambridge, UK) [42]. This profile consists of several models, weighted differently and combined into a single score by a probabilistic scoring algorithm. For reference, scores range from 0 to 1, with 0 suggesting extremely non–drug-like and 1 suggesting the perfect drug.
Figure 5.

(A) Plot showing the StarDrop (Optibrium, Ltd., Cambridge, UK)–derived logS versus a multimetric oral non-CNS profile score. Importance for the score: logS = 0.9, HIA = 0.85, logP = 0.6, logD = 0.6, hERG pIC50 = 0.4, 2D6 affinity category = 0.3, 2C9 pKi = 0.3, P-gp category = 0.3, PPB90 category = 0.2, BBB category = 0.11, BBB log(brain:blood) = 0.11 (B) P-gp category, (C) PPB90, Plasma protein binding >90%, (D) HIA, human intestinal absorption category, and probability of prediction of FTC-2, TD-1a and PF-74. The size of the circles correlates with the corresponding score or probability.
Optibrium’s oral non-CNS drug profile is composed of the following metrics: logS (intrinsic aqueous solubility); classification for human intestinal absorption; logP (octanol/water); hERG (human ether-à-go-go-related gene) pIC50 (mammalian cells); cytochrome P450 CYP2D6 classification; cytochrome P450 CYP2C9 pKi values; classification of P-glycoprotein transport; classification of blood-brain barrier (BBB) penetration; and predicted BBB penetration value. The models and their respective importance to the profile are shown in Figure 5A. As can be seen, FTC-2 and TD-1a display a four- and five-fold improved score compared to PF74. TD-1a has also improved aqueous solubility compared to PF74, as judged by the logS, which improves overall bioavailability. Moreover, PF74 and FTC-2 are not a substrate of the P-Glycoprotein (P-gp), and TD-1a is predicted to be a substrate; however, one with a lower probability (Figure 5B). Overall, FTC-2 and TD-1a show improved oral non-CNS drug profile scores, primarily due to improved solubility (logS and logD) and lower probability for plasma protein binding (Figure 5C), which increases bioavailability and is predicted to be absorbed by the human intestine (Figure 5D).
A significant hurdle for PF74′s development is its poor metabolic stability [20]. Orally administered drugs must first pass the intestinal wall, followed by the portal circulation to the liver before reaching the bloodstream. Both sides are locations for first-pass metabolism and can adversely metabolize drugs before adequate plasma concentrations are reached. Therefore, we next sought to investigate whether or not our compounds had improved predicted metabolic stability. We employed a computational analysis first demonstrated to be an accurate indicator of metabolic stability by the Cocklin group [19,43,44]. This analysis uses the P450 module in StarDrop V7 (Optibrium, Ltd., Cambridge, UK) to predict each compound’s major metabolizing Cytochrome P450 isoforms using the WhichP450™ model, followed by affinity prediction to that isoform using the HYDE function in SeeSAR (BioSolveIT Gmbh, Sankt Augustin, Germany) [44,45,46]. The results of this analysis are shown in Figure 6.
Figure 6.

Computational prediction of metabolic stability. (A) Prediction of the major metabolizing CYP isoforms. All compounds are predicted to be metabolized primarily by the 3A4 isoform, including PF74. (B) Overall composite site lability (CSL) score and number of labile sites within the compound (for metabolism). A lower CSL score indicates a more stable molecule. The prediction was achieved using the StarDrop (version 7) P450 module. (C) Predicted affinity of docked FTC-2, TD-1a, and PF74 to Cytochrome P450 3A4 (PDB ID: 4D78). The lower boundary for predicted 3A4 affinity was used utilizing the hydrogen bond and dehydration scoring function (HYDE) implemented in SeeSAR12.1.
All compounds, including PF74, are predicted to be primarily metabolized by the CYP3A4 isoform (Figure 6A). Moreover, and in agreement with our computational prediction, a recent study also experimentally verified that PF74s poor metabolic stability is attributed mainly to its poor resistance to cytochrome P450 3A4 [18]. We therefore, analyzed the predicted metabolic lability of our compounds and PF74 by the CY3A4 isoform by comparing the overall composite site lability (CSL) score and the number of labile sites. The CSL score reflects the overall efficiency of metabolism of the molecule by combining the labilities of individual sites within the compound. The number of labile sites for TD-1a is comparable to PF74, while FTC-2 displays three labile sites primarily at the secondary amine, the hydroxyl group, and the carbon between the two electronegative fluorine atoms, which apply an electron-withdrawing effect on this carbon atom.
The slight nuances in the CSL score between those three indicated increased metabolic stability in the following order FTC-2, TD-1a, and PF74 (Figure 6B), with PF74 displaying the lowest CSL score and thereby indicating higher metabolic stability.
In addition to the CSL score and the number of labile sites, which assume that all compounds bind with similar affinity to the CYP3A4 isoform, other factors such as compound reduction rate and actual binding affinity to the CYP3A4 isoform can infer metabolic stability. Moreover, binding affinity can also be influenced by intrinsic compound properties such as size and lipophilicity. We, therefore, performed predictive binding affinity calculations using the HYdrogen bond and DEhydration (HYDE) energy scoring function in SeeSAR 12.1 (BioSolveIT Gmbh, Sankt Augustin, Germany). For this analysis, we used the structure of the human CYPA4 bound to an inhibitor (PDB ID 4D78). The HYDE scoring function in SeeSAR provides a range of affinities, spanning an upper and lower limit. Therefore, we used the lower limit as the affinity parameter to compare FTC-2, TD-1a, and PF74 (Figure 6C), which indicated low nM-affinity for PF74 to CYP3A4, while FTC-2 and especially TD-1a have significantly lower predicted CYP3A4 affinity. Combining the results from these predictions (CSL scores, labile sites, and predicted CYP3A4 affinity), this analysis indicates that compound TD-1a should have improved metabolic stability than PF74, primarily due to the significantly lower CYP3A4 affinity.
Next, we evaluated potential toxicities associated with our compounds to identify possible issues and address them early in our drug development pipeline. We, therefore, include genotoxicity and hepatotoxicity endpoints in our multiparameter optimization for FTC-2, TD-1a, and PF74 using the Derek Nexus module within Stardrop V7. Derek Nexus is a knowledge- and rule-based expert system for semi-quantitatively estimating DNA reactive moieties within molecules. Based on this prediction, none of our compounds, including PF74, show any concerning likelihood of genotoxicity or hepatotoxicity (Figure 7). In contrast, and used as positive controls in our prediction, ethyl methanesulfonate (EMS) and Lumiracoxib are known to have in vivo genotoxic and hepatotoxic issues.
Figure 7.

Genotoxicity and hepatotoxicity endpoints for selected compounds. The highlighted table shows the StarDrop V7 (Optibrium, Ltd., Cambridge, UK)–derived toxicity endpoints using the Derek Nexus module based on a knowledge-based prediction. Structural alerts within the molecule with a level of likelihood for concerns are based on precedence from experimental data. Likelihood level 0 = No report; 1 = Inactive; 2 = Equivocal; 3 = Plausible; 4 = Probable. Ethyl methanesulfonate (EMS) was used as a positive control for genotoxicity [47], and Lumiracoxib [48] as a positive control for hepatotoxicity endpoints.
4. Conclusions
In this study, a novel series of 2-pyridone-bearing phenylalanine derivatives have been designed, synthesized, and evaluated as HIV capsid modulators. Most of the newly synthesized compounds displayed anti-HIV activity, in which FTC-2 is the most potent anti-HIV-1 compound (EC50 = 5.36 ± 0.98 μM), while TD-1a is more potent at blocking the replication of HIV-2 (EC50 = 4.86 ± 1.71 μM). Moreover, both compounds showed acceptable cytotoxicity (FTC-2 selectivity index HIV-1 > 49.57; HIV-2 > 17.08, TD-1a selectivity index HIV-1 = 5.15; HIV-2 = 17.81), comparable to the control compound PF74. The water solubility test found that TD-1a showed a small increase or equivalent water solubility compared with PF74, while the water solubility of FTC-2 was improved by a factor of hundreds. Although molecular docking showed that FTC-2 had an ideal binding mode, some key functions were still missing, causing a decrease in antiviral potency. Notably, the orientation of the carbonyl on the 2-pyridone moiety could contribute to a lower potency by orienting the ring unfavorably for cation-π interactions by Lys70 and Arg173. Future modifications of this series could account for this. For example, stronger electron-donating groups, such as alkyl groups, could increase electronegativity within the ring system and the probability of cation-π interactions for potency improvement.
The water solubility test found that TD-1a showed a moderately increased water solubility compared with PF74, while the water solubility of FTC-2 was improved by a factor of hundreds. Furthermore, using a computational approach, we predicted drug-like properties and metabolic stability for FTC-2 and TD-1a, of which TD-1a showed an improved overall drug-likeness score and metabolic stability. These predictions, although previously demonstrated as indicators of drug-likeness and metabolic stability, have to be experimentally verified in future studies. Although the anti-HIV-1 activity of the compounds was reduced, the anti-HIV-2 activity of TD-1a was improved relative to PF74 and TC-2 (a non-fluorine-substituted FTC-2). This result shows that the modification of the terminal amino group of TC-2 is tolerable. We believe this study positively contributes to the repertoire of novel inhibitor chemotypes targeting the HIV capsid protein and increases the likelihood of advancing to synthetically accessible, potent antivirals.
5. Experimental Section
5.1. Chemistry
1H NMR and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer or Bruker AV-600 spectrometer (Bruker, Billerica, MA, USA) using solvents as indicated (DMSO-d6). Chemical shifts were reported in δ values (ppm) with tetramethylsilane (TMS) as the internal reference, and J values were reported in hertz (Hz). Melting points (mp) were determined on a micromelting point apparatus (Tianjin Optics, Tianjin, China) and were uncorrected. TLC was performed on Silica Gel GF254 for TLC (Merck KGaA, Darmstadt, Germany), and spots were visualized by iodine vapor or irradiation with UV light (λ = 254 nm). Flash column chromatography was performed on a column packed with Silica Gel60 (200–300 mesh). Thin-layer chromatography was performed on pre-coated HUANGHAI_HSGF254 (Yantai, Shandong, China), 0.15–0.2 mm TLC-plates. Solvents were of reagent grade and were purified and dried by standard methods when necessary. The concentration of the reaction solutions involved the use of a rotary evaporator at reduced pressure. The solvents of dichloromethane, TEA and methanol etc., were obtained from Sinopharm Chemical Reagent Co., Ltd. (SCRC, Shanghai, China), and were of AR grade. The key reactants, including 4-methoxy-N-methylaniline, N-(tert-butoxycarbonyl)-L-phenylalanine etc. were purchased from Bide Pharmatech Co., Ltd. (Shanghai, China) or Shanghai Haohong Scientific Co., Ltd. (Shanghai, China) The purity of the final representative compounds was checked by HPLC and was >95%.
5.1.1. General Procedure for the Synthesis of 2a and 2b
To a solution of Boc-L-phenylalanine or Boc-3,5-difluoro-L-phenylalanine (1 eq.) in 20 mL dichloromethane, HATU (1.5 eq.) was added at 0 °C, and the mixture was stirred for 0.5 h. Subsequently, DIEA (2 eq.) and 4-methoxy-N-methylaniline (1.2 eq.) were added to the mixture and then stirred at room temperature for another 2 h (monitored by TLC). The reaction solution was initially washed with saturated sodium bicarbonate, extracted with DCM (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford a corresponding crude product, purified by flash column chromatography to afford intermediates 2a and 2b.
tert-butyl (S)-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (2a)
Yellow oil, yield: 81%. 1H NMR (400 MHz, DMSO-d6): δ 7.22 (d, J = 8.3 Hz, 2H, Ph-H), 7.20–7.11 (m, 3H, Ph-H), 7.09 (d, J = 8.2 Hz, 1H, Ph-H), 7.03 (d, J = 8.6 Hz, 2H, Ph-H), 6.79 (d, J = 7.3 Hz, 2H, Ph-H), 4.27–4.06 (m, 1H, CH), 3.81 (s, 3H, OCH3), 3.13 (s, 3H, NCH3), 2.75 (dd, J = 13.4, 3.8 Hz, 1H, PhCH), 2.61 (dd, J = 13.3, 10.3 Hz, 1H, PhCH), 1.30 (s, 9H, C(CH3)). ESI-MS: m/z 385.4 (M + 1)+, 407.5 (M + 23)+. C22H28N2O4 [384.48].
tert-butyl (S)-(3-(3,5-difluorophenyl)-1-((4-methoxyphenyl) (methyl)amino)-1-oxopropan-2-yl)carbamate (2b)
Yellow oil, yield: 89%. 1H NMR (400 MHz, DMSO-d6): δ 7.31 (d, J = 8.4 Hz, 2H, Ph-H), 7.13–7.04 (m, 3H, Ph-H), 7.01 (d, J = 9.5 Hz, 1H, Ph-H), 6.44 (d, J = 8.3 Hz, 2H, Ph-H), 4.20–4.10 (m, 1H, CH), 3.81 (s, 3H, OCH3), 3.14 (s, 3H, NCH3), 2.82–2.61 (m, 2H, PhCH2), 1.29 (s, 9H, C(CH3)). ESI-MS: m/z 421.07 (M + 1)+, 443.17 (M + 23)+. C22H26F2N2O4 [420.46].
5.1.2. General Procedure for the Synthesis of 3a and 3b
Trifluoroacetic acid (5.0 eq.) was added dropwise to the corresponding substituted intermediate 2a or 2b (1.0 eq.) in 30 mL dichloromethane and stirred at room temperature for 1 h (monitored by TLC). The resulting mixture solution was then alkalized to pH ~7 with saturated sodium bicarbonate solution and then extracted with dichloromethane (3 × 30 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude products 3a and 3b.
(S)-2-amino-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (3a)
Yellow oil, yield: 75%. 1H NMR (400 MHz, DMSO-d6): δ 7.29–7.13 (m, 3H, Ph-H), 7.03–6.75 (m, 6H, Ph-H), 3.77 (s, 3H, OCH3), 3.44–3.35 (m, 1H, CH), 3.06 (s, 3H, NCH3), 2.75 (dd, J = 12.8, 6.7 Hz, 1H, PhCH), 2.45 (dd, J = 12.9, 7.1 Hz, 1H, PhCH), 1.87 (s, 2H, NH2). ESI-MS: m/z 285.05 (M + 1)+. C17H20N2O2 [284.36].
(S)-2-amino-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (3b)
Yellow oil, yield: 77%. 1H NMR (400 MHz, DMSO-d6): δ 7.10–6.93 (m, 5H, Ph-H), 6.57 (h, J = 4.1 Hz, 2H, Ph-H), 3.78 (s, 3H, OCH3), 3.35 (dd, J = 7.6, 5.9 Hz, 1H, CH), 3.09 (s, 3H, NCH3), 2.74 (dd, J = 13.1, 5.8 Hz, 1H, PhCH), 2.54–2.45 (m, 1H, PhCH), 1.82 (s, 2H, NH2). ESI-MS: m/z 321.11 (M + 1)+, m/z 343.25 (M + 23)+. C17H18F2N2O2 [320.34].
5.1.3. General Procedure for the Synthesis of 4a and 4b
Bromoacetic acid (1.2 eq.) and HATU (1.5 eq.) were mixed in 15 mL dichloromethane and stirred in an ice bath for 0.5 h. The corresponding substituted intermediate 3a or 3b (1 eq.) and DIEA (2 eq.) were then slowly added to the above solution at 0 °C. The reaction system was then stirred at room temperature for an additional 0.5 h (monitored by TLC). The resulting mixture was initially washed with saturated sodium bicarbonate and extracted with DCM (3 × 20 mL) and dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford a corresponding crude product, purified by flash column chromatography to afford intermediates 4a and 4b.
(S)-2-(2-bromoacetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (4a)
White oil, yield: 73%. 1H NMR (600 MHz, DMSO-d6): δ 8.62 (d, J = 7.9 Hz, 1H, NH), 7.22–7.16 (m, 3H, Ph-H), 7.05 (d, J = 8.3 Hz, 2H, Ph-H), 6.96 (d, J = 9.2 Hz, 2H, Ph-H), 6.88 (d, J = 6.2 Hz, 2H, Ph-H), 4.44 (td, J = 8.4, 5.6 Hz, 1H, CH), 3.82 (d, J = 2.4 Hz, 2H, CH2), 3.79 (s, 3H, OCH3), 3.10 (s, 3H, NCH3), 2.87 (dd, J = 13.5, 5.5 Hz, 1H, PhCH), 2.65 (dd, J = 13.5, 8.7 Hz, 1H, PhCH). ESI-MS: m/z 405.4 (M + 1)+. C19H21BrN2O3 [405.29].
(S)-2-(2-bromoacetamido)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (4b)
White solid, yield: 78%. 1H NMR (400 MHz, DMSO-d6): δ 8.70 (d, J = 8.0 Hz, 1H, NH), 7.22 (d, J = 8.8 Hz, 2H, Ph-H), 7.04 (d, J = 8.6 Hz, 3H, Ph-H), 6.52 (d, J = 6.3 Hz, 2H, Ph-H), 4.44 (qd, J = 8.6, 4.6 Hz, 1H, CH), 3.81 (s, 2H, BrCH2), 3.80 (s, 3H, OCH3), 3.13 (s, 3H, NCH3), 2.89 (dd, J = 13.7, 4.6 Hz, 1H, PhCH), 2.69 (dd, J = 13.7, 9.3 Hz, 1H, PhCH). ESI-MS: m/z 443.15 (M + 2)+, m/z 463.16 (M – 1 + 23)+. C17H18F2N2O2 [441.27].
5.1.4. General Procedure for the Synthesis of FTC-1 and TC-1
Under ice bath, tert-butyl (2-oxo-1,2-dihydropyridin-4-yl)carbamate (1.2 eq.) was dissolved in the solution of tetrahydrofuran (6 mL), and then the NaH (2 eq) was added slowly. The resulting mixture was then stirred for 30 min. The corresponding intermediate 4a or 4b was then added, and the resulting mixture was then stirred for 2 h at room temperature (monitored by TLC). The reaction mixture was then concentrated under reduced pressure, and the remains were resoluted with ethyl acetate. The resulting mixture was initially washed with saturated sodium bicarbonate, extracted with ethyl acetate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the corresponding crude product, which was purified by flash column chromatography to result in the products FTC-1 and TC-1.
tert-butyl (S)-(1-(2-((3-(3,5-difluorophenyl)-1-((4-methoxyphenyl)(methyl)amino)-1-oxopropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)carbamate (FTC-1)
Yellow solid, yield: 83%.
tert-butyl (S)-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)carbamate (TC-1)
Yellow solid, yield: 74%. 1H NMR (600 MHz, DMSO-d6) δ 9.55 (s, 1H, NH), 8.51 (d, J = 7.8 Hz, 1H, NH), 7.29 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.20 (d, J = 7.2 Hz, 2H, Ph), 7.00 (s, 2H, Ph), 6.90 (m, 5H, Ph), 6.47 (d, J = 2.3 Hz, 1H, 2-Pyridone), 6.29–6.26 (m, 1H, 2-Pyridone), 4.48 (d, J = 15.8 Hz, 1H, CH2), 4.42 (q, J = 8.0 Hz, 1H, CH), 4.35 (d, J = 15.8 Hz, 1H, CH2), 3.76 (s, 3H, OCH3), 3.08 (s, 3H, NCH3), 2.87 (dd, J = 13.5, 5.8 Hz, 1H, PhCH), 2.66 (dd, J = 13.5, 8.4 Hz, 1H, PhCH), 1.47 (s, 9H, Boc). 13C NMR (150 MHz, DMSO-d6) δ 171.30 (C=O), 167.19 (C=O), 162.43 (C=O), 158.96 (C=O), 152.63, 149.71, 140.13, 137.78, 135.93, 129.40 (2 × C), 129.05 (2 × C), 128.62 (2 × C), 126.91, 115.06 (2 × C), 101.88, 98.94, 80.69, 55.85, 51.86, 49.91, 38.09, 37.74, 28.41 (3 × C). ESI-MS: m/z 535.4 (M + 1)+. C29H34N4O6 [534.25].
5.1.5. General Procedure for the Synthesis of FTC-2 and TC-2
Trifluoroacetic acid (5.0 eq.) was added dropwise to the corresponding substituted intermediate FTC-1 or TC-1 (1.0 eq.) in 30 mL dichloromethane and stirred at room temperature for 1 h (monitored by TLC). The resulting mixture solution was then alkalized to pH ~7 with saturated sodium bicarbonate solution and then extracted with dichloromethane (3 × 30 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude products, which was purified by flash column chromatography to afford products FTC-2 and TC-2. (MS, 1H NMR and 13C NMR spectra are shown in Supplementary Materials)
(S)-2-(2-(4-amino-2-oxopyridin-1(2H)-yl)acetamido)-3-(3,5-difluorophenyl)-N-(4-methoxyphenyl)-N-methylpropanamide (FTC-2)
Yellow solid, yield: 86%. m.p.: 63–65 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.44 (d, J = 7.8 Hz, 1H, NH), 7.18 (d, J = 8.3 Hz, 2H, Ph), 7.06 (d, J = 7.4 Hz, 1H, 2-Pyridone), 7.02 (tt, J = 9.3, 2.5 Hz, 1H, Ph), 6.99–6.95 (m, 2H, Ph), 6.57–6.51 (m, 2H, Ph), 5.97 (d, J = 5.1 Hz, 2H, NH2), 5.62 (dd, J = 7.4, 2.4 Hz, 1H, 2-Pyridone), 5.21 (d, J = 2.5 Hz, 1H, 2-Pyridone), 4.42 (td, J = 8.1, 4.9 Hz, 1H, CH), 4.39–4.23 (m, 2H, CH2), 3.78 (s, 3H, OCH3), 3.11 (s, 3H, NCH3), 2.86 (dd, J = 13.7, 4.9 Hz, 1H, PhCH), 2.69 (dd, J = 13.7, 8.8 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 170.86 (C=O), 168.03 (C=O), 162.68 (C=O), 162.55 (dd, 1JCF = 245.8, 3JCF = 13.3 Hz, 2 × C), 159.12, 157.72, 142.41 (t, 3JCF = 9.3 Hz), 139.52, 135.84, 129.10 (2 × C), 115.21 (2 × C), 112.42 (dd, 2JCF = 19.8, 4JCF = 4.9 Hz, 2 × C), 102.47 (t, 2JCF = 25.5 Hz), 98.71, 92.91, 55.89, 51.42, 49.48, 37.71, 37.38. ESI-MS: m/z 471.4 (M + 1)+, 493.5 (M + 23)+. C24H24F2N4O4 [470.18].
(S)-2-(2-(4-amino-2-oxopyridin-1(2H)-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (TC-2)
Yellow solid, yield: 79%. m.p.: 164–168 ℃. 1H NMR (600 MHz, DMSO-d6) δ 8.39 (d, J = 7.7 Hz, 1H, NH), 7.25–7.13 (m, 4H, Ph, NH2), 7.03 (d, J = 7.4 Hz, 2H, Ph), 6.93–6.86 (m, 5H, Ph), 5.96 (d, J = 5.0 Hz, 1H, 2-Pyridone), 5.61 (dd, J = 7.4, 2.4 Hz, 1H, 2-Pyridone), 5.21 (d, J = 2.4 Hz, 1H, 2-Pyridone), 4.42 (td, J = 7.7, 5.6 Hz, 1H, CH), 4.30 (dd, J = 86.7, 15.8 Hz, 2H, CH2), 3.76 (s, 3H, OCH3), 3.08 (s, 3H, NCH3), 2.85 (dd, J = 13.5, 5.7 Hz, 1H, PhCH), 2.64 (dd, J = 13.5, 8.5 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.34 (C=O), 167.89 (C=O), 162.70 (C=O), 158.96, 157.70, 139.57, 137.81, 135.96, 129.41 (2 × C), 129.07 (2 × C), 128.61 (2 × C), 126.89, 115.07 (2 × C), 98.68, 92.93, 55.86, 51.78, 49.44, 38.11, 37.74.ESI-MS: m/z 435.5 (M + 1)+. C24H36N4O4 [434.20].
5.1.6. General Procedure for the Synthesis of TD-1a–1l
Under an ice bath, the key intermediate TC-2 (1 eq.), corresponding substituted benzoyl chloride (1.5 eq.), TEA (2 eq.) were dissolved in the solution of dichloromethane (10 mL). The resulting mixture was then stirred at room temperature (monitored by TLC). The reaction mixture was then extracted with dichloromethane (20 mL), and the combined organic phase was washed with saturated NaCl solution (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the corresponding crude product, which was purified by recrystallization or preparation thin layer chromatography to result in TD-1a–1l.
(S)-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1a)
Yellow solid, yield: 87%. m.p.: 102–107 ℃. 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H, NH), 8.60 (d, J = 7.7 Hz, 1H, NH), 7.92 (d, J = 7.2 Hz, 2H, Ph), 7.63 (t, J = 7.2 Hz, 1H, Ph), 7.55 (t, J = 7.5 Hz, 2H, Ph), 7.40 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.21 (d, J = 6.9 Hz, 3H, Ph (2H), 2-Pyridone (1H)), 7.07–6.98 (m, 2H, Ph), 6.95–6.87 (m, 5H, Ph), 6.58 (dd, J = 7.5, 2.4 Hz, 1H, 2-Pyridone), 4.54 (d, J = 15.8 Hz, 1H, CH2), 4.44 (d, J = 8.1 Hz, 1H, CH), 4.40 (d, J = 15.9 Hz, 1H, CH2), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.88 (dd, J = 13.6, 5.5 Hz, 1H, PhCH), 2.67 (dd, J = 13.4, 8.6 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.31 (C=O), 167.14 (C=O), 167.06 (C=O), 162.55 (C=O), 158.97, 149.26, 140.22, 137.80, 135.94, 134.73, 132.59, 129.41 (2 × C), 129.07 (2 × C), 128.94 (2 × C), 128.64 (2 × C), 128.33 (2 × C), 126.93, 115.08 (2 × C), 104.77, 100.10, 55.86, 51.90, 50.08, 38.09, 37.75.ESI-MS: m/z 539.5 (M + 1)+, m/z 561.4 (M + 23)+. C31H30N4O5 [538.22].
(S)-4-fluoro-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1b)
Yellow solid, yield: 83%. m.p.: 124–126 ℃. 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H, NH), 8.60 (d, J = 7.7 Hz, 1H, NH), 8.06–7.97 (m, 2H, Ph), 7.44–7.35 (m, 3H, Ph, Ph (2H), 2-Pyridone (1H)), 7.21 (d, J = 6.9 Hz, 3H, Ph (2H), 2-Pyridone (1H)), 7.07–6.98 (m, 2H, Ph), 6.95–6.87 (m, 5H, Ph), 6.56 (dd, J = 7.5, 2.4 Hz, 1H, 2-Pyridone), 4.57–4.36 (m, 3H, CH2, CH), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.91–2.84 (m, 1H, PhCH), 2.67 (dd, J = 13.4, 8.6 Hz, 1H, PhCH). 13C NMR (151 MHz, DMSO-d6) δ 171.31 (C=O), 167.12 (C=O), 165.90 (C=O), 164.86 (d, 1JCF = 250.0 Hz), 162.52 (C=O), 158.97, 149.18, 140.25, 137.80, 135.94, 131.21, 131.15, 129.41 (2 × C), 129.07 (2 × C), 128.64 (2 × C), 126.92 (2 × C), 115.92 (d, 2JCF = 22.0 Hz, 2 × C), 115.08 (2 × C), 104.82, 100.06, 55.85, 51.90, 50.08, 38.09, 37.75.ESI-MS: m/z 557.3 (M + 1)+, m/z 579.4 (M + 23)+. C31H29FN4O5 [556.21].
(S)-3-fluoro-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1c)
Yellow solid, yield: 85%. m.p.: 118–120 ℃. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H, NH), 8.61 (d, J = 7.7 Hz, 1H, NH), 7.77 (dd, J = 14.3, 8.8 Hz, 2H, Ph), 7.61 (td, J = 8.0, 5.8 Hz, 1H, Ph), 7.49 (td, J = 8.5, 2.6 Hz, 1H, Ph), 7.41 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.26–7.18 (m, 3H, Ph (2H), 2-Pyridone (1H)), 7.07–6.99 (m, 2H, Ph), 6.96–6.88 (m, 5H, Ph), 6.57 (dd, J = 7.5, 2.4 Hz, 1H, 2-Pyridone (1H)), 4.59–4.38 (m, 3H, CH2, CH), 3.77 (s, 3H, OCH3), 3.10 (s, 3H, NCH3), 2.88 (dd, J = 13.7, 5.5 Hz, 1H, PhCH), 2.68 (dd, J = 13.4, 8.6 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.31 (C=O), 167.10 (C=O), 165.66 (C=O), 162.49 (C=O), 162.37 (d, 1JCF = 244.8 Hz), 158.97, 149.00, 140.32, 137.80, 136.95 (d, 3JCF = 6.6 Hz), 135.94, 131.17 (d, 2JCF = 8.0 Hz), 129.41 (2 × C), 129.07, 128.64 (2 × C), 126.93, 124.58 (d, 3JCF = 2.8 Hz), 115.18 (d, 2JCF = 22.7 Hz), 115.08 (2 × C), 105.03, 100.03, 55.86, 51.90, 50.09, 38.09, 37.75. ESI-MS: m/z 557.3 (M + 1)+, m/z 579.4 (M + 23)+. C31H29FN4O5 [556.21].
(S)-2-fluoro-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1d)
Yellow solid, yield: 86%. m.p.: 96–99 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H, NH), 8.59 (d, J = 7.7 Hz, 1H, NH), 7.66 (td, J = 7.4, 1.8 Hz, 1H, Ph), 7.60 (td, J = 7.6, 2.0 Hz, 1H, Ph), 7.42–7.36 (m, 2H, Ph), 7.33 (d, J = 7.6 Hz, 1H, 2-Pyridone), 7.20 (d, J = 6.8 Hz, 3H, Ph (2H), 2-Pyridone (1H)), 7.07–6.97 (m, 2H, Ph), 6.91 (dd, J = 7.4, 4.9 Hz, 4H, Ph), 6.84 (d, J = 2.3 Hz, 1H, Ph), 6.44 (dd, J = 7.5, 2.4 Hz, 1H, 2-Pyridone), 4.57–4.37 (m, 3H, CH2, CH), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.91–2.84 (m, 1H, PhCH), 2.67 (dd, J = 13.4, 8.6 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.30 (C=O), 167.08 (C=O), 164.28 (C=O), 162.49 (C=O), 159.36 (d, 1JCF = 249.4 Hz), 158.97, 148.73, 140.56, 137.79, 135.93, 133.54 (d, 3JCF = 8.4 Hz), 130.37 (d, 4JCF = 2.6 Hz), 129.41 (2 × C), 129.07 (2 × C), 128.64 (2 × C), 126.93, 125.11 (d, 3JCF = 3.4 Hz), 124.76 (d, 2JCF = 14.8 Hz), 116.71 (d, 2JCF = 21.6 Hz), 115.07 (2 × C), 104.53, 99.65, 55.85, 51.90, 50.12, 38.09, 37.75. ESI-MS: m/z 557.3 (M + 1)+, m/z 579.4 (M + 23)+. C31H29FN4O5 [556.21].
(S)-4-chloro-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1e)
Yellow solid, yield: 80%. m.p.: 121–125 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H, NH), 8.61 (d, J = 7.8 Hz, 1H, NH), 7.95 (d, J = 8.6 Hz, 2H, Ph), 7.63 (d, J = 8.5 Hz, 2H, Ph), 7.40 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.21 (d, J = 6.8 Hz, 3H, Ph (2H), 2-Pyridone (1H)), 7.02 (d, J = 6.0 Hz, 2H, Ph), 6.94–6.87 (m, 5H, Ph), 6.56 (dd, J = 7.5, 2.4 Hz, 1H, 2-Pyridone), 4.59–4.36 (m, 3H, CH2, CH), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.88 (dd, J = 13.5, 5.6 Hz, 1H, PhCH), 2.67 (dd, J = 13.4, 8.6 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.30 (C=O), 167.11 (C=O), 165.94 (C=O), 162.50 (C=O), 158.97, 149.09, 140.29, 137.79, 137.50, 135.94, 133.40, 130.31 (2 × C), 129.41 (2 × C), 129.06 (2 × C), 129.03 (2 × C), 128.64 (2 × C), 126.92, 115.08 (2 × C), 104.93, 100.04, 55.86, 51.90, 50.09, 38.09, 37.75. ESI-MS: m/z 573.4 (M + 1)+, m/z 595.4 (M + 23)+. C31H29ClN4O5 [572.18].
(S)-4-bromo-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1f)
Yellow solid, yield: 88%. m.p.: 112–116 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H, NH), 8.60 (d, J = 7.7 Hz, 1H, NH), 7.88 (d, J = 8.5 Hz, 2H, Ph), 7.77 (d, J = 8.5 Hz, 2H, Ph), 7.40 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.21 (d, J = 6.8 Hz, 3H, Ph (2H), 2-Pyridone (1H)), 7.07–6.98 (m, 2H, Ph), 6.95–6.87 (m, 5H, Ph), 6.56 (dd, J = 7.5, 2.4 Hz, 1H, 2-Pyridone), 4.57–4.37 (m, 3H, CH2, CH), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.88 (dd, J = 13.5, 5.6 Hz, 1H, PhCH), 2.67 (dd, J = 13.5, 8.6 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.30 (C=O), 167.10 (C=O), 166.07 (C=O), 162.50 (C=O), 158.97, 149.08, 140.29, 137.79, 135.93, 133.76, 132.16, 131.98 (2 × C), 131.74, 130.46 (2 × C), 129.41 (2 × C), 129.07, 128.64 (2 × C), 126.92, 115.08 (2 × C), 104.94, 100.04, 55.86, 51.90, 50.09, 38.09, 37.75. ESI-MS: m/z 617.4 (M + 1)+. C31H29BrN4O5 [616.13].
(S)-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)-4-methylbenzamide (TD-1g)
Yellow solid, yield: 71%. m.p.: 118–120 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H, NH), 8.60 (d, J = 7.7 Hz, 1H, NH), 7.84 (d, J = 8.0 Hz, 2H, Ph), 7.39 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.35 (d, J = 8.0 Hz, 2H, Ph), 7.21 (d, J = 6.8 Hz, 3H, Ph (2H), 2-Pyridone (1H)), 7.05–6.98 (m, 2H, Ph), 6.89 (s, 5H, Ph), 6.58 (dd, J = 7.5, 2.4 Hz, 1H, 2-Pyridone), 4.57–4.36 (m, 3H, CH2, CH), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.88 (dd, J = 13.5, 5.6 Hz, 1H, PhCH), 2.67 (dd, J = 13.4, 8.6 Hz, 1H, PhCH), 2.39 (s, 3H, PhCH3). 13C NMR (150 MHz, DMSO-d6) δ 171.31 (C=O), 167.15 (C=O), 166.82 (C=O), 162.55 (C=O), 158.97, 149.33, 142.79, 140.16, 137.80, 135.93, 131.83, 129.47 (2 × C), 129.41 (2 × C), 129.07 (2 × C), 128.64 (2 × C), 128.39 (2 × C), 126.93, 115.07 (2 × C), 104.66, 100.12, 55.85, 51.90, 50.06, 38.09, 37.75, 21.50. ESI-MS: m/z 553.5 (M + 1)+, m/z 575.5 (M + 23)+. C32H32N4O5 [552.24].
(S)-4-methoxy-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1h)
Yellow solid, yield: 86%. m.p.: 109–112 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H, NH), 8.57 (d, J = 7.7 Hz, 1H, NH), 7.93 (d, J = 8.7 Hz, 2H, Ph), 7.37 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.24–7.17 (m, 3H, Ph (2H), 2-Pyridone (1H)), 7.07 (d, J = 8.9 Hz, 2H, Ph), 7.01 (d, J = 8.3 Hz, 2H, Ph), 6.95–6.87 (m, 5H, Ph), 6.58 (dd, J = 7.5, 2.3 Hz, 1H, 2-Pyridone), 4.56–4.36 (m, 3H, CH2, CH), 3.84 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.88 (dd, J = 13.7, 5.5 Hz, 1H, PhCH), 2.70–2.63 (m, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.31 (C=O), 167.16 (C=O), 166.30 (C=O), 162.88 (C=O), 162.57, 158.97, 149.45, 140.09, 137.80, 135.94, 130.40 (2 × C), 129.41 (2 × C), 129.07 (2 × C), 128.64 (2 × C), 126.92, 126.71, 115.08 (2 × C), 114.21 (2 × C), 104.51, 100.15, 55.99, 55.85, 51.90, 50.05, 38.10, 37.75. ESI-MS: m/z 569.5 (M + 1)+, m/z 591.5 (M + 23)+. C32H32N4O6 [568.23].
methyl (S)-4-((1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)carbamoyl)benzoate (TD-1i)
Yellow solid, yield: 75%. m.p.: 98–100 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H, NH), 8.59 (d, J = 7.7 Hz, 1H, NH), 8.10 (d, J = 8.2 Hz, 2H, Ph), 8.04 (d, J = 9.2 Hz, 2H, Ph), 7.41 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.25–7.15 (m, 3H, Ph (2H), 2-Pyridone (1H)), 7.02 (d, J = 8.3 Hz, 2H, Ph), 6.95–6.87 (m, 5H, Ph), 6.57 (dd, J = 7.5, 2.3 Hz, 1H, 2-Pyridone), 4.58–4.38 (m, 3H, CH2, CH), 3.90 (s, 3H, COOCH3), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.88 (dd, J = 13.6, 5.6 Hz, 1H, PhCH), 2.71–2.64 (m, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.31 (C=O), 167.10 (C=O), 166.25 (C=O), 166.08 (C=O), 162.50 (C=O), 158.97, 149.00, 140.35, 137.79, 135.93, 130.02, 129.77, 129.68 (2 × C), 129.41 (2 × C), 129.07 (2 × C), 128.75 (2 × C), 128.64 (2 × C), 126.92, 115.08 (2 × C), 105.08, 100.03, 55.85, 52.90, 51.91, 50.11, 38.09, 37.75. ESI-MS: m/z 597.4 (M + 1)+. C33H32N4O7 [596.23].
(S)-4-cyano-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1j)
Yellow solid, yield: 83%. m.p.: 134–136 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1H, NH), 8.58 (d, J = 7.7 Hz, 1H, NH), 8.07 (d, J = 8.3 Hz, 2H, Ph), 8.03 (d, J = 8.2 Hz, 2H, Ph), 7.41 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.20 (d, J = 6.6 Hz, 3H, Ph (2H), 2-Pyridone (1H)), 7.01 (d, J = 8.2 Hz, 2H, Ph), 6.91 (dd, J = 5.7, 3.2 Hz, 5H, Ph), 6.55 (dd, J = 7.5, 2.3 Hz, 1H, 2-Pyridone), 4.59–4.37 (m, 3H, CH2, CH), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.88 (dd, J = 13.6, 5.4 Hz, 1H, PhCH), 2.67 (dd, J = 13.4, 8.5 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.30 (C=O), 167.07 (C=O), 165.73 (C=O), 162.46 (C=O), 158.97, 148.87, 140.43, 138.67, 137.79, 135.93, 132.98 (2 × C), 129.41 (2 × C), 129.19 (2 × C), 129.06 (2 × C), 128.64 (2 × C), 126.93, 115.08 (2 × C), 105.17, 105.10, 99.98, 99.90, 55.86, 51.91, 50.12, 38.09, 37.75. ESI-MS: m/z 564.5 (M + 1)+, m/z 586.4 (M + 23)+. C32H29N5O5 [563.22].
(S)-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)-2-naphthamide (TD-1k)
Yellow solid, yield: 81%. m.p.: 113–117 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H, NH), 8.59 (d, J = 8.0 Hz, 1H, NH), 8.57 (s, 1H, Naphthalene), 8.12–7.96 (m, 4H, Naphthalene), 7.70–7.60 (m, 2H, Naphthalene), 7.42 (d, J = 7.4 Hz, 1H, 2-Pyridone), 7.26–7.15 (m, 3H, Ph (2H), 2-Pyridone (1H)), 7.07–6.99 (m, 2H, Ph), 6.98 (d, J = 2.3 Hz, 1H, Ph), 6.95–6.88 (m, 4H, Ph), 6.63 (dd, J = 7.5, 2.3 Hz, 1H, 2-Pyridone), 4.59–4.38 (m, 3H), 3.76 (s, 3H), 3.09 (s, 3H), 2.88 (dd, J = 13.3, 5.8 Hz, 1H), 2.68 (dd, J = 13.5, 8.4 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 171.32 (C=O), 167.15 (C=O), 167.09 (C=O), 162.58 (C=O), 158.98, 149.32, 140.27, 137.80, 135.94, 134.98, 132.47, 132.00, 130.96, 129.52 (2 × C), 129.42 (2 × C), 129.08 (2 × C), 128.92, 128.65 (2 × C), 128.63 (2 × C), 128.18, 127.46, 126.93, 124.83, 115.09 (2 × C), 104.81, 100.13, 55.86, 51.91, 50.11, 38.10, 37.76. ESI-MS: m/z 589.4 (M + 1)+, m/z 611.4 (M + 23)+. C35H32N4O5 [588.24].
(S)-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)-4-nitrobenzamide (TD-1l)
Yellow solid, yield: 81%. m.p.: 134–136 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.61 (s, 1H, NH), 8.58 (d, J = 7.7 Hz, 1H, NH), 8.39–8.36 (m, 2H, Ph), 8.18–8.14 (m, 2H, Ph), 7.43 (d, J = 7.4 Hz, 1H, 2-Pyridone), 7.24–7.17 (m, 3H, Ph (2H), 2-Pyridone (1H)), 7.02 (s, 2H, Ph), 6.94–6.89 (m, 5H, Ph), 6.57 (dd, J = 7.5, 2.4 Hz, 1H, 2-Pyridone), 4.59–4.40 (m, 3H, CH2, CH), 3.76 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.88 (dd, J = 13.6, 5.6 Hz, 1H, PhCH), 2.68 (dd, J = 13.5, 8.5 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.30 (C=O), 167.07 (C=O), 165.47 (C=O), 162.46 (C=O), 158.97, 149.95, 148.84, 140.45, 140.27, 137.79, 135.93, 129.92 (2 × C), 129.41 (2 × C), 129.07 (2 × C), 128.64 (2 × C), 126.93, 124.04 (2 × C), 115.08 (2 × C), 105.27, 99.99, 55.86, 51.91, 50.13, 38.09, 37.75. ESI-MS: m/z 584.4 (M + 1)+, m/z 606.4 (M + 23)+. C31H29N5O7 [583.21].
5.1.7. Procedure for the Synthesis of TD-1m
TD-1l and 10% Pd/C (10% w/w) were dissolved in methanol (5 mL) and dichloromethane (5 mL), and the solution degassed and was stirred at room temperature for 2h under H2. The mixture was filtered and concentrated, and the resulting residue were purified by recrystallization or preparation thin layer chromatography to provide the target compounds TD-1m.
(S)-4-amino-N-(1-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)-2-oxo-1,2-dihydropyridin-4-yl)benzamide (TD-1m)
Yellow solid, yield: 65%. m.p.: 137–139 °C. 1H NMR (600 MHz, DMSO-d6) δ 9.79 (s, 1H, NH), 8.55 (d, J = 7.7 Hz, 1H, NH), 7.69 (d, J = 8.6 Hz, 2H, Ph), 7.33 (d, J = 7.5 Hz, 1H, 2-Pyridone), 7.24–7.17 (m, 3H, Ph (2H), 2-Pyridone (1H)), 7.01 (s, 2H, Ph), 6.94–6.88 (m, 5H, Ph), 6.61 (d, J = 8.6 Hz, 2H, Ph), 6.59 (dd, J = 7.5, 2.3 Hz, 1H, 2-Pyridone), 5.85 (s, 2H, NH2), 4.55–4.35 (m, 3H, CH2, CH), 3.76 (s, 3H, OCH3), 3.10 (s, 3H, NCH3), 2.87 (dd, J = 12.1, 4.4 Hz, 1H, PhCH), 2.67 (dd, J = 13.4, 8.4 Hz, 1H, PhCH). 13C NMR (150 MHz, DMSO-d6) δ 171.32 (C=O), 167.22 (C=O), 166.51 (C=O), 162.63 (C=O), 158.96, 153.19, 149.84, 139.85, 137.79, 135.93, 130.23 (2 × C), 129.41 (2 × C), 129.07, 128.64 (2 × C), 126.92, 120.68, 115.08 (2 × C), 113.04 (2 × C), 103.91, 100.23, 55.85, 51.88, 50.00, 38.09, 37.75. ESI-MS: m/z 554.5 (M + 1)+, m/z 576.4 (M + 23)+. C31H31N5O5 [553.23].
5.2. In Vitro Anti-HIV Assay in MT-4 Cells
Evaluation of the antiviral activity of the compounds against HIV in MT-4 cells was performed using the MTT assay as described below. Stock solutions (10 × final concentration) of test compounds were added in 25 µL volumes to two series of triplicate wells to allow for the simultaneous evaluation of their effects on mock- and HIV-infected cells at the beginning of each experiment. Serial five-fold dilutions of test compounds were made directly in flat-bottomed 96-well microtiter trays using a Biomek 3000 robot (Beckman Instruments, Fullerton, CA, USA). Untreated HIV- and mock-infected cell samples were included as controls. HIV stock (50 µL) at 100–300 CCID50 (50% cell culture infectious doses) or culture medium was added to either the infected or mock-infected wells of the microtiter tray. Mock-infected cells were used to evaluate the effects of the test compound on uninfected cells to assess the test compounds’ cytotoxicity. Exponentially growing MT-4 cells were centrifuged for 5 min at 220 g, and the supernatant was discarded. The MT-4 cells were resuspended at 6 × 105 cells/mL, and 50 µL volumes were transferred to the microtiter tray wells. Five days after infection, the viability of mock-and HIV-infected cells was examined spectrophotometrically using the MTT assay. The MTT assay is based on the reduction of yellow colored 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Acros Organics, Belgium) by mitochondrial dehydrogenase activity in metabolically active cells to a blue-purple formazan that can be measured spectrophotometrically. The absorbances were read in an eight-channel computer-controlled photometer (Infinite M1000, Tecan, Swiss) at two wavelengths (540 and 690 nm). All data were calculated using the median absorbance value of three wells. The 50% cytotoxic concentration (CC50) was defined as the concentration of the test compound that reduced the absorbance (OD540) of the mock-infected control sample by 50%. The concentration achieving 50% protection against the cytopathic effect of the virus in infected cells was defined as the 50% effective concentration (EC50).
5.3. Solubility Studies
We weighed about 1mg of the compound, dissolved it with 1ml of methanol, and diluted it in gradient (twice each time) to obtain 10 concentrations. The peak area of the compound was tested by HPLC, and the peak area- concentration standard curve was drawn. In addition, about 1 mg, 0.6 mg and 0.3 mg of the compound were weighed and dissolved in 1 mL of pH 2, pH 7 and pH 7.4 phosphate buffer to obtain the saturated solution of the compound. HPLC was used to test the peak area under different pH, and the water solubility of the compound was obtained through the standard curve.
5.4. Molecular Docking Studies
All molecules were prepared using LigPrep (Maestro, Schrödinger, LLC, New York, NY, USA, 2021), and the hexameric HIV-1 CA (PDB ID: 5HGL) protein was used as a receptor for the docking calculations. The Glide application was used for all docking studies with default parameters. The docking results were visualized by PyMOL v2.3.0 (Schrödinger, New York, NY, USA).
5.5. Computational Assessment of Drug-like Properties, Metabolic Stability, and Toxicity
Drug-like properties, metabolic stability, and toxicity were assessed using the P450 and Derek Nexus module within Stardrop V7 [42] (Optibrium, Ltd., Cambridge, UK).
For metabolic stability evaluation, representative compounds were docked into the human CYP3A4 isoform (PDB ID 4D78). CYP3A4 was prepared using Autodock tools [49,50,51,52], where essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added. The grid box for the docking search was centered around the catalytic center of CYP3A4 [49,50,51]. Docking calculations were performed using AutoDock via DockingServer [52]. The docked poses were further evaluated with SeeSAR 12.1 (BioSolveIT Gmbh, Germany) utilizing the predictive binding affinity calculations using the HYdrogen bond and DEhydration (HYDE) energy scoring function [44,45,46]. The lower boundary of the affinity prediction for the poses with acceptable torsion angles and clash scores were used in combination with composite site lability (CSL), and the number of labile sites within the molecule to predict metabolic stability.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/molecules27217640/s1. Supplementary material includes MS, 1H NMR and 13C NMR spectra for target compounds and standard curves of representative compounds in solubility assay.
Author Contributions
Conceptualization, X.L. and P.Z.; methodology, E.D.C., C.P., A.D., X.L. and P.Z.; software, X.Z., S.C. and A.D.; validation, X.Z.; formal analysis, X.Z.; investigation, X.Z., L.S., X.S., Z.L., D.D., X.J. and S.Z.; resources, E.D.C., C.P., A.D., P.Z. and X.L.; data curation, X.Z., L.S., A.D., X.L. and P.Z.; writing—original draft preparation, X.Z. and L.S.; writing—review and editing, X.Z., S.X., S.Z., S.C., A.D., X.L. and P.Z.; visualization, X.Z., S.Z. and A.D.; supervision, P.Z., X.L. and A.D.; project administration, P.Z. and X.L.; funding acquisition, P.Z., X.L. and S.C. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Sample Availability
Samples of the compounds are available from the authors.
Funding Statement
We gratefully acknowledge financial support from National Natural Science Foundation of China (NSFC Nos. 82173677, 81773574, 82204196), the Key Project of NSFC for International Cooperation (No. 81420108027), Science Foundation for Outstanding Young Scholars of Shandong Province (ZR2020JQ31), Qilu Young Scholars Program of Shandong University, the Taishan Scholar Program at Shandong Province, Shandong Provincial Natural Science Foundation (ZR2022QH015) and NIH/NIAID grant R01AI150491 (Loll, PI, Salvino, Co-I).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Maartens G., Celum C., Lewin S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet. 2014;384:258–271. doi: 10.1016/S0140-6736(14)60164-1. [DOI] [PubMed] [Google Scholar]
- 2.Campbell-Yesufu O.T., Gandhi R.T. Update on human immunodeficiency virus (HIV)-2 infection. Clin. Infect. Dis. 2011;52:780–787. doi: 10.1093/cid/ciq248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nyamweya S., Hegedus A., Jaye A., Rowland-Jones S., Flanagan K.L., Macallan D.C. Comparing HIV-1 and HIV-2 infection: Lessons for viral immunopathogenesis. Rev. Med. Virol. 2013;23:221–240. doi: 10.1002/rmv.1739. [DOI] [PubMed] [Google Scholar]
- 4.Ray M., Logan R., Sterne J.A., Hernández-Díaz S., Robins J.M., Sabin C., Bansi L., van Sighem A., de Wolf F., Costagliola D., et al. The effect of combined antiretroviral therapy on the overall mortality of HIV-infected individuals. Aids. 2010;24:123–137. doi: 10.1097/QAD.0b013e3283324283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z., Cherukupalli S., Xie M., Wang W., Jiang X., Jia R., Pannecouque C., De Clercq E., Kang D., Zhan P., et al. Contemporary medicinal chemistry strategies for the discovery and development of novel HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2022;65:3729–3757. doi: 10.1021/acs.jmedchem.1c01758. [DOI] [PubMed] [Google Scholar]
- 6.Du J., Guo J., Kang D., Li Z., Wang G., Wu J., Zhang Z., Fang H., Hou X., Huang Z., et al. New techniques and strategies in drug discovery. Chin. Chem. Lett. 2020;31:1695–1708. doi: 10.1016/j.cclet.2020.03.028. [DOI] [Google Scholar]
- 7.Rossi E., Meuser M.E., Cunanan C.J., Cocklin S. Structure, Function, and Interactions of the HIV-1 Capsid Protein. Life. 2021;11:100. doi: 10.3390/life11020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu S., Sun L., Zalloum W.A., Zhang X., Huang T., Ding D., Tao Y., Zhao F., Gao S., Kang D., et al. From design to biological mechanism evaluation of phenylalanine-bearing HIV-1 capsid inhibitors targeting a vital assembly interface. Chin. Chem. Lett. 2022 doi: 10.1016/j.cclet.2022.06.034. [DOI] [Google Scholar]
- 9.Sun L., Zhang X., Xu S., Huang T., Song S., Cherukupalli S., Zhan P., Liu X. An insight on medicinal aspects of novel HIV-1 capsid protein inhibitors. Eur. J. Med. Chem. 2021;217:113380. doi: 10.1016/j.ejmech.2021.113380. [DOI] [PubMed] [Google Scholar]
- 10.Dick R.A., Mallery D.L., Vogt V.M., James L.C. IP6 Regulation of HIV Capsid Assembly, Stability, and Uncoating. Viruses. 2018;10:640. doi: 10.3390/v10110640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gruenke P.R., Aneja R., Welbourn S., Ukah O.B., Sarafianos S.G., Burke D.H., Lange M.J. Selection and identification of an RNA aptamer that specifically binds the HIV-1 capsid lattice and inhibits viral replication. Nucleic Acids Res. 2022;50:1701–1717. doi: 10.1093/nar/gkab1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X., Xu S., Sun L., Ding D., Tao Y., Kang D., Liu X., Zhan P. HIV-1 capsid inhibitors: A sword to destroy the virus. Future Med. Chem. 2022;14:605–607. doi: 10.4155/fmc-2022-0008. [DOI] [PubMed] [Google Scholar]
- 13.Rihn S.J., Wilson S.J., Loman N.J., Alim M., Bakker S.E., Bhella D., Gifford R.J., Rixon F.J., Bieniasz P.D. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog. 2013;9:e1003461. doi: 10.1371/journal.ppat.1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L., Dick A., Meuser M.E., Huang T., Zalloum W.A., Chen C.H., Cherukupalli S., Xu S., Ding X., Gao P., et al. Design, synthesis, and mechanism study of benzenesulfonamide-containing Phenylalanine derivatives as novel HIV-1 capsid inhibitors with improved antiviral activities. J. Med. Chem. 2020;63:4790–4810. doi: 10.1021/acs.jmedchem.0c00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X., Sun L., Meuser M.E., Zalloum W.A., Xu S., Huang T., Cherukupalli S., Jiang X., Ding X., Tao Y., et al. Design, synthesis, and mechanism study of dimerized phenylalanine derivatives as novel HIV-1 capsid inhibitors. Eur. J. Med. Chem. 2021;226:113848. doi: 10.1016/j.ejmech.2021.113848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu S., Sun L., Dick A., Zalloum W.A., Huang T., Meuser M.E., Zhang X., Tao Y., Cherukupalli S., Ding D., et al. Design, synthesis, and mechanistic investigations of phenylalanine derivatives containing a benzothiazole moiety as HIV-1 capsid inhibitors with improved metabolic stability. Eur. J. Med. Chem. 2022;227:113903. doi: 10.1016/j.ejmech.2021.113903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vernekar S.K.V., Sahani R.L., Casey M.C., Kankanala J., Wang L., Kirby K.A., Du H., Zhang H., Tedbury P.R., Xie J., et al. Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules. Viruses. 2020;12:452. doi: 10.3390/v12040452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L., Casey M.C., Vernekar S.K.V., Sahani R.L., Kirby K.A., Du H., Zhang H., Tedbury P.R., Xie J., Sarafianos S.G., et al. Novel PF74-like small molecules targeting the HIV-1 capsid protein: Balance of potency and metabolic stability. Acta Pharm. Sin. B. 2021;11:810–822. doi: 10.1016/j.apsb.2020.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meuser M.E., Reddy P.A.N., Dick A., Maurancy J.M., Salvino J.M., Cocklin S. Rapid optimization of the metabolic stability of a human immunodeficiency virus type-1 capsid inhibitor using a multistep computational workflow. J. Med. Chem. 2021;64:3747–3766. doi: 10.1021/acs.jmedchem.0c01810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J.P., Francis A.C., Meuser M.E., Mankowski M., Ptak R.G., Rashad A.A., Melikyan G.B., Cocklin S. Exploring modifications of an HIV-1 capsid inhibitor: Design, synthesis, and mechanism of action. J. Drug Des. Res. 2018;5:1070. [PMC free article] [PubMed] [Google Scholar]
- 21.Chin C.R., Perreira J.M., Savidis G., Portmann J.M., Aker A.M., Feeley E.M., Smith M.C., Brass A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015;13:1717–1731. doi: 10.1016/j.celrep.2015.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharya A., Alam S.L., Fricke T., Zadrozny K., Sedzicki J., Taylor A.B., Demeler B., Pornillos O., Ganser-Pornillos B.K., Diaz-Griffero F., et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA. 2014;111:18625–18630. doi: 10.1073/pnas.1419945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price A.J., Jacques D.A., McEwan W.A., Fletcher A.J., Essig S., Chin J.W., Halambage U.D., Aiken C., James L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014;10:e1004459. doi: 10.1371/journal.ppat.1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rasaiyaah J., Tan C.P., Fletcher A.J., Price A.J., Blondeau C., Hilditch L., Jacques D.A., Selwood D.L., James L.C., Noursadeghi M., et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price A.J., Fletcher A.J., Schaller T., Elliott T., Lee K., KewalRamani V.N., Chin J.W., Towers G.J., James L.C. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012;8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee K., Ambrose Z., Martin T.D., Oztop I., Mulky A., Julias J.G., Vandegraaff N., Baumann J.G., Wang R., Yuen W., et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010;7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marini B., Kertesz-Farkas A., Ali H., Lucic B., Lisek K., Manganaro L., Pongor S., Luzzati R., Recchia A., Mavilio F., et al. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015;521:227–231. doi: 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 28.Matreyek K.A., Yücel S.S., Li X., Engelman A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013;9:e1003693. doi: 10.1371/journal.ppat.1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di Nunzio F., Fricke T., Miccio A., Valle-Casuso J.C., Perez P., Souque P., Rizzi E., Severgnini M., Mavilio F., Charneau P., et al. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology. 2013;440:8–18. doi: 10.1016/j.virol.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lelek M., Di Nunzio F., Henriques R., Charneau P., Arhel N., Zimmer C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc. Natl. Acad. Sci. USA. 2012;109:8564–8569. doi: 10.1073/pnas.1013267109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaller T., Ocwieja K.E., Rasaiyaah J., Price A.J., Brady T.L., Roth S.L., Hué S., Fletcher A.J., Lee K., KewalRamani V.N., et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011;7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matreyek K.A., Engelman A. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 2011;85:7818–7827. doi: 10.1128/JVI.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bichel K., Price A.J., Schaller T., Towers G.J., Freund S.M., James L.C. HIV-1 capsid undergoes coupled binding and isomerization by the nuclear pore protein NUP358. Retrovirology. 2013;10:81. doi: 10.1186/1742-4690-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dick A., Meuser M.E., Cocklin S. Clade-Specific Alterations within the HIV-1 Capsid Protein with Implications for Nuclear Translocation. Biomolecules. 2022;12:695. doi: 10.3390/biom12050695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dick A., Cocklin S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules. 2020;25:1687. doi: 10.3390/molecules25071687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blair W.S., Pickford C., Irving S.L., Brown D.G., Anderson M., Bazin R., Cao J., Ciaramella G., Isaacson J., Jackson L., et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010;6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rankovic S., Ramalho R., Aiken C., Rousso I. PF74 Reinforces the HIV-1 Capsid To Impair Reverse Transcription-Induced Uncoating. J. Virol. 2018;92:e00845-18. doi: 10.1128/JVI.00845-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacques D.A., McEwan W.A., Hilditch L., Price A.J., Towers G.J., James L.C. HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature. 2016;536:349–353. doi: 10.1038/nature19098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Link J.O., Rhee M.S., Tse W.C., Zheng J., Somoza J.R., Rowe W., Begley R., Chiu A., Mulato A., Hansen D., et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature. 2020;584:614–618. doi: 10.1038/s41586-020-2443-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bester S.M., Wei G., Zhao H., Adu-Ampratwum D., Iqbal N., Courouble V.V., Francis A.C., Annamalai A.S., Singh P.K., Shkriabai N., et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science. 2020;370:360–364. doi: 10.1126/science.abb4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margot N.A., Naik V., VanderVeen L., Anoshchenko O., Singh R., Dvory-Sobol H., Rhee M.S., Callebaut C. Resistance analyses in Highly Treatment-Experienced People with HIV Treated with the Novel Capsid HIV Inhibitor Lenacapavir. J. Infect. Dis. 2022:jiac364. doi: 10.1093/infdis/jiac364. [DOI] [PubMed] [Google Scholar]
- 42.Segall M., Champness E., Obrezanova O., Leeding C. Beyond profiling: Using ADMET models to guide decisions. Chem. Biodivers. 2009;6:2144–2151. doi: 10.1002/cbdv.200900148. [DOI] [PubMed] [Google Scholar]
- 43.Tuyishime M., Danish M., Princiotto A., Mankowski M.K., Lawrence R., Lombart H.G., Esikov K., Berniac J., Liang K., Ji J., et al. Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorg. Med. Chem. Lett. 2014;24:5439–5445. doi: 10.1016/j.bmcl.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karadsheh R., Meuser M.E., Cocklin S. Composition and orientation of the core region of novel HIV-1 entry inhibitors influences metabolic stability. Molecules. 2020;25:1430. doi: 10.3390/molecules25061430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tyzack J.D., Hunt P.A., Segall M.D. Predicting regioselectivity and lability of cytochrome P450 metabolism using quantum mechanical simulations. J. Chem. Inf. Model. 2016;56:2180–2193. doi: 10.1021/acs.jcim.6b00233. [DOI] [PubMed] [Google Scholar]
- 46.Hunt P.A., Segall M.D., Tyzack J.D. WhichP450: A multi-class categorical model to predict the major metabolising CYP450 isoform for a compound. J. Comput. Aided Mol. Des. 2018;32:537–546. doi: 10.1007/s10822-018-0107-0. [DOI] [PubMed] [Google Scholar]
- 47.Sega G.A. A review of the genetic effects of ethyl methanesulfonate. Mutat. Res. 1984;134:113–142. doi: 10.1016/0165-1110(84)90007-1. [DOI] [PubMed] [Google Scholar]
- 48.Pillans P.I., Ghiculescu R.A., Lampe G., Wilson R., Wong R., Macdonald G.A. Severe acute liver injury associated with lumiracoxib. J. Gastroenterol. Hepatol. 2012;27:1102–1105. doi: 10.1111/j.1440-1746.2011.07036.x. [DOI] [PubMed] [Google Scholar]
- 49.Huey R., Morris G.M., Olson A.J., Goodsell D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 50.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morris G.M., Huey R., Olson A.J. Current Protocols in Bioinformatics. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2008. Using AutoDock for ligand-receptor docking. Chapter 8, Unit 8.14. [DOI] [PubMed] [Google Scholar]
- 52.Bikadi Z., Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009;1:15. doi: 10.1186/1758-2946-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.