

Summary
With the continuous emergence of highly transmissible SARS-CoV-2 variants, the comparison of their infectivity has become a critical issue for public health. However, a direct assessment of the viral characteristic has been challenging because of the lack of appropriate experimental models and efficient methods. Here, we integrated human alveolar organoids and single-cell transcriptome sequencing to facilitate the evaluation. In a proof-of-concept study with four highly transmissible SARS-CoV-2 variants, including GR (B.1.1.119), Alpha (B.1.1.7), Delta (B.1.617.2), and Omicron (BA.1), a rapid evaluation of the relative infectivity was possible. Our system demonstrates that the Omicron variant is 5- to 7-fold more infectious to human alveolar cells than the other SARS-CoV-2 variants at the initial stage of infection. To our knowledge, for the first time, this study measures the relative infectivity of the Omicron variant under multiple virus co-infection and provides new experimental procedures that can be applied to monitor emerging viral variants.
Subject areas: Public health, Virology
Graphical abstract
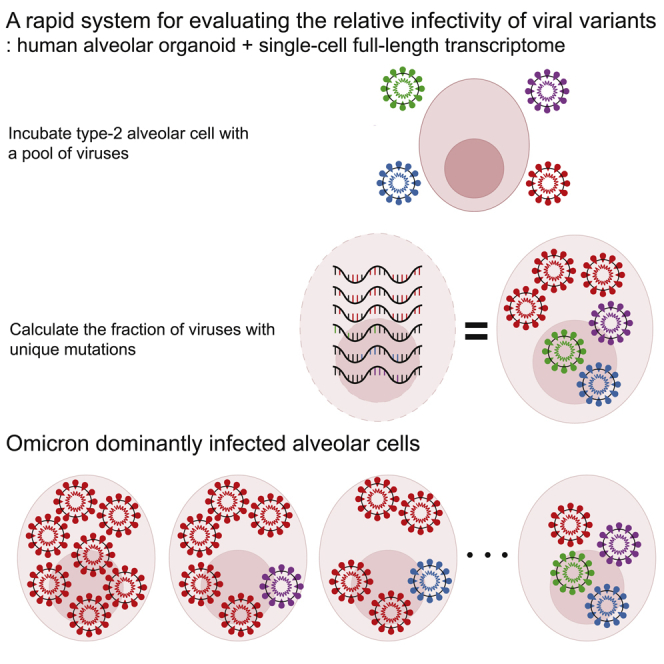
Highlights
-
•
An efficient system for evaluating the relative infectivity of SARS-CoV-2 variants
-
•
Human alveolar organoids are host cells for comparing viral infectivity against lungs
-
•
Full-length single-cell RNA-seq identifies viral variants in each infected cell
-
•
The Omicron variant shows 5 to 7 times higher infectivity in human alveolar cells
Public health; Virology
Introduction
During the global spread of the coronavirus disease 2019 (COVID-19), many novel SARS-CoV-2 variants of concern (VOC) have emerged, posing an increased risk to global public health and of quarantine.1,2,3 International communities, such as GISAID,4 PANGO,5 and Nextstrain,6 have been monitoring and assessing the evolution of SARS-CoV-2 using periodic genomic sequencing of viral samples. The sequencing results have identified a few major SARS-CoV-2 variants, including GR (B.1.1.119) with the D614G variant,7 Alpha (B.1.1.7) (first detected in UK), and Delta (B.1.617.2) (first detected in India) (WHO). In Nov 2021, the Omicron variant (BA.1), characterized with 32 mutations in the spike protein, emerged from South Africa and is currently the dominant variant in many countries.8
To understand the functional impacts and pathological characteristics of each VOC, various approaches have been conducted including epidemiological studies,9 spike binding affinity assay,10,11,12 experimental model studies,12,13,14 and genetically engineered virus comparison studies. The epidemiological studies illustrate characteristics of viral transmission and clinical severity, but their underlying cellular and molecular mechanisms cannot be investigated. The spike binding assay measures the affinity between the virus spike protein and human receptor, but its biological impact is cryptic. For the experimental model studies, including animal models15 and cell lines, the issue of viral tropism is inherent. Often, genetically engineered viruses with a specific mutation of interest, rather than natural viral variants (e.g., D614G (GR, Alpha, Delta, and Omicron),16 N501Y (Alpha and Omicron)17 or P681R (Delta)18,19), are used in infection studies, but these engineered viruses may not reflect the full characteristics of natural VOCs.
Despite all these efforts, the direct measurement of the relative infectivity of multiple VOCs, particularly the impact of the natural virus on physiological human tissues, has not been investigated. Here, we developed a rapid, fully controlled virus competition system by integrating normal human type-2 alveolar cell (hAT2 cell) organoid and single-cell full-length transcriptome sequencing. We successfully trace the viruses responsible for an infected alveolar cell. Furthermore, we compared the relative infectivity of viral variants under multiple virus co-infection at the single-cell level.
Results
A rapid, fully controlled virus competition system
First, hAT2 cells in alveolar organoids are single-cell dissociated and then exposed to a mixture of SARS-CoV-2 variants (Figure 1A). After minutes to days long culture, the full-length transcriptomes of the infected cells are sequenced at the single-cell resolution (adopting the SMART-seq3 technique)20 to capture the viral genomic mutations. These mutations are used to identify which VOCs are responsible for an individual cell’s infection.
Figure 1.

Integration of alveolar organoids and full-length single-cell RNA-seq techniques to understand the relative infectivity of SARS-CoV-2 variants
(A) Schematic diagram of the experimental procedures of the virus competition assay with the expected experimental time shown in parentheses.
(B) Clonal viral genomic mutations (VAF >99%) in the four viral variants that are used for viral tracing with the viral genomic mutations from GISAID reference shown in parentheses.
(C) Normalized expression levels of host genes (UMI counts) in the infected single-cells (Yaxis values of dots are normalized expression +1).
(D) The number of reads (1 sequencing read is 150 base pairs) from full-length single-cell transcriptome sequencing for each infected cell. Dark blue, the number of total reads; orange, the number of viral reads.
(E) The genomic location of the viral genomic mutations (the top four panels). The normalized coverage of viral transcripts in infected cells by two different single-cell transcriptomic methods (the bottom panel).
(F) Comparison of two different viral variants decomposition methods (average VAF method and non-negative matrix factorization method). The dashed line represents the median (0.99).
(G) Number of unique viral mutations covered by transcriptome sequencing and the accuracy of viral variant decomposition.
As a proof-of-concept study, we selected four SARS-CoV-2 variants, the GR clade virus (B.1.1.119), Alpha (B.1.1.7), Delta (B.1.617.2), and Omicron (BA.1), which are known to have been highly transmissible viruses during the pandemic. For our infection experiments, we used viral stocks which were collected from Korean patients and maintained by the Korea Disease Control and Prevention Agency (KDCA). RNA sequencing of these viral variants identified 118 clonal genomic alterations for tracing viral variants (Figure 1B; Table S1), 94% of which (n = 125) are known to be present in the standard genome sequences of GR, Alpha, Delta, and Omicron variants (Figure S1A).
In our optimized infection experiments, viral incubation of alveolar cells was conducted at a multiplicity of infection (MOI) of 10 collectively, with each of the SARS-CoV-2 variants equally allocated for the incubation (STAR Methods). On average, an alveolar cell interacted with 10 viral plaque-forming units (PFU), and each viral variant had an equal chance of cellular infection. We checked the number of infective (viable) viruses used for viral variants mixture with plaque assay (Figure S1B). The relative proportion of infective viruses was substantially balanced within 1.5-fold among the four viral variants (Figure S1C).
Furthermore, the viral incubation time was mostly 5 min (86%; the others were incubated for 60 min), which was sufficient for infection of hAT2 cells. Then, single alveolar cells were isolated in a microchip after checking the number of cells under the brightfield and fluorescence microscopy (Figures 1A and S1D).
Robust infection in alveolar cells
The full-length transcriptome for SARS-CoV-2 infected single cells by SMART-seq3 was sequenced by short-read paired-end sequencing with ∼314Mb of sequencing throughput per cell, or ∼2.1 M reads with 150 bp per cell, on average. The transcriptional profiles of the host genes confirmed that the infected human cells are hAT2 cells (Figure 1C).
In the single-cell transcriptome of 244 infected cells that passed the quality check and threshold of the infection criteria (STAR Methods), the proportion of viral sequences over total sequences ranges from 0.08 to 60% (Figure 1D). For the infected alveolar cells, the expression profile of viral RNA transcripts is consistent with the previous report21 that the 3′ genomic regions of the viral genome showed much higher RNA expression levels (Figure 1E). Of note, the full length transcriptome method, SMART-seq3 has more uniform coverage than the 3′ enriched transcriptome method, such as 10X Chromium.22 Therefore, SMART-seq3 can detect more viral genomic mutations which are missed by 10X Chromium.
Decomposition of viral variants in infected alveolar cells
Considering the 118 clonal viral genomic mutations as viral variant barcodes (Figures 1B and 1E, Table S1), we decomposed the fraction of each viral variant responsible for an individual cell’s infection by using variant allele fraction (VAF). We used two algorithms, the average VAF method and the non-negative matrix factorization (NMF) (STAR Methods). The results of the two algorithms, the fraction of viral variants in infected alveolar cells, were overall concordant with each other (Figure 1F). Only eight cells (3.3%) showed an insufficient cosine similarity (<80%). In these cells, a large fraction of viral mutation loci was stochastically uncovered in the full-length single-cell transcriptome sequencing (Figure 1G).
The Omicron variant dominantly infected alveolar cells
Of the 244 infected cells, 97 (39.8%), 92 (37.7%), and 52 (21.4%) cells were dominantly infected by single, double, and multiple viral variants, respectively, suggesting that multiple viral entries are possible in the experimental condition. For the remaining 3 cells (1.2%), unique viral variants could not be assigned.
Despite the equal chance of infection, each SARS-CoV-2 variant showed strikingly different frequencies in the infected cells (Figure 2A). For instance, of the 97 cells with a single variant infection, 63 (65.0%) were caused by the Omicron variant, followed by Alpha (n = 20; 20.6%), GR (n = 13; 13.4%), and Delta (n = 1; 1.0%). The Omicron variant was 2.60-fold more frequently observed than the random expectation (95% confidence interval = [2.18, 2.97]; p = 3.0 × 10−19), implying an ∼7.4-fold higher infectivity than the other viruses by odds ratio under the same infectivity among viruses (STAR Methods).
Figure 2.

A higher infectivity of the Omicron variant in human type-2 alveolar cells
(A) The proportion of each viral variant in each infected cell. Experimental conditions (post infection time and viral incubation time) are shown at the bottom. The order of infected cells (Xaxis) is identical to the one in Figure 1D. The distance between two adjacent big ticks is 10 cells. The distance between one big tick and one small tick is 5 cells.
(B) The proportion of each viral variant over 12 different experimental batches in this study. Only cells with a viral incubation time of 5 min are shown.
(C) The proportion of the Omicron variant increases in the batch with a longer viral incubation time.
A similar conclusion was robustly drawn from a parallel analysis with all 244 cells, including the ones infected by two or more variants. Here, the Omicron variant was found in 199 cells (81.6%), followed by 114 (46.7%), 75 (30.7%), and 56 (23.0%) for the Alpha, GR and Delta variants, respectively, which is also biased toward the Omicron variant (p = 5.26 × 10−43) (Figure 2A). Taking into consideration the relative viral burden of each variant in an infected cell (a weighted average), the Omicron variant involved 143.2 cells (58.7%), out-competing the other variants. This result means that the Omicron variant was observed 2.34-fold more frequently than the random expectation (95% confidence interval = [2.09, 2.60]; p = 1.37 × 10−33) and showed a 5.6-times higher infectivity than the other viruses. This result was concordant with the results drawn from the cells with the single variant infection.
In the pairwise comparison with the other variants by odds ratio, the Omicron variant showed ∼4.8 (against the Alpha), ∼9.7 (against the GR), and ∼31.6 (against the Delta), times higher infectivity in the assay. We believe that the dominance of the Omicron is robust because the trend was replicated in 9 independent batches (Figure 2B). Of note, in an experiment with a longer viral incubation time (60 min), the predominance of the Omicron variant was even higher (Figure 2C).
Our calculation of the relative infectivity is conducted under an assumption of equal infection chance among the four viral variants. However, in our plaque assays (Figure S1C), the fraction of infective Omicron variant in the source was ∼20.5% on average, slightly lower than 25%. The higher infectivity of the Omicron variant will be further enhanced if we take its original fraction into consideration.
Viral mutations during virus stock preparation
Through the VAF of the mutation, we are able to trace the probable stage when the mutation was acquired. Because we passaged one more time to make viral stock for the virus competition experiment after receiving the passage 3 viral stock which originated from the patient sample, we can distinguish whether mutations were acquired before passage 3 or between passage 3 and passage 4. Using a total of 48 viral stock (passage 4) RNA sequencing with 4 viral variants and 12 experimental batches, we analyzed the mutations compared to the references from GISAID.
We found a total of 34 mutations (GR: 8; Alpha: 10; Delta: 5; Omicron: 11) compared to the GISAID reference (Figure S1A). 18 mutations (GR: 3; Alpha: 5; Delta: 2; Omicron: 8) are highly likely to be accumulated between passage 3 and passage 4 because the VAF difference between the GISAID and viral stocks (passage 4) is low (<0.5). On the other hand, 16 mutations (GR: 5; Alpha: 5; Delta: 3; Omicron: 3) might be acquired before passage 3 because the VAF difference between the GISAID and viral stocks (passage 4) is high (>0.8).
Of note, because the experimental batches were aliquots of the viral stock (passage 4), the VAFs across batches are similar. However, in the Omicron variant, some mutations had heterogeneous VAFs among batches. The viruses with these mutations might result from the subclonal viral population of the passage 3 viral stock, considering the relatively lower number of the Omicron for sequencing compared to other variants.
Discussion
In this study, we developed a new system to measure and directly compare the relative infectivity of SARS-CoV-2 variants. Our system suggested that the Omicron (BA.1) variant is 5-to 7- times more infectious than the other viral variants, including the GR (B.1.1.119), Alpha (B.1.1.7), and Delta (B.1.617.2), against human alveolar cells.
Our system evaluates the infectivity of respiratory viruses directly against their target cell types, hAT2 cells in contrast with other approaches based on animal models15 or 2D cell lines.23 Using human alveolar organoids, our assay is free from the issue of viral tropism and recapitulates normal human tissue physiology. Furthermore, this strongpoint allows our system to be expanded to any virus if organoids have the main target cell of the virus.
Moreover, this system also has technical advantages. The system can trace viral variants with high sensitivity because full-length single-cell RNA sequencing captures much information, which is viral genomic mutations, compared to 3′ enriched single-cell RNA sequencing. In addition, the relative infectivity of viral variants can be quickly determined in a fully controlled condition because the turn-around time of our assay is ∼3 days.
Although our study has many advantages, the data should be interpreted carefully because the experimental condition is different from the condition of epidemiological studies. An epidemiology study showed that the Delta transmits faster than the Alpha.9 However, in our results and spike binding affinity study,24 the Alpha is more infectious than Delta at the initial stage of infection. Presumably, the Alpha is more infective at the early phase of infection, but the Delta has replicated more in the late stage and/or during the clinical course. Likewise, our data should be interpreted cautiously because four different viral variants competed in the series of experiments, and the small number of infected cells in experimental batches. To accurately measure the relative infectivity of the two variants specifically, another set of experiments, using the Alpha and Delta only, may be necessary.
Of note, recent studies showed that the Omicron depends more on an endocytic pathway for cellular entry rather than membrane fusion entry by TMPRSS2 in the alveolar cells,12,14 speculating the Omicron variant is less infective than the other variant in alveolar cells. Though our alveolar organoids express TMPRSS2 robustly as in the research by Meng et al. (Figure 1B), our data indicates that the Omicron variant much more rapidly infected hAT2 cells than the other variants.
We expect that this system could further investigate transcriptome changes of human genes at the single-cell resolution level. To do so, infected cells should be incubated for a longer time, ideally for at least 24 h, as such a duration is necessary for alveolar cells to reprogram their transcription against viral infection.21,22 Furthermore, this assay can also be applied to an organoid co-culture infection model, which may allow us to investigate the response of immune cells to different viral variants. In the future, together with other complementary approaches, our method will help to reveal the functional characteristics of emerging viral variants, especially for comparison among variants.
Limitations of the study
Technically, calculating the number of infective viruses before mixing each viral variant is required, because the fraction of infective viruses can differ among viral variants (Figure S1C). Moreover, because the fraction of defective viruses are imbalanced, infected cells should be sufficiently washed to remove floating viral RNA transcripts. Furthermore, the manual selection of the infected single cells is the rate-limiting step in our assay. However, the process can be readily scalable by using a fluorescence-activated cell sorter.25
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD31 APC | Biolegend | Cat#303116; RRID:AB_187751 |
| Anti-human CD45 APC | Biolegend | Cat#368512; RRID:AB_2566372 |
| Anti-human EpCAM FITC | Biolegend | Cat#324204; RRID:AB_756078 |
| Mouse anti-HTII-280 IgM | Terrace Biotech | Cat#TB-27AHT2-280; RRID:AB_2832931_ |
| PE goat anti-mouse IgM | Thermo-Fisher | Cat#12-5790-81; RRID:AB_465939 |
| Bacterial and virus strains | ||
| SARS-CoV-2 GR clade (B.1.1.119) | KCDC | GISAID Number: EPI_ISL_812963 |
| SARS-CoV-2 Alpha variant (B.1.1.7) | KCDC | GISAID Number: EPI_ISL_738139 |
| SARS-CoV-2 Delta variant (B.1.617.2) | KCDC | GISAID Number: EPI_ISL_2887353 |
| SARS-CoV-2 Omicron variant (B.1.1.529) | KCDC | GISAID Number: EPI_ISL_6959993 |
| Biological samples | ||
| Human lung tissue samples | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase/Dispase | Sigma-Aldrich | Cat#10269638001 |
| Dispase II | Sigma-Aldrich | Cat#4942078001 |
| DNase I | Sigma-Aldrich | Cat#D4527-10KU |
| RBC lysis solution | Roche | Cat#11814389001 |
| Accutase | STEMCELL Technologies | Cat#07920 |
| Modified Eagle Medium | Gibco | Cat#11935-046 |
| HI FBS | Gibco | Cat#10082-147 |
| Serum-free DMEM | Gibco | Cat#41966-029 |
| FBS | Gibco | Cat#16000-044 |
| Growth Factor Reduced Matrigel | CORNING | Cat#356231 |
| Y-27632 | Sigma-Aldrich | Cat#Y0503 |
| Advanced DMEM/F12 | Thermo-Fisher | Cat#12634010 |
| B27 supplement | Thermo-Fisher | Cat#17504044 |
| FGF 7 | PEPROTECH | Cat#100-19 |
| FGF 10 | PEPROTECH | Cat#100-26 |
| Noggin | PEPROTECH | Cat#120-10C |
| EGF | PEPROTECH | Cat#100-15 |
| N-Acetylcysteine | Sigma-Aldrich | Cat#A9165 |
| Nicotinamide | Sigma-Aldrich | Cat#N0636 |
| SB431542 | Calbiochem | Cat#616461 |
| CHIR99021 | TOCRIS | Cat#4423 |
| Recombinant Human R-Spondin 1 protein | R&D Systems | Cat#4645-RS |
| HEPES | Gibco | Cat#15140-122 |
| Penicillin/Streptomycin | Gibco | Cat#15630-080 |
| Glutamax-I | Gibco | Cat#35050-061 |
| Amphotericin B | Sigma-Aldrich | Cat#A2942 |
| Gentamicin | Sigma-Aldrich | Cat#G1397 |
| Recovery solution | Corning | Cat#354253 |
| CellTracker Green CMFDA Dye | Thermo-Fisher | Cat#C2925 |
| BSA solution | Sigma-Aldrich | Cat#A8412s |
| Recombinant RNAse Inhibitor | Takarabio | Cat#2313A |
| dNTP Set. 100 mM Solutions | Thermo-Fisher | Cat#R0181 |
| RIGAKU REAGENTS PEG 8000, 50%(w/v) | Rigaku reagents | Cat#25322-68-3 |
| Maxima H Minus Reverse Transcriptase | Thermo-Fisher | Cat#EP0753 |
| GTP(Tris bufferered solution 100 mM) | Thermo-Fisher | Cat#R1461 |
| KAPA HiFi Hotstart PCR Kit | Roche | Cat#KK2502 |
| Triton™ X-100 solution 10% | Sigma-Aldrich | Cat#93443-100ML |
| Sodium Chloride (5M) | Invitrogen | Cat#AM9760G |
| Magnesium Chlrode (1M) | Invitrogen | Cat#AM9530G |
| NN-Dimethylformamide | Sigma-Aldrich | Cat#D4551 |
| Tris-HCl pH 7.5(1M) | Tech & Innovation | Cat#BTH-9183-001L |
| Tris-HCl pH 8.3(1M) | Tech & Innovation | Cat#BTH-9175-001L |
| Phusion™ High-Fidelity DNA Polymerase | Thermo-Fisher | Cat#F-530S |
| Nextera XT DNA library prep kit | Illumina | Cat#20015963 |
| Critical commercial assays | ||
| C-chip Neubauer improved | iNCYTO | Cat#DHC-01 |
| QIAamp viral mini kit | Qiagen | Cat#52904 |
| NEBNext ARTIC SARS-CoV-2 FS kit | NEB | Cat#E7658S |
| Smart aliquotor CE chip | iBioChip | Cat#H2-SACE-5PK |
| Deposited data | ||
| Single-cell bam files of infected alveolar cells aligned by BWA and RNA sequencing of viral variants stock | EGA | EGA: EGAS00001006730 |
| All scripts for calculating fraction of each SARS-CoV-2 variant | Zenodo | Zenodo: https://doi.org/10.5281/zenodo.7275406 |
| Additional metadata for analysis | Mendeley | Mendeley data: https://doi.org/10.17632/j26ht6sy3p.1 |
| Experimental models: Cell lines | ||
| VeroE6 cell | Kim et al.21 | RRID:CVCL_0059 |
| Oligonucleotides | ||
| OligoT30VN /5Biosg/ACGAGCATCAGCAGCATACGAT TTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN |
Hagemann-Jensen et al.20 | IDT |
| TSO /5Biosg/AGAGACAGATTGCGCAATGNN NNNNNNrGrGrG |
Hagemann-Jensen et al.20 | IDT |
| Forward primer TCGTCGGCAGCGTCAGATGTGTATAAGA GACAGATTGCGCAA∗T∗G |
Hagemann-Jensen et al.20 | IDT |
| Reverse primer ACGAGCATCAGCAGCATAC∗G∗A |
Hagemann-Jensen et al.20 | IDT |
| Software and algorithms | ||
| FACSDiva software version (ver. 6.1.3) | BD Biosciences | |
| R (ver. 4.0.3) | Comprehensive R Archive Network | https://cran.r-project.org |
| Python (ver. 2.7.16) | Python Software Foundation | https://www.python.org/ |
| Samtools (ver. 1.9) | Li et al.26 | http://www.htslib.org; RRID:SCR_002105 |
| Varscan2 (ver. 2.4.2) | Koboldt et al.27 | http://dkoboldt.github.io/varscan/; RRID:SCR_006849 |
| HaplotypeCaller | Poplin et al.28 |
https://gatk.broadinstitute.org/hc/en-us/articles/360037225632-HaplotypeCaller RRID:SCR_001876 |
| Integrated Genomics Viewer | Thorvaldsdóttir et al.29 |
http://software.broadinstitute.org/software/igv/ RRID:SCR_011793 |
| STAR (ver. 2.6.4) | Dobin et al.30 |
https://github.com/alexdobin/STAR;RRID:SCR_015899 RRID:SCR_004463 |
| zUMIs | Parekh1 et al.31 |
https://github.com/sdparekh/zUMIs RRID:SCR_016139 |
| BWA-MEM | Li32 |
http://bio-bwa.sourceforge.net/bwa.shtml RRID:SCR_010910 |
| iVar | Grubaugh et al.33 | https://github.com/andersen-lab/ivar |
| Fastp | Chen et al.34 |
https://github.com/OpenGene/fastp RRID:SCR_016962 |
| Kraken | Wood et al.35 | https://github.com/DerrickWood/kraken |
| Seurat | Stuart et al.36 |
https://github.com/satijalab/seurat RRID:SCR_007322 |
| Scran | Lun et al.37 |
https://github.com/elswob/SCRAN RRID:SCR_016944 |
| Scanpy | Wolf et al.38 |
https://github.com/scverse/scanpy RRID:SCR_018139 |
| Gggenes | https://github.com/wilkox/gggenes | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Young Seok Ju (ysju@kaist.ac.kr).
Materials availability
All 3D models generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experiment model and subject details
Alveolar organoids establishment from human lung tissues
Human normal lung tissues were acquired from lung cancer patients with lobectomy surgery at SNUH with informed consent (IRB approval no. C-1809-137-975). From the human lung surgical samples, alveolar organoids were established as previously described.22 To remove cell-free RNA from RSPO-1 conditioned media, we used lyophilized RSPO-1 (80 ng/mL) (R&D systems 4645-RS) instead of the RSPO-1 conditioned media.
Virus stock preparation of each viral variant used for the competition assay
VeroE6 cells were infected with a 0.01 MOI and grown in DMEM with 2% FBS and 1% P/S for 48 h at 37°C with 5% CO2 as previously described for the virus stock preparation.22 A purified viral stock was used to calculate the number of live viruses by plaque assay. The passage of all the viral stocks we used for the competition assay was 4. We counted passage 1 as the virus acquired after the first infection of the patient sample to VeroE6.
Method details
Virus competition system among VOCs: single cell infection with multiple VOCs
First, human alveolar organoids were recovered by depolymerizing the Matrigel (Corning 354,230) with Recovery solution (Corning 354,253) at 4°C for 20 min. Furthermore, to remove the remaining Matrigel and dissociate the organoids into single cells, the organoids were incubated in Accutase (Stem Cell Technologies 07,920) at 37°C for 5 min with additional mechanical pipetting. After washing, the cells were manually counted with the iNCYTO chip (iNCYTO DHC-N01). The cells were resuspended in Advanced DMEM/F12 (Thermo-fisher 12,634,010) with 1 U/mL Penicillin/Streptomycin (Gibco 15,630-080), 10 mM HEPES (Gibco 15,140-122), and 1% Glutamax (Gibco 35,050-061) (v/v) (hereafter referred to as ADF+++). 5,000 cells in 175 ul ADF +++ were aliquoted into Protein LoBind tubes (Eppendorf 0,030,108,116).
For the preparation of the viral variants mixture, based on the PFU concentration of each viral variant, we diluted each viral variant stock to contain equal amounts of viable viruses before mixing viral variants (MOI ∼2.5; 12,500 PFU each respectively). Then each diluted viral variants were mixed in a Protein LoBind tube with a final volume of 175 ul. After adding the viral variants mixture viruses to alveolar cells, the cell-virus solution was thoroughly mixed by pipetting by 20 times. The tubes were then incubated for 5 min at 37°C and then, the cell-virus mix was washed for a total of 400,00X to remove the viable virus at the cell surface. Washed cells were embedded in Matrigel and cells were incubated for different post-infection times. Post-infection times were divided into 5 min, 4, 24, 48, and 72 h. Without batch O, the viral incubation time of all batches is 5 min. Two populations of infected cells with different viral incubation times of 5 and 60 min consist of batch O. This information is shown as viral incubation time with a heatmap (Figure 2A).
Virus competition system among VOCs: Single-cell isolation
Fluorescence microscopy was used to increase the contrast of the live cells and decrease the single-cell capture time. Furthermore, a microchip was used to isolate a single cell, and pictures were taken to store the cell status (Figure S1D).
After various post-infection times, infected alveolar cells were recovered from Matrigel same as above. Recovered alveolar cells were washed at least 400,00X to reduce the number of free-floating viral RNA transcripts. The cells were incubated with a 1,000X CMFDA cell tracker (Thermo-Fisher C2925) for 10 min at 37°C. Next, the cells were loaded with 0.5% BSA (Sigma-Aldrich A8412) to decrease cell attachment on the smart aliquotor CE chip (iBioChip H2-SACE-5PK), and a single cell was picked up using 1 ul pipette after manual curation of the cell number using a fluorescence and phase contrast microscope.
Virus competition system among VOCs: Library preparation for isolated single cells
For the SMART-seq3 library preparation, the minimal lysis buffer amount was first optimized for the cell in 1 ul PBS. It was determined that a 4 times volume in every step before the first bead cleanup, compared to the original SMART-seq3 paper, was enough to successfully amplify the cDNA from the RNA.
The lysis buffer was added to a captured cell, snap frozen, and stored at −80°C until lysis and reverse transcription. Then the cells were thawed with the lysis buffer followed by the SMART-seq3 library protocol.20 The pooled library was sequenced by Illumina NovaSeq by paired-end sequencing with an average of 314 Mb per cell.
The fraction of infective viruses and viral transcripts in a virus mixture
We conducted a plaque assay to count the number of infective (viable) viruses at each diluted viral variant and amplicon sequencing to calculate the fraction of viral RNA transcripts, at the viral mixture over experimental batches H, I J, and K (Figure S1B). For plaque assay, diluted viruses were used to calculate the PFU of each viral variant from each experimental batch. Ideally, each viral variant has an equal 12,500 PFU. For viral RNA transcripts, we extracted RNA from the viral variant mixture and conducted RNA sequencing of the amplicon library with the same method in each viral variant’s RNA sequencing above. After the same alignment method with the viral genomic mutations, we calculated the fraction of viral RNA transcript from each viral variant. Ideally, the viral RNA transcript of each viral variant consists of 25%.
Viral genomic mutations
To confirm the mutations of each virus that were used for the viral barcodes, we first conducted RNA sequencing of the viral stocks of each variant. Viral RNA was extracted by the QIAamp viral mini kit (Qiagen 52,904) from a virus stock containing >10,000 viral PFU. Then, a sequencing library was constructed with the NEBNext ARTIC SARS-CoV-2 FS kit (NEB E7658S).
For each viral variant, paired-end sequencing was conducted by Illumina NovaSeq. Adapter sequences were removed with fastp.34 Then, the adapter-removed fastq files were aligned to the SARS-CoV-2 reference genome sequence, wuhCor1.fa (NC_045512v2) with BWA-MEM.32 Primer sequences in the amplicons were removed using iVar.33 Any human contamination reads were removed by Kraken.35
The mutation sites were called using VarScan pileup2snp and pileip2indel27 and GATK HaplotypeCaller.28 Candidate calls were manually inspected using Integrative Genomic Viewer29 and in total, 118 clonal mutations (VAF>99.5%) were finally obtained (Table S1) and compared among the four viral variants (Figures 1B and S1A). For indel, we didn’t count the reads which can’t span the homopolymer region right next to the indel position.
Since some loci were stochastically not covered by RNA sequencing of viral stock, we rescued the mutations. For example, the mutation with the genomic position 6,954 from the Alpha variant batch B and the mutation with the genomic position 23,948 from the Omicron variant batch D were manually rescued (Figure S1A).
Data processing of the full-length single-cell transcriptome sequencing
From the pooled fastq file, we counted reads and UMIs of gene expression using zUMIs.31 This condition is the same as the SMART-seq3 paper.20 To align single-cell transcriptome sequences from infected cells, a joint reference genome sequence was established by concatenating the human genome (GRCh38.p13) and SARS-CoV-2 genome (wuhCor1.fa). A joint gene annotation file (gtf) was also generated by merging the primary annotation gtf of GRCh38.p13 and ncbiGenes.gtf downloaded from the human GENCODE site and SARS-CoV-2 UCSC site, respectively. From the gtf file, we removed the nested exons of ORF1ab for calculating the gene expression of the viral transcripts. The UMI count matrix for exons was used for the transcriptome analysis.
For the quality control of the single-cell RNA sequencing data, any cells with a mitochondria percentage above 40, the number of genes expressed below 1,250, or a total UMI count below 1,250 were excluded. To normalize expression levels of infected alveolar cells, we divided the UMI count of each gene by the total UMI count of a cell, multiply 10,000 and add 1 for plotting as a log scale (Figure 1C). Single-cell RNA sequencing analysis was done using Scanpy, if not otherwise stated.38
Decomposition of the viral variants that infected a single-cell
We used the variant allele fraction (VAF) of each variant locus to calculate the proportion of the viral variant that infected a specific cell. Because not all 118 loci were covered by single-cell transcriptome sequencing, specific consideration was necessary for an accurate decomposition. To this end, we used two different methods which were finally proven to be concordant with each other.
The first method is to utilize the average VAF of the multiple mutation loci. For each of the four viral variants, we calculated the average VAF of all covered unique mutation sites of a viral variant as a proxy of their fraction in a cell (Falpha, Fdelta, Fomicron, and FGR). For cells with the sum of the average VAFs (Fsum; Falpha + Fdelta + Fomicron + FGR) smaller than 1, cellular infection was explained by an average of ∼0.95 quite well. For most cells with Fsum> 1, we normalized each F value with the Fsum value. For cells explicitly infected but having Falpha + Fdelta + Fomicron = 0, FGR was explicitly assigned to 1 because the GR variant has only 3 specific mutations (Figure 1B), and the viral variant is more likely to be unexposed in the transcriptome sequencing by chance. Lastly, for cells with more than two viral variants which are not covered for every unique mutation of that (NA), 1 - (Fsum without NA) is assigned as unknown.
The second method is using non-negative matrix factorization (NMF). Given a sample VAF matrix of size 118 × number of samples, the linear combination of the VOC genotypes (0 or 1) that best explains each VAF column of the sample matrix was calculated. This process can be summarized as follows where the VOC matrix has a size of 118 × 4, and the weight matrix has a size of 4 × number of samples:
| VAF matrix = Geno-type matrix × Weight matrix |
Because the two methods showed a high linear correlation, we used and showed the results obtained from the first method in (Figure 1F). Cosine similarity was calculated after NA converting to zero. Even if we removed infected cells with NA fraction, the median and pattern of the cosine similarity were similar to converting the NA to 0 in infected alveolar cells.
Criteria for infection at a single cell
In practice, some extracellular virus RNA may contribute to the viral reads in single-cell transcriptome sequencing although a cell is not infected. From empty wells in the single-cell transcriptome sequencing, we set a threshold for the viral infection as 4 or more viral UMI in more than 2 different viral genes except for the N gene. Although alveolar organoids were washed at least 40,000, loading washed cell solution multiple times into a microchip leads to a higher number of free viral RNA transcripts.
Normalized coverage of virus transcripts
We drew the gene plot above the coverage plot by gggenes. For two single-cell transcriptome methods, we used the current paper’s data using the full-length transcriptome method and the previous paper’s data, infected alveolar cells at 3 days post-infection with MOI 1, using 3′ enriched method.22 The read-depth of deduplicated bam files was analyzed by SAMtools26 and normalized for each infected cell.
Quantification and statistical analysis
Statistical analysis
To calculate the Omicron’s dominancy for single and total infected cells, we adopted the proportion test with a random expectation of 0.25. Furthermore, to compare each variant’s infectivity, we calculated the odds ratios for each pair of viruses.
Acknowledgments
We thank Myung Jin Yang (GSMSE KAIST) for his valuable technical help and comments. This project was supported in part by GENOME INSIGHT Inc., the National Research Foundation of Korea (NRF-2020R1A3B2078973), the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health& Welfare, Republic of Korea (HI14C1277). This research was also supported by the Research Program 2020funded by the Seoul National University College of Medicine Research Foundation (800-20200516). This research was supported by KNIHfund (2022-NI-043-00, 6634-325-210).
Author contributions
Conceptualization, Y.S.J.; Infection, T.K. and K.I.M.; Virus preparation J.W.K and J.C.; Virus decomposition, T.K. and K.I.M.; Specimen preparation K.J.N. and Y.H.K.; Writing, T.K., K.I.M., and Y.S.J; Manuscript finalization, all authors; Supervision Y.S.J., J.-Y.L., and K.J.N.
Declaration of interests
Young Seok Ju is a co-founder and Chairperson of Genome Insight. Jeong Seok Lee is a co-founder and chief executive officer of Genome Insight. Kwon Joong Na is a co-founder and chief medical officer of Portrai, Inc.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105571.
Contributor Information
Kwon Joong Na, Email: npeter1@snu.ac.kr.
Joo-Yeon Lee, Email: ljyljy@nih.go.kr.
Young Seok Ju, Email: ysju@kaist.ac.kr.
Supplemental information
Data and code availability
Single-cell bam files of SMART-seq3 are uploaded to the European Genome-Phenome Archive. Furthermore, metadata of infected alveolar cells, expression of alveolar cell markers, normalized depth of viral transcripts by two different methods, and raw data of the number of viruses by two different methods are uploaded on Mendeley. Accession numbers are listed in the key resources table. The data will be fully available as soon as the administration process completes.
All original code and additional files have been deposited at Zenodo is publicly available. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J.D., Pearson C.A.B., Russell T.W., Tully D.C., Washburne A.D., et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Sci New York N Y. 2021;372:eabg3055. doi: 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Twohig K.A., Nyberg T., Zaidi A., Thelwall S., Sinnathamby M.A., Aliabadi S., Seaman S.R., Harris R.J., Hope R., Lopez-Bernal J., et al. Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: a cohort study. Lancet Infect. Dis. 2022;22:35–42. doi: 10.1016/s1473-3099(21)00475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheikh A., McMenamin J., Taylor B., Robertson C., Collaborators P.H.S., the E.I. SARS-CoV-2 Delta VOC in Scotland: demographics, risk of hospital admission, and vaccine effectiveness. Lancet. 2021;397:2461–2462. doi: 10.1016/s0140-6736(21)01358-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khare S., Gurry C., Freitas L., Schultz M.B., Bach G., Diallo A., Akite N., Ho J., Lee R.T., Yeo W., et al. GISAID’s role in pandemic response. China CDC Wkly. 2021;3:1049–1051. doi: 10.46234/ccdcw2021.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Toole Á., Hill V., Pybus O.G., Watts A., Bogoch I.I., Khan K., Messina J.P., Tegally H., Lessells R.R., et al. COVID-19 Genomics UK (COG-UK) consortium. Network for Genomic Surveillance in South Africa (NGS-SA) Brazil-UK CADDE Genomic Network; Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2. Wellcome Open Res. 2021;6:121. doi: 10.12688/wellcomeopenres.16661.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandes J.D., Hinrichs A.S., Clawson H., Gonzalez J.N., Lee B.T., Nassar L.R., Raney B.J., Rosenbloom K.R., Nerli S., Rao A.A., et al. The UCSC SARS-CoV-2 genome browser. Nat. Genet. 2020;52:991–998. doi: 10.1038/s41588-020-0700-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hart W.S., Miller E., Andrews N.J., Waight P., Maini P.K., Funk S., Thompson R.N. Generation time of the alpha and delta SARS-CoV-2 variants: an epidemiological analysis. Lancet Infect. Dis. 2022;22:603–610. doi: 10.1016/s1473-3099(22)00001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCallum M., Walls A.C., Sprouse K.R., Bowen J.E., Rosen L.E., Dang H.V., Marco A.D., Franko N., Tilles S.W., Logue J., et al. Molecular basis of immune evasion by the Delta and Kappa SARS-CoV-2 variants. Science. 2021;374:1621–1626. doi: 10.1126/science.abl8506. [DOI] [PubMed] [Google Scholar]
- 11.McCallum M., Bassi J., Marco A.D., Chen A., Walls A.C., Iulio J.D., Tortorici M.A., Navarro M.-J., Silacci-Fregni C., Saliba C., et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science. 2021;373:648–654. doi: 10.1126/science.abi7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meng B., Abdullahi A., Ferreira I.A.T.M., Goonawardane N., Saito A., Kimura I., Yamasoba D., Gerber P.P., Fatihi S., Rathore S., et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts tropism and fusogenicity. Nature. 2022;603:706–714. doi: 10.1038/s41586-022-04474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hui K.P.Y., Ho J.C.W., Cheung M., Ng K., Ching R.H.H., Lai K., Kam T.T., Gu H., Sit K.-Y., Hsin M.K.Y., et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature. 2022;603:715–720. doi: 10.1038/s41586-022-04479-6. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki R., Yamasoba D., Kimura I., Wang L., Kishimoto M., Ito J., Morioka Y., Nao N., Nasser H., Uriu K., et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature. 2022;603:700–705. doi: 10.1038/s41586-022-04462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ulrich L., Halwe N.J., Taddeo A., Ebert N., Schön J., Devisme C., Trüeb B.S., Hoffmann B., Wider M., Fan X., et al. Enhanced fitness of SARS-CoV-2 variant of concern Alpha but not Beta. Nature. 2022;602:307–313. doi: 10.1038/s41586-021-04342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou B., Thao T.T.N., Hoffmann D., Taddeo A., Ebert N., Labroussaa F., Pohlmann A., King J., Steiner S., Kelly J.N., et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature. 2021;592:122–127. doi: 10.1038/s41586-021-03361-1. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y., Liu J., Plante K.S., Plante J.A., Xie X., Zhang X., Ku Z., An Z., Scharton D., Schindewolf C., et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature. 2022;602:294–299. doi: 10.1038/s41586-021-04245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saito A., Irie T., Suzuki R., Maemura T., Nasser H., Uriu K., Kosugi Y., Shirakawa K., Sadamasu K., Kimura I., et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature. 2022;602:300–306. doi: 10.1038/s41586-021-04266-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y., Liu J., Johnson B.A., Xia H., Ku Z., Schindewolf C., Widen S.G., An Z., Weaver S.C., Menachery V.D., et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv. 2021 doi: 10.1101/2021.08.12.456173. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagemann-Jensen M., Ziegenhain C., Chen P., Ramsköld D., Hendriks G.-J., Larsson A.J.M., Faridani O.R., Sandberg R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020;38:708–714. doi: 10.1038/s41587-020-0497-0. [DOI] [PubMed] [Google Scholar]
- 21.Kim D., Lee J.-Y., Yang J.-S., Kim J.W., Kim V.N., Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Youk J., Kim T., Evans K.V., Jeong Y.-I., Hur Y., Hong S.P., Kim J.H., Yi K., Kim S.Y., Na K.J., et al. Three-dimensional human alveolar stem cell culture models reveal infection response to SARS-CoV-2. Cell Stem Cell. 2020;27:905–919.e10. doi: 10.1016/j.stem.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peacock T.P., Brown J.C., Zhou J., Thakur N., Newman J., Kugathasan R., Sukhova K., Kaforou M., Bailey D., Barclay W.S. The SARS-CoV-2 variant, Omicron, shows rapid replication in human primary nasal epithelial cultures and efficiently uses the endosomal route of entry. bioRxiv. 2022 doi: 10.1101/2021.12.31.474653. Preprint at. [DOI] [Google Scholar]
- 24.Han P., Li L., Liu S., Wang Q., Zhang D., Xu Z., Han P., Li X., Peng Q., Su C., et al. Receptor binding and complex structures of human ACE2 to spike RBD from Omicron and Delta SARS-CoV-2. Cell. 2022;185:630–640.e10. doi: 10.1016/j.cell.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hagemann-Jensen M., Ziegenhain C., Sandberg R. Scalable full-transcript coverage single cell RNA sequencing with Smart-seq3xpress. bioRxiv. 2021 doi: 10.1101/2021.07.10.451889. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Subgroup, 1000 Genome Project Data Processing the sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poplin R., Ruano-Rubio V., DePristo M.A., Fennell T.J., Carneiro M.O., Van der Auwera G.A., Kling D.E., Gauthier L.D., Levy-Moonshine A., Roazen D., et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2018 doi: 10.1101/201178. Preprint at. [DOI] [Google Scholar]
- 29.Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinfrom. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parekh1 S., Ziegenhain C., Vieth B., Enard W., Hellmann I. zUMIs - a fast and flexible pipeline to process RNA sequencing data with UMIs. GigaScience. 2018;7:giy059. doi: 10.1093/gigascience/giy059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Arxiv. 2013 doi: 10.48550/arxiv.1303.3997. Preprint at. [DOI] [Google Scholar]
- 33.Grubaugh N.D., Gangavarapu K., Quick J., Matteson N.L., Jesus J.G.D., Main B.J., Tan A.L., Paul L.M., Brackney D.E., Grewal S., et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lun A.T.L., Bach K., Marioni J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016;17:75. doi: 10.1186/s13059-016-0947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell bam files of SMART-seq3 are uploaded to the European Genome-Phenome Archive. Furthermore, metadata of infected alveolar cells, expression of alveolar cell markers, normalized depth of viral transcripts by two different methods, and raw data of the number of viruses by two different methods are uploaded on Mendeley. Accession numbers are listed in the key resources table. The data will be fully available as soon as the administration process completes.
All original code and additional files have been deposited at Zenodo is publicly available. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.