

Abstract
OBJECTIVES:
Four peer-reviewed publications have reported results from randomized controlled trials of convalescent plasma for coronavirus disease 2019 infection; none were conducted in the United States nor used standard plasma as a comparator. To determine if administration of convalescent plasma to patients with coronavirus disease 2019 increases antibodies to severe acute respiratory syndrome coronavirus 2 and improves outcome.
DESIGN:
Double-blind randomized controlled trial.
SETTING:
Hospital in New York.
PATIENTS:
Patients with polymerase chain reaction documented coronavirus disease 2019 infection.
INTERVENTIONS:
Patients were randomized (4:1) to receive 2 U of convalescent plasma versus standard plasma. Antibodies to severe acute respiratory syndrome coronavirus 2 were measured in plasma units and in trial recipients.
MEASUREMENTS AND MAIN RESULTS:
Enrollment was terminated after emergency use authorization was granted for convalescent plasma. Seventy-four patients were randomized. At baseline, mean (sd) Acute Physiology and Chronic Health Evaluation II score (23.4 [5.6] and 22.5 [6.6]), percent of patients intubated (19% and 20%), and median (interquartile range) days from symptom onset to randomization of 9 (6–18) and 9 (6–15), were similar in the convalescent plasma versus standard plasma arms, respectively. Convalescent plasma had high neutralizing activity (median [interquartile range] titer 1:526 [1:359–1:786]) and its administration increased antibodies to severe acute respiratory syndrome coronavirus 2 by 14.4%, whereas standard plasma administration led to an 8.6% decrease (p = 0.005). No difference was observed for ventilator-free days through 28 days (primary study endpoint): median (interquartile range) of 28 (2–28) versus 28 (0–28; p = 0.86) for the convalescent plasma and standard plasma groups, respectively. A greater than or equal to 2 point improvement in the World Health Organization scale was achieved by 20% of subjects in both arms (p = 0.99). All-cause mortality through 90 days was numerically lower in the convalescent plasma versus standard plasma groups (27% vs 33%; p = 0.63) but did not achieve statistical significance. A key prespecified subgroup analysis of time to death in patients who were intubated at baseline was statistically significant; however, sample size numbers were small.
CONCLUSIONS:
Administration of convalescent plasma to hospitalized patients with coronavirus disease 2019 infection increased antibodies to severe acute respiratory syndrome coronavirus disease 2 but was not associated with improved outcome.
Keywords: convalescent plasma, coronavirus disease 2019, infection, randomized
The global coronavirus disease 2019 (COVID-19) pandemic has resulted in more than 1.5 million deaths worldwide (1, 2). Historical data, mostly from nonrandomized studies, suggest that convalescent plasma (CP) may be a useful tool in the treatment of some viral illnesses with limited therapeutic alternatives (3, 4). Early in the COVID-19 pandemic, several case series (5–9) and matched cohort studies (10–12) reported encouraging preliminary results for CP. An uncontrolled national expanded access program (EAP) spanning 2,747 U.S. sites with over 85,000 patients transfused (13) reported a favorable safety profile through the first 5,000 (14) and subsequent 20,000 patients (15). More recently, an exploratory analysis of 35,000 hospitalized patients from this uncontrolled EAP reported encouraging but weak relationships between 7-day mortality and both 1) time from diagnosis to treatment and 2) CP immunoglobulin G (IgG) levels (16). Based in part on these emerging data and the large unmet medical need, the U.S. Food and Drug Administration (FDA) granted an emergency use authorization (EUA) for CP in the treatment of COVID-19 on August 23, 2020 (17).
Notwithstanding the above, to our knowledge, there have been only four peer-reviewed publications reporting the results from randomized controlled trials (RCTs) of CP for COVID-19 infection (18–20). Three of these trials, which took place in China (19), India (18), and Argentina (20), reported no significant benefit to CP. A second trial from Argentina involving early administration (within 72 hr after symptom onset) of CP showed benefit (21). Two of these trials were not blinded (18, 19), and none were conducted in the United States, where the deployment of care may be different. In addition, none of these trials used standard plasma (SP) as a control arm, which could affect the results.
Herein, we report the results of a double-blind RCT, comparing CP versus SP, initiated in the New York Metropolitan area during the “spring 2020” COVID-19 surge.
MATERIALS AND METHODS
After review and approval by the FDA (Investigational New Drug No. 19823) and Stony Brook University’s Institutional Review Board (2020-00209), the trial was registered prior to enrollment of the first subject (ClinicalTrials.gov NCT04344535).
Convalescent Plasma and Standard Plasma
Potential CP donors were recruited beginning on April 8, 2020. Details of this process are described elsewhere (22) and summarized in Supplemental Detailed Methods (http://links.lww.com/CCM/G335). CP was collected by Blood Bank staff using standard procedures and stored in our hospital’s Blood Bank. SP collected prior to January 2020 was administered to patients randomized to the SP group.
Plasma Recipient Eligibility and Equity of Invitation to Participate
The trial was open to enrollment on April 8, 2020.
Inclusion criteria were adult patients hospitalized with a confirmed diagnosis of COVID-19 infection from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reverse transcription polymerase chain reaction (PCR) testing.
Exclusion criteria are described in Supplemental Detailed Methods (http://links.lww.com/CCM/G335).
Randomization, Allocation Concealment, and Blinding
Patients were randomized 4:1 to CP or SP using permuted block randomization lists generated using SAS software (version 9.4; SAS Institute, Cary NC) and implemented using an interactive web response randomization tool in Research Electronic Data Capture (REDCap). With a planned enrollment of 500, the 4:1 ratio was anticipated to allow for potential benefit in a high percentage of patients (n = 400, 80%) but still allow for a control group (n = 100, 20%) to assess safety and efficacy of this unproven therapy. Randomization was stratified by nonintubated versus intubated patients.
Team M study members, who were unblinded, randomized subjects as above. The only other individuals who were unblinded were Blood Bank personnel since they needed to label and dispense the masked CP or SP. The bags of CP or SP had an identical label, stating “CP or SP” to preserve blinding.
All study personnel who collected data were blinded to study assignment at all times.
Main Interventions
Subjects received a single “dose” of 2 U of either CP or SP (total volume approximately 480 mL). Each unit of plasma (approximately 240 mL) was administered over 1–4 hours, using standard hospital procedures.
Blood was obtained on day 0 (baseline prior to plasma infusion) and then on days 1, 7, 14, and 21 (only while still hospitalized) for determination of SARS-CoV-2 antibody levels.
Safety Monitoring and Data and Safety Monitoring Board
A blinded Safety Monitor (S.N. coauthor), not otherwise involved in the study, reviewed all reportable adverse events, The trial’s independent Data and Safety Monitoring Board (DSMB) including an unblinded DSMB statistician operated under an approved charter.
Endpoints, Data Management, and Statistics
Primary Endpoint.
The total number of ventilator-free days from randomization to day 28 with ventilator-free days was defined as the total number of calendar days or proportions of calendar days during the first 28 days after randomization. Subjects never requiring intubation were assigned a time of 28 days and those who died by day 28 were assigned 0 ventilator-free days.
Secondary Endpoints.
Death. All-cause mortality through 90 days postrandomization was assessed as time to event (number of days from randomization to death).
World Health Organization ordinal scale.
Consistent with many other clinical trials related to COVID-19, we recorded the World Health Organization (WHO) ordinal scale on each day from randomization through 28 days.
Immune response.
Antibodies to SARS-CoV-2 were measured in several ways: 1) neutralizing antibodies (plaque reduction and pseudovirus) were measured in a random subset of plasma bags, defined as the neutralizing titer (NT50) to achieve a 50% reduction in infectivity consistent with previous publications and 2) blood was obtained from plasma recipients on day 0 (baseline prior to plasma infusion), and then on days 1, 7, 14, and 21 (only while still hospitalized) for determination of SARS-CoV-2 antibody levels using a rapid immunochromatographic test.
Data Management, Sample Size, and Statistical Methods
Data for this study were collected by blinded study personal and managed using the REDCap electronic capture tool.
The analysis population for the primary and key secondary endpoints was all patients randomized, that is, “intent-to-treat” Additional “per protocol” analyses were performed for study endpoints on subjects who were randomized and received both units of plasma they were assigned to.
The original sample size would have provided ample participants to detect a 2.5 days (sd = 6 d) difference for CP group in ventilator-free days with 90% power and a two-sided significant level of α equals to 0.05. To account for likely heterogeneity and allow for additional exploratory subset analyses, we targeted a sample size of 500 participants, randomized at a 4:1 ratio of CP to regular plasma randomization.
See Supplemental Detailed Methods for additional statistical details (http://links.lww.com/CCM/G335).
RESULTS
Details regarding the results from our CP donor selection and collection process are reported elsewhere (22). Briefly, initiation of screening for CP donors began on April 8, 2020. Overall, 865 U of CP (mean 240 mL/U, total volume 207,824 mL) were collected from 262 convalescent donors. In a randomly selected subset of units, CP had high levels of neutralizing antibodies to SARS-CoV-2. NT50 were median (interquartile range [IQR]) 1:334 (1:192–1:714) in a pseudotype assay (23) and 1:526 (1:359–1:786) in a plaque neutralization assay (gold standard) using SARS-CoV-2 (Fig. 1).
Figure 1.
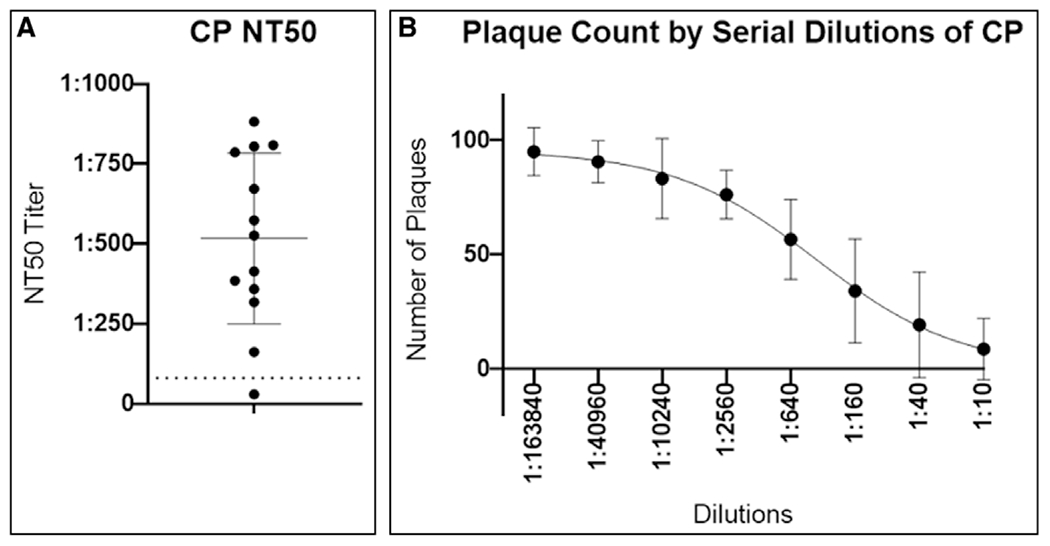
Convalescent plasma (CP) neutralizing titer (NT50) against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). A, The median (interquartile range) NT50 for randomly selected units tested in a plaque neutralization assay using SARS-CoV-2 virus (n = 13). Each dot represents the calculated NT50 value (titer) for an individual plasma sample. The dashed line at 1:80 corresponds with the minimum suggested titer recommended by the Food and Drug Administration for CP. B, The average (sd) number of plaques of SARS-CoV-2 for serial dilutions of CP.
Overall, 82 hospitalized patients, that is, recipients, were enrolled. Anecdotally, there was large interest in the trial by families of patients who had been ventilator dependent for several weeks but who were beyond the enrollment window. In contrast, many patients with mild COVID-19 infection were reluctant to try a blood product transfusion experimental therapy.
Enrollment in the trial was stopped on August 24, 2020, after FDA granted EUA for CP since it seemed unlikely that patients would wish to participate in a randomized trial for an “approved therapy.” At this time, 74 patients had been randomized (Supplemental Fig. 1, http://links.lww.com/CCM/G337; legend, http://links.lww.com/CCM/G339). Group assignment was consistent with the planned 4:1 target (n = 59 CP, n = 15 SP). Only two patients were randomized and did not receive plasma (one in each study arm). One patient (CP arm) experienced a serious transfusion reaction after a small volume was administered and thus did not complete the 2 U administration. No patients were lost to follow-up through the 90-day study period.
Study groups were balanced with regard to most baseline characteristics, for example, age, sex, body mass index (Table 1). Importantly, the percent of patients intubated at baseline (19% vs 20%), and median (IQR) days from symptom onset to randomization of 9 (6–18) versus 9 (6–15), were similar in the CP versus SP arms, respectively. In addition, Acute Physiology and Chronic Health Evaluation II scores mean (sd), calculated to provide a composite index of comorbidities and acute physiologic status, were well balanced in the two groups, CP 23.4 (5.6) versus SP 22.5 (6.6). There were some imbalances at baseline, for example, more patients receiving CP received remdesivir and were on supplemental oxygen (Table 1).
TABLE 1.
Characteristics of the Patients at Baseline
| Characteristics | Convalescent Plasma, n = 59 | Standard Plasma, n = 15 |
|---|---|---|
| Age, mean (sd) | 67 (15.8) | 64 (17.4) |
|
| ||
| Male sex, n (%) | 36 (61.0) | 8 (53.3) |
|
| ||
| Race White, n (%) | 42 (71.2) | 8 (53.3) |
|
| ||
| Body mass index, kg/m2, median (IQR) | 28.9 (24.0–33.6) | 27.8 (23.1–30.2) |
|
| ||
| Diabetes, n (%) | 19 (32.2) | 6 (40.0) |
|
| ||
| Hypertension, n (%) | 40 (67.8) | 11 (73.3) |
|
| ||
| Chronic obstructive pulmonary disease, n (%) | 7 (11.9) | 2 (13.3) |
|
| ||
| Chronic heart failure, n (%) | 11 (18.6) | 2 (13.3) |
|
| ||
| Chronic renal insufficiency, n (%) | 7 (11.9) | 0 (0) |
|
| ||
| Coronary artery disease, coronary artery bypass graft surgery, percutaneous coronary intervention, myocardial infarction, n (%) | 12 (20.3) | 3 (20.0) |
|
| ||
| Cerebrovascular disease, n (%) | 9 (15.3) | 2 (13.3) |
|
| ||
| Receiving immunosuppressant medication, n (%) | 4 (6.8) | 2 (13.3) |
|
| ||
| Polymerase chain reaction+ documented COVID-19 infection, n (%) | 59 (100) | 15 (100) |
|
| ||
| Days symptom start to randomization, median (IQR) | 9 (6–18) | 9 (6–15) |
|
| ||
| Days admission to randomization, median (IQR) | 4 (2–10) | 4 (3–6) |
|
| ||
| In ICU at randomization, n (%) | 17 (28.8) | 3 (20.0) |
|
| ||
| Type of oxygen supplementation, n (%) | ||
| Nasal cannula or mask | 30 (50.8) | 4 (26.7) |
| High-flow nasal cannula or continuous positive airway pressure)/bilevel positive airway pressure | 3 (5.1) | 2 (13.3) |
| Intubated | 11 (18.6) | 3 (20.0) |
|
| ||
| Any FDA COVID-19 sign or symptoms*, n (%) | 46 (78.0) | 13 (86.7) |
|
| ||
| Any FDA severe COVID-19 sign/symptom, n (%) | 44 (74.6) | 13 (86.7) |
| Shortness of breath | 38 (64.4) | 7 (46.7) |
| Respiratory rate ≥ 30 | 14 (23.7) | 2 (13.3) |
| o2 saturation ≤ 93% | 32 (54.2) | 9 (60.0) |
| Pao2/Fio2 < 300 | 15 (25.4) | 3 (20.0) |
| Bilateral lung infiltrates | 39 (66.1) | 12 (80.0) |
|
| ||
| Acute Physiology and Chronic Health Evaluation II score, mean (sd) | 23.4 (5.6) | 22.5 (6.6) |
|
| ||
| Laboratory values, median (IQR) | ||
| Serum creatinine, mg/dL | 0.82 (0.64–1.11) | 0.64 (0.56–0.86) |
| d-dimer, ng/mL | 746 (407–1,524) | 424 (294–631) |
| C-reactive protein, mg/mL | 3.1 (0.9–9.3) | 1.9 (0.7–8.1) |
| Lymphocyte count, ×103/cmm | 0.64 (0.305–1.265) | 0.95 (0.49–1.1) |
|
| ||
| Putative COVID-19 treatments during trial, n (%) | ||
| Glucocorticoids | 37 (62.7) | 8 (53.3) |
| Remdesivir | 16 (27.1) | 2 (13.3) |
| Hydroxychloroquine | 20 (33.9) | 3 (20.0) |
| Tocilizumab | 13 (22.0) | 3 (20.0) |
| Sarilumab | 1 (1.7) | 0 (0) |
COVID-19 = coronavirus disease 2019, FDA = U. S. Food and Drug Administration, IQR = interquartile range.
IgG and immunoglobulin M antibodies to SARS-CoV-2 were already present at baseline in most subjects (Fig. 2, A and B). Time (min) from randomization to initiation of treatment was (median [IQR]) 96.5 (82–134) in the SP arm and 105 (85–117) in the CP arm. Administration of the 2 U of CP resulted in a 14.4% increase in IgG antibodies to SARS-CoV-2 from baseline to day 1, whereas administration of SP resulted in an 8.6% decrease during this period (CP vs SP Wilcoxon rank-sum p = 0.005; Fig. 2C).
Figure 2.
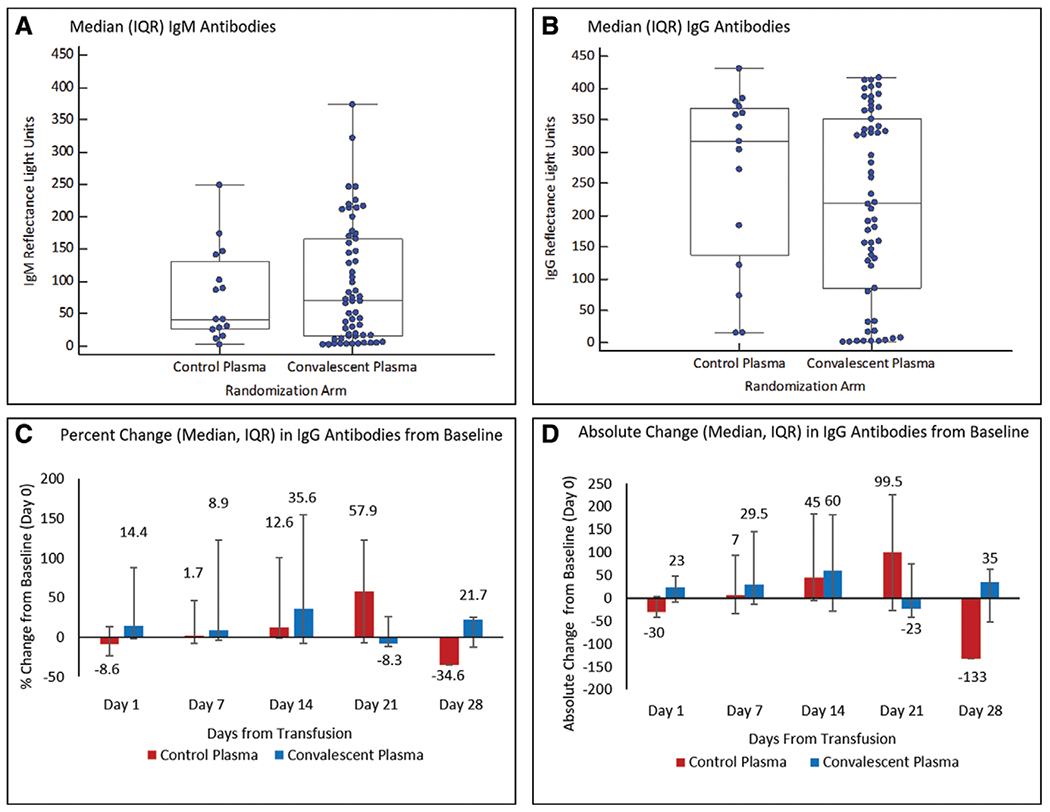
Antibodies to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in plasma recipients. Presence of immunoglobulin M (IgM) (A) and immunoglobulin G (IgG) (B) antibody levels to the nucleocapsid protein (NP) of SARS-CoV-2 in trial participants at baseline. Humans without exposure to SARS-CoV-2 typically exhibit less than 25 reflectance light units, with several hundred reflectance light units indicating a very strong antigen/antibody band. Percent (C) and absolute (D) changes in IgG antibody levels to the NP of SARS-CoV-2 at 1, 7, 14, 21, and 28 d after administration of 2 U of convalescent versus standard plasma (on day 0–baseline). Antibody levels were not measured after hospital discharge/death. IQR = interquartile range.
No significant difference between study groups was observed for the primary endpoint (ventilator-free days through 28 d): median (IQR) of 28 (2–28) versus 28 (0–28), Wilcoxon rank-sum (p = 0.86), for the CP and SP groups, respectively. A greater than or equal to 2 point improvement in the WHO ordinal scale was achieved by 20% of subjects in both of the study arms (Fisher p = 0.99). All-cause mortality (time to event) through 28 and 90 days postrandomization are shown in Figure 3, A and B, respectively. Mortality at 90 days was numerically lower in the CP versus SP groups (27% vs 33%; p = 0.63) but did not achieve statistical significance. Subgroup analysis for time to death in patients who were intubated at baseline was statistically significant (Fig. 3C); however, sample size numbers were small. No difference was detected in patients not intubated at baseline (Fig. 3D).
Figure 3.
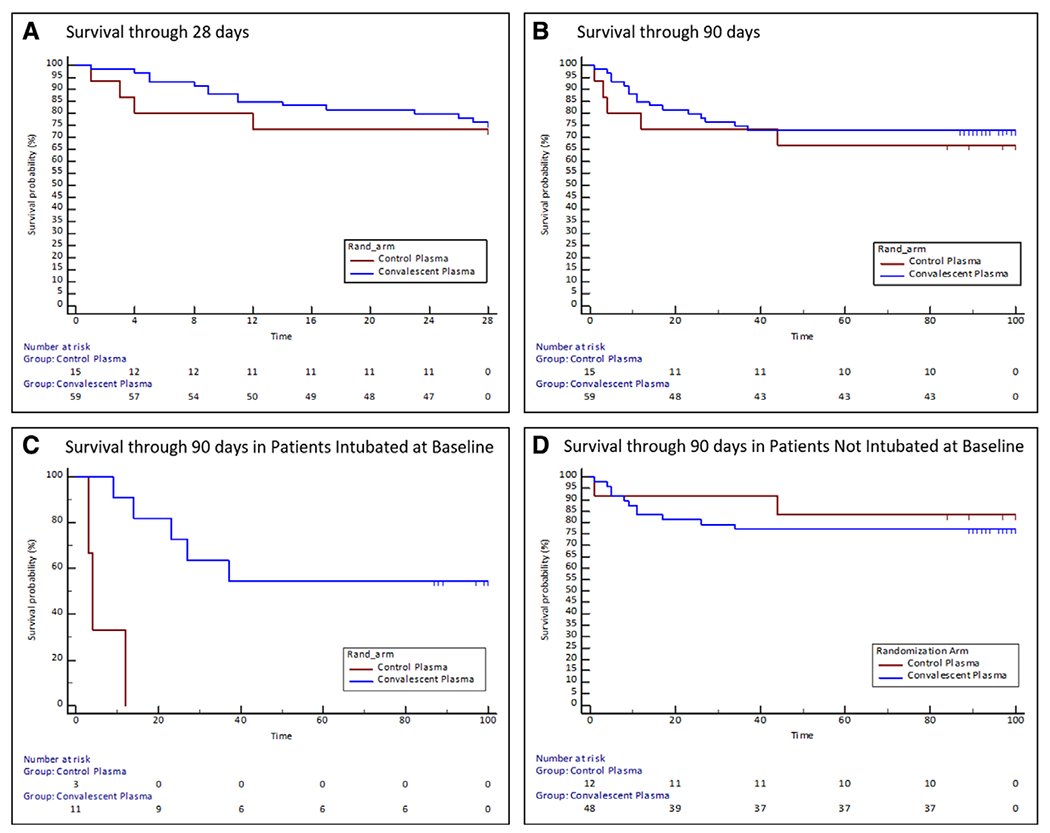
Time to all-cause mortality from randomization to 28 and 90 d. The Kaplan-Meier failure estimates of the time from intervention (administration of convalescent plasma or standard plasma) to death through 28 d (A) and 90 d (B) for all randomized subjects and for patients intubated (C) and not intubated (D) at baseline (prespecified subset analyses). The 90-d study window was defined as 90 ± 10 d.
Per protocol analyses, excluding the three subjects who did not receive the entire 2 U of plasma yielded similar results. Short Form-36 at 90 days was collected in only 25 subjects, due in large part to only 20 patients being discharged to home in the 90-day window. Therefore, these data were not analyzed. Changes in body temperature over time were similar between groups (Supplemental Fig. 2, http://links.lww.com/CCM/G338; legend, http://links.lww.com/CCM/G339). Frequency of adverse events was similar in both study groups during the first 28 days after randomization (Table 2).
TABLE 2.
Plasma Administration, Adverse Events/Safety, Follow-Up, and Outcomes
| Characteristics | Convalescent Plasma, n = 59 | Standard Plasma, n = 15 | Wilcoxon Rank-Sum p or Hazard Ratio (95% CI) |
|---|---|---|---|
| Received any amount of assigned plasma, n (%) | 58 (98) | 14 (93) | – |
|
| |||
| Did not receive any plasma, n (%) | 1 (2) | 1 (7) | – |
|
| |||
| IgG antibody to severe acute respiratory syndrome coronavirus 2, reflectance light unit, median (IQR) | – | ||
| IgG plasma bag number 1 | 285 (240–331) | 7 (2–11) | – |
| IgG plasma bag number 2 | 292 (250–335) | 6 (3–14) | – |
| IgM plasma bag number 1 | 75 (38–129) | 5 (4–12) | – |
| IgM plasma bag number 2 | 67 (51–122) | 6 (5–11) | – |
|
| |||
| Infusion-related event, n (%) | 1 (2) | 0 (0) | – |
|
| |||
| Any SAE in the first 28 d, n (%) | 16 (30) | 4 (27) | – |
| Any SAE in the first 24 hr | 2 (4) | 1 (7) | – |
| Any SAE in days 2–7 | 8 (15) | 2 (13) | – |
| Any SAE in days 8–28 | 10 (19) | 1 (7) | – |
|
| |||
| Lost to follow-up at study completion (90 ± 10 d), n (%) | 0 (0) | 0 (0) | – |
|
| |||
| Outcomes | |||
| Primary outcome, ventilator-free days, median (IQR) | 28 (2–28) | 28 (0–28) | p = 0.86 |
| Ventilator-free days in patients who were intubated at baseline | 16 (0–23) | 0 (0–0) | p = 0.10 |
| Ventilator-free days in patients who were not intubated at baseline | 28 (27.5–28) | 28 (28) | p = 0.49 |
| Secondary outcomes, n (%) | |||
| 90-d all-cause mortality | 16 (27) | 5 (33) | 0.74 (0.27–2.03) |
| 90-d mortality in patients who were intubated at baseline | 5 (45) | 3 (100) | 0.05 (0.01–0.46) |
| 90-d mortality in patients who were not intubated at baseline | 11 (23) | 2 (17) | 1.43 (0.32–6.46) |
| 28-d all-cause mortality | 14 (24) | 4 (27) | 0.80 (0.26–2.44) |
| World Health Organization ordinal scale 2 point change | 12 (20) | 3 (20) | 0.75 (0.21–2.68) |
IgG = immunoglobulin G, IgM = immunoglobulin M, IQR = interquartile range, SAE = serious adverse event.
Reflectance light units of antigen/antibody band in immunochromatographic test to severe acute respiratory syndrome coronavirus 2 (Fig. 1 for neutralizing antibody activity results).
SAE including death, grade 4 or greater organ failure, serious infusion reaction warranting termination of plasma infusion.
Dashes indicate no statistic for these variables.
DISCUSSION
To our knowledge, this is the first report from a double-blind RCT of CP for COVID-19 infection conducted in the United States. CP collected by our Blood Bank had high neutralizing antibodies and its administration resulted in a statistically significant increase in antibodies to SARS-CoV-2 compared with SP. No significant differences, however, were observed for the primary endpoint (ventilator-free days through 28 d) or secondary endpoints (death, WHO ordinal scale). All-cause mortality through 28 and 90 days was numerically lower in the CP versus SP groups but did not achieve statistical significance. Therefore, administration of CP to our hospitalized patients with COVID-19 infection increased antibodies to SARS-CoV-2 but was not associated with improved outcome.
During the early phases of the pandemic, CP was suggested as a potential therapy for COVID-19 infection. CP is an attractive potential therapy since there is some, albeit weak, evidence of efficacy in previous infectious outbreaks (3, 4). Importantly, it can be deployed within weeks of a new infectious outbreak since it is easy to identify individuals who have recovered from infection and the process for collection of CP is a routine procedure that blood banks and most hospitals can provide. Therefore, CP can theoretically be used within a month of a disease outbreak, in contrast to more “synthetic” and targeted pharmaceuticals, which require more time to develop. CP can also be deployed in countries with fewer resources since pharmaceuticals are often prohibitively expensive in these settings. Finally, an attractive feature of CP is that one does not need to identify the most effective antibodies in CP since the large pool of antibodies presumably contains a subset that can neutralize the pathogen.
In light of the above, during the Spring 2020 “surge,” when our hospital had over 500 COVID-19 patients (135 intubated), we felt compelled to investigate CP as a potential treatment. Our hope that CP might be effective in our patients was tempered by the lack of strong data supporting the use of CP in other outbreaks. For example, in one of the few published RCTs of CP therapy (the National Institutes of Health [NIH] funded multicenter randomized double-blind RCT of CP for treatment of influenza), 2 U of CP was not effective compared with 2 U of SP (24). Nevertheless, given our hope that CP might be effective, we opted for 4: 1 randomization, where 80% of patients would receive CP while a small (20%) control group would be used to allow for assessment of efficacy and safety.
Several other aspects of our trial and results warrant discussion. First, we agreed with experts who speculated early in the pandemic that CP might be more efficacious early in the course of infection before patients can mount an adequate antibody response. Unfortunately, given the known delays in testing, we had limited access to enroll PCR positive outpatients, and more importantly, did not have mechanisms in place to do outpatient CP transfusions, which are highly regulated. Therefore, we focused on hospitalized patients where there was a large unmet need. We considered limiting enrollment to only those within the first few days after symptom onset but discarded this idea since it was not clear how we could accurately determine the exact date symptoms started. Therefore, we used hospital admission date as an objective measure for inclusion in the trial. Our concern that we might treat many patients “late” in their infection appears to have been valid, since the duration of estimated symptom onset to randomization was 9 days (median) in both study arms, and consistent with this fact, most patients had already generated antibodies to SARS-CoV-2 at baseline (Fig. 2, A and B). Indeed, 80% and 87% of patients in our CP and SP arms, respectively, had detectable IgG antibodies to SARS-CoV-2 at baseline. Our results are consistent with Agarwal et al (18) who found that 83% of patients in their CP COVID-19 RCT had detectable antibodies to SARS-CoV-2 at baseline. Results from an RCT involving early administration (within 72 hr after symptom onset) of CP vs saline showed benefit (21), which is in contrast to the apparent ineffectiveness of CP for “late” treatment of COVID-19 infection we and others (18–20) have reported.
The CP we collected met FDA’s suggested minimum titer (1:80) for neutralizing activity. All CP units exhibited strong antibody levels using an immunochromatographic test (Table 2). A randomly selected subset of units was also tested for neutralizing activity. CP units had high neutralizing activity with a median NT50 of 1:334 in the pseudotype virus assay (23) and a median NT50 of 1:526 in the “gold standard” plaque neutralizing assay, which uses SARS-CoV-2 virus in a BioSafety Level-3 laboratory (Fig. 1). Administration of our high titer CP to trial participants resulted in a small but statistically significant increase in antibodies (Fig. 2, C and D), whereas antibody levels decreased acutely after administration of the 450–550 mL of SP, likely due to hemodilution. The “small” increase in antibody level we observed is consistent with Agarwal et al (18) and is not surprising given that the 450–550 mL of administered CP is diluted in an approximate 5,000 mL circulating volume. Li et al (19) did not report antibody levels, and Simonovich et al (20), whose trial did not show CP to be protective, reported increases of median titers from 1:50 at baseline to 1:400 on day 2 in both patients receiving CP as well as the control group receiving saline.
We terminated enrollment in the trial on August 24, 2020, after the FDA granted EUA for CP. At this time, 82 patients had been consented and 74 patients had been randomized. We considered continuing the trial but concluded it would be too challenging to consent patients/healthcare proxies for an “experimental treatment,” with many potential risks listed in the consent form such as “HIV,” “anaphylaxis,” “transfusion-related acute lung injury,” and “antibody-mediated enhancement of infection,” when they could instead request the CP as an FDA “approved” therapy. We were concerned that many patients and their families did not understand the difference between a conditionally approved product (EUA) and one that is fully approved.
Our study has several limitations. It is a single-center study, so the results may not be generalizable to other hospitals. Given the smaller sample size than planned, it is underpowered to rule out a potential benefit that might exist (type 2 error). All-cause mortality through 28 and 90 days postrandomization was numerically lower in the CP versus SP groups, and there was separation between arms in the time to event analyses (Fig. 3, A and B), but neither analysis achieved statistical significance. We cannot know if enrollment to the target sample size would have shown this mortality difference to be significant. An exploratory analysis for time to death in the prespecified subset of patients who were intubated at baseline was statistically significant (Fig. 3C and Table 2); however, sample size numbers were small. Of note, other endpoints such as ventilator-free days and improvement in the WHO ordinal scale were almost identical in the two study groups, tempering enthusiasm for this intervention. Our results are largely consistent with the three previously published peer-reviewed RCTs that were conducted in China (n = 103) (19), India (n = 464) (18), and Argentina (n = 334) (20). The RCTs in China and India were not blinded and used a control group of “standard treatment.” The trial in Argentina used saline as the comparator with the bag and infusion tubing covered with an opaque sleeve; it is not clear how effective blinding was maintained with this strategy. We also do not know whether use of SP as the comparator in our trial affected our results. Many other RCTs of CP for infectious diseases have chosen SP as the comparator, for example, 2 U of SP were used in an NIH-funded multicenter RCT of influenza infection (24). Use of SP ensures blinding and allows one to determine if there is efficacy of the antibodies to the pathogen in question, that is, does “CP” confer incremental benefit over SP? Studies that do not use SP as the comparator cannot know if a benefit observed is due to the antibodies or if it is due to some other element within plasma.
Our study has several strengths. It was a double-blind RCT, which is the gold standard for evidence-based medicine. It was a pragmatic design that focused on the types of patients that overwhelmed our hospital early in the pandemic. We enrolled patients with mild and severe COVID-19 infection, which introduced more variability but also represented the “real world setting,” where both patients with mild and severe infection might benefit from CP. In addition, more than 20% of trial participants were non-White, which is similar to nationally published vaccine trials. Another strength of this study is the rigorous quality control and data management systems used to record and track patient outcomes. Finally, while the FDA did not require antibody testing, we only qualified donors who had a robust antibody response by an immunochromatographic test. Of note, a random subset of units tested had high neutralizing antibody titers using a pseudotype assay (23), as well as the gold standard SARS-CoV-2 plaque naturalization assay (Fig. 1).
In summary, in this double-blind RCT, administration of 2 U of CP to patients hospitalized in New York with COVID-19 infection increased antibodies to SARS-CoV-2 but was not associated with improved outcome. Results from this and previous trials (18–20) do not support the use of CP for the treatment of hospitalized patients with COVID-19 infection.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank the Apheresis/Blood Bank staff and Stony Brook Medicine Information Technology for their significant assistance in helping us to operationalize the trial. We would also like to thank Kenneth Kaushansky, MD (Dean and Senior Vice President for Health Sciences, Renaissance School of Medicine) who provided invaluable financial and administrative support for this trial.
Supported, in part, by Stony Brook Medicine.
Dr. Fries received support for article research from the National Institutes of Health. The remaining authors have disclosed that they do not have any potential conflicts of interest.
APPENDIX 1
Stony Brook Medicine COVID Plasma Trial Group are as follows: Investigators: Elliott Bennett-Guerrero (Principal Investigator, Critical Care), Tahmeena Ahmed (Pathology/Blood Bank), Bettina C. Fries (Infectious Disease), Sharon Nachman (Safety Monitor), Jamie Romeiser (Biostatistics), Huda Salman (Hematology), Lisa Senzel (Blood Bank), and Eric Spitzer (Pathology, Laboratory Services). Team 1 (Online Survey/In Person Scheduling): Giuseppina Caravella (Team Leader), Laura Harper, Diana Kaell, Melanie Keister, David Komatsu, Jessica Lamb, Deidre Lee, Jane O’Keefe, Ajish Pallai, Elizabeth Roemer, William Scherl, Sandra Skinner, and Leah Smith-McAllister. Team 2 (In Person Screening Visits): Molly Rago (Team Leader), Margaret Brand, Andrew Bryan, Lauren Festa, Susan Fiore, Shannen Harbourne, Audrey Hecker-Crawford, Ann Lavorna, Caryn McKenna, Robert Repetti, Curtis Roggemann, Haseena Sahib, Margaret Shevik, Sunitha Singh, Ruth Stein, and Kathy Vivas. Team 3 (Patient/Recipient Screening, Plasma Administration, Data Capture): Margaret Andrew (Team Leader), Audrey Anderson, Joan Arata, Marlene Baumeister, Susan Boudreau, Patricia Brill, Noelle Daley, Christine Gearwar, Laura Generale, Darcy Halper, and Erin J. Healy; Coleen Letscher, Dawn Madigan, Katherine Markarian-Askinazi, Ana Mavarez-Martinez, Sebastian Munoz, Christine Pol, Grace Propper, and Dishaw Holiprosad; and Rajeev Fernando, Nandini Seshan, and Sophia Pham. Team M (Antibody Testing/Randomization): Lillian Talbot (Team Leader), Nicholas Browne, Jason Carter, Megan Cosgrove, Alex Freedenberg, and Andrew Sisti. Plaque Reduction Neutralization Assay: Janet Hearing. Regulatory (Investigational New Drug [IND] and Institutional Review Board [IRB] support): Suman Grewal (IND support), Caterina Vacchi-Suzzi (IRB support), and Angie Wong. Data and Safety Monitoring Board (DSMB): Robert Califf, Nicholas Bandarenko, and Timothy McMahon (Chair) and Wei Hou (unblinded DSMB statistician).
Footnotes
Stony Brook Medicine COVID Plasma Trial Group is listed in Appendix 1.
Trial registration: ClinicalTrials.gov NCT04344535.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (http://journals.lww.com/ccmjournal).
REFERENCES
- 1.Dong E, Du H, Gardner L: An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis 2020; 20:533–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanders JM, Monogue ML, Jodlowski TZ, et al. : Pharmacologic treatments for coronavirus disease 2019 (COVID-19): A review. JAMA 2020; 323:1824–1836 [DOI] [PubMed] [Google Scholar]
- 3.Mair-Jenkins J, Saavedra-Campos M, Baillie JK, et al. ; Convalescent Plasma Study Group: The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: A systematic review and exploratory meta-analysis. J Infect Dis 2015; 211:80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marano G, Vaglio S, Pupella S, et al. : Convalescent plasma: New evidence for an old therapeutic tool? Blood Transfus 2016; 14:152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duan K, Liu B, Li C, et al. : Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci U S A 2020; 117:9490–9496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin C, Gu J, Yuan Y, et al. : Treatment of six COVID-19 patients with convalescent plasma. medRxiv 2020.05.21.20109512 [Google Scholar]
- 7.Salazar E, Perez KK, Ashraf M, et al. : Treatment of coronavirus disease 2019 (COVID-19) patients with convalescent plasma. Am J Pathol 2020; 190:1680–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen C, Wang Z, Zhao F, et al. : Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA 2020; 323:1582–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye M, Fu D, Ren Y, et al. : Treatment with convalescent plasma for COVID-19 patients in Wuhan, China. J Med Virol 2020; 92:1890–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu STH, Lin HM, Baine I, et al. : Convalescent plasma treatment of severe COVID-19: A propensity score-matched control study. Nat Med 2020; 26:1708–1713 [DOI] [PubMed] [Google Scholar]
- 11.Rogers R, Shehadeh F, Mylona E, et al. : Convalescent plasma for patients with severe COVID-19: A matched cohort study. Clin Infect Dis 2020. Oct 10. [online ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salazar E, Christensen PA, Graviss EA, et al. : Treatment of COVID-19 patients with convalescent plasma reveals a signal of significantly decreased mortality. Am J Pathol 2020; 190:2290–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joyner M: Expanded Access to Convalescent Plasma for the Treatment of Patients with COVID-19 protocol. 2020. Available at: https://www.uscovidplasma.org. Accessed May 27, 2020
- 14.Joyner MJ, Wright RS, Fairweather D, et al. : Early safety indicators of COVID-19 convalescent plasma in 5000 patients. J Clin Invest 2020; 130:4791–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joyner MJ, Bruno KA, Klassen SA, et al. : Safety update: COVID-19 convalescent plasma in 20,000 hospitalized patients. Mayo Clin Proc 2020; 95:1888–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joyner M, Senefeld JW, Klassen SA, et al. ; US EAP COVID-19 Plasma Consortium: Effect of convalescent plasma on mortality among hospitalized patients with COVID-19: Initial three-month experience. medRxiv 2020.08.12.20169359 [Google Scholar]
- 17.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research: Investigational COVID-19 Convalescent Plasma Guidance for Industry. 2021. Available at: https://www.fda.gov/media/136798/download. Accessed April 9, 2021
- 18.Agarwal A, Mukherjee A, Kumar G, et al. ; PLACID Trial Collaborators: Convalescent plasma in the management of moderate Covid-19 in adults in India: Open label phase II multicentre randomised controlled trial (PLACID Trial). BMJ 2020; 371:m3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Zhang W, Hu Y, et al. : Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: A randomized clinical trial. JAMA 2020; 324:460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simonovich VA, Burgos Pratx LD, Scibona P, et al. ; PlasmAr Study Group: A randomized trial of convalescent plasma in COVID-19 severe pneumonia. N Engl J Med 2021; 384:619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Libster R, Pérez Marc G, Wappner D, et al. ; Fundación INFANT–COVID-19 Group: Early high-titer plasma therapy to prevent severe COVID-19 in older adults. N Engl J Med 2021; 384:610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter JA, Freedenberg AT, Romeiser JL, et al. ; Stony Brook Medicine COVID Plasma Trial Group: Impact of serological and PCR testing requirements on the selection of COVID-19 convalescent plasma donors. Transfusion 2021. Feb 8. [online ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freedenberg AT, Pan C-H, Diehl WE, et al. : Neutralizing activity to SARS-CoV-2 of convalescent and control plasma used in a randomized controlled trial. Transfusion 2021. Jan 15. [online ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beigel JH, Aga E, Elie-Turenne MC, et al. ; IRC005 Study Team: Anti-influenza immune plasma for the treatment of patients with severe influenza A: A randomised, double-blind, phase 3 trial. Lancet Respir Med 2019; 7:941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.